大気圧窒素・空気中 ArF エキシマーレーザーによる
N
2O、NO
2の光分解処理プロセスの開発
1:九州大学先導物質化学研究所 2:九州大学総合理工学府 量子プロセス理工学専攻
3: (独)科学技術振興機構 CREST
辻 正治
1-3,迫 寛之
2,野田 健二
2,仙田 慎
2,辻 剛志
1-3Development of Photochemical Removal Processes of N
2O and NO
2by ArF excimer Laser in N
2or Air at Atmospheric Pressure
Masaharu Tsuji*, Hiroshi Sako, Kenji Noda, Makoto Senda, and Takeshi Tsuji
1: Institute for Materials Chemistry and Engineering, Kyushu University
2: Department of Applied Science of Electronics and Materials, Graduate School of Engineering
Sciences, Kyushu University
3: CREST, Japanese Science and Technology
Abstract
Photochemical removal of N2O and NO2 by 193 nm ArF excimer laser has been investigated in N2 or air at
atmospheric pressure. The residual amounts of N2O and NO2 and formation ratios of N2, O2, NOx, and O3
were measured as a function of laser irradiation time and N2O or NO2 concentration. N2O was nearly
completely converted to N2 and O2 without NOx emission in N2 and air at a low N2O concentration of 100
ppm after laser irradiation for 15 min at a laser power of 100 mJ/pulse. The decomposition model of N2O
in N2 or air was simulated by possible photochemical processes from known photochemical and gas-kinetic
data. A reasonable agreement was found between experimental results and calculated values, indicating that our model calculation was valid. The decomposition process of NO2 depended strongly on the NO2
concentration. NO2 was efficiently converted into N2, O2, and NO in N2 or air in the NO2 concentration
range of 240-3000 ppm at a laser power of 80 mJ/pulse, and the relative formation ratios of N2:NO in N2 and
air were 1:0.38~0.79 and 1:0.41~0.63, respectively. The major decomposition process in N2 and air in this
NO2 pressure range was attributed to 193 nm photolysis of NO2 into NO*(X2Π:v”≥1) + O followed by
second absorption of 193 nm light of NO*(X2Π:v”≥1) leading to N(4S) + O(3P) dissociation. The N(4S) +
NO → N2 + O(3P) reaction finally converts NO to N2. NO2 was completely oxidized into N2O5 in air at low
NO2 pressure of 100 ppm. The major conversion process in air in this low NO2 pressure range was
oxidation of NO2 into N2O5 via NO3 due to effects of O3.
Key Words : N2O, NO2, Vacuum ultraviolet, ArF excimer laser, FTIR
1.はじめに 地球上の資源供給源(ソース)と公害物質吸収源(シンク)の有限性に起因する地球の扶養力限 界からの逸脱が、化石燃料枯渇や地球温暖化の形で急速に顕在化している。特に東アジア地域にお いては、中国の急速な経済発展に伴い、これまでに人類が経験したことのない速度と規模で工業化 と人口爆発が同時進行している。その結果、幾何級数的に増大しつつあるエネルギー消費量、炭酸 ガス排出量、さらに環境汚染は、全人類の生存に関わる国際的かつ喫緊の問題となっている。 環境汚染は大気・水圏・土壌などに及んでいるが、今後、我が国への深刻な影響が懸念されるの は大気環境汚染問題である。その理由は中国を初めとする東アジアの火力発電所では窒素や硫黄分 が高い石炭が使用されているが、排煙処理装置はほとんど設置されていないためである。そのため にNOx, SOx などの大気環境汚染物質が放出され、酸性雨やオゾン層破壊による地球温暖化の原因
*連絡先:[email protected], ホームページ:http://133.5.181.45/TSUJI/
となっている。今後、中国を始めとする東アジア諸国の急激な経済発展が進めば、電力需要が大幅 に増大する関係で、大気汚染や地球温暖化問題は益々深刻化するものと思われる。大気汚染物質の 処理技術は触媒、燃焼、吸着法など、既に一部工業化されている1)。しかし、これらの従来の技術 は高コストなために、経済力が乏しい東アジアの発展途上国の発電所では処理装置が装備されてい ないのが実情であり、これらの発展途上国でも容易に使用可能な廉価で高効率な環境汚染物質の処 理技術の早急な開発が切望されている。
窒素酸化物NOx には NO, NO2, N2O などがある。そのうち亜酸化窒素(N2O)は、対流圏中の濃度
は約310 ppb 程度と微量ではあるが、約 150 年という長い寿命を持つことや温暖化係数が CO2の310
倍であるために、温暖化やオゾン層破壊への寄与が大きいことが知られている。N2O は主に土壌や
海洋における微生物活動により発生する。しかし近年、病院や自動車、化学工場などから排出され
る人為的な発生源が全排出量の約半分を占めるようになり、N2O の対流圏濃度は年々増加傾向にあ
る2)。N2O は SOx 存在下では NOx, CO, 炭化水素の分解に使用される白金、パラジウム、ロジウム
を主成分とする三元触媒は被毒され分解処理が困難なため、N2O 除去法の開発は現在、NOx 除去プ ロセス研究の中心課題となっている。一方、NO2は自動車や発電所、工場などの排ガスとして放出 され、呼吸器疾患、光化学スモッグ、酸性雨などを引き起こす代表的 NOx である。現在はアンモ ニアなどの還元剤を用いるSCR 法(選択的触媒還元法)や三元触媒などが NOx 処理に使用されて いるが、酸素過多の条件ではN2O が生成することや、SOx や O2の存在により触媒が被毒されると いう問題がある。このような状況下、高価な触媒を使用せず東アジアの発展途上国でも使用可能な N2O や NO2などのNOx 分解処理プロセスの開発が期待されている。 近年、低温プラズマを用いた排煙処理法が発展途上国でも使用可能な新しい低コスト排煙処理法 として注目され、種々の新技術の開発研究が世界中で活発に行われ、その一部は既に実用化段階に 到達している3)。これまでの低温プラズマ法による排煙処理技術として最も研究開発が進んでいる のは、大気圧で作動するコロナ放電法とバリア放電法である。低温プラズマの最大の問題点は大気 圧空気中ではN2, O2の反応でNOx が発生し、NOx の完全除去が困難なことである4)。これは低温 プラズマ中での主要な活性種が高速電子であり、高速電子はNOx, N2, O2と(1)-(3)のように非選択的 に反応し、後続する2 次反応(4), (5)で NOx が発生するためである。 反応例 (*は電子的励起状態を示す。) e- + NO → N + O + e- (1) 電子衝突解離 e- + N 2 → N* + N + e- (2) 電子衝突解離 e- + O 2 → O + O + e- (3) 電子衝突解離 N* + O 2 → NO + O (4) 2 分子反応 NO + O + N2 → NO2 + N2 (5) 3 体再結合 このことは空気中で低温プラズマのみで NOx の発生を抑制し、かつ有害物質を分解除去すること は原理的に極めて困難なことを示唆するもので、低温プラズマに代わる NOx などの有害物質を選 択的に分解除去可能な新たな発想に基づく技術の開発が望まれている。 筆者らは電子に比べて光が分子に対する高い反応選択性を有することやプロセスの制御性に優 れていることに着目して、NOx の光プロセスによる分解処理装置の開発研究を行っている 5-8)。例 えばN2O, NO2分子は真空紫外領域の光を吸収して分解することが知られているが、N2は150 nm よ
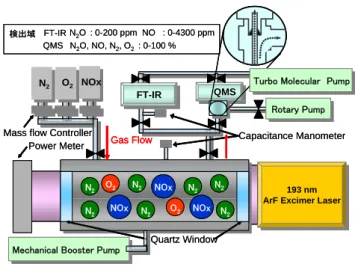
193 nm ArF Excimer Laser
193 nm ArF Excimer Laser Power Meter N2 O2 NOx FT-IR FT-IR Rotary Pump Rotary Pump N2O : 0-200 ppm NO : 0-4300 ppm FT-IR QMS N2O, NO, N2, O2: 0-100 % QMS Gas Flow NOx NOx NOx N2 N2 N2 N2 O2 O2 N2 N2 N2 Capacitance Manometer
Mechanical Booster Pump
Mechanical Booster Pump
Turbo Molecular Pump
Turbo Molecular Pump 検出域
Mass flow Controller
Quartz Window
193 nm ArF Excimer Laser
193 nm ArF Excimer Laser Power Meter N2 O2 NOx FT-IR FT-IR Rotary Pump Rotary Pump N2O : 0-200 ppm NO : 0-4300 ppm FT-IR QMS N2O, NO, N2, O2: 0-100 % QMS Gas Flow Gas Flow NOx NOx NOx N2 N2 N2 N2 O2 O2 N2 N2 N2 NOx NOx NOx N2 N2 N2 N2 O2 O2 N2 N2 N2 Capacitance Manometer
Mechanical Booster Pump
Mechanical Booster Pump
Turbo Molecular Pump
Turbo Molecular Pump 検出域
Mass flow Controller
Quartz Window
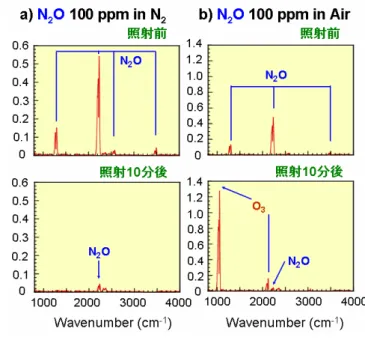
Fig. 1. ArF エキシマーレーザーを用いる NOx 分解 処理装置 Fig. 2. レーザー照射前後の質量スペクトル り長波長に吸収がなく、また127 nm 以下の波長でないと N 原子に分解できない。よって 150 nm 以 上の光照射ではNOx の発生源となる N2の分解によるN*原子の生成は無視できる。そこで筆者らは、 新たなN2O, NO2除去法として、波長193 nm では N2が分解しないことやN2O, NO2の光吸収係数が O2と比べて、それぞれ約300 倍、900 倍大きいことに注目し、ArF エキシマーレーザーを用いた N2O, NO2の大気圧窒素・空気中での選択的な光分解処理プロセスの開発研究を行っている。本稿では最 近の主要な研究成果を報告する。 2.実験 Fig. 1 に実験装置図を示す。N2O または NO2を N2または空気(N2:O2 = 4:1)で希釈 後、反応セル内に導入し、系内を大気圧に 保った後、ArF エキシマーレーザー(波長: 193 nm、繰り返し周波数:5 Hz、照射時間: 0.5~45 分)を照射した。レーザーのエネルギ ーはN2O の実験では 100 mJ/pulse、NO2の実 験では80 mJ/pulse を使用した。実験は全て バッチ式で行い、光照射後のN2O または NO2 の残留率、N2, O2, NOx の生成率を、高濃度 (5%) の 場 合 は ガ ス 分 析 用 質 量 分 析 計 (ANELVA 社製 QMS)を、低濃度(100-3000 ppm)の場合はフーリエ変換赤外吸収スペク トル分析計(HORIBA 社製 FTIR)を用いて決 定した。なお生成率は各生成物の生成濃度の 初期NOx 濃度に対する比を表す。 3.結果と考察 3―1 N2O の光照射処理 5%という高 N2O 濃度での質量スペクトル の測定結果をFig. 2 に示す。a)は窒素中、b) は空気中でレーザーを 10 分照射前後のスペ クトルである。窒素中、空気中ともに N2O+ ピークがレーザー照射後、それぞれ16, 12% に減少している。窒素中、空気中ともにレー ザー照射前に図中に黒丸で示した NO+ピークが出現しているが、これは N 2O の電子衝撃によるフ ラグメントイオンでありNO 由来のピークではない。注目される点は N2/O2混合ガスの放電プラズ マではNO 生成により強く出現する NO+ピークがFig. 2b)の空気中のスペクトルでも観測されない ことである。このことは 193 nm 光プロセスを用いると空気中でも放電プラズマ中とは異なり NO の発生を抑制した状態で窒素中と同様にN2O を効率よく分解処理可能なことを示唆している。 N2O 濃度 1%以下では質量分析計の検出感度が低いためにガス分析が困難であった。そこで ppm
Fig. 3. レーザー照射前後の FTIR スペクトル ppm の N2O にレーザーを 10 分照射前後の FTIR スペクトルである。窒素中及び空気中と もにレーザー照射後には N2O ピークが大き く減少し、N2O が効率よく分解されるととも にNO, NO2が全く生成していないことがわか る。また空気中では主に雰囲気ガスの酸素に 由来する高濃度のオゾンの生成が観測され た。以上のことから100 ppm 程度の低濃度条 件下では N2O は N2とO2または O3に変換さ れると考えられる。上記のような質量スペク トル、FTIR スペクトルの解析から N2O の残 留率とN2, O2, NO の生成率を様々な実験条件 下で決定した。 Fig. 4, 5 に、大気圧窒素及び空気中で N2O を5%含む系における N2O の残留率と N2, O2, NO の生成率のレーザー照射時間依存性をそれぞれ示 す。実線が実験値、点線はN2O, O2, NO, NO2, O3の光吸収とN(4S), O(3P,1D), O3などの活性化学種の 2 次、3 次反応過程として全部で 27 個の気相素反応過程の吸収係数と反応速度定数9-19)を考慮して 得たモデル計算値である。モデル計算結果は実測結果をほぼ再現することができ、N2O 光分解モデ ルの妥当性が示唆された。Fig. 4, 5 より N2O はレーザー照射後 5 分で窒素中では約 53%、空気中で は約50%分解し、N2O の光分解に対する酸素の影響は少ないことがわかった。また窒素中では照射 20 分後、空気中では照射 30 分後に N2O をほぼ完全に無害な N2とO2へ分解できた。一方NO の生 成に関しては、窒素中で15%以下に抑制できたのに対し、空気中では 7%以下にさらに抑制可能で あった。この実験結果は酸素を導入することでNO の生成をさらに抑制できることを示唆している。 空気中の方がNO 濃度が低い理由の一つは空気中では NO は次の O2やO3との反応により消失する ためと考えられる。 0 20 40 60 80 100 0 5 10 15 20 25 30 Resid ual amo u nt of N 2 O an d fo rm a ti o n r a ti os o f N2 , O 2 , a n d NO / % N2O N2 O2 NO
Irradiation time (min)
Fig. 5. 大気圧窒素中での 5 % N2O 光分解におけ るN2O の残留率、N2, O2, NO 生成率のレーザー 照射時間依存性。破線はモデル計算値。 0 20 40 60 80 100 0 5 10 15 20 25 30 Re s idu al amo unt o f N 2 O a n d fo rm a tio n r a tio s o f N 2 , O 2 , a n d N O / % N 2O N 2 O2 NO
Irradiation time (min)
Fig. 4. 大気圧空気中での 5 % N2O 光分解にお
けるN2O 残留率、N2, O2, NO 生成率のレーザー
0 20 40 60 80 100 0 5 10 15 20 25 30 Re sid ual am ou n t of N 2 O an d fo rm at io n r a ti o s o f N2 , O 2 , a n d NO / %
Irradiation time (min)
N2O N2 NO O2
Fig. 7.
大気圧空気中での100 ppm N2O の光 分解におけるN2O 残留率, N2, NO 生成率の レーザー照射時間依存性。破線はモデル計 算値。2NO + O2 → 2NO2 (6) k = 1.93×10-38 cm6molecule-2s-1 [18,19]
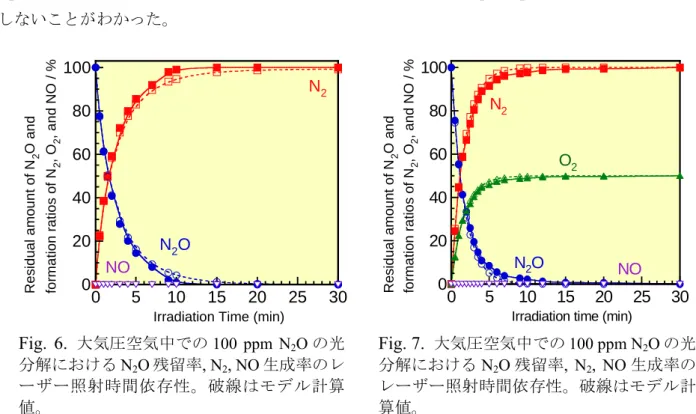
NO + O3 → NO2+ O2 (7) k = 1.8×10-14 cm3molecule-1s-1 [18,19] これらの反応で生成するNO2は以下のNO2の節で述べるようにNO + 1/2O2解離以外に1/2N2 + O2 にも解離すると考えられる。 低N2O 濃度での実験結果の例として窒素及び空気中で N2O を 100 ppm 含む系に対するレーザー 照射時間依存性の測定結果をFig. 6, 7 に示す。図から明らかなように N2O を 100 ppm 含む系では、 N2O は、いずれもレーザー照射約 15 分後に、ほぼ完全に無害な N2とO2に分解し、NO は全く生成 しないことがわかった。 NO の生成率の N2O 濃度との関係の例として、大気圧空気中で N2O 濃度を 0.01% (100 ppm)から 5% まで変化させ、193 nm 光を 5 分間照射後の N2O 濃度依存性を Fig. 8 に示す。Fig. 8 より N2O 濃度 が低くなるにつれて、N2O の残留率 14%まで減少し、同時に NO の濃度も減少し、濃度 1%でほぼ ゼロになることがわかった。N2O の ArF エキシマーレーザー照射下での分解において NO はレーザ ー光分解(8)で生成する O(1D)と N 2O との反応(9b)で生成すると考えられる。 N2O + hν → O(1D) + N2 (8) σ = 8.95×10-20 cm2 molecule-1 [9] O(1D) + N 2O → N2 + O2 (9a)
k = 4.4×10-11 cm3molecule-1s-1 [18,19] → 2NO (9b)
k = 7.2×10-11 cm3molecule-1s-1 [18,19] 空気中においてO(1D)の反応(9a), (9b)は下記の衝突電子緩和過程(10), (11)と競争する。 O(1D) + N 2 → O(3P) + N2 (10) k = 2.6×10-11 cm3molecule-1s-1 [18,19] O(1D) + O 2 → O(3P) + O2 (11) k = 4.0×10-11 cm3molecule-1s-1 [18,19] 式(9)-(11)から NO の生成速度は次式で与えられる。 0 20 40 60 80 100 0 5 10 15 20 25 30 Re s idu a l am o unt o f N2 O a n d fo rm a tion r a tio s o f N2 , O 2 , a n d NO / % N2O N2 NO
Irradiation Time (min)
Fig. 6.
大気圧空気中での100 ppm N2O の光分解におけるN2O 残留率, N2, NO 生成率のレ
ーザー照射時間依存性。破線はモデル計算 値。
0 20 40 60 80 100 0 5 10 15 20 25 30 R e s idu al am oun t o f N O2 an d fo rm at io n r a ti os o f N2 , O 2 , a n d N O / % NO2 N2 NO
Irradiation time (min) O2 Fig. 9. 大気圧窒素中での5% NO2の光分解に おけるNO2残留率、N2, O2, NO 生成率のレー ザー照射時間依存性 0 20 40 60 80 100 0 5 10 15 20 25 30 Re s id u al a m oun t o f NO 2 an d fo rm at io n r a ti os o f N2 , O 2 , an d N O / % NO2 N2 NO
Irradiation time (min) O2 Fig. 10. 大気圧窒素中での200 ppm NO2の光 分解におけるNO2残留率、N2, O2, NO 生成率 のレーザー照射時間依存性 N2O 濃度が低下するにつれて NO 濃度が減少するの は 、 低 濃 度 に な る に つ れ て(12) 式 の 分 母 中 の
]
O
[
]
N
[
2 10 2 9k
k
+
項の寄与が増大し、NO 生成速度が 減少するためと考えられる。この結果から本手法が 特に約 1%以下の低濃度 N2O の完全分解無害化処理 法として有効であることがわかった。 3―2 NO2の光照射処理 NO2はNO と並ぶ代表的 NOx であり酸性雨の原因 物質である。筆者らはNO2のArF エキシマー光分解 についても研究した。Fig. 9, 10 に窒素中、濃度 5%と 200 ppm の NO2をレーザー光分解した場合のNO2残 留率およびN2, O2, NO 生成率のレーザー照射時間依 存性を示す。濃度5%の場合には、NO2はN2, O2, NO に分解し、残留率は照射時間30 分で 77%に留まった。主な窒素含有生成物は NO, N2でありNO:N2 生成率は分解率がほぼ一定となった照射15 分後には、それぞれ約 10, 6%であった。実際のプロセ ス中でのNO2の排出濃度に近い200 ppm という低濃度では照射時間 3 分で残留率は窒素中では 10% まで急激に減少した。注目される点はNO2濃度200 ppm での NO, N2生成比が照射3 分後に 25, 33% となり、N2/NO 生成比が NO2濃度5%の場合の 2.2 倍に増大している点である。 NO2は紫外光によりNO + O(3P,1D)に分解することは良く知られている11)。筆者らのNO2の193 nm レーザー光分解で一番注目される結果は、NO2は有害な NO への分解に留まらず、無害な N2まで ] O [ ] N [ ] O N [ 2 ] O N [ ] O N [ 2 ] O )][ D ( O [ ] N )][ D ( O [ ] O N )][ D ( O [ 2 ] O N )][ D ( O [ ] O N )[ D ( O [ 2 ] NO [ 2 10 2 9 2 8 2 8 2 8 2 1 10 2 1 9 2 1 8 2 1 8 2 1 8 k k k k k k k k k k dt d b a b b a b + + + = + + + ∝(12)
0 20 40 60 80 100 0 1 2 3 4 5 Re sid u al am ou n t o f N2 O a nd for m a tion r a tios o f N2 , O 2 , a n d NO / % N2 N2O NO N2O concentration (%) Fig. 8. 大気圧空気中での 100 ppm N2O の光 分解におけるN2O 残留率、N2, NO 生成率の N2O 濃度依存性:レーザー照射 5 分後。破 線はモデル計算値。0 20 40 60 80 100 0 5 10 15 20 25 30 Re s id u al am oun t o f N O2 an d fo rm a tion r a tio s o f N 2 , O 2 , a n d N O / % NO2 N2 NO
Irradiation time (min) O2 Fig. 12. 大気圧空気中での200 ppm NO2の光分 解におけるNO2残留率、N2, O2, NO 生成率のレ ーザー照射時間依存性 0 20 40 60 80 100 0 5 10 15 20 25 30 Re sid u al am ou n t o f N O2 an d fo rm a tion r a tio s of N2 , O 2 , an d NO / % NO2 N2 NO
Irradiation time (min) O2 Fig. 11. 大気圧空気中での5% NO2の光分解にお けるNO2残留率、N2, O2, NO 生成率のレーザー照 射時間依存性 変換されることを見出したことである。この予期せぬ実験結果はNO2がレーザー照射下の光プロセ スでN2とO2へ還元可能なことを示す初めての結果である。 NO2 + hν(193 nm) → NO → 1/2N2 (窒素化合物の変化) (13) NO2の193 nm 一光子分解で生成する NO, O(3P,1D)と原料である NO2間の反応でNO2をN2まで変換 する反応は存在しない18,19)。またNO の結合解離エネルギーは 6.496 eV であり11)、193 nm (6.42 eV) と比べて高いので193 nm 光を用いて基底振動状態の NO を N + O へ分解することはできない。よ ってNO が N2, O2 まで分解するのは主として反応(15a)で生成する振動励起 NO(X2Πi:v”=1~16)が 193 nm 光を再吸収し12,20)、高励起状態へ励起後N + O へ前期解離し、さらに N(4S)と NO の反応(16)に よりN2へ変換されるためと考えられる。 NO(X2Π : v” = 1~16) + hν → NO(A2Σ+,D2Σ+,E2Σ+) (14)
NO(D2Σ+,E2Σ+) → NO(X2Π,A2Σ+) + hν (蛍光) (15a)
→ N(4S) + O (3P) (前期解離) (15b) N(4S) + NO → N 2 + O (16) 実際の NO2排ガス中には酸素も含まれるので、酸素存在下でも効率よく NO2を分解する必要が ある。Fig. 11, 12 は空気中における 5%, 200 ppm NO2の分解実験でのレーザー照射時間依存性であ る。NO2濃度5%での残留率は照射 30 分後で約 90%であり窒素中と比べて約 10%の増加が見られる が、NO2濃度200 ppm では残留率は照射 3 分後で 57%と窒素中の 10%と比べて大幅に増加した。一 方、NO 生成率は窒素中と同様に 10%以下であり N2(最大 20%)や O2(最大 44%)の生成率と比べて低 い。この結果から大気圧空気中でも200 ppm という低濃度の NO2では193 nm レーザー照射下で 40% 程度の分解効率で主にN2, O2へ変換可能なことがわかった。 Fig. 13, 14 はレーザー照射 30 秒後の窒素、空気中での NO2の残留率とN2, O2, NO 生成率の NO2 濃度依存性を約200 ppm から 3000 ppm の範囲で調べた結果である。NO2の残留率は窒素中ではNO2
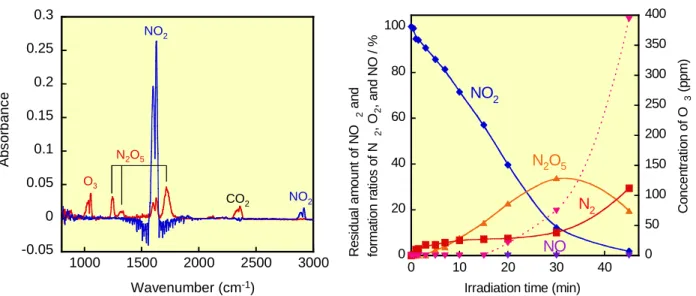
0 10 20 30 40 50 60 70 0 1000 2000 3000 Res id ual am o u n t o f N O2 an d fo rm a ti on r a ti o s o f N2 , O 2 , and N O / % NO2 N2 NO O2 NO2 concentration (ppm) Fig. 13. 大気圧窒素中での NO2光分解における N2O 残留率、N2, O2, NO 生成率の NO2濃度依存 性:レーザー照射時間30 秒 0 10 20 30 40 50 60 70 0 1000 2000 3000 Re s idu al am oun t o f NO 2 an d fo rm a tio n r a tio s o f N2 , O 2 , a n d N O / % NO2 N2 NO O2 NO2 concentration (ppm) Fig. 14. 大気圧空気中での NO2光分解における N2O 残留率、N2, O2, NO 生成率の NO2濃度依存 性:レーザー照射時間30 秒 濃度の増加とともに40%から 45%へ 5%増加するのに対して NO の生成率は 17%から 9%へ減少し た。一方空気中ではNO2の残留率はNO2濃度が240 ppm から 500 ppm へ増加するに従い 70%から 47%へ大きく減少するが、1000-3000 ppm では 52%までのわずかな増加に留まった。これに対して NO の生成率は 240 ppm から 1000 ppm へ増加するにつれて 7%から 12%へ増加するが、その後は 若干減少する傾向があり、3000 ppm では 8%であった。これらの結果は NO2濃度1000-3000 ppm 領 域では窒素中と空気中で NO2残留率や NO 生成率に大差はなく、酸素存在下でも NO2を主として N2, O2へ変換可能なことを示している。 NO2の193 nm 光分解は以下の(17)式の NO と O(1D,3P)への分解と、それによって生じる O(1D,3P) とNO2との反応(18), (19)により進行すると考えられる。 NO2 + hν(193 nm) → NO + αO(1D) + βO(3P) (α/β = 1.2) (17) O(1D) + NO 2 → NO + O2 (18) O(3P) + NO 2 → NO + O2 (19) 一般にO2存在下でNO2の分解が抑制される傾向があるのは、一旦生成したNO から下記の酸化反 応によりNO2が再生する反応が起こるためであろう。 NO + O + O2 → NO2 + O2 (20) 2NO + O2 → 2NO2 (21) NO2レーザー光分解過程のNO2濃度の影響をさらに調べるために100 ppm NO2/air の条件下でも 研究した。その結果、Fig. 15 に示すように低 NO2濃度では生成物が大きく変化し、200 ppm 以上の 高濃度では観測されなかった N2O5,O3の生成も確認された。一方、この濃度では NO は全く観測 されなかった。Fig. 16 に NO2の残留率とN2O5, N2, NO 生成率と O3濃度のレーザー照射時間依存性
-0.05 0 0.05 0.1 0.15 0.2 0.25 0.3 1000 1500 2000 2500 3000 Wavenumber (cm-1) Ab s o rb anc e N2O5 CO2 NO2 NO2 O3
Fig. 15. 200 ppm NO2/air 中(青)、100 ppm NO2/air
中(赤)での照射 30 分後の FTIR スペクトル 0 20 40 60 80 100 0 50 100 150 200 250 300 350 400 0 10 20 30 40 R e s id u al am oun t o f N O 2 an d fo rm a ti on r a ti os of N 2 , O 2 , a n d N O / %
Irradiation time (min)
NO2 N2 NO N2O5 C o nc en tr a tio n of O 3 (p p m ) Fig. 16. 100 ppm NO2/air 中での NO2 残留率と N2O5, N2, NO 生成率および生成 O3濃度の照射時 間依存性 を示す。NO2は照射時間の増加とともに緩やかに減少し、照射45 分でほぼ消失した。200 ppm NO2/air ではO3は検出されなかったが、実際は200, 100 ppm どちらの場合でも空気中の O2の分解反応(22) で生成するO(3P)と O 2の三体再結合反応(23), (24)によりほぼ同一濃度の O3が生成すると予想される。 O2 + hν(193 nm) → 2O(3P) (22) O(3P) + O 2 + N2 → O3 +N2 (23) O(3P) + O 2 + O2 → O3 + O2 (24) それにもかかわらず200 ppm NO2/air 中では O3が検出されないのは、反応(17)で生成する NO 濃度 が高いためにO3が反応(25)で消費されるためと考えられる。 NO + O3 → NO2 + O2 (25) 長時間照射では O3が触媒的役割を果たし、NO2生成と NO2光分解が平衡に達するために NO2残留 率とN2, O2, NO 生成率が一定になると考えられる。一方 100 ppm NO2/air では、NO2光分解によっ て生成するNO 濃度が低く、O3は NO2と反応してNO3となり、さらにNO2との再結合反応により 分解処理が容易なN2O5が生成する。 O3 + NO2 → NO3 + O2 (26) NO3 + NO2 → N2O5 (27) NO2が完全に消費された後はO3は蓄積され、その濃度は急激に増大すると考えられる。 4.まとめ 大気圧窒素・空気中でのArF エキシマーレーザーによる N2O, NO2の光分解プロセスの研究開発 を行った。その結果、高価な触媒を用いることなく、N2O を大気圧窒素・空気中で無害な N2とO2
に高効率、高分解処理が可能なことがわかった。また実用に有利な低濃度(1%以下)の N2O の光 分解を行った。その結果、193 nm 光照射 15 分でほぼ完全に N2O を分解することができ、放電法と は異なり、空気中でも有害なNO は全く生成しないことがわかった。本研究により、実際に排出さ れるppm レベルの低濃度 N2O の光分解処理に対して 193 nm 光を用いた光プロセスがより有効なこ とが示唆された。 また大気圧でのArF エキシマーレーザーによる NO2の分解プロセスは NO2濃度に依存すること が判明した。窒素中ではNO2濃度を200-3000 ppm まで低くすることで、有害な NO の生成を抑制 し、無害なN2, O2まで分解可能なことを見出した。またNO2濃度100 ppm の空気中では、O3の生 成も確認された。そのO3の酸化力を用いて、NO2をNO3を経てN2O5へと転換可能であることが判 明した。水に不溶なNO2とは異なり、N2O5は水に溶けて硝酸として処理可能なために、193 nm 光 を用いた本手法は、新たな選択的NO2除去法として期待できる。 謝辞:本研究の一部は文部省科研費基盤研究B(課題番号 15310059) 並びに 2002 年度から開始され た科学技術振興機構CREST 「ナノ環境触媒」と「ナノエネルギー材料」の援助により行われたも のであり、これらの助成に感謝の意を表します。 参考文献 1) 環境触媒、実際と展望、日本表面科学会編、共立出版 (1997).
2) Japan’s Third National Communication under the United Nations Framework Convention on Climate
Change, The Government of Japan, 2002. (http://unfccc.int/resource/docs/natc/japnc3.pdf)
3) M. Tsuji, K. Nakano, J. Kumagae, T. Matsuzaki, T. Tsuji, Surface and Coating Technology, 165, 296 (2003).
4) プラズマの生成と診断 ―応用への道― プラズマ・核融合学会編、6 章、エネルギー・環境
工学へのプラズマ応用 (2004).
5) M. Tsuji, J. Kumagae, T. Tsuji, T. Hamagami, J. Hazardous Materials, 108, 189 (2004). 6) M. Tsuji, K. Noda, H. Sako, T. Hamagami, T. Tsuji, Chemistry. Letters, 34, 496 (2005).
7) M. Tsuji, H. Sako, K. Noda, M. Senda, T. Hamagami, and T. Tsuji, Chemistry Letters, 34, 812 (2005). 8) M. Tsuji, H. Sako, K. Noda, M. Senda, and T. Tsuji, Fuel, submitted for publication.
9) A. A. Turnipseed, G. L. Vaghjian, T. Gierczak, J. E. Thompson, A. R. Ravishankara, J. Chem. Phys. 95, 3244.(1991).
10) Y. Niwa, A. Matsuzaki, A. Nishio, H. Sato, I. Tanaka, J. Phys. Chem. A, 101, 668 (1997). 11) H. Okabe, Photochemisty of Small Molecules, John Wiley & Sons, New York (1978). 12) J. Zavelovich, M. Rothschild, W. Gornik, C. K. Rhodes, J. Chem. Phys., 74, 6787 (1981).
13) S. Nishida, K. Takahashi, Y. Matsumi, N. Taniguchi, S. Hayashida, J. Phys. Chem. A, 108, 2451 (2004). 14) T. Nakayama, K. Takahashi, Y. Matsumi, N. Taniguchi, S. Hayashida, J. Geophys. Res.-Atmospheres
108, 4668 (2003).
15) F. F. Marmo, J. Opt. Soc. Am., 43, 1186 (1953).
16) B. L. Bakker, D. H. Parker, J. Chem. Phys., 112, 4037 (2000). 17) F. Sun, G. P. Glass, R. F. Curl, Chem. Phys. Lett., 337, 72 (2001).
18) R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson Jr., J. A. Kerr, M. J. Rossi, J. Troe, J. Phys. Chem.
Ref. Data 26, 1329 (1997). Updated data were obtained from NIST Chemical Database on the Web, Public Beta Release 1.2, Standard Reference Database 17, Version 7.0 (http://kinetics.nist.gov/
index.php).
19) IUPAC Gas Kinetic Data Evaluation, Summary Table of Kinetic Data, November 2003 (http://www.iupac-kinetic.ch.cam.ac.uk/).