平 成 1 5 年 度
1,3-ジヒドロ-2-イミダゾロン体を 反応素子とする効率不斉合成
薬科学専攻 薬品資源学講座 分子薬化学研究室
片平 智和
Asymmetric Synthesis Based on Chiral Functionalization of 1,3-dihydro-2-imidazolone Tomokazu Katahira
Diamines are a biologically, medicinally and synthetically, important class of compounds. The 1,2-diamine skeleton is a structural unit found in a number of bioactive compounds of medicinal interest such as peptidic antibiotics, vitamin H, antitumor agents and opioid receptor agonists and also functions as a chelating ligand for metal catalysts in asymmetric synthesis. The simple heterocycle, 1,3-dihydro-2-imidazolone, which is susceptible to various modes of addition reactions, represents a potential building block for the synthesis of 1,2-diamines.
1.Synthesis of Optically Active Diaminocarboxylic Acids of Biological Interest
Intramolecular Ru(II)[RuCl2(PPh3)3]-catalyzed cycloaddition of the trichloroacetyl pendant group to an 2-imidazolone moiety resulted in the exclusive formation of the 12-membered cycloadduct with complete diastereoselectivity, thus providing a new synthetic route to enantiomerically pure 1,2-diamines.
C l
C l N N
O O
N O N
O
O
Ac Cl Cl
Cl
O O
N O N
O
O
Ac Cl Cl
N O N
O Ac
O O O Cl X
Cl S
N O N
O O O O
Cl Cl Ac
N NH OM e
O HOCl Cl
Tos
N NH OM e
O Tos
HO S
NH HN
O
Ru(II)
Ru(II)
R
3
R R
6
R
4
7 2
5
X=Cl,OMe
X=OMe 1
R R
R
(a)
( b )
Thus, the compound (2 ) underwent intramolecular cyclization in the presence of Ru(II) complexs to the
12-membered cycloadduct (3 ) with complete ragio- and diastereo-selectivity and good yield. Reductive cleavage of the optically pure cycloaddcut (3 ) with LiBH4-MeOH(1:2) gave 4-methoxy-2-imidazolidinone (4 ) which served as a chiral synthon for a variety of 1,2-diamines. This versatility was demonstrated by a facile conversion to the amino analog (8 ) of (3S,4S)-4-amino-3-hydroxy-6-methylheptanoic acid (statine), a key structural component of amastatine (route a).
This methodology was applied to the chiral synthesis of the diaminocarboxylic acid with three contiguous stereogenic centers, which is the amino analog (9 ) of (2S,3R,4R,6E)-3-hydroxy-4-methyl-2-(methylamino)-6- octenoic acid (Me-Bmt), an unusual and key amino acid component of cyclosporin (route b).
2.Catalytic Dissymmetrization of m e s o-2-Imidazolidinones
(i) Preparation of versatile chiral synthons for the 1,2-diamines, (4S,5S)- and (4R,5R)-DMIm
N N O
O O
Ac Ac
NH2 OH
THF BH3S(Me)2
HN N O
O O
HN N O
O O
Cbz
N NH O
O O
Cbz
HN NH O MeO OM e
HN NH O MeO OM e HN N
O HO
O Cbz
N NH O O OH
Cbz MeO
OM e
(R, R)
(S, S) DMIm 10
11
12
ent-12 ent-13
13 14
ent-14 Ac
>99%ee R
R
R R
R
R
The enantioselective monodeacetylation of a series of meso-4,5-dialkoxy-2-imidazolidinones (1 0) by an oxazaborolidine-catalyzed borane reduction proceeded smoothly to give the kinetically discriminated monoacetyl derivatives (1 1) in excellent enantioselectivity. Both enantiomers of the 4,5-dimethoxy-2-imidazolidinones (1 4 and ent-1 4) were synthesized by the straightforward manipulation, including facile conversion to enantiomeric
mono-N-Cbz derivatives (1 2 and ent-1 2), followed by stepwise methanolysis.
Both methoxy groups of (4S,5S)- and (4R,5R)-4,5-dimethoxy-2-imidazolidinones (DMIm) (1 4 and ent-1 4) can readily be replaced with primary to tertiary alkyl groups and aryls with full retention of configuration in a stepwise manner.
Subsequent ring-opening provides a versatile route to optically active threo-1,2-diamines.
(ii) Application of (4S,5S)- and (4R,5R)-DMIms
Chiral DMIm synthon (1 4) was served as a good precurser for bis(imidazoline) auxiliaries (1 8), which were obtained by the regioselective replacement of both 4- and 5-methoxy group with organo cuprates/BF3·O Et2 with full retention of configuration via 1 6 and 1 7.
N N N N
R R
R R
To s To s
18 HN N
O MeO
Ac OMe
(4 R, 5R)
4 5
HN N O R
Ac OMe
N NH O
R R
To s NH NH2
R R
To s
14 15 16 17 R= Ph, tB u, B n
3 . C o n c l u s i o n
This study demonstrates high synthetic potential of chiral functionalization of dihydro-2-imidazolidinone, leading to 1,2-diamine compounds of biological and synthetic interest. Further applications as chiral building blocks are apparent.
熊 本 大 学 学 位 論 文
1,3-ジヒドロ-2-イミダゾロン体を 反応素子とする効率不斉合成
2 0 0 4
片 平 智 和
Asymmetric Synthesis Based on Chiral Functionalization of 1,3-dihydro-2-imidazolone
2 0 0 4
Tomokazu Katahira
目 次
第1章 緒論
1
1−1 本研究の背景 1
1−2 本研究の目的 3
第2章 ラジカル環化反応を基軸としたキラル合成
7
2−1 はじめに(分子間ラジカル付加反応) 7
2−2 分子内ラジカル環化反応による 1,2-ジアミン類の合成
−隣接不斉中心構造の構築− 8
2−2−1 トリクロロアセチル基含有2-イミダゾロン誘導体の合成 9
2−2−2 分子内ラジカル環化反応 10
2−2−3 隣接不斉ジアミン構造の構築 10
2−2−4 スタチン類似体への誘導 14
2−3 分子内ラジカル環化反応による 1,2-ジアミン類の合成
−3連続不斉中心構造の構築− 15
2−3−1 α,α-ジクロロアジル基含有2-イミダゾロン誘導体の合成 15
2−3−2 分子内ラジカル環化反応 17
2−3−3 3連続不斉中心構造の構築 18
2−3−4 MeBmt類似体への誘導 21
2−3 小活 22
第3章 多目的キラル合成子
DMIm
の効率合成法の開発と利用22
3−1 はじめに 24
3−2 メソ型2-イミダゾリドン化合物の触媒的不均斉化 24
3−2−1 メソ型ジアセチル化合物の合成 25
3−2−2 触媒的不均斉化 26
3−2−3 絶対配置の決定 30
3−2−4 エナンチオ選択性発現に対する考察 30
3−2−5 (S,S)-DMIm への変換 32
3−2−6 (R,R)-DMIm への変換 34
3−2−7 メソ-1,2-ジオール類の不均性化 35
3−3 ビスイミダゾリン系不斉配位子の合成 36
3−3−1 DMIm の 4,5 位メトキシ部位へのアルキル基の導入 36
3−3−2 ビスイミダゾリドン体の合成 37
3−3−3 アルドール反応への適用 38
3−4 小活 39
第4章 総括
42
X 線結晶解析
45
実験の部
48
引用文献
91
謝辞
93
第1章 緒論
1−1 本研究の背景
1,2-ジアミン構造は、生物学的にも合成化学上も興味深い重要な構成骨格である。これら骨格の高選択 的且つ汎用性合成法の開発および確立は、幅広い可能性を秘めた重要な化学的課題である。1,2-ジアミン 骨格をその構造を内包するものとして、カルボキシラーゼの補酵素であるビオチン1)やペプチド性抗生物 質の構成ジアミノ酸等の天然生理活性物質の他、金属との複合体である坑腫瘍薬のオキサリプラチン2)や オピオイド受容体に作用する化合物3)等の非天然生理活性物質等数多く挙げられる。また、化学量論的及 び触媒的に不斉源を利用した不斉合成の研究は近年活発に行われており、下図に例示したように1,2-ジア ミン構造も広範囲にわたり研究されている。4) さらに、数多くの医薬品や生理活性物質に内包されている 2−アミノアルコール構造のアミノ類似体としての生理活性についても興味ある点である(Figure 1)。
N( Me)2 NH- Tos
NH HN
Pt O O
O O
N
NM e Cl
Cl O
C COOH CH2NH2 H2N H C
COOH C H NH2
COOH H2N H
Ph Ph
NH2 NH2
NH2 H2N
X Y
NH2 HO
X Y
S
COOH NH
HN O
H2N
NH2
COOH
Antitumor Agent s
Opioi d Rec eptor Type кAgonist Component of Viom ycin
Amino A cids
Chiral A uxiliar ies
Amino A lcohol Ana logs Vitamin H
bi ot ine
Peptidic Antibi ot ics
Figure 1. 1,2-Diamines as Chiral Auxiliaries and Compounds of Biological Interest.
R2 R1 O2N
R2 R1 X X
NR NR R2 R1
NH2 H2N
R2 R1 COOH
R1 H2N
ZN R
R2 OH R1 HO
-N R1R2 -N R1R2
R1R2N-
+ 1,2-diammines
-N R1R2
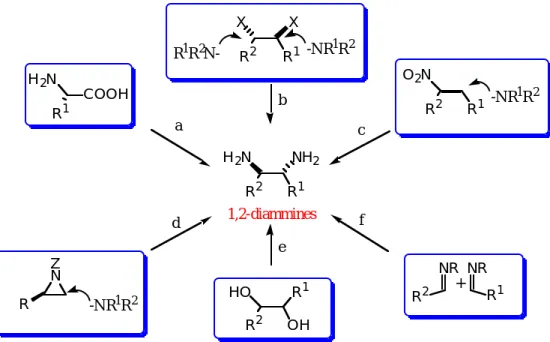
Figure 2 . Methods for the Synthesis of 1,2-Disubstituted 1,2-Diamines.
a
b
c
f e
d
1,2-ジアミン類の合成法としてFigure 2に示すようなさまざまな手法が確立されている。即ち、アミ ノ酸やアミノアルデヒドからの誘導(a)5)、ハロアルカン等への窒素求核種の置換導入(b)6)、窒素求核種の ニトロアルケンやアジリジン環への共役付加反応(c,d)7)、1,2-ジオール体のジアミノ化(e)8)、イミン同士の カップリング反応(f)9)などが挙げられる。その多くはラセミ体の合成に終わっており、選択性、収率、汎 用性などの観点をふまえた簡便かつ緻密な構築手法の開発が早急に求められている。核酸やタンパク質に 見られるように、生体を構成している物質の多くはキラル分子から成り立っており、薬物を投与した場合、
その一方のエナンチオマーのみが薬物の標的部位と特異的に相互作用を起こすことが通例である。このよ うにエナンチオマー間で生体におよぼす生理活性が異なるため、一方の異性体が有害な作用を持つ場合、
ラセミ体であることは致命的な欠陥となる。したがって、100%の光学純度で光学活性物質を効率良く合 成する手法の確立は創薬の観点から極めて重要である。10)
HN NH O
X
HN NH O
Y R1
HN NH O
Y R1
H2N NH2 R2
5
*
4
4
* * *
*
Scheme 1
1 2 3
当研究室では以前より、エチレン尿素から大量合成可能な簡単な複素5員環である2-イミダゾロンを構 築材とした1,2-ジアミン骨格の効率的キラル構築法の開発を行ってきた(S c h e m e 1)。11) 即ち、2-イミダ
ゾロン(1 )の4, 5位二重結合部位への位置及び立体選択的反応性置換基X、Yの不斉選択的導入によるキ
ラル合成子 (2 )の開発及び、これら置換基の異なる反応性を利用した段階的変換を経た1,2-ジアミン類 (4 ) の合成についての方法論を展開してきた。しかし、医薬品の合成を始めとする有機合成化学の分野におい ては、複雑な不斉骨格の簡便且つ緻密な構築手法の開発が現在も尚早急に求められている。
1 - 2
本研究の目的以上のような背景のもとに本研究では、簡単な複素環である1,3- ジヒドロ-2-イミダゾロン(1 )を構築材 とするキラル合成研究を以下の二観点から展開した。即ち1)ラジカル付加環化反応を鍵とした1,2-ジア ミン類のキラル合成、2)キラル合成子4,5-Dimethoxy-2-imidazolidinone(DMIm)の効率合成法の開発とその 利用研究である。
1)ラジカル付加環化反応を鍵とした1,2-ジアミノカルボン酸類のキラル合成
キラルなアシル基を持つ2-イミダゾロン(5 )とCCl4などのポリハロ化合物とのジアステレオ選択的分 子間ラジカル付加反応を検討し、成績体(6 a or 6 b)が高い選択性で得られた場合にはこれらをキラル中間 体とした合成手法を展開する(S c h e m e 2)。結果は後述しているとおり、予想した位置および立体選択性 が得られなかった。
N N
O R2 R1
X
N N
O R
R1 R2
Ru (II)
X
N N
O R
R1 R2
* *
+ * *
Scheme 2
5 6a 6b
R - X
そこで、キラルなアシル基としてカンファー骨格を含むものを採用し、ラジカル付加反応を分子内に適 用することにより、カンファー骨格の立体障害により位置及び立体選択性を完全なものにし、光学的に純 粋なキラル合成子を得、新規ジアミン合成への適用を考えた。
O O N
O N
O CCl3
O R
N O N
O R
O O O
Cl Cl
Cl Ru Cl2(PPh3)3
NH HN
O HO Cl Cl OMe
R1 NH2
NH2 R2
*
* *
*
* *
Scheme 3
7 8
9 10
まず、トリクロロアセチル基をペンダントグループとする2-イミダゾロン(7 )を出発物質としたラジカ ル環化反応によって、隣接する不斉中心を持つジアミン(1 0)への誘導が期待できる(S c h e m e 3)。この手 法はスタチン12)やシクロへキシルスタチン13)のアミノ類似体の合成への適用が考えられる。
この方法論は、3連続不斉中心を持つジアミンのキラル合成へさらなる応用発展が可能である(S c h e m e 4 )。
O O
N O N
O
O R1 Cl Cl R
N O N
O O O O
Cl R1
R Ru Cl2(PPh3)3
NH HN
O OMe R1
R2
R2 R3
NH2
NH2 R1
* *
* *
*
Scheme 4
11 12
13 14
* *
*
*
即 ち 、 免 疫 抑 制 剤 シ ク ロ ス ポ リ ン 中 の C9ー 異 常 ア ミ ノ 酸 (2S, 3R, 4R,6E)-3-Hydroxy-4- methyl-2-(methylamino)-6-octenoic acid [MeBmt]14)のアミノ類似体合成への適用である。
2)多様なキラル合成子4,5-Dimethoxy-imidazolidinone(DMIm)の効率合成法の開発とその利用研究
2-イミダゾロンから当グループにより合成された、キラル合成子4,5-Dimethoxy-2-imidazolidinone(DMIm)
(1 5)は、両方のメトキシ部位が高い反応性をもっており、アルキル基をはじめフェニル基、ビニル基など
様々な置換基へ立体保持のまま段階的な置換基の導入が可能であるため、1,2-ジアミン類(1 8)の合成にお いて汎用性に優れた合成子であるといえる(S c h e m e 5)。
HN NH O
OMe
HN N O R1 OMe
N NH O R1 R2
To s Tos-HN NH- Boc
R1 R2 MeO
15 16 17 18
* * * *
* *
R O
* *
S c h e m e 5
本化合物は不斉補助基の導入による光学分割法(Scheme 6 path a)によって合成されていたため、汎用 性、操作性の点で問題があった。今回、メソ型イミダゾリドン体の触媒的不均斉化プロセスを鍵反応とし た効率よい新規合成法の開発を目的とした(Scheme 6 path b)。
NH2 OH
N N
O
O O
Ac
Ac HN N
O
O O
Ac
N N
O
Ac Ac HN N
O MeO OMe
Ac
BH3-SMe2
HN N O
MeO OMe
MAC N NH
O
MeO OMe
Ac
R R R R
MAC- Cl Cl
O OMe
me so-form
* *
(S,S)- and (R,R)-Ac-DM Im
catalytic approach
Scheme 6
a
b
19
20 21
22 23
2 4
更に、DMImの利用としてビスイミダゾリン系不斉配位子を設計合成し、その有用性を実証することを 目指した(S c h e m e 7)。
N N N N
R R
R R
To s To s
NH NH2
R R
To s HN N
O MeO OMe
Ac
触媒的不斉反応 への適用
Scheme 7
2 4 2 5 2 6
第2章 ラジカル環化反応を基軸としたキラル合成
2−1 はじめに(分子間ラジカル付加反応)
1,3-ジヒドロ-2-イミダゾロンとCCl4やCCl3CNなどのポリハロ化合物との分子間ラジカル付加反応は トランス選択性よく、二重結合部位である4位、5位に容易に付加するという知見に基づき、位置選択性 を確実なものにするための検討を行った(Table 1)。
Table 1 . Intermolecular Radical Additions of Polyhalomethyl Compounds
N N
O R1 Ac
X
N N
O Cl
Ac R1
Cl
N N
O X
Ac R1
MAC
To s
X
CCl3 CCl3 CCl4
CCl4
O OCH3 -OC
OCH3 -OC
MAC
MEAC CCl3CN CCl2CN RuCl2(P Ph3)3
Entry R1 1
3
61
44 Solvent
* complex mixture.
2 14
28a : 28b 1 : 1
4 : 1 - * Solvent
reflux
MEAC
Yield (%) * *
* * 27
28a
2 8 b
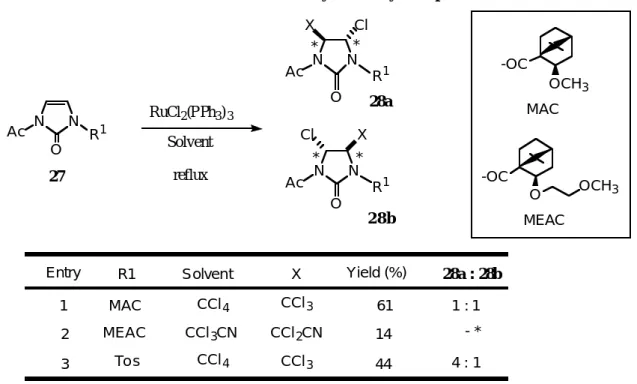
2-イミダゾロンのN-置換基としてアセチル基、そして嵩高いケトピン酸由来のMAC基及びその誘導体 MEAC基をもつジアシル-2-イミダゾロン(2 7)体については、位置選択性のみならずジアステレオ選択性 も全く見られなかった。CCl3CNを用いた時は、収率も低く10数%に過ぎなかった。位置選択性について は電子的な要因が影響を及ぼしていると考えられたため、一方をトシル基にし同様の反応を行ったところ、
位置選択性が完全ではないが見られた。しかし、どの反応においても錯体の配位場内の反応であるので、
高い不斉誘起を期待したが、ジアステレオ選択性は全く認められず、ほぼ 1 : 1であった。これら付加体 の4位の Cl 基は、種々のアルキル基などへ 容易に置換変換できるの で、合成子としての利用も考えられ るが低いジアステレオ選択性と低収率のため、これ以上の検討は行なわず、分子内での付加反応を検討す ることにした。
2−2 分子内ラジカル環化反応による
1 , 2 -
ジアミン類の合成−隣接不斉中心構造の構築−
2−2−1 トリクロロアセチル基含有2 -イミダゾロン誘導体の合成
分子内ラジカル環化反応ルートを検討するため、D-ケトピン酸(2 9)を出発原料としてトリクロロアセ チル基をペンダントグループとするキラル補助試薬(3 5)の合成を行った(S c h e m e 8)。
O OH
tBuO
O
OH O
tBuO
Br
O O
tBuO
O O HO
THF
O O
tBuO
O CCl3
O CH2Cl2
O O
tBuO
O O
HO
O CCl3
O ,NaH
THF 84%
30
L-Selectride
MeOH O3, NaBH4
94%
CCl3COCl, Et3N
75%
29 31
32
34
Scheme 8 , H+
98%
CH2Cl2
CF3COOH CH2Cl2
33
35 98%
99%
即ち、ケトピン酸(2 9)のtBuエステル化(3 0) 後、L-Selectrideによる還元(3 1)、アリル化(3 2)、オゾン酸 化及び NaBH4還元を順次行い2-exo-hydroxyethoxy体(3 3)を得た15)。
この末端水酸基にトリクロロアセチルクロリドを反応させて、エステル体(3 4)とした後、トリフルオ ロ酢酸による脱tBu化によりトリクロロアセチル基をペンダントグループとするアポカンファンカルボン
酸誘導体(3 5)を得た。
N Ac N
O NH Ac
HN O
NH N
O Ac DMAP
IPA 76%
19 37
Scheme 9 e- / MeOH
pyridi ne / Ac2O 3 6
一方、エチレン尿素(3 6)から陽極電解酸化16)、Ac2O / pyridineによる脱メタノール化を経て1,3-ジアセ チル-2-イミダゾロン(1 9)を合成し、イソプロパノール溶媒中DMAPの添加により収率よくモノアセチル 体(3 7)を得た(S c h e m e 9)。
O O
HO
O CCl3
O
NH N
O A c
O O
N O N
O CCl3
O SOCl2 Ac
reflux Et3N,DMAP 93%
35 38
Scheme 10 3 7
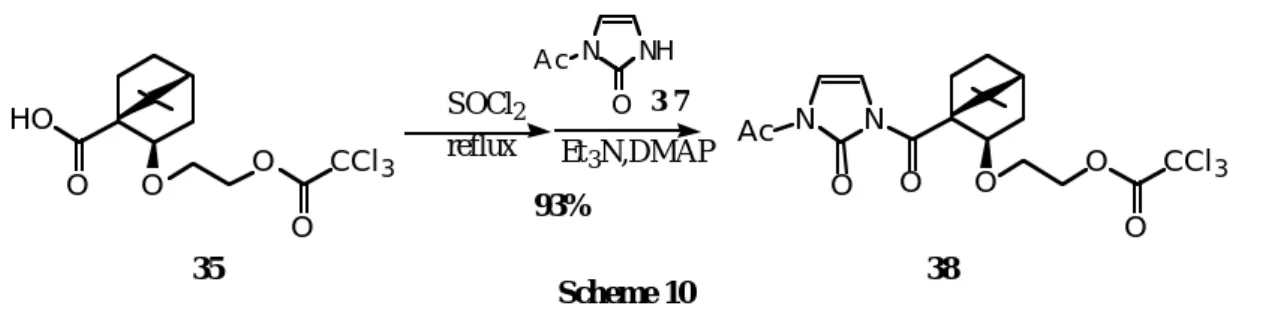
アポカンファンカルボン酸(3 5)を SOCl2により酸クロリドとした後、モノアセチルイミダゾロン(3 6) と縮合させることにより、1,3位を保護した2-イミダゾロン体(3 8)を収率よく得た(S c h e m e 1 0)。
化合物(3 8)は、分子模型を組み立ててみると図に示したように、ペンダント側鎖の長さもトリクロロ
エチル基が反応する二重結合の近くにくるために適切であり、分子模型上無理のない閉環が可能であると
予想した(Figure 3)。つまり、両アシ
ルカルボニル基と環内カルボニル基と の双極子反発とカンファー骨格の立体 障害のために、トリクロロアセチル基 が4,5位二重結合部位に容易に接近で
きるtransoid-コンホメーションが極め
て安定であると考えられる。
N
N Cl
Cl
Cl
Figure 3. Perspective View of 38.
O O
N O N
O CCl3
O Ac
2−2−2 分子内ラジカル環化反応
N-アセチル-2-イミダゾロン体(3 8)のRuCl2(PPh3)3
17)による分子内ラジカル付加環化反応は、完全な位置 及びジアステレオ選択性で進行し、12員環マクロライド(4R,5S)体(3 9)のみが生成した(Table 2)。この 極めて高い立体制御は前項の安定コンホメーション(Figure 3)がベンゼン還流下でも完璧に保持された ためと思われる。
O O
N O N
O CCl3
O Ac
N O N
O Ac
O O O
Cl Cl
Cl
Entry
Conditions
Ru Cl2(PPh3)3 (e q.) Additive (eq.)
Time (h) Yield (%)
2 3
6
1 0.1 - 93 48
0.1 La(OTf)3 ( 0.05) 1 76
0.05 La(OTf)3 ( 0.025) 7 72
0.1 ZnCl2 (0.1) 1 85
5 0.1 ZnCl2 (0.05 ) 1 82
4 0.1 La(OTf)3 ( 0.1) 1 57
Table 2 . Ru(II)-Catalyzed Intramolecular Radical Additions of N-Acyl-2-imidazolones with a Trichloroacetyl pendant group
conditions
Benzene 4R 5S
38 3 9
RuCl2(PPh3)3による本環化反応は、長時間のベンゼン還流を必要とする(Entry 1 )が、添加物として
La(OTf)3を触媒量用いると劇的な反応加速が観察された(Entry 2)。添加物については、La(OTf)3以外のラ
ンタノイド系金属トリフラートでも同様の高い活性を示した。希土類金属塩以外では ZnCl2を用いた時が よい結果を与えた(Entries 5,6)。RuCl2(PPh3)3、La(OTf)3の当量に関してはどちらも少ないと反応速度の 低下を招き、多すぎると反応生成物及び原料の分解につながる(Entries 3,4)。RuCl2(PPh3)3 (0.1eq.)に対 し、金属塩(0.05〜0.1eq.)が最適であった。本反応中、脱 Ac 化が微量ではあるが観察された。RuCl2(PPh3)3
のみで反応を行った時にはほとんどみられなかったので、これは添加物として加えた金属塩(La(OTf)3な ど)の影響であると考えられる。そこで比較的切れにくいベンゾイル体(4 2)について試みた(S c h e m e 1 1)。
O OH N
O HN
O
O O
N O HN
O CCl3
O
O O
N O N
O CCl3
O ph
O DMAP
MeO H
O O
N O N
O CCl3
O Ac
Scheme 11
CH2Cl2 Et3N CCl3COCl
89%
CH2Cl2 PhCOCl Et3N
74%
38 40
41 42
80%
N-ベンゾイル体(4 2)は、N-アセチル体(3 8)から DMAP/MeOH による両アシル基の切断後(4 0)、トリク ロロアセチル基の再導入(4 1)、ベンゾイル化を経て収率よく得た。
分子内ラジカル環化反応を行ったところ、閉環収率92%へと Ac 体の76%に比べ向上が見られた (S c h e m e 1 2)。
O O
N O N
O CCl3
O
N O N
O O O O
Cl Cl
Ph Cl
O Ph
O RuCl2(P Ph3)3 (0. 1eq. )
Benzene
4R 5S
La(OTf)3 (0.05eq.)
1h 92%
Scheme 12
4 2 4 3
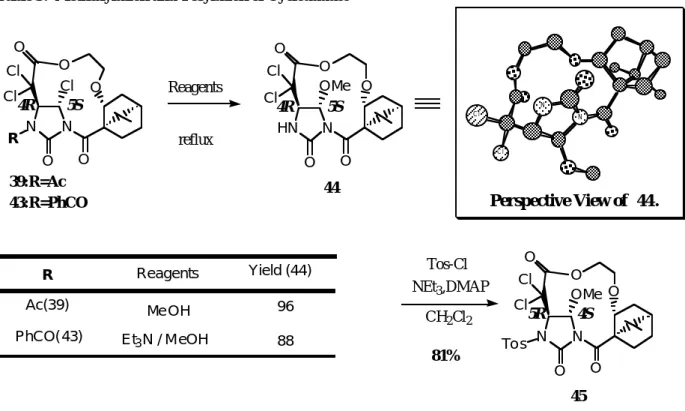
付加環化体(3 9)や(4 3)の構造については、メタノール処理により容易に得られる5-メトキシ体(4 4)の X線結晶解析により絶対配置を含め決定した。
2−2−3 隣接不斉ジアミン構造の構築
C l C l N
N
N O HN
O
O O O Cl OMe
Cl
N O N
O O O O Cl Cl
Cl
R
N O N
O
O O O Cl OMe
Cl
To s Me OH
Ac(39) PhCO( 43)
Yield (44)
Et3N / MeOH
96 88 Reagents
Table 3 . Methoxylation and Tosylation of Cycloadduct
reflux
4R 5S 4R 5S
Reagents
39:R=Ac 43:R=PhCO
44
CH2Cl2 Tos-Cl NEt3,DMAP
81%
5R 4S
45
Perspective View of 44 .
R
アセチル体(3 9)はMeOH中還流のみでメトキシ化と同時に、脱アセチル化が進行した。ベンゾイル体
(4 3)については、メトキシ化後ベンゾイル基の切断という2段階の過程を経て化合物(4 4)を得、N-保護
基としてトシル基を導入(4 5)した(Table 3)。
N O N
O
O O O Cl OMe
Cl
To s
NH N
O OMe To s
HO Cl Cl
O OH
MeOH HO Li BH4
THF
5R 4S
90%
5R 4S
Scheme 13
45 46
91%
化合物(4 5)をLiBH4-MeOH(1:2)により還元的に脱アシル化して、対応するアルコール体(4 6)を得た18)。
副生する不斉補助剤はジオール体として回収した(S c h e m e 1 3)。化合物(4 6)は4位メトキシ部位、5位ア ルコール部位に多様な置換基への変換が期待できるためキラル合成子としての効率良い利用が示唆される。
NH N
O OMe To s
TB DM SO Cl Cl NH
N O
OMe To s
HO Cl Cl
NH N
O R
To s TB DM SO Cl Cl
Entry R-X (eq.) Time(h) Yield(%)a) Recovery(%) 1
2
PhLi (4.0) n-BuLi (4.0)
1.0 2.0
46 51
26 20
3 n-BuMgBr (4.0) 3.0 55 40
4 iso -BuMgBr (4.0) 3.0 51 41
5 iso -BuMgBr (8.0) 3.0 61 33
Product 48a 48b 48b 48c 48c 6 cyclohexyl-MgBr (8.0) 3.0 48d
Table 4. The BF3-Pro moted Substitution of 4-Methoxy-2-imidazolidinones(47) by Alkyl and Phenyl Cuprates
58 35
5R 4S
imidazole 5R 4S tBuSiMe2-Cl
DMF
Li-Cl,CuCN R-Li,BF3OEt2
THF
48b: R=Bu
4S 5R
48a: R=Ph
48c: R=
a) Conversion of 47 to 48
46 47
48d: R=
86%
キラル合成子(4 6)の水酸基をtert-ブチルジメチルシリルで保護した後(4 7)、4位メトキシ基はルイス 酸(BF3•OEt2) 存在下各種銅試薬19)によりフェニル基(4 8 a)、ブチル基(4 8 b)、イソブチル基(4 8 c)、シクロ ヘキシル基(48d)などの炭素置換基に完全な立体保持で変換することができた(Table 4)。
本反応はイミニウムカチオン経由で進行すると考えられ、5位置換基の立体的影響の小さい方向から一 方的に銅試薬が接近するため立体配置は保持されると思われる。
2−2−4 スタチン類似体への誘導
キラル合成子(4 8)の汎用性を示す一環として、4位にイソブチル基を導入し、スタチンの類似体(5 2) への変換を行った(Scheme14)。
NH N
O To s HO Cl Cl
NH N
O To s MeOOC Cl Cl
NH N
O To s TB DM SO Cl Cl
NH N
O To s MeOOC Bu3SnH
AI BN
HN To s NH
MeOOC
Boc Boc2O CH2N2
PCC Na OAc CH2Cl2 TBAF
THF
5R CH2N2
4S
100%
4S 5R
4S 5R
4S 5S
Benzene
4S
Ba(OH)2·8H2O 5S
Et OH, H2O
48c 49
50
79%
51 84%
73% 52
S c h e m e 1 4
即ち、化合物(4 8 c)の5位の水酸基をTBAFを用いてアルコール体(4 9)とし、PCCによる酸化20)後、ジ アゾメタン処理によりメチルエステル(5 0)を得た。Bu3SnHによる還元的脱クロル化(5 1)、Ba(OH)2·8H2O によるイミダゾリジノン環の開環、ジアゾメタン処理を経て、スタチンのジアミノアナログ体(5 2)の合 成に成功した。
2−3 分子内ラジカル環化反応による
1 , 2 -
ジアミン類の合成−3連続不斉中心構造の構築−
2−3−1 α,α-ジクロロアシル基含有2 -イミダゾロン誘導体の合成
分子内環化手法のさらなる応用をはかるために、3連続不斉中心を有するジアミン類への適用について 検討した。シクロスポリンに内包されるMeBmt14)のジアミノ類似体を標的物質として、逆合成ルート (S c h e m e 1 5)を考えた。これに基づき2,2-Dichloro-(4E)-hexenoic acid (5 4)を得るため、2-butyn-1-ol (5 9)を 出発原料として合成を行った(S c h e m e 1 6)。
CO2H Me
NH2 NH- Me N
O HN
O
O O
O
OMe
O O
N O N
O
O Cl Cl Ac
55
Scheme 15
*
* * a
56 58 NH N
O Ac
37 +
+ 53
54
O OH
HO O
COOH Cl Cl
NH HN
O OMe HO
5 7
Me C C CH2OH OH Cl
COOtBu Cl Cl
COOH Cl Cl LAH, NaOMe
THF (CCl3)2CO
PPh3
81%
HC Cl2COOtBu LDA
THF 61%
TFA CH2Cl2
86%
59 60
54
61
62
Scheme 16
61%
即ち、2-butyn-1-ol (5 9)をLiAlH4 -NaOMe / THFによりトランスアルケン(6 0)へと還元し、PPh3 / (CCl3)2CO によりクロル体(6 1)に変換した。LDA存在下、ジクロロ酢酸のtert-ブチルエステルにブテニル 基 を 導入 し (6 2)、 トリ フ ル オ ロ 酢 酸 処 理 に よ り 脱 tert-ブ チ ル エ ス テ ル 化 を 行 っ て 、 目 的 の 2,2-Dichloro-(4E)-hexenoic acid (5 4)を得た。
次 に 、分子内 ラジカル環化反応を検討するため、α,α-ジクロロアシル基をペンダントグループとする N-アポカンファンカルボニル-2-イミダゾロン誘導体(5 5)の合成を行った(S c h e m e 1 7)。
O OH
tBuO
O
OH O
tBuO
Br
O O
tBuO
O O HO
THF O
O
tBuO
Cl COClCl
O O
tBuO
O
O Cl Cl
NH N
O Ac
O O
N O N
O
O Cl Cl Ac
O O
HO O
O Cl Cl ,NaH
THF 84%
64
L-Selectride
MeOH O3, NaBH4
87%
65
66 , H+ 99%
CH2Cl2
CF3COOH CH2Cl2
67
69 ,Et3N DMAP
CH2Cl2 71%
68
SOCl2
reflux Et3N,DMAP 79%
r.t air
55 63
Scheme 17
98%
96%
L-ケトピン酸(6 3)を出発原料として前項の2−2−1と同様の操作を経て、アルコール体(6 7)を得5)、 2,2-ジクロロへキセン酸クロリドと縮合させカルボン酸エステル(6 8)とした後、トリフルオロ酢酸による 脱tBu化によりα,α-ジクロロアシル基をペンダントグループとするアポカンファンカルボン酸誘導体
(6 9)を得た。モノアセチルイミダゾロンとの縮合によりN-アシル-2-イミダゾロン体(5 5)を得た。
2−3−2 分子内ラジカル環化反応
得られたN-アシル-2-イミダゾロン体(5 6)のRuCl2(PPh3)3
7)によるラジカル環化反応をベンゼンあるいは トルエン還流下で検討した(Table 5)。
O O
N O N
O
O Cl Cl Ac
N O N
O O O O
Cl Cl
Ac
Toluene Solvent Entry
Conditions Ru Cl2(PPh3)3 (e q.) La(OTf)3 (eq.)
Time (h) Yield(%)
2 3 4
0.05
0.05 48 16 (61)
0.3 188 55 (0)
0.3 24 34 (36)
0.3 24 29 (44)
5 0.3 48 53 (0)
0.05 - 0.05
1 0.1 Benzene
Benzene
Toluene Toluene
Toluene
6 0.2 0.05 48 63 (0)
Toluene*
7 0.2 0.05 72 66 (0)
* Performed at 100˚C.
(Recovery(%)) Table 5 . Ru(II)-Catalyzed Intramolecular Radical Additions of N-Acyl-2-imidazolones with
a Dichloroacyl Pendant Group
Conditions
55 70
R R
reflux Solvent
本反応は原料(5 5)と付加環化体(7 0)とをシリカゲルカラムクロマトで分離することができなかったた め、可及的完全に反応を進行させることが必要であった。しかし、トリクロロアセチル基をペンダントグ ループとする2-イミダゾロン(3 8)の時と比べて、付加反応の進行は遅く、RuCl2(PPh3)3の当量を増やし、
長時間、トルエン還流というシビアな条件が必要であった。添加物のLa(OTf)3の効果も小さかった。
本ラジカル環化反応においては、12員環マクロライド体としてジアステレオマー体の2種類が生成した。
短時間で反応を終了させるとNMRにより原料及びジアステレオマー2種類の混合物が観測された。しか し、シビアな条件下(高温、長時間)で原料の消失まで反応を進行させると、ほぼ1種類の付加体のみが 観測された。二種類のジアステレオマーはカラムクロマトにて分離することができたので、メジャー、マ イナーともに付加環化条件での挙動を観察したところ、メジャージアステレオマーは定量的に回収するこ とができたが、マイナージアステレオマーは12時間で22%、24時間で8%と時間をかけるにつれて分解す ることが分かった。
2−3−3 3連続不斉中心構造の構築
得られた付加環化体(7 0)をMeOH中還流しその後Cs2CO3処理することにより、5-メトキシ体へと変換 し(7 1)、N-トシル化(7 2)後、還元的脱クロル化(7 3)を行った(S c h e m e 1 8)。
N O N
O O O O
Cl
Ac N
O HN
O
O O O Cl OMe
N O N
O
O O O Cl OMe
N O N
O
O O O
OMe To s To s
Cl
Entry Yield(%)
2
61 92 1
Et3B, (TMS)3SiH / Toluene, -78˚C AIBN, (TMS)3SiH / Benzene , reflux
Conditions
Scheme 18 MeOH
Cs2CO3 MeOH 84%
R R
Conditions
R R R R
R
CH2Cl2 Tos-Cl NEt3,DMAP
84%
70 71
72 73
R R
n-Bu3SnH / AIBNを用いベンゼン還流下の強い条件で反応を行うと化合物(7 3)のジアステレオマー比は
19:1であった(Entry 1)。(Me3Si)3SiH と Et3B を用いて-78°と緩和な条件下で反応を行うと99% d.e.以上と ほぼ完全な選択性で光学活性還元体(7 3)を得た。この構造については NOE スペクトル解析により決定 した(Figure 4)。
N N
O OMe
O O O O
Cl
N N
O O
Hb Cl O
O O Ha
OMe
Tos Tos
NOE positive
Figure 4. NOE of 73 7 3
n-Bu3SnH及び(Me3Si)3SiHによるハロゲン化アルキルの還元は、S c h e m e 1 9に示す経路によりラジカル 機構で進行していると考えられている21)。
(M e3Si)3SiH H• + (M e3Si)3Si•
(M e3Si)3Si• + RX (M e3Si)3SiX + R•
R• + (M e3Si)3SiH RH + (M e3Si)3Si • hn
S c h e m e 1 9
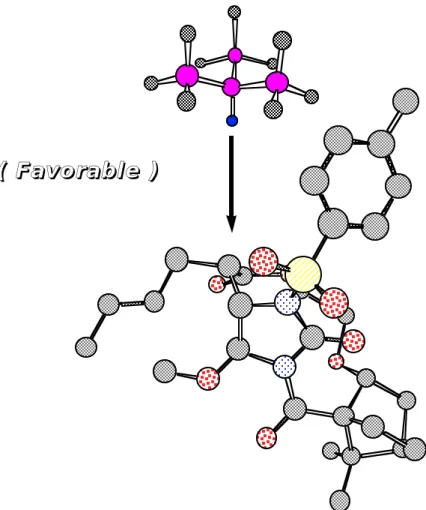
付加環化体(7 1)の還元的脱クロル化も同様の経路で進行すると考えられる。即ち、クロル基がシリル ラジカルによりラジカル的に引き抜かれ、生成した炭素ラジカルに(Me3Si)3SiHが立体障害の少ないsi-面 方向からH·を与えると考えられる (Figure 5)。従って、立体的に嵩高い(Me3Si)3SiHによる還元がより高 いジアステレオ選択性で進行し、(R)体を与える理由が理解できる。
Figure 5. Plausible Mechanism for Reductive Dechlorination.
( F a v o r a b l e ) ( F a v o r a b l e )
光学活性付加環化体(7 3)をLiBH4-MeOH(1:2)により還元的に脱アシル化して、アルコール体(7 4)を得 た18)。ここに3連続の不斉中心(R , R , R)配置を有するキラル合成子の立体選択的構築に成功した(S c h e m e 2 0)。
NH N
O OMe To s
HO
N O N
O
O O O
OMe To s
LiBH4, MeOH (1:2) THF
86%
R R
R R
R R
Scheme 20
73 7 4
2−3−4 MeBmt 類似体への誘導
3連続不斉中心(R , R , R)配置を有する光学活性2−イミダゾリジノン体(7 4)から、シクロスポリンに内 包されている水酸化アミノ酸MeBmtのジアミノ類似体(8 1)への誘導を行った(S c h e m e 2 1)。
NH N
O OMe To s
HO
NH N
O OMe To s
MsO
NH N
O OMe
To s CH2I
NH N
O OMe
To s Me
N N
O CN
To s Me
Me
N N
O OMe To s
Me
Me
N N
O
COOMe
To s Me
Me
NH HN
COOMe To s
Me
Me CH2N2
R R
R MsCl, Et3N CH2Cl2
82%
R R
R
NaI THF
84%
R R R
THF 92%
R R
R MeI, NaH
THF 93%
TMSCN BF3·OEt2
CH2Cl2 R
R R
R R
100%
S K2CO3
MeOH
2N HCl
83%
Et OH, H2O
Ba(OH)2·8H2O R
R
R R
S
S
71%
74 75 76
77 78
79 80
81 Scheme 21 Bu3SnH, Et3B
即ち、2-イミダゾリジノン体(7 4)の水酸基をメシル化(7 5)し、NaI / n-Bu3SnHを用いるラジカル還元22) によりメチル体(7 7)に変換した。N-メチル化(7 8)後、4位のメトキシ基をTMSCN / BF3·OEt2によるシア
ノ化(7 9)、引き続きEtOH中K2CO3によるアルカリ処理により中間体としてイミノエーテルを経て、塩酸
で処理することによりエステル体(8 0)を収率よく得た。Ba(OH)2·8H2Oによるイミダゾリジノン環の開環
により、MeBmt のジアミノ類似体(8 1)への誘導に成功した。
2−3 小括
2−イミダゾロン環の4位、5位二重結合部位の効率よい不斉機能化手法として、分子内ラジカル環化 反応を適用して、隣接及び3連続の不斉中心を有する1,2-ジアミン類のキラル合成に応用し、その有用性 を実証することができた(S c h e m e 2 2)。
O O
N O N
O
O
RCO Cl Cl
Cl
NH N
O OMe To s
HO Cl Cl
HN NH MeOOC
To s N
O N
O RCO
O O O
Cl Cl
Ru (II) Cl
Boc
Scheme 22
99% >d.e 38 R=C H3
42 R=Ph
39 R=CH3 43 R=Ph
46
* *
5 2
即ち、隣接する不斉中心をもつ1,2-ジアミン類の合成については、トリクロロアセチル基をペンダント グループとする2-イミダゾロン体(3 8, 4 2)のRuCl2(PPh3)3による分子内ラジカル環化反応により、完全な ジアステレオ選択性で12員環マクロライド体(3 9,4 3)を得ることができた。本反応はLa(OTf)3などルイス 酸性化合物の添加により劇的に加速されるとの興味ある知見を得たが、加速機構は不明である。得られた 付加環化体を隣接する不斉中心を有するキラル合成子(4 6)に誘導し、スタチンのアミノ類似体(5 2)等の キラル合成に利用した。
N O N
O Ac
O O O
Cl Cl
NH N
O OMe To s
HO
NH- Me NH
COOMe To s
O O
N O N
O
O
Ac Cl Cl Ru (II)
(TMS)3SiH
N O N
O To s
O O O
OMe Li BH4-MeOH
Scheme 23
99% >d.e 56
73
70
74
*
* *
8 1
一方、本方法論の汎用性を示すために、3連続不斉中心を有するジアミン類としてシクロスポリンに内
包されるMeBmtのアミノ類似体を標的として、2,2-ジクロロ-4-ヘキセノイル基をペンダントグループとす
る2-イミダゾロン(5 6)から、本ラジカル環化反応を利用し、立体選択的還元を経てほぼ完全な立体選択 性で3連続不斉中心を有するキラル合成子(7 3)の構築に成功し、3連続不斉中心構造のMeBmtのアミノ 類似体(8 1)を光学的に純粋に合成した(S c h e m e 2 3)。
ここに、本方法論の更なる有用性を実証することができた。また、本研究により開発したキラル合成子
(4 6)や(7 4)の4位には1級、2級、3級のアルキル基やアリール基など多彩な炭素置換基を立体保持で導
入できるので、多岐にわたるジアミン類のキラル合成が可能である。この線に添った、本方法論の幅広い 応用展開が期待できる。
第3章 多目的キラル合成子 DMIm の効率合成法の開発と利用
3−1 はじめに
N, N'-ジアシル2-イミダゾリジノン体は容易に脱モノアセチル化反応を起こし、定量的にモノアセチル 体を与えることは既に明らかとなっている。この知見を踏まえ、当研究室ではジアセチルイミダゾリジノ
ン体(8 2 , 8 5)のエナンチオトピックな左右のアセチル基を立体的嵩高さと配座固定の点で特徴のあるアミ
ノアルコール(8 3)を触媒量を用いて選択的に除去することにより、一段階で光学活性なモノアセチルイ ミダゾリジノン体(8 4 , 8 6)を得ることに成功している(S c h e m e 2 4)。23)
N
N O
Ac
Ac
NH
N O
Ac R
R R R
N
N O
Ac
Ac
NH
N O
Ac NH2
OH
> 99 %ee
BH3 · SMe2
98 %ee
Thexylborane
(10 mol%) + BH3
82 84
85
83
86 S c h e m e 2 4
従来、DMIm(8 9 a , 8 9 b)は、MAC基(8 7)を用いた光学分割により合成していた24)が、煩雑で大量合成
に適しておらず、簡易合成法の確立は重要な課題だった(S c h e m e 2 5)。
N N
O
Ac Ac
Cl
O OMe
MAC -Cl HN N
O MeO
MAC
OMe BnOH
HN N O MeO
MAC OMe
HN N O MeO
MAC H2 OMe
19
87 BF3·OEt2
Pd-Black
89b 89a (4S, 5S)
(4R, 5R)
4 5
4 5
Scheme 25 8 8
そこで、メソ型4,5-ジアルコキシ-2-イミダゾリジノン体のボランによるエナンチオ選択的脱アセチル化 反応を鍵としたDMImの効率良い合成法の開発を目指した。