諸 言 近年の分子遺伝学の急速な発展は,さまざまな先天 性心疾患の成因を分子レベルで解明しつつある.また, 不整脈や動脈硬化などこれまで後天的な疾患と考えら れてきたもののなかにも,遺伝子の関与が存在するこ とが明らかになった.疾患遺伝子や SNPS,エピジェネ ティクスの研究が進歩するなか,今後は先天性心疾患 の分野でも,疾患遺伝子の同定や分子細胞学的病態の 解明が加速度的に進むであろう.本稿では,先天性心 疾患と遺伝子異常と題して,遺伝要因が考えられる先 天性心疾患について概説し,診療上の留意点などにつ いて述べる. 先天性心疾患と遺伝要因について 日本小児循環器学会疫学委員会の調査(Table 1)にお ける松岡らの報告1, 2)によれば,先天性心疾患において, 染色体異常(古典的染色体異常および微細欠失症候群) が 8%,単一遺伝子病など遺伝要因によるものが 5%, 催奇形因子や環境要因によると考えられるものが 1% 以下で,残る 85%は多因子遺伝である.すなわち,遺
先天性心疾患と遺伝子異常
Keywords:congenital heart disease, genetics, chromosome, environment factor
稲井 慶
東京女子医科大学循環器小児科
Genetic Abnormality and Congenital Heart Disease
Kei Inai
Department of Pediatric Cardiology, Heart Institute of Japan, Tokyo Women's Medical University, Tokyo, Japan
Congenital heart disease is the most common type of birth defect. It is well known that congenital heart disease can occur in the setting of multiple birth defects as part of various chromosomal and/or genetic disorders. Even in isolated cardiac defects without chromosomal or genetic abnormalities, familial cases have also been described. Therefore, pediatric cardiologists should have thorough knowledge of these disorders and should be required to have some familiarity with the genetic backgrounds to congenital heart disease.
I present, in this review, the clinical features and genetic backgrounds of various chromosomal and gene abnormalities with congenital heart disease. And also, I would like to highlight interaction of certain genetic and environmental factors known to be involved with some congenital heart disease. We should fully recognize that the interaction between genetic backgrounds and environmental factors makes clinical features of the congenital heart disease in both a syndromic disorder and isolated defects with no syndromic association. This recognition is very important in both the clinical settings and basic research.
要 旨 多くの染色体異常や遺伝子異常において,先天性心疾患がしばしば合併することはよく知られている.明らかな 遺伝子異常がつきとめられてはいなくても,遺伝的背景が濃厚な心疾患に遭遇することも稀ではなく,これらの疾 患に対する知識は小児循環器科医にとって,非常に重要である.また,診療にあたっては十分な遺伝学的知識を備え ておかなければならないことはいうまでもない. 本稿では,先天性心疾患と遺伝子異常と題して,遺伝的要因を持つ先天性心疾患の臨床的特徴と遺伝学的背景や 診療上の留意点などを示した.ただし,先天性心疾患においては,遺伝的要因と環境要因が相互に作用しあって疾患 が出現し,表現型が形作られる.胎内および出生後の環境要因によって疾患関与遺伝子の表現型に与える影響が多 種多様に変化しているともいえるため,診療にあたっては,両方の要因をバランスよく考えていくことが臨床上も 基礎研究上も大切である.
【教育講演】遺伝子異常
伝的要因と環境要因が相互に作用しあって疾患が出現 し,表現型が形成される.胎内および出生後の環境要 因によって疾患関与遺伝子の表現型に与える影響が多 種多様に変化していると考えられる.遺伝的背景また は遺伝子異常が考えられる先天性心疾患の診療にあた る場合,このようなことを十分認識したうえで,診療 を始めるべきである.発端者の家系内の心疾患発生状 況を詳細に問診して,一般的な素因の家系か家族性が 濃厚な家系かを明らかにする必要がある.詳細な家系 図を書くことは遺伝性疾患の診断の第一歩である.参 考までに Fig. 1 に家系図に用いる主な記号を示した. 第一子が先天性心疾患の場合,次子の経験的再発率 は 2 ~ 5%である.同胞内に二人の先天性心疾患患者 がいる場合再発率は 7 ~ 10%となり,三人以上だと 50%を超える.また,母親からの再発率が高く,母親が 先天性心疾患の場合,経験的再発率は 2 ~ 18%で,父 親の場合と比べて 2 ~ 5 倍高い.この事実は胎内にお ける環境因子の重要性を示唆している可能性がある. これらの家族性が明らかでなければ心疾患の発生率は 一般集団と変わらないと考えてよい. 染色体異常や単一遺伝子疾患であれば,再発率は 50%となる.ただし,このようなケースであっても,胎 内環境などの要因が影響を与えるため,先天性心疾患 の発症や表現型が必ずしも発端者と同じにはならな い.また,同一遺伝子異常であっても疾患そのものが 全く違って発症する可能性もあり,遺伝子異常が多様 Fig. 1 Human pedigree nomenclature.
Male, Female, normal
Dizygotic twins Monozygotic twin Spontaneous abortion, abortion Pregnancy, gender-undetermined Proband Male, Female, patient
Death, stillborn
Marriage or marital relation Divorce
Consanguineous marriage
P SB
Japanese Society of Pediatric Cardiology and Cardiac Surgery
Committee of Epidemiology1)
1990.4 〜 1999.6 (2,654 pts)
Department of Pediatric Cardiology Tokyo Women’s Medical University
In-patients2)
1976(422pts) Congenital heart disease patients(No.) % % ・monogenic disease ・chromosomal abnormalities ・teratogen ・multiple factors inheritance 124 217 13 2,300 4.7 8.2 0.5 86.7 87.2 1.4 3.1 95.5 1)Matsuoka R, et al:JSPCCS 2003;19:606-621 2)Andoh M:Perinatal medicine 1978;8:991-999
で全身的な影響を持つものであるという認識が重要で ある3). したがって,診療にあたっては,発端者の妊娠時に 何らかの環境要因の関与を疑わせるものがなかったか どうかの聴取も忘れてはならない.催奇形因子として, 薬物では炭酸リチウム,アンフェタミン,ヒダントイ ンがよく知られている.胎児性アルコール症候群も先 天性心疾患を合併する.感染症では風疹が最も有名で あり,ワクチン後進国であるわが国では未だに重要で ある.母体の全身疾患としては SLE で房室ブロックが, 糖尿病で心筋症や完全大血管転位症の発生が有名であ る4).また,嗜好品の聴取も行うべきである.日本小児 循環器学会疫学委員会報告によれば,母親の妊娠中の 喫煙率が,PDA(16%),TOF(12%),ASD(12%),VSD (12%)と一般の喫煙率(8%)と比較して高率であった.
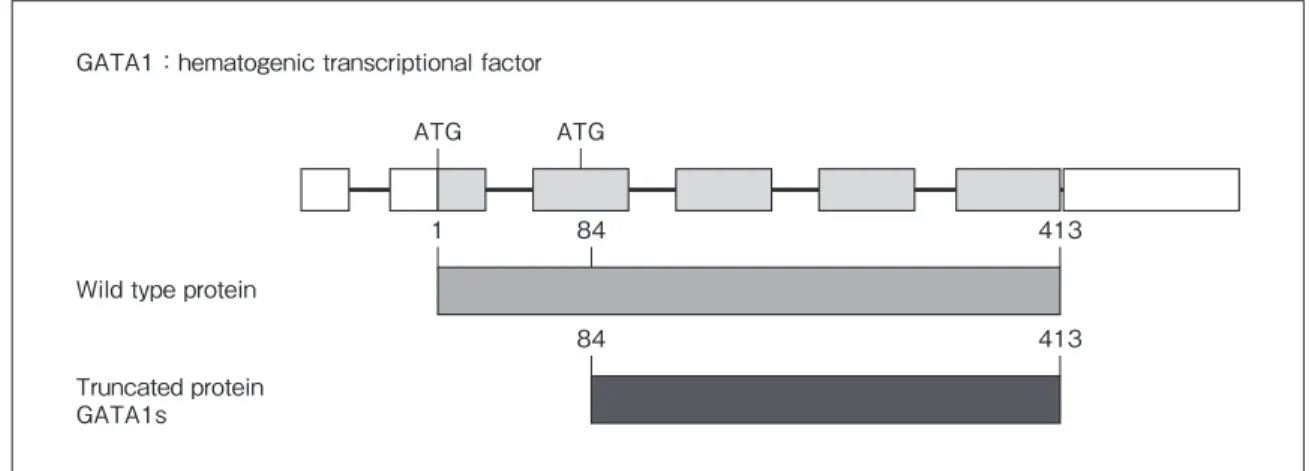
妊 娠 中 の 飲 酒 頻 度 も TGA(28 %),ASD(24 %),TOF (23%)において高率であった.これらの嗜好品は児の 胎内発育に影響を与え,心疾患の発生に促進的に働い ている可能性がある.このような環境因子に関する問 診は両親(特に母親)に罪悪感を与えることのないよう に十分な配慮をしつつ行わなければならない.また, それは次子の再発についての相談を受けた時に重要な 情報となる5). 次に,遺伝要因の明らかな先天性心疾患・症候群の 主だったものについておのおの具体的に述べる. 古典的染色体異常症候群 1.Down 症候群 21 番染色体の trisomy によって起こり,出生 1,000 人 に 1 人認める.その主な責任領域は 21q22.2-3 と考えら れている.筋緊張低下,特異的顔貌(内眼角贅皮,鼻根 部平定,巨舌など),知能発達障害などを伴う.約半数 で心疾患を合併する.房室中隔欠損,心室中隔欠損な ど肺高血流性心疾患が多い.肺高血圧症が発生しやす く,進行も早いため,肺高血圧を伴う左右シャント疾 患の場合,乳児期早期(4 ヵ月以内)の心内修復術が必 要になる.乳児期から甲状腺機能異常の合併が高頻度 であることと,白血病の罹患率が高いことに留意して おく必要がある.Down 症候群の新生児の 10%に一過 性骨髄異形成症が生じ,そのうち 20 ~ 30%で急性骨 髄性白血病を発症するとされる.その成因はいまだ不 明だが,一過性骨髄異形成症を発症する患者の多くに GATA1 遺伝子の N 末端の変異が認められる(Fig. 2). 長じては肥満,高尿酸血症,糖尿病などの問題を生じ やすい.再発率は一般に 1 ~ 2%だが,出産時の母の年 齢によって違ってくる.Robertson 転座では 5 ~ 15%と 上昇するので注意が必要である. 2.18trisomy 18 番染色体の trisomy によって起こり,出生 4,000 ~ 6,000 に 1 人の頻度である.責任領域は 18 番染色体長 腕 8q11.2 付近であり,5%がモザイクである.子宮内発 育不全があり,特異的顔貌(後頭部の突出した逆三角形 の顔),短い胸骨,揺り椅子様足底,関節拘縮などを主 徴とする.ほとんどの患者で心疾患を合併する.最も 特徴的なのは房室弁や半月弁の肥厚や結節などの polyvalvular disease である.心室中隔欠損,心房中隔欠 損,動脈管開存の合併率も高く,多くは肺高血流型心 疾患である.10%には大血管転位,両大血管右室起始 や左心低形成などチアノーゼ性複合心疾患が生じる. 生命予後は極めて不良で,侵襲的検査や外科治療には 倫理的な議論があり,施設間で対応の違いがある.再 発率は 1%程度と考えられる.
Fig. 2 GATA1 gene mutation of Down syndrome. GATA1:hematogenic transcriptional factor
Wild type protein
Truncated protein GATA1s ATG 1 84 413 84 413 ATG
Ⅹ染色体の欠失に起因し,低身長,性腺機能不全を 主徴とする症候群である.1768 年に Giovanni Morgagni によって初めて記載され,1930 年代に Henry Turner に よって疾患単位として確立された.染色体異常のなか では最もよくみられる症候群のひとつで,2,500 出生に 1 人とされるが,多くは胎児期に死亡し,自然流産の 7 ~ 10%を占めると考えられている6). (1)心血管病変 23 ~ 40%に心疾患を合併する.最も多い疾患は大動 脈二尖弁であり,石灰化が進んで大動脈弁狭窄や閉鎖 不全が発症することがある.患者の 10%に大動脈縮窄 症を合併し,高血圧の主因となる.特に翼状頸を伴う 患者に多いとされる.その他では部分肺静脈環流異常 や僧帽弁逸脱がある.成人すると大動脈拡大が出現す ることがあり(8 ~ 42%)大動脈解離の発症もみられ, 突然死の原因になるため大動脈径の経過観察が必要で ある.高血圧の発症リスクは健常女性の 3 倍とされ, 虚血性心疾患の発生リスクも高い.約半数の患者が耐 糖能異常を持っており,脂肪代謝異常も高率であるた め,成人後も心疾患の定期フォローは患者の予後に とって極めて重要である. (2)その他の臨床像 低身長と性腺機能不全はほぼ全ての患者にみられ る.知能は一般に正常であるが,運動能力や認知能力 に障害を持つことがある.小顎,毛髪線低位,短頸,高 口蓋もしばしばみられる.翼状頸,四肢のリンパ浮腫, 爪の異常,脊椎側彎も特徴的である.自然に二次性徴 を来して月経のみられる患者は 12 ~ 16%であり,多 くの患者は女性ホルモンの補充が必要である.甲状腺 疾患の合併率が高く,特に橋本病を 20 ~ 30%に合併 する.当初は臨床症状にも乏しいので,思春期以後は 定期的な甲状腺機能や抗甲状腺抗体の検査は欠かせな い.その他,腎奇形や難聴の合併頻度が高いことも知 られている.Turner 症候群の女性はこれまで述べてき た低身長,性成熟の遅延や認知機能の障害などからう まく社会で人間関係を築きにくいことがあり,時に心 理社会的な問題を抱えることがあるため,適切なカウ ンセリングが必要となることもある. (3)遺伝的背景 自然妊娠率は低い.最近の生殖補助医療の発展に よって,妊娠も可能となり,他の原因による不妊女性 と同等の体外受精による妊娠率となっているが,流産, 死産,染色体異常症の発症率は高い. 染色体微細欠失症候群 1.22q11.2 欠失症候群 染色体 22q11.2 の部分欠失により引き起こされる疾 患である.DiGeorge 症候群,円錐動脈幹異常顔貌症候 群 conotruncal anmaly face syndrome(CAFS),軟 口 蓋 心 臓顔貌症候群 velo-cardio-facial syndrome(VCFS)の 3 症 候群を含包する(ただし,DiGeorge 症候群には 22q11.2 の部分欠失を伴わない場合もある).本症候群は 1976 年に東京女子医科大学の高尾らが CAFS を提唱し,臨 床診断を確立したことに始まる.出生 3,000 ~ 4,000 人 に 1 人とされ,先天性心疾患の 1 ~ 2%を占める7). (1)心血管病変 80%以上に何らかの心血管病変を合併する(心疾患 を持たない患者も存在する).最も多い合併心疾患は Fallot 四徴症であり,特に肺動脈閉鎖を伴う Fallot 四徴 症の約半数は本症候群である.これに心室中隔欠損(特 に膜様部欠損)が続く.次に本症候群と極めて関連が強 いのは,大動脈離断症で,特に typeB をみた場合,本症 候群を強く疑うべきである.また高位大動脈弓,内頸 動脈や鎖骨下動脈の起始異常,左上大静脈遺残など血 管の異常も多く,約 70%に合併する.稀にはこのよう な血管異常によって血管輪を形成することにもあるた め注意が必要である. (2)顔貌の特徴 CAFS にみられる顔貌の特徴とは,眼間開離,眼裂狭 小,鼻翼と鼻屋の接合異常,小顎,小さい口,鼻根部扁 平,扁平な頬,耳介下方付着,口蓋裂などである.特に 鼻翼と鼻屋の接合異常は最も高頻度にみられる特徴で ある.口蓋裂や鼻咽喉不全による鼻声や構音障害もし ばしば聞かれる. (3)成長と発育 軽度から中等度の知能発達障害を伴うことが多く, 学習障害もしばしばみられる.性格は一般に明朗で, 対人関係はむしろよいことが多い.乳幼児期は発達の 遅れは目立たないこともあるが,小学生になると算数 などで学習についていけなくなることが多く,小学校 高学年になると特別支援学級に転入する患者が増え る.なかには大学まで進学できるケースもあり,就業 や結婚を経て通常の家庭生活を営める場合もある(こ のような場合,心疾患がなく,顔貌や声が典型的でな ければ成人まで診断されていない患者も存在する).思 春期以降,統合失調症や双極性障害などの精神疾患を 発症する確率が一般的頻度より高いことが知られてい る.特に統合失調症は 10 ~ 20%の患者で発症すると 報告されており,一般人口における罹患率と比較して
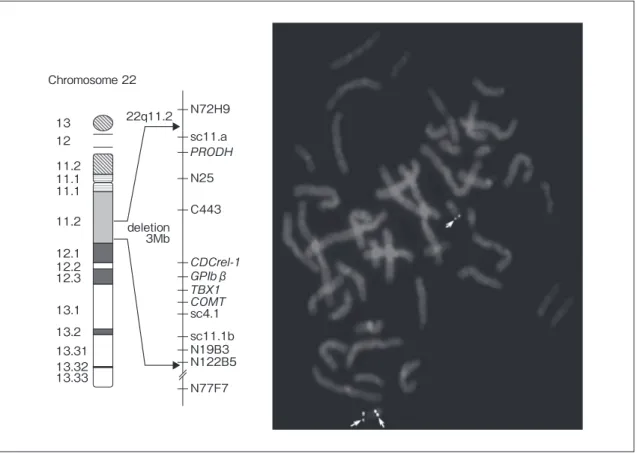
際立って高い.統合失調症患者の 1 ~ 6%で 22 番染色 体の部分欠失を認める. (4)その他の臨床的特徴 胸腺の低形成または無形成による易感染性がみられ ることがあるが,重篤な免疫不全はみられない.副甲 状腺の低形成または無形成に伴う低 Ca 血症も頻度の 高い合併症である.新生児期や乳児期早期にテタニー を伴う低 Ca 血症で,Ca の補充やビタミン D の投与が 必要となることが多い.多くは成長とともに改善する が,思春期以降さまざまなストレス等をきっかけに再 発することがあり,定期フォローが必要である. (5)遺伝子異常と遺伝様式 1981 年に DGS 患者の染色体 22 番の長腕 q11 の転座 による部分欠失が報告されたのが最初である.その後 1992 年に DGS のみならず,VCFS で,さらに 1995 年 に CAFS においても同様の欠失が報告された.欠失領 域は 1.5 ~ 3.0Mb で,ここに約 30 個の遺伝子が確認さ れている.欠失領域の両端には DNA 反復配列があり, 組み換え時のミスマッチが起こった結果と考えられる (Fig. 3). 遺伝様式のほとんどは孤発例で,松岡らの報告によ ると,CAFS180 例中家族性を認めるのは 13%のみで あった.次世代での発生率は 50%で,診断の段階から 遺伝相談が必須である.家族性 CAFS では,世代を経 るごとに表現型が重篤になる傾向がある.また,母子 例のほうが多いことも知られている.このことは,本 症候群自体には性差はないため,男性患者が生殖適応 度が低いか社会的要因で家族を持ちにくい可能性を示 唆するものである. 2.Williams 症候群 1961 年に Williams らによって報告された.染色体 7q11.23 領域の半接合体欠失があり(Fig. 4),小妖精様 顔貌と大動脈弁上狭窄や末梢性肺動脈狭窄などの心疾 患を主徴とする症候群である.出生 20,000 ~ 30,000 人 に 1 人でみられる8). (1)心血管病変 80%に何らかの心疾患が認められる.最も特徴的な のは大動脈弁上狭窄や末梢性肺動脈狭窄であり,これ に続いて肺動脈弁狭窄,心室中隔欠損,僧帽弁逸脱な どがある.肺動脈狭窄の多くは自然軽快するが,大動 脈弁上狭窄は軽症例から重症例までさまざまであるも のの,進行性であることが多い.年長患者の 30%に高 血圧を認める.
Fig. 3 Typical deletion of del22q11.2 syndrome. 22q11.2 deletion 3Mb 13 Chromosome 22 12 11.2 11.1 11.1 11.2 12.1 12.2 12.3 13.1 13.2 13.31 13.32 13.33 N72H9 sc11.a N25 C443 β sc4.1 sc11.1b N19B3 N122B5 N77F7
(2)その他の臨床像 小妖精様顔貌は厚い唇,長い仁中,大きな口,鼻根部 平低,腫れぼったい上眼瞼,小顎などの特徴をもつ.歯 の異常を認めることが多い.性格は一般に陽気で社交 的かつ多弁である.大部分の患者で中等度の知的発達 障害を伴う.また,視空間認知障害を伴いやすく,全体 的な空間把握能力が低い.その反面,優れた記憶力や 豊かな音楽感受性を示す患者もいる.音に対して敏感 なことがあり,突発的な騒音に神経質になる患者もい る.心疾患を伴わない場合,診断されないまま,注意欠 陥障害や学習障害としてフォローされていることもあ る.排尿障害,脱肛,関節の異常(浅臼蓋など)の合併も 多い.不安やうつなど精神疾患も時にみられる. (3)遺伝子異常と遺伝学的問題 染色体 7q11.23 の欠失領域には約 20 個の遺伝子が確 認されている.大動脈弁上狭窄に関連する遺伝子はエ ラスチン遺伝子 ELN である.LIMK1 遺伝子と視空間認 知障害との関連を示唆する報告がある.その他,本症 候群の多様な表現形に対する責任遺伝子が同定されつ つある. 大部分の患者は孤発例である.次世代の発生率は CAFS 同様 50%であるが,一般に CAFS よりも知的発 達障害が重いことが多いので,家族をなすのは難しい 場合が多い. 単一遺伝子異常 1.Marfan 症候群 Marfan 症 候 群 は,1896 年 に フ ラ ン ス の 小 児 科 医 Marfan によって報告された結合織疾患である.高身長, 長い四肢,脊椎彎曲,漏斗胸,蜘蛛指,関節過伸展など の骨格異常に水晶体脱臼,近視などの眼症状と心血管 疾患を伴う.5,000 人に 1 人の頻度で,性差はない.常 染色体優性遺伝を示し,15 番染色体上(15Q21.1)にある fibrillin 遺伝子(FBN1)が原因遺伝子である.Fibrillin は 分子量 350Kd の大きな糖蛋白で,細胞外間質に存在す る microfibril や弾性繊維の成分であり,全身結合織に 広く存在している.ただし遺伝子診断はまだ一般化に 広く行われてはいないため,診断は 2010 年に改訂され た国際的診断基準(Ghent nosology)によって臨床的に行 われることが多い9). (1)心血管病変 Valsalva 洞,大動脈弁輪,上行大動脈の拡大(annulo-aortic ectasia)と大動脈閉鎖不全が最も特徴的である. 僧帽弁逸脱と僧帽弁閉鎖不全,肺動脈拡大と肺動脈閉 鎖不全もみられる.大動脈の拡大に伴う解離や破裂の 予防と管理が極めて重要である.一般に小児期には僧 帽弁閉鎖不全のみがみられる場合が多いが,思春期以 降大動脈弁輪拡張と大動脈閉鎖不全が進行する. (2)その他の臨床症状 ほとんどの患者は高身長で,側彎などの脊椎変形や 鳩胸,漏斗胸などの胸郭,胸骨の異常を伴っている.妊 娠や体重増加では説明できない皮膚線条をしばしば認 める.近視の合併率は高くしばしば小児期から進行す る.65 ~ 75%で水晶体脱臼を発症し,眼内レンズ逢着 術が必要になる.高口蓋,歯牙の形成異常や難聴もみ られる.腰椎,仙骨部など下部脊椎の硬膜拡張が CT や MRI でみられることがあるが,多くは無症状である. (3)遺伝的問題など 臨床的な類縁疾患が多数みられることに留意する必 要がある.Fibrillin1 遺伝子の変異は Sprintzen-Goldberg 症候群(Marfan 様体型に精神運動発達遅滞,頭蓋骨癒 合や腹部の筋弛緩を伴う症候群)や家族性大動脈瘤, Congenital contractual arachnodactyly などでも存在し,臨 床的に Marfan 症候群の診断基準を満たさなくても水 晶体脱臼や僧帽弁逸脱のある患者でみられることがあ る.また,TGFBR1 および TGFBR2 遺伝子変異を持つ Loeys-Dietz 症候群は Marfan 症候群と極めて類似した 症候を持っているが,頸部動脈の tortuosity,眼間解離, 口蓋垂裂を伴うのが特徴で,上行大動脈以外のみ拡大 や瘤を認める.いずれも Marfan 症候群の診断でなくと Fig. 4 Typical deletion of Williams syndrome.
β Chromosome 7
も同様の心血管病変を発生する可能性があると考えて 慎重なフォローアップをするべきである. 前述のように,母親が本症候群の場合,次世代への 影響は 50%であるが,流産率は一般女性とかわらない. ただし,心血管病変を持つことから妊娠中や分娩時に は産科と循環器の医師の連携による十分なケアが必要 である.両親ともに本症候群の場合は次世代の発生率 は 75%になる. 2.Noonan 症候群 1963 年 Noonan らによってはじめて報告された.特 異的顔貌に低身長,外反肘,リンパ浮腫,心血管疾患を 伴う.以前は male Turner とも呼ばれ,女児の場合は Turner 症候群との鑑別が必要となる.これまで複数の 原因遺伝子が同定されている.チロシンリン酸化酵素 SHP-2 をコードする,12 番染色体 q24.1 上の PTPN11 が最初に同定され,本症候群の約半数にその変異が認 められる.12p12.1 上の KRAS 遺伝子,SOS 遺伝子,RAF1 遺伝子の変異も報告されている.出生 1,000 ~ 2,500 に 1 人の発生率で,性差はみられない10). (1)心血管病変 約 80%に何らかの心血管疾患を合併する.肺動脈狭 窄が最も多く,狭小弁輪や弁の異形成が原因である. 分枝部狭窄を伴うことも多い.その他,心房中隔欠損, 不完全房室中隔欠損,大動脈縮窄,Fallot 四徴症などが みられる.肥大型心筋症を伴う患者が 20 ~ 30%にみ られ,RAF1 遺伝子の関与が示唆されている. (2)その他の臨床像 眼間隔離,内眼角贅皮,眼瞼裂斜下に低位耳介を伴 う特徴的顔貌と翼状頸や後髪線低位がみられる.ただ し長じるにつれてこれらの特徴ははっきりしなくな り,顔貌だけでは判断がつかなくなることがある.身 長は低く,約半数は 3 パーセンタイル以下で,成長ホ ルモン治療を受ける患者もいるが,一方で正常身長の 患者も存在する.乳頭間離解や停留睾丸もしばしばみ られる.乳児期には哺乳障害が強く,体重増加は不良 だが,幼児期にはこのような摂食障害は自然軽快する. 精神運動発達遅滞や学習障害は 25 ~ 30%にみられ, 知能発達が正常な患者も少なくない.白血病,骨髄異 形成や凝固障害などの血液疾患の発症率も高い.生命 予後は心疾患や血液疾患の重症度によるが一般に良好 である. (3)遺伝的問題など 常染色体優性遺伝形式をとり,次世代の発生率は 50%である.男性では精子形成不全がみられることが 多いが,女性の場合妊孕性は正常である.現在,遺伝子 変異と表現型についての検討が進んでいるが,心疾患 や血液疾患の重症度の推定はいまだ困難である. 3.Holt-Oram 症候群 1960 年に Holt と Oram が記載した心房中隔欠損に上 肢(特に檮骨)の骨形成異常を伴う症候群である.出生 10万人に1人の頻度で,心臓⊖上肢症候群と同義である. 心血管病変としては,心房中隔欠損(二次孔欠損)と房 室ブロックが最も特徴的である.他には心室中隔欠損, Fallot 四徴症や房室中隔欠損を伴う場合もある.上肢の 異常としては,檮骨欠損や低形成,母指欠損,合指,ア ザラシ肢などがみられ,左側に多く発症する.同様の 上肢骨の異常は VACTERL 連合でもみられるが,合併 する心疾患に違いがあり,椎骨の異常,消化管奇形や 腎奇形などによって鑑別される.1994 年に連鎖解析に よって,責任遺伝子座が 12q21.3 ~ q22 に特定され,後 に TBX5 が責任遺伝子として同定された.TBX5 および その周辺の転写因子活性に応じて,さまざまな表現型 を示すものと考えられている. 4.心房内臓錯位症候群 心房内臓錯位症候群は無脾症,多脾症を含む幅広い スペクトラムを包括する疾患概念であり,胸腹部内臓 の位置異常や形成不全と複合型心血管奇形を伴う.無 脾症は一般に右側相同を原則とし,共通心房,共通房 室弁口,総肺動脈還流異常,単心室などの重篤なチア ノーゼ性心疾患を認めることが多い.多脾症は左側相 同を原則とし,下大静脈欠損,両側上大静脈や心房中 隔の位置異常による部分肺静脈還流異常を特徴とする 心奇形を認める.総じて無脾症のほうが心疾患として 重篤なことが多い.これまで家族性の内臓錯位におい て 3 つの遺伝子変異が同定されている.Xp26.2 の ZIC3, 3p22 ~ p21.3 の ACVR2B,1q42.1 の LEFTY A であるが, いずれも検出頻度は低く,多くの患者において責任遺 伝子は不明である. 結 語 先天性心疾患と遺伝子異常と題して,遺伝要因を伴 う先天性心疾患について概説した.先天性心疾患では 大部分が多因子遺伝であるため,成因が明らかでない ものが多く,表現型も極めて多彩であるため,染色体 異常や責任遺伝子の同定のみならず,環境因子の研究 や遺伝因子と環境因子がどのように作用し合っている のかについての知見も重要である.そのためには,分 子生物学のみならず,疫学や公衆衛生学からのアプ
師による詳細な臨床情報や知見の集積がなくてはなら ず,小児循環器科医にとって,遺伝や疫学に対する十 分な知識は必須のものであるといえよう. 【 参 考 文 献 】 1) 日本小児循環器学会疫学委員会:松岡瑠美子,森 克 彦,安藤正彦:先天性心血管疾患の疫学調査- 1990年 4 月~1999年 7月.2,645家系の報告-.日小児循環器会誌 2003;19:606-621 2) 日本小児循環器学会心血管疾患の遺伝子疫学委員会: 松岡瑠美子,南沢 亨,秋元 馨,ほか:家族内発症心血 管疾患(心室中隔欠損,心房中隔欠損,ファロー四徴,動 脈管開存)に関する疫学調査報告- 1999年 8月~2002年 7月-.日小児循環器会誌 2003;19:622-628
3) Pierpoint ME, Basson CT, Benson DW Jr, et al:Genetic basis for congenital heart defects:current knoeledge.:a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young endorsed by the
3015-3038
4) Jenkins KJ, Correa A, Feinstein JA, et al:Noninherited risk factors and congenital cardiovascular defects:Current knowledge.:a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young:endorsed by the American Academy of Pediatrics. Circulation 2007;115:2995-3014
5) Nora JJ:From generational studies to a multilevel genetic-environmental interaction. J Am Coll Cardiol 1994;23: 1468-1471
6) Elsheikh M, Dunger DB, Conway GS, et al:Turnerʼs syndrome in adulthood. Endocrine Rev 2002;23:120-140 7) Yagi H, Furutani Y, Hmada H, et al:Role of TBX1 in human
del22q11.2 syndrome. Lancet 2003;362:1366-1373 8) Committee on genetics American Academy of Pediatrics
AP:Health care supervision for children with Williams syndrome. Pediatrics 2001;107:1192-1204
9) Bolar N, Van Laer L, Loeys BL:Marfan syndrome:from gene to therapy. Curr Opin Pediatr 2012;24:498-504 10) Roberts AE, Allanson JE, Tartaglia M, et al:Noonan