2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 1
目次
1. 医薬品への曝露 ...4 1.1 総括的安全性評価計画及び安全性試験の記述... 4 1.1.1 デノスマブの臨床開発計画の概要 ... 4 1.1.1.1 骨巨細胞腫を対象とした臨床開発... 4 1.1.1.2 骨巨細胞腫以外の疾患を対象とした臨床開発... 6 1.1.2 安全性評価計画の概要... 7 1.1.3 試験結果の要約... 10 1.2 全般的な曝露状況 ... 10 1.3 治験対象集団の人口統計学的特性及びその他の特性... 12 1.3.1 被験者の内訳... 12 1.3.2 人口統計学的及び基準値の特性... 14 2. 有害事象 ...17 2.1 有害事象の解析 ... 17 2.1.1 比較的よく見られる有害事象... 18 2.1.2 死亡... 23 2.1.3 その他の重篤な有害事象... 23 2.1.4 その他の重要な有害事象... 27 2.1.4.1 治験薬の投与中止又は試験中止に至った有害事象... 27 2.1.4.2 重要な有害事象 ... 28 2.1.5 器官別又は症候群別有害事象の解析 ... 37 2.1.6 長期曝露の安全性... 38 2.1.6.1 骨巨細胞腫を対象とした臨床試験... 38 2.1.6.2 120 日安全性アップデート ... 39 2.2 個別有害事象の文章による説明 ... 39 3. 臨床検査値の評価...40 3.1 アルブミン補正血清カルシウム値 ... 40 3.2 リン ... 42 3.3 抗デノスマブ抗体形成 ... 44 3.4 その他の全臨床検査パラメータ ... 44 4. バイタルサイン、身体的所見及び安全性に関連する他の観察項目...44 4.1 バイタルサイン ... 44 4.2 心電図評価 ... 45 5. 特別な患者集団及び状況下における安全性 ...46 5.1 内因性要因 ... 46 5.1.1 部分集団解析の安全性の結果... 462.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 5.1.2 デノスマブの用量選択の評価... 47 5.1.3 その他の疾患でのデノスマブの安全性 ... 47 5.1.3.1 骨粗鬆症患者 ... 47 5.1.3.2 がん患者 ... 48 5.1.4 腎機能障害患者での安全性解析... 51 5.2 外因性要因 ... 51 5.3 薬物相互作用 ... 51 5.4 妊娠及び授乳時の使用 ... 51 5.5 過量投与 ... 53 5.6 薬物乱用 ... 53 5.7 離脱症状及び反跳現象 ... 53 5.8 自動車運転及び機械操作に対する影響又は精神機能の障害... 54 6. 市販後データ...55 7. 付録 ...57
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
3 略語一覧
略語 略していない表現(英) 略していない表現(日)
CDS core data sheet 企業中核データシート
CSR clinical study report 治験総括報告書
CTCAE Common Terminology Criteria for
Adverse Effects 有害事象共通用語規準
CYP cytochrome p450 enzyme チトクローム P450
GCTB giant cell tumor of bone 骨巨細胞腫
ICH International Conference on
Harmonisation 日米 EU 医薬品規制調和国際会議
MedDRA Medical Dictionary for Regulatory Activities ICH 国際医薬用語集
NCI National Cancer Institute 米国国立がん研究所
ONJ osteonecrosis of the jaw 顎骨壊死
OPG osteoprotegerin オステオプロテゲリン
PSUR periodic safety update report 定期的安全性最新報告
Q6M every 6 months 6 ヵ月に 1 回
Q4W every 4 weeks 4 週に 1 回
RANKL RANK ligand RANK リガンド
SMQ standard MedDRA query MedDRA 標準検索式
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
1.
医薬品への曝露
骨巨細胞腫(giant cell tumor of bone: GCTB)は、増殖が速いまれな原発性骨腫瘍である。男 性に比べ女性に好発し、通常、骨格が成熟した 20~50 歳の成人に発症する(Zheng et al, 2001)。 米国、欧州連合(EU)、カナダ及びオーストラリアで新たに診断される症例数は、それぞれ毎 年約 800 名、800 名、80 名、及び 30 名である。国内における発症報告数は、1972 年~2003 年で 2126 名(電子化集計前)、2006 年~2008 年で 415 名(電子化集計)であり、1972 年~2008 年(2004 年及び 2005 年を除く)で合計 2541 名が報告されている。2008 年の報告は単年で 142 名であった(全国骨腫瘍登録一覧表, 2008)。 GCTB の臨床像は一般に偏心性の溶骨性病変であり、通常は長骨骨端に認められ、病変の約 50%は膝部位に発症する(Szendroi et al, 2003、Szendroi, 2004)。根治療法は外科的手術である が、約 10~75%の患者が切除後に再発を来す(Malawer et al, 2008)。GCTB の治療薬として現 在承認されている医薬品はない。ビスフォスフォネートや放射線療法などが使用されているが、 安定した治療効果は報告されていない。再発病変の治療では、根治的切除が繰り返されること が多い(Szendroi, 2004)。切断又は罹患関節の切除を伴う手術の施行により、生活の質の低下、 持続的な骨喪失、及び二次性関節炎に至ることがある。無治療のまま放置した場合や十分な治 療成績が得られなかった場合は、GCTB が進行して骨喪失及び骨痛を来し、時には死に至る。
GCTB は RANK を発現しており、その増殖は RANK リガンド(RANK ligand: RANKL)に 依存している(Szendroi et al, 2003)。デノスマブは、RANKL に結合してその活性を中和する ヒト型モノクローナル IgG2 抗体であり、RANK の活性化を阻害し、破骨細胞の形成、機能、 及び生存を抑制する。GCTB 患者では、デノスマブによる RANKL の阻害により、破骨細胞・ 腫瘍関連巨細胞が消失し、骨吸収及び病勢の進行が抑制される(試験 20040215、20062004、 及び AMG162-B-J201)。
1.1
総括的安全性評価計画及び安全性試験の記述
1.1.1
デノスマブの臨床開発計画の概要
1.1.1.1 骨巨細胞腫を対象とした臨床開発 アムジェン社は、GCTB 被験者 304 名(合計)を対象とした 2 つの国際共同第 II 相試験を実 施した。このうち試験 20040215 には、被験者 37 名が組み入れられた。進行中の試験 20062004 では、データカットオフ日(2011 年 3 月 25 日)の時点で被験者 281 名(14 名は試験 20040215 から移行)の安全性データが得られている。さらに、本邦では第一三共株式会社が試験 AMG162-B-J201 を実施しており、12 ヵ月カットオフ解析のデータカットオフ日(20 年 月 日)時点で被験者 17 名の安全性データが得られている。2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 5 ブの安全性を評価することを目的とした。本試験では、デノスマブ 120 mg の 4 週間に 1 回 (Q4W)皮下投与に、第 8 日及び第 15 日の 120 mg 負荷投与を加えた投与スケジュールとし た。デノスマブ投与は、腫瘍の完全切除、病勢の進行、治験責任医師又は治験依頼者による中 止の判断、被験者による中止の申し出、ビスフォスフォネート製剤、カルシトニンもしくはイ ンターフェロン α-2a 投与、又は試験 20062004 への移行のいずれかに至るまで継続した。ただ し、デノスマブ投与が有効であった被験者には、再発した場合などにデノスマブの再投与を可 能とした。デノスマブの最終投与後の安全性データは、6 ヵ月ごとに最長 2 年間収集した。 本モジュールでは、試験 20040215 に組み入れられた被験者 37 名の安全性データを示す。試 験 20040215 の主要解析(最初の被験者の組み入れ日[2006 年 7 月 日]から主要解析のデ ータカットオフ日[20 年 月 日]まで)の結果は主要解析の治験総括報告書にまとめ、 最終解析(主要解析データカットオフ日以降の最長 2 年間の安全性追跡調査データを含む)の 結果は最終解析の治験総括報告書にまとめた。両治験総括報告書は本承認申請資料に含めた。 試験20062004(外国第 II 相臨床試験) 試験 20062004 は、成人及び骨格が成熟した未成年(18 歳未満)の GCTB 患者を対象とした 進行中の非盲検、単一群、第 II 相試験であり、デノスマブの安全性、並びに治験責任医師が 判定した抗腫瘍効果を評価するためにデザインされた。試験 20040215 と同様に、デノスマブ 120 mg Q4W の皮下投与に、第 8 日及び第 15 日の 120 mg 負荷投与を加えた投与スケジュール とした。本試験には、以下の 3 つのコホートを組み入れた: • コホート 1: 切除不能な GCTB 患者(例: 仙骨又は脊椎の GCTB、あるいは肺転移など の多発性病巣を有する患者) • コホート 2: 切除可能な GCTB を有し、重度の後遺症を残す手術(例: 関節切除、手足 の切断、又は片側骨盤切断)の実施が試験中に予定されている患者 • コホート 3: 試験 20040215 から移行した被験者 コホート 2 では、デノスマブ投与の抗腫瘍効果及び安全性に加え、手術の種類及び実施の有 無を集計し、手術可能な被験者に対するデノスマブの有効性を評価した。 試験 20040215 に参加した 3 名は、治験薬投与中止後 5~20 ヵ月で再発を来したため、その 後、試験 20062004 に組み入れられた。この 3 名は、コホート 3 ではなくコホート 1 に組み入 れられた。 コホート 1 の被験者には、臨床的有益性が認められる限り、デノスマブ 120 mg の Q4W 投 与を継続した。コホート 2 の被験者には、腫瘍の完全切除後 6 回までデノスマブ投与を継続し た。試験 20040215 からコホート 3 に移行し、かつ移行時にデノスマブ投与を受けていた被験 者には、移行時の Q4W スケジュールに従ってデノスマブ 120 mg の皮下投与を継続した。試 験 20040215 の安全性追跡調査から移行した被験者については、試験 20062004 で安全性追跡調 査を継続し、試験 20040215 の試験終了時の来院から最長 2 年間にわたり追跡されている。 本モジュールでは、試験 20062004 に組み入れられた 281 名の安全性データを示す。あらか じめ計画した第 3 回中間解析の結果(カットオフ日[2011 年 3 月 25 日]までのデータを含む)
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg を治験総括報告書にまとめ、本承認申請資料に含めた。 試験AMG162-B-J201 試験 AMG162-B-J201 は、成人及び骨格が成熟した未成年(20 歳未満)の GCTB 患者を対象 とした進行中の非盲検、単一群、第 II 相試験であり、デノスマブの客観的抗腫瘍効果のプロ スペクティブな評価、並びに薬物動態及び安全性の評価を目的とした。試験 20040215 及び 20062004 と同様、デノスマブ 120 mg Q4W の皮下投与に、第 8 日及び第 15 日の 120 mg 負荷 投与を加えた投与スケジュールとした。切除可能な被験者には、腫瘍の完全切除後 6 回までデ ノスマブ投与を継続した。他のすべての被験者では、病勢の進行の確認、治験責任医師又は治 験依頼者による中止の判断、被験者による中止の申し出、治験責任医師による臨床的有益性な しの判断、あるいは併用禁止療法の実施のいずれかに至るまでデノスマブ投与を継続した。た だし、デノスマブ投与が有効であった被験者には、再発した場合などにデノスマブの再投与を 可能とした。 本モジュールでは、試験 AMG162-B-J201 に組み入れられた 17 名の安全性データを示す。あ らかじめ計画した 6 ヵ月カットオフ解析の結果(カットオフ日[20 年 月 日])及び 12 ヵ月カットオフ解析の結果(カットオフ日[20 年 月 日])を治験総括報告書にまとめ、 本承認申請資料に含めた。 1.1.1.2 骨巨細胞腫以外の疾患を対象とした臨床開発
デノスマブ(120 mg Q4W 皮下投与)は XGEVA の販売名で、preventing or reducing the risk of skeletal-related events in patients with bone metastases from solid tumors を効能・効果として、欧米 を含めた世界各国で広く承認されている。日本ではランマークの販売名で、多発性骨髄腫によ る骨病変及び固形癌骨転移による骨病変を効能・効果として承認されている。骨転移を有する 進行がん患者を対象としたデノスマブの承認申請の妥当性は、主として 3 つの国際共同、無作 為化、二重盲検、実薬対照、ピボタル第 III 相臨床試験により示されており、デノスマブ投与 を受けた 2841 名及びゾレドロン酸投与を受けた 2836 名のデータが含まれている(癌骨転移患 者における骨関連事象[skeletal-related event: SRE]の抑制に関する承認申請[ランマーク SRE 申請]資料のモジュール 2.7.4[試験 20050103、20050136、及び 20050244 の延長盲検投与期の 結果を含む])。なお、試験 20050103 及び 20050136 では二重盲検投与期終了時、予定された評 価を継続中のすべての被験者は、2 年間又はデノスマブが上市されるまでのいずれか早い時点 まで、非盲検下でデノスマブ 120 mg Q4W の皮下投与を受けられることとした(非盲検投与期)。 本承認申請には、これらの非盲検投与期の治験総括報告書を参考資料として含めた。
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 7 患者又は多発性骨髄腫患者を対象とした実薬対照第 III 相試験 また、試験 20050147(合計 1432 名)では、骨転移の発現リスクが高い去勢抵抗性前立腺癌 患者を対象に、プラセボを対照としてデノスマブによる無骨転移生存期間の延長効果を検討し た。なお、本承認申請には、試験 20050147(主要盲検投与期及び延長盲検投与期)の治験総 括報告書を参考資料として含めた。
1.1.2
安全性評価計画の概要
本モジュールでは、外国 2 試験 20040215 及び 20062004 の安全性データの併合解析(以下、 外国 2 試験併合解析)と試験 AMG162-B-J201(6 ヵ月カットオフ解析)の安全性データを中心 にまとめ、これらの類似性を検討した。また、試験 AMG162-B-J201 の 12 ヵ月カットオフ解析 の結果及び上記 3 試験を対象とした 3 試験併合解析の結果も提示した。結果は、併合した結果 と試験ごとの結果の両方を示した。 外国 2 試験併合解析における安全性併合解析対象集団には、試験 20040215 の投与期間中、 又は試験 20062004 の第 3 回中間解析のカットオフ日(2011 年 3 月 25 日)までに治験薬投与 を 1 回以上受けた全被験者を含めた。試験 20062004 への参加前に試験 20040215 に参加してい た被験者については、試験 20040215 での投与開始から試験 20062004 における投与終了までの 全データを解析対象とした。いずれの安全性解析でも、試験 20062004 については 3 つのコホ ートのデータを併合した。なお、外国 2 試験併合解析における安全性解析に、安全性追跡調査 データは含めなかった。 試験 AMG162-B-J201 の安全性解析対象集団には、6 ヵ月カットオフ解析のカットオフ日 (20 年 月 日)までに治験薬投与を 1 回以上受けた全被験者を含めた。3 試験併合解析 では、外国 2 試験併合解析及び試験 AMG162-B-J201 の安全性解析対象集団を併合し、安全性 併合解析対象集団とした。また、12 ヵ月カットオフ解析のカットオフ日(20 年 月 日) までに治験薬を 1 回以上受けた全被験者に基づく結果を第 2.1.6.1 項に示す。 本モジュールでは必要に応じ、参考として進行がん患者を対象としたデノスマブの 4 試験 (試験 20050103、20050136、20050244、及び 20050147[第 1.1.1.2 項])のデータも示した。治験薬の曝露状況の解析
GCTB を対象とした臨床試験でデノスマブ投与を 1 回以上受けた被験者数及び曝露期間につ いて、外国 2 試験併合解析又は 3 試験併合解析の結果と個々の試験の結果を示した。また、以 下の解析を実施した。 • 試験日数、治験薬の投与回数、及び治験薬の曝露量 • 年齢、性別、及び人種別の曝露状況有害事象の解析
外国 2 試験併合解析における有害事象の解析では、日米 EU 医薬品規制調和国際会議(ICH) 国際医薬用語集(MedDRA、第 14.1 版)を用い、「ICH 活動で作成された MedDRA ユーザー2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
のためのガイド 医薬品副作用/有害事象および医学的・社会的背景、適応症に対する適用」 の「MedDRA 用語選択: 考慮事項」に記載のコーディングガイドライン(MedDRA MSSO, 2011) に基づいて事象を分類した。本モジュールでは、試験 20040215 及び 20062004 の有害事象に MedDRA 第 14.1 版を用いたため、試験 20040215 については、一部の有害事象(基本語)は、 試験 20040215 の主要解析の治験総括報告書(MedDRA 第 11.0 版を使用)及び試験 20040215 の最終解析の治験総括報告書(MedDRA 第 13.1 版を使用)に報告された有害事象(基本語) とわずかに異なる可能性がある。なお、試験 20062004 の第 3 回中間解析治験総括報告書では、 有害事象の解析に MedDRA 第 14.1 版を使用した。 試験 AMG162-B-J201 及び 3 試験併合解析では MedDRA 第 15.1 版を用いたため、有害事象 (基本語)は外国 2 試験併合解析で報告されたものと異なるものがある。 試験 20040215、20062004、及び AMG162-B-J201 の安全性解析対象集団、並びに外国 2 試験 併合解析及び 3 試験併合解析の安全性併合解析対象集団を用い、次の安全性データを集計した。 • 有害事象、重篤な有害事象、及び死亡の発現率(治験薬との関連性があるもの及び全体) • 米国国立がん研究所(National Cancer Institute: NCI)の有害事象共通用語規準(Common
Terminology Criteria for Adverse Effects: CTCAE)第 3.0 版(外国 2 試験)又は第 4.0 版 JCOG 版(試験 AMG162-B-J201)のグレード 3、4、又は 5 に該当する有害事象及び治験薬と の関連性がある有害事象の発現率
• MedDRA 標準検索式(standard MedDRA query: SMQ)「肝障害(hepatic disorders)」を用 いた包括的検索に基づく薬剤に関連する肝障害の発現率
• SMQ「重症皮膚副作用(severe cutaneous adverse reactions)」を用いた包括的検索に基づ く重症皮膚副作用の発現率
その他の重要な有害事象の解析
試験 20040215、20062004、及び AMG162-B-J201 の安全性解析対象集団、並びに外国 2 試験 併合解析及び 3 試験併合解析の安全性併合解析対象集団を用い、治験薬の投与中止又は試験中 止に至った有害事象の発現率(治験薬との関連性があるもの及び全体)を集計した。
さらに、低カルシウム血症、顎骨壊死(osteonecrosis of the jaw: ONJ)と判定された事象、過 敏症と関連する可能性のある有害事象、感染症、悪性新生物、心血管系事象について、有害事 象及び重篤な有害事象(該当する場合)の発現率を、試験ごと、外国 2 試験併合解析、及び 3 試験併合解析で示した。また、各試験で認められた悪性骨腫瘍の各事象について叙述した。こ れらの解析方法を表 2.7.4.1-1に示す。また、参考として、進行がん患者を対象としたデノスマ ブの 4 試験(試験 20050103、20050136、20050244、及び 20050147)で発現した主な有害事象
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 9 語を更新した。感染症、心臓障害、及び血管障害については MedDRA バージョンの更新のみ であった。本モジュールでは、この新たな MedDRA 検索用語を使用した結果を示した。その ため、試験 20040215 については、本モジュールと主要解析治験総括報告書及び最終解析治験 総括報告書との間で有害事象の件数及び種類がわずかに異なる場合がある。 表 2.7.4.1-1 重要な有害事象を対象に実施した解析 有害事象 解析 低カルシウム血症 MedDRA(第 14.1 版又は第 15.1 版a)基本語の検索用語(付録 2)により特定した 低カルシウム血症の有害事象及び重篤な有害事象の発現率、並びにこれらの事象 を発現した被験者の血清カルシウム値。 血清カルシウムの測定値の要約統計量及び変化率の時間推移(第 3.1 項)。 ONJ 独立判定委員会により ONJ と判定された事象の発現率。独立判定委員会に判定を 依頼した ONJ の可能性のある事象の特定に用いた MedDRA(第 14.1 版又は第 15.1 版a)基本語を、付録 2に示す。 過敏症と関連する可 能性がある有害事象 MedDRA(第 14.1 版又は第 15.1 版a)基本語の検索用語(付録 2)により特定した 過敏症と関連する可能性がある有害事象及び重篤な有害事象の発現率。 感染症 感染症の有害事象及び重篤な有害事象の発現率(MedDRA 器官別大分類の「感染
症および寄生虫症 infections and infestations」)。
悪性腫瘍
MedDRA 器官別大分類の「良性、悪性および詳細不明の新生物(嚢胞およびポリ ープを含む)neoplasms benign, malignant and unspecified (incl cysts and polyps)」に該
当する基本語(第 14.1 版又は第 15.1 版a)を目視で抽出することにより特定した
悪性腫瘍の発現率。
心臓・血管障害 器官別大分類の「心臓障害 cardiac disorders」及び「血管障害 vascular disorders」
に該当する有害事象及び重篤な有害事象の発現率。 a 外国 2 試験併合解析では第 14.1 版、試験 AMG162-B-J201 及び 3 試験併合解析では第 15.1 版を用いた。
臨床検査値の解析
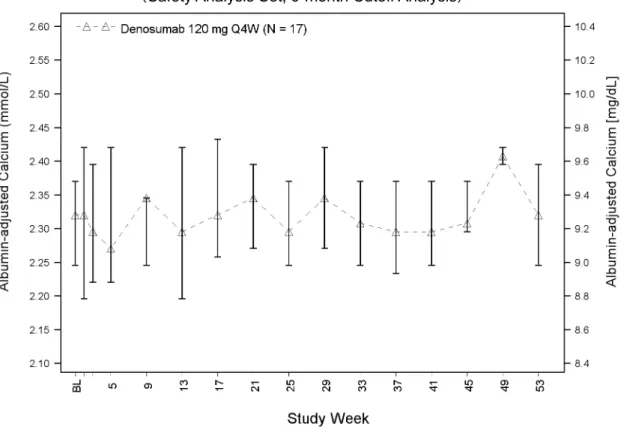
グレード 3 及び 4 の臨床検査値異常、グレード 2 以上の血清カルシウム値の低下、及び血清 カルシウム値のベースラインからの 2 グレード以上低下の発現率を、試験ごと、外国 2 試験併 合解析、又は 3 試験併合解析で示した。NCI CTCAE 第 3.0 版(外国 2 試験)又は第 4.0 版 JCOG 版(試験 AMG162-B-J201)による臨床検査値のグレード評価にあたっては、各被験者の臨床 検査値に関するすべてのグレードを集計するのではなく、各被験者で認められた最も高いグレ ード(「最も悪い」)の臨床検査値を集計した(例えば、試験期間中にグレード 3 とグレード 4 の両方の値が認められた被験者は、グレード 4 が認められた被験者として 1 回のみ集計した)。 各臨床検査項目のグレード別の NCI CTCAE 第 3.0 版分類を付録 3 の表 2.7.4.7-7に、第 4.0 版 JCOG 版を付録 3 の表 2.7.4.7-8に示す。 血清カルシウム濃度の解析では、血清アルブミン値が 40 g/L 未満の場合、次の補正式を用 いてカルシウム濃度をアルブミンで補正した。 アルブミン補正血清カルシウム濃度(mmol/L)= 血清カルシウム濃度(mmol/L)+[40 g/L – 血清アルブミン濃度(g/L)]× 0.02 本式適用の有無によりカルシウム濃度にはアルブミン補正値と非補正値の場合があるが、こ れらをまとめて「アルブミン補正血清カルシウム値」という用語を用いた。 抗デノスマブ抗体の解析では、電気化学発光ブリッジング免疫測定法により、検体中の結合2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 抗体の有無をスクリーニングした。抗体陽性の検体は、細胞を用いた mRNA 発現の化学発光 法による測定で、中和抗体の確認を行った。免疫原性測定法に関する詳細な手順及び個々の試 験結果は、各試験の治験総括報告書(試験 20040215 主要解析、20062004 第 3 回中間解析、及 び20050147)及びランマーク SRE 申請資料のモジュール 5 第 5.3.5.3 項の免疫原性に関する併 合報告書に示した。
1.1.3
試験結果の要約
試験 20040215、20062004、及び AMG162-B-J201 の結果の要約は、モジュール 2.7.3 に示し た。参考として、他の試験(20050103、20050136、20050244、及び 20050147)の結果の要約 を第 5.1.3.2.1 項及び第 5.1.3.2.2 項に示す。1.2
全般的な曝露状況
本モジュールでは、試験 20040215 及び 20062004 でデノスマブ 120 mg Q4W 投与を受けた GCTB 患者 304 名の安全性データを、外国 2 試験併合解析の結果として示す。デノスマブの投 与を受けた 304 名のうち、投与期間 1 年以上は 147 名、2 年以上は 46 名、3 年以上は 15 名で あった(表 2.7.4.1-2)。投与回数の中央値(Q1、Q3)は 14(8.0、21.5)回で、範囲は 1~60 回であった(表 2.7.4.1-3)。試験期間の中央値(Q1、Q3)は 11.17(5.36、18.23)ヵ月であっ た。試験期間の中央値は、試験 20040215 で 19.4 ヵ月、試験 20062004 で 10.4 ヵ月であった。 年齢別及び性別の曝露状況はTable tias5-2.1、人種別の曝露状況はTable tias5-2.2に示す。試験 AMG162-B-J201(6 ヵ月カットオフ解析)では、デノスマブの投与を受けた 17 名のう ち、投与期間 1 ヵ月以上は 17 名、6 ヵ月以上は 13 名であった(表 2.7.4.1-2)。投与回数の中央 値(Q1、Q3)は 11(9、14)回で、範囲は 6~16 回であった(表 2.7.4.1-3)。試験期間の中央 値(Q1、Q3)は 7.56(6.21、10.45)ヵ月であった。3 試験併合解析の結果はTable 1tias2-1.1 に示す。 市販後の曝露状況は第 6 項に示す。 表 2.7.4.1-2 骨巨細胞腫を対象とした試験でデノスマブ投与を受けた被験者数(累積曝露期 間及び試験別) Denosumab
≥ 1 Dose ≥ 1 Month ≥ 6 Months ≥ 1 Year ≥ 2 Years ≥ 3 Years
Study 20040215 and Study 20062004
304 296 226 147 46 15
Study AMG162-B-J201 (6-month Cutoff Analysis)
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 11 表 2.7.4.1-3 治験薬投与の要約 Study 20040215 Denosumab 120 mg Q4W Study 20062004 Denosumab 120 mg Q4W Overallb Denosumab 120 mg Q4W Study AMG162-B-J201c Denosumab 120 mg Q4W
Number of subjects enrolled 37 282 305 17
Number of months on studya
n 37 282 305 17 Mean 22.13 11.50 13.42 8.14 SD 16.28 7.57 10.71 3.02 Median 19.38 10.40 11.17 7.56 Q1, Q3 7.69, 38.90 5.32, 16.72 5.36, 18.23 6.21, 10.45 Min, Max 2.0, 48.9 0.0, 29.1 0.0, 54.1 3.4, 12.4
Number of subjects receiving
≥1 dose 37 281 304
17
of investigational product
Number of doses received
n 37 281 304 17 Mean 24.3 14.3 16.2 11.5 SD 17.0 7.9 10.9 3.3 Median 21.0 13.0 14.0 11.0 Q1, Q3 9.0, 42.0 7.0, 20.0 8.0, 21.5 9.0, 14.0 Min, Max 4, 54 1, 33 1, 60 6, 16
a Defined as the time period from the first dose of investigational product, or enrollment date if subjects did not take any dose, to the end of study date or the analysis cutoff date, whichever comes first.
bIntegrated Analysis (20040215/20062004); GCTB Full Analysis Set, Treatment Analysis Phase. Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from Study 20040215 and end at Study 20062004.
Four subjects from study 20040215 were rolled over to the safety follow up phase only, never received investigational product in study 20062004, and were excluded from the treatment phase analysis in study 20062004.
c CSR of Study AMG162-B-J201; Enrolled Subjects (6-month Cutoff Analysis)
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg
1.3
治験対象集団の人口統計学的特性及びその他の特性
1.3.1
被験者の内訳
試験 20040215 及び 20062004 には合計 305 名の被験者が組み入れられ、304 名がデノスマブ 投与を 1 回以上受けた(表 2.7.4.1-4)。 外国 2 試験併合解析では、試験 20040215 の終了時又は試験 20062004 のデータカットオフ日 (2011 年 3 月 25 日)までに 65 名(21.3%)がデノスマブ投与を中止した(表 2.7.4.1-4)。比 較的よく見られたデノスマブ投与の中止理由は、治験実施計画書に規定された基準(完全切除 術)(7.2%)、管理上の決定(3.3%)、有害事象(3.3%)、及び病勢の進行(2.0%)であった。 試験 20040215 は、被験者 37 名が組み入れられ、全員がデノスマブ投与を 1 回以上受けた。 試験 20040215 の終了時にデノスマブの投与中止に至っていなかった被験者は、すべて試験 20062004 に移行した(投与中止として取り扱った)。投与中止理由として多かったのは、試験 20062004 への移行(12 名[32.4%]、このうち 1 名は試験 20062004 ではデノスマブ投与を受け ず安全性の追跡調査期間のみに移行し、残り 11 名は試験 20062004 でデノスマブ投与が継続さ れた)、及び治験実施計画書に規定された基準(完全切除術 10 名[27.0%])であった(表 2.7.4.1-4)。 なお、試験 20040215 では、完全切除術を受けた被験者又は病勢の進行が認められた被験者は、 デノスマブ投与を中止する規定であった。 試験 20062004 は、被験者 282 名が組み入れられ、281 名がデノスマブ投与を 1 回以上受け た。試験 20040215 から試験 20062004 に移行した 12 名以外に、試験 20040215 を中止した後、 試験 20062004 に組み入れられた被験者が 3 名存在した。試験 20062004 では、第 3 回中間解析 のデータカットオフ日時点までに、43 名がデノスマブ投与を中止した。中止理由は、治験実 施計画書に規定された基準(完全切除術)13 名(4.6%)、管理上の決定 9 名(3.2%)、有害事 象 9 名(3.2%)、病勢の進行 3 名(1.1%)であった。完全切除術を受けた被験者(コホート 2) では、その後 6 回以内で投与を中止し、その他の被験者では、病勢の進行が認められた場合に 投与を中止する規定であった。 試験 20040215 では試験完了時に全被験者が試験を中止し、試験 20062004 では 2011 年 3 月 25 日の時点までに 14.5%の被験者が試験を中止した。2 試験全体では、21.0%の被験者が試験 を中止した(Table tias1-1.1)。多く認められた試験中止理由は、デノスマブ投与の中止理由と 同様であった。 試験 AMG162-B-J201 では、被験者 17 名が組み入れられ、全員がデノスマブ投与を 1 回以上 受けた。6 ヵ月カットオフ解析のデータカットオフ日までに、デノスマブ投与を中止した被験 者は認められなかった。2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 13 表 2.7.4.1-4 治験薬の投与中止理由 Study 20040215 Denosumab 120 mg Q4W Study 20062004 Denosumab 120 mg Q4W Overallb Denosumab 120 mg Q4W Study AMG162-B-J201d Denosumab 120 mg Q4W n (%) n (%) n (%) n (%) Enrolled 37 282 305 17 (100) Received IP 37 (100.0) 281 (99.6) 304 (99.7) 17 (100) IP ongoing 0 (0.0) 238 (84.4) 238 (78.0) 17 (100) Discontinued IP 36 (97.3) 43 (15.2) 65 (21.3) 0 (0.0) Protocol-specified criteriaa 10 (27.0) 13 (4.6) 22 (7.2) 0 (0.0) Administrative decision 1 (2.7) 9 (3.2) 10 (3.3) 0 (0.0) Adverse event 1 (2.7) 9 (3.2) 10 (3.3) 0 (0.0) Disease progression 3 (8.1) 3 (1.1) 6 (2.0) 0 (0.0) Consent withdrawn 2 (5.4) 3 (1.1) 5 (1.6) 0 (0.0) Other 4 (10.8) 1 (0.4) 3 (1.0) 0 (0.0)
Requirement for alternative
therapy 1 (2.7) 2 (0.7) 3 (1.0) 0 (0.0) Noncompliance 2 (5.4) 0 (0.0) 2 (0.7) 0 (0.0) Lost to follow-up 0 (0.0) 2 (0.7) 2 (0.7) 0 (0.0) Rollover to study 20062004 12 (32.4) 0 (0.0) 1 (0.3) 0 (0.0) Pregnancy 0 (0.0) 1 (0.4) 1 (0.3) 0 (0.0)
Missing end of IP reasonc 1 (2.7) 0 (0.0) 1 (0.3) 0 (0.0)
Never received IP 0 (0.0) 1 (0.4) 1 (0.3) 0 (0.0)
IP = Investigational product
Percentages based on number of subjects enrolled a Complete resection.
b
Integrated Analysis (20040215/20062004); Enrolled Subjects, Treatment Analysis Phase, Integrated Analysis of Safety. Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
c Site closed, unable to resolve end of IP reason
Four subjects from study 20040215 were rolled over to the safety follow up phase only, never received IP in study 20062004, and were excluded from the treatment phase analysis in study 20062004. For these 4 subjects, the reason for study discontinuation in study 20040215 is counted as the reason for discontinuation in the overall column. For Subject (corresponding 20062004 Subject ID = ), the end of study reason was listed as rollover to study 20062004; the other 3 subjects had reasons for discontinuation listed as administrative decision (n=1) and other (n=2).
d
CSR of Study AMG162-B-J201; Enrolled Subjects (6-month Cutoff Analysis)
Source: Table tias 1-1.2, Table 15.1-5 of Study AMG162-B-J201 CSR
136* 136*
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg
1.3.2
人口統計学的及び基準値の特性
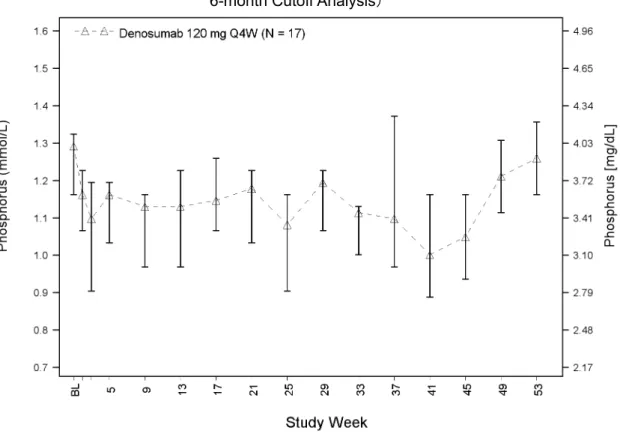
外国 2 試験併合解析における安全性併合解析対象集団の年齢の中央値(範囲)は 33(13~ 83)歳であり(表 2.7.4.1-5)、被験者の 96.7%が 65 歳未満、99.0%が 75 歳未満であった。未成 年被験者(外国では 18 歳未満)10 名はすべて、試験 20062004 の被験者であった。また、日 本の未成年の定義(20 歳未満)に合致する被験者数は、試験 20040215 で 2 名及び試験 20062004 で 19 名であった。女性の割合は、全被験者の 58.4%であった。被験者の多くは白人(80.3%) であり、次いでヒスパニック系又はラテン系(6.2%)、黒人(5.9%)、アジア系(4.9%)であ った。ベースライン時の身体的特性に関する要約統計量はTable tias2-2.1に示した。 被験者の約 35%は初発又は再発の切除可能な GCTB を有し、約 65%は初発又は再発の切除 不能な GCTB を有していた(表 2.7.4.1-6)。標的病変部位として多かったのは、骨盤(29.5%)、 下肢(25.9%)、及びその他(22.6%)であった。試験 20040215 で初発の切除可能な GCTB を 有する被験者が認められなかったことを除き、GCTB の種類及び部位に試験間で差はみられな かった(試験 20040215 では治験実施計画書に従い、初発の切除可能な GCTB を有する被験者 が除外された)。 また、試験 20040215 及び 20062004 の全体で、ベースライン時のアルブミン補正血清カルシ ウム濃度の中央値(範囲)は 2.39(2.1~3.7)mmol/L(9.58[9.30~9.81]mg/dL)、リン濃度 の中央値(範囲)は 1.16(0.6~1.9)mmol/L(3.60[3.22~4.00]mg/dL)であった(Table tias2-3.1)。 試験 AMG162-B-J201 の安全性解析対象集団では、年齢の中央値(範囲)は 30(18~66)歳 であり(表 2.7.4.1-5)、被験者の 17 名中 16 名(94.1%)が 65 歳未満で、17 名全員が 75 歳未 満であった。未成年被験者(20 歳未満)は 1 名であった。女性は 9 名(52.9%)であった。ベ ースライン時の身体的特性に関する要約統計量は試験 AMG162-B-J201 治験総括報告書の Table 15.1-11に示した。被験者の約 24%は初発又は再発の切除可能な GCTB を有し、約 76% は初発又は再発の切除不能な GCTB を有していた(表 2.7.4.1-6)。治験責任医師が選択した標 的病変で最も多かったのは骨盤(35.3%)であり、次に多かったのは「その他」(23.5%)であ った。「その他」に含まれた標的病変部位は、主に肺であった。また、ベースライン時の中央 値(範囲)として、アルブミン補正血清カルシウム濃度は 2.32 (2.1~2.5) mmol/L(9.30[8.6 ~10.2]mg/dL)、リン濃度は 1.29 (0.9~1.4) mmol/L(4.00[2.7~4.4]mg/dL)であった (Table 15.1-25)。以上より、試験 AMG162-B-J201 の人口統計学的特性及び基準値の特性(人 種を除く)は外国 2 試験併合解析と同様であった。 ベースライン時の人口統計学的及び基準値の特性の 3 試験併合解析は、Table 1tias3-1.1、 Table 1tias3-1.2、Table 1tias3-2.1、Table 1tias3-3.1、及びTable 1tias3-3.2に示す。2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 15 表 2.7.4.1-5 ベースラインの人口統計学的特性 Study 20040215 Denosumab 120 mg Q4W (N = 37) Study 20062004 Denosumab 120 mg Q4W (N = 282) Overalla Denosumab 120 mg Q4W (N = 305) Study AMG162-B-J201b Denosumab 120 mg Q4W (N = 17) Sex - n (%) Female 20 (54.1) 164 (58.2) 178 (58.4) 9 (52.9) Male 17 (45.9) 118 (41.8) 127 (41.6) 8 (47.1)
Ethnic group / race - n (%)
White or Caucasian 27 (73.0) 230 (81.6) 245 (80.3) 0 (0.0)
Black or African American 2 (5.4) 16 (5.7) 18 (5.9) 0 (0.0)
Hispanic or Latino 5 (13.5) 15 (5.3) 19 (6.2) 0 (0.0)
Asian 3 (8.1) 12 (4.3) 15 (4.9) 0 (0.0)
Japanese 0 (0.0) 0 (0.0) 0 (0.0) 17 (100.0)
American Indian or Alaska Native 0 (0.0) 1 (0.4) 0 (0.0) 0 (0.0)
Native Hawaiian or Other Pacific
Islander 0 (0.0) 1 (0.4) 1 (0.3) 0 (0.0) Other 0 (0.0) 7 (2.5) 7 (2.3) 0 (0.0) Age (years) n 37 282 305 17 Mean 33.9 35.5 35.3 36.3 SD 12.3 13.3 13.2 15.6 Median 30.0 33.0 33.0 30.0 Q1, Q3 25.0, 40.0 25.0, 44.0 25.0, 44.0 24.0, 47.0 Min, Max 19, 63 13, 83 13, 83 18, 66
Age 65 years or over at enrollment - n (%)
< 65 years 37 (100.0) 272 (96.5) 295 (96.7) 16 (94.1)
≥ 65 years 0 (0.0) 10 (3.5) 10 (3.3) 1 (5.9)
Age 75 years or over at enrollment - n (%)
< 75 years 37 (100.0) 279 (98.9) 302 (99.0) 17 (100.0)
≥ 75 years 0 (0.0) 3 (1.1) 3 (1.0) 0 (0.0)
N = Number of subjects enrolled
aIntegrated Analysis (20040215/20062004); Integrated Safety Analysis Set. Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
All baseline information was based on 20040215 for roll over subjects in overall column.
Four subjects from study 20040215 were rolled over to the safety follow up phase only, never received IP in study 20062004, and were excluded from the treatment phase analysis in study 20062004.
b CSR of Study AMG162-B-J201; Safety Analysis Set (6-month Cutoff Analysis)
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 表 2.7.4.1-6 ベースラインの疾患特性 Study 20040215 Denosumab 120 mg Q4W (N = 37) Study 20062004 Denosumab 120 mg Q4W (N = 282) Overalla Denosumab 120 mg Q4W (N = 305) Study AMG162-B-J201c Denosumab 120 mg Q4W (N = 17) GCTB disease type - n(%) Primary resectable 0 (0.0) 63 (22.3) 63 (20.7) 2 (11.8) Primary unresectable 13 (35.1) 50 (17.7) 61 (20.0) 5 (29.4) Recurrent resectable 6 (16.2) 38 (13.5) 44 (14.4) 2 (11.8) Recurrent unresectable 18 (48.6) 131 (46.5) 137 (44.9) 8 (47.1)
Longest dimension of target lesion (mm)b
n 36 282 304 17 Mean 58.4 65.8 65.7 70.6 SD 37.8 45.0 44.4 58.6 Median 50.5 58.5 58.0 44.0 Q1, Q3 31.0, 79.5 33.0, 89.0 33.9, 89.5 28.4, 98.3 Min, Max 6, 170 3, 308 6, 308 15, 228
Location of target lesion - n(%)
Pelvis 9 (24.3) 85 (30.1) 90 (29.5) 6 (35.3) Lower extremities 8 (21.6) 72 (25.5) 79 (25.9) 3 (17.6) Other 11 (29.7) 62 (22.0) 69 (22.6) 4 (23.5) Upper extremities 5 (13.5) 29 (10.3) 32 (10.5) 2 (11.8) Spine 3 (8.1) 27 (9.6) 27 (8.9) 1 (5.9) Head/neck 0 (0.0) 7 (2.5) 7 (2.3) 1 (5.9) Missing 1 (2.7) 0 (0.0) 1 (0.3) 0 (0.0)
N = Number of subjects enrolled
aIntegrated Analysis (20040215/20062004); Integrated Safety Analysis Set. Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
b Target lesions were selected and evaluated by the investigator.
All baseline information was based on 20040215 for roll over subjects in overall column.
Four subjects from study 20040215 were rolled over to the safety follow up phase only, never received IP in study 20062004, and were excluded from the treatment phase analysis in study 20062004.
c
CSR of Study AMG162-B-J201; Safety Analysis Set (6-month Cutoff Analysis)
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 17
2.
有害事象
2.1
有害事象の解析
外国 2 試験併合解析では、治験薬を 1 回以上投与された安全性併合解析対象集団の被験者 304 名のうち、試験期間中に有害事象を 1 件以上発現した被験者は 259 名(85.2%)であった (表 2.7.4.2-1)。重篤な有害事象は 34 名(11.2%)、治験薬との関連性があると判定された重篤 な有害事象は 3 名(1.0%)に認められた。2 名(0.7%)の死亡が報告された。死因は、被験者 試験 20040215)が悪性新生物 neoplasm malignant、被験者 試験 20062004) が呼吸不全 respiratory failure であった(Table tias6-9.2)。いずれの死亡もデノスマブ投与との 関連性はないと判定された。 試験 AMG162-B-J201(6 ヵ月カットオフ解析)では、治験薬を 1 回以上投与された安全性解 析対象集団の被験者 17 名のうち、データカットオフ日までに有害事象を 1 件以上発現した被 験者は 16 名(94.1%)であった(表 2.7.4.2-1)。重篤な有害事象は 2 名(11.8%)、治験薬との 関連性があると判定された重篤な有害事象は認められなかった。試験 AMG162-B-J201(6 ヵ月 カットオフ解析)では、データカットオフ日までに死亡は認められなかった。有害事象の 3 試験併合解析結果の概要は、Table 1tias4-1.1及びTable 1tias4-1.2に示す。 各試験の有害事象及びグレード 3 以上の有害事象の発現状況を付録 6に示す。
097* 105*
2.7.4 臨床的安全性の概要 デノスマブ ランマーク皮下注 120mg 表 2.7.4.2-1 有害事象発現率の要約 Study 20040215 Denosumab 120 mg Q4W (N=37) n (%) Study 20062004 Denosumab 120 mg Q4W (N=281) n (%) Overallb Denosumab 120 mg Q4W (N=304) n (%) Study AMG162-B-J201c Denosumab 120 mg Q4W (N = 17) n (%)
Adverse events regardless of relationship
All 33 (89.2) 236 (84.0) 259 (85.2) 16 (94.1)
Serious 9 (24.3) 25 (8.9) 34 (11.2) 2 (11.8)
Fatal 1 (2.7) 1 (0.4) 2 (0.7) 0 (0.0)
Leading to study discontinuation 2 (5.4) 13 (4.6) 15 (4.9) 0 (0.0)
Leading to investigational product
discontinuation 2 (5.4) 14 (5.0) 16 (5.3) 0 (0.0)
CTCAE Grade 3, 4, or 5 10 (27.0) 50 (17.8) 59 (19.4) 2 (11.8)
Adverse events related to investigational producta
All 12 (32.4) 140 (49.8) 149 (49.0) 9 (52.9)
Serious 0 (0.0) 3 (1.1) 3 (1.0) 0 (0.0)
Fatal 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)
Leading to study discontinuation 1 (2.7) 2 (0.7) 3 (1.0) 0 (0.0)
Leading to investigational product
discontinuation 1 (2.7) 2 (0.7) 3 (1.0) 0 (0.0)
CTCAE Grade 3, 4, or 5 1 (2.7) 15 (5.3) 16 (5.3) 0 (0.0)
N = Number of subjects who received ≥ 1 active dose of investigational product CTCAE version 3.0
Includes only treatment-emergent adverse events a
Includes only events for which the investigator indicated there was a reasonable possibility they may have been caused by investigational product
b
Integrated Analysis (20040215/20062004); Integrated Safety Analysis Set. Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
c CSR of Study AMG162-B-J201; Safety Analysis Set (6-month Cutoff Analysis)
Source: Table tias6-1.1, Table 15.3-2.1of Study AMG162-B-J201 CSR
2.1.1
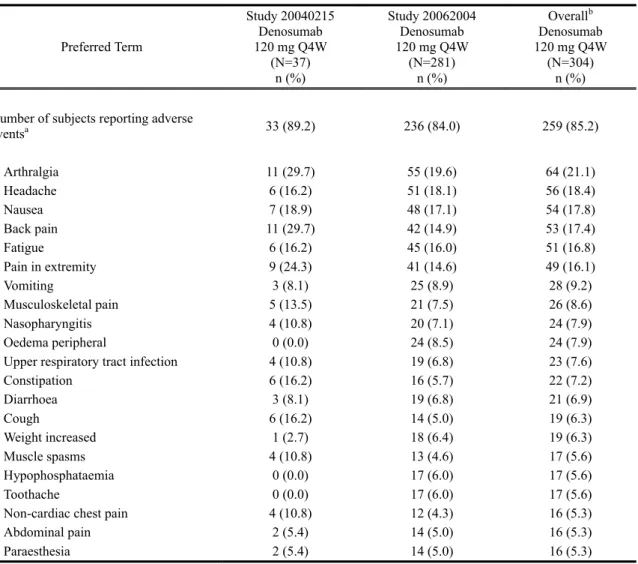
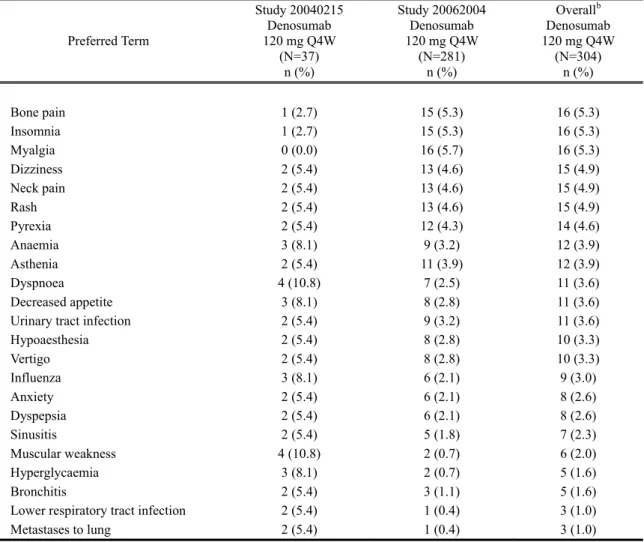
比較的よく見られる有害事象
試験 20040215 又は 20062004 で発現率が 5%以上であった有害事象の一覧(試験別の発現率 及び外国 2 試験併合解析での発現率)を表 2.7.4.2-2に示す。比較的よく見られた有害事象は、 外国 2 試験併合解析の発現率で、関節痛 arthralgia(21.1%)、頭痛 headache(18.4%)、悪心 nausea (17.8%)、背部痛 back pain(17.4%)、疲労 fatigue(16.8%)、及び四肢痛 pain in extremity(16.1%) であった。
外国 2 試験併合解析で、MedDRA の器官別大分類として有害事象発現率が高かったのは、 筋骨格系および結合組織障害 musculoskeletal and connective tissue disorders(54.6%)、胃腸障害
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
19
験薬との関連性があると判定された有害事象で、両試験を通じて比較的よく見られたのは、疲 労 fatigue(9.9%)、頭痛 headache(9.9%)、悪心 nausea(7.6%)、及び低リン酸血症
hypophosphatemia(4.6%)であった。
グレード 3、4、又は 5 の有害事象は、試験 20040215 で 27.0%、試験 20062004 で 17.8%の被 験者に認められた(Table tias6-23.1及びTable tias6-23.2)。両試験を通じて比較的よく見られた グレード 3、4、又は 5 の有害事象は、低リン酸血症 hypophosphatemia(3.0%)、背部痛 back pain (1.3%)、四肢痛 pain in extremity(1.3%)、うつ病 depression(1.0%)、筋骨格痛 musculoskeletal pain(1.0%)、及び貧血 anemia(1.0%)であった(Table tias6-23.2)。
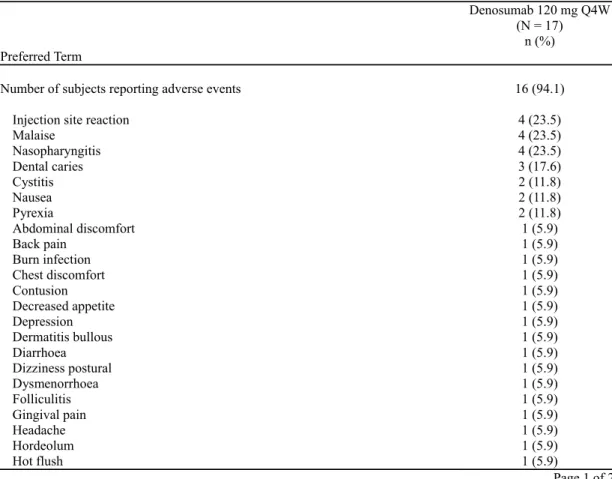
治験責任医師により治験薬との関連性があると判定されたグレード 3、4、又は 5 の有害事 象は、試験 20040215 で 2.7%(1 名)、試験 20062004 で 5.3%(15 名)の被験者に認められた (Table tias6-24.2)。治験薬との関連性があるグレード 3、4、又は 5 の有害事象で、両試験を 通じて比較的よく見られたのは、低リン酸血症 hypophosphatemia(2.6%)、骨髄炎 osteomyelitis (0.7%)、及び ONJ(0.7%)であった。治験薬との関連性があるその他のグレード 3、4、又は 5 の有害事象はすべて、各 1 名のみであった。 試験 AMG162-B-J201 で発現した全有害事象の一覧を表 2.7.4.2-3に示す。2 名以上認められ た有害事象は、注射部位反応 injection site reaction 4 名(23.5%)、倦怠感 malaise 4 名(23.5%)、 鼻咽頭炎 nasopharyngitis 4 名(23.5%)、齲歯 dental caries 3 名(17.6%)、膀胱炎 cystitis 2 名 (11.8%)、悪心 nausea 2 名(11.8%)、及び発熱 pyrexia 2 名(11.8%)であった。MedDRA の 器官別大分類として有害事象発現率が高かったのは、感染症および寄生虫症 infections and infestations 10 名(58.8%、第 2.1.4.2.4 項)、一般・全身障害および投与部位の状態 general disorders and administration site conditions 9 名(52.9%)、胃腸障害 gastrointestinal disorders 7 名(41.2%)、 及び筋骨格系および結合組織障害 musculoskeletal and connective tissue disorders 4 名(23.5%) であった(Table 15.3-2.2)。治験責任医師により治験薬との関連性があると判定された有害事 象は 9 名(52.9%)に認められ(Table 15.3-2.5)、2 名以上認められた事象は、注射部位反応 injection site reaction 4 名(23.5%)、倦怠感 malaise 2 名(11.8%)、及び発熱 pyrexia 2 名(11.8%) であった。
試験 AMG162-B-J201 では、グレード 3、4、又は 5 の有害事象は 2 名の被験者に認められ、 疼痛 pain 及び気胸 pneumothorax が各 1 名であった(Table 15.3-2.29及びTable 15.3-2.30)。こ れらの有害事象は、治験責任医師により治験薬との関連性は否定された(Table 15.3-2.31及び Table 15.3-2.32)。
比較的よく見られた有害事象に関する 3 試験併合解析結果は、Table 1tias4-2.1、 Table 1tias4-3.1、Table 1tias4-3.2、Table 1tias4-4.1、及びTable 1tias4-5.1に示す。
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
表 2.7.4.2-2 いずれかの試験で発現率が 5%以上であった有害事象(基本語):外国 2 試験併 合解析(Safety Subjects, Treatment Analysis Phase, Integrated Analysis of Safety.)
Preferred Term Study 20040215 Denosumab 120 mg Q4W (N=37) n (%) Study 20062004 Denosumab 120 mg Q4W (N=281) n (%) Overallb Denosumab 120 mg Q4W (N=304) n (%)
Number of subjects reporting adverse
eventsa 33 (89.2) 236 (84.0) 259 (85.2) Arthralgia 11 (29.7) 55 (19.6) 64 (21.1) Headache 6 (16.2) 51 (18.1) 56 (18.4) Nausea 7 (18.9) 48 (17.1) 54 (17.8) Back pain 11 (29.7) 42 (14.9) 53 (17.4) Fatigue 6 (16.2) 45 (16.0) 51 (16.8) Pain in extremity 9 (24.3) 41 (14.6) 49 (16.1) Vomiting 3 (8.1) 25 (8.9) 28 (9.2) Musculoskeletal pain 5 (13.5) 21 (7.5) 26 (8.6) Nasopharyngitis 4 (10.8) 20 (7.1) 24 (7.9) Oedema peripheral 0 (0.0) 24 (8.5) 24 (7.9)
Upper respiratory tract infection 4 (10.8) 19 (6.8) 23 (7.6)
Constipation 6 (16.2) 16 (5.7) 22 (7.2) Diarrhoea 3 (8.1) 19 (6.8) 21 (6.9) Cough 6 (16.2) 14 (5.0) 19 (6.3) Weight increased 1 (2.7) 18 (6.4) 19 (6.3) Muscle spasms 4 (10.8) 13 (4.6) 17 (5.6) Hypophosphataemia 0 (0.0) 17 (6.0) 17 (5.6) Toothache 0 (0.0) 17 (6.0) 17 (5.6)
Non-cardiac chest pain 4 (10.8) 12 (4.3) 16 (5.3)
Abdominal pain 2 (5.4) 14 (5.0) 16 (5.3)
Paraesthesia 2 (5.4) 14 (5.0) 16 (5.3)
Page 1 of 2 N = Number of subjects who received ≥ 1 active dose of investigational product
n = Number of subjects reporting ≥ 1 event Includes only treatment-emergent adverse events
Preferred terms are sorted by descending order of frequency in the overall denosumab group and coded using MedDRA Version 14.1.
a Includes all adverse events, not only those occurring with ≥ 5% frequency b
Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
21
表 2.7.4.2-2 いずれかの試験で発現率が 5%以上であった有害事象(基本語):外国 2 試験併 合解析(Safety Subjects, Treatment Analysis Phase, Integrated Analysis of Safety)
Preferred Term Study 20040215 Denosumab 120 mg Q4W (N=37) n (%) Study 20062004 Denosumab 120 mg Q4W (N=281) n (%) Overallb Denosumab 120 mg Q4W (N=304) n (%) Bone pain 1 (2.7) 15 (5.3) 16 (5.3) Insomnia 1 (2.7) 15 (5.3) 16 (5.3) Myalgia 0 (0.0) 16 (5.7) 16 (5.3) Dizziness 2 (5.4) 13 (4.6) 15 (4.9) Neck pain 2 (5.4) 13 (4.6) 15 (4.9) Rash 2 (5.4) 13 (4.6) 15 (4.9) Pyrexia 2 (5.4) 12 (4.3) 14 (4.6) Anaemia 3 (8.1) 9 (3.2) 12 (3.9) Asthenia 2 (5.4) 11 (3.9) 12 (3.9) Dyspnoea 4 (10.8) 7 (2.5) 11 (3.6) Decreased appetite 3 (8.1) 8 (2.8) 11 (3.6)
Urinary tract infection 2 (5.4) 9 (3.2) 11 (3.6)
Hypoaesthesia 2 (5.4) 8 (2.8) 10 (3.3) Vertigo 2 (5.4) 8 (2.8) 10 (3.3) Influenza 3 (8.1) 6 (2.1) 9 (3.0) Anxiety 2 (5.4) 6 (2.1) 8 (2.6) Dyspepsia 2 (5.4) 6 (2.1) 8 (2.6) Sinusitis 2 (5.4) 5 (1.8) 7 (2.3) Muscular weakness 4 (10.8) 2 (0.7) 6 (2.0) Hyperglycaemia 3 (8.1) 2 (0.7) 5 (1.6) Bronchitis 2 (5.4) 3 (1.1) 5 (1.6)
Lower respiratory tract infection 2 (5.4) 1 (0.4) 3 (1.0)
Metastases to lung 2 (5.4) 1 (0.4) 3 (1.0)
Page 2 of 2 N = Number of subjects who received ≥ 1 active dose of investigational product
n = Number of subjects reporting ≥ 1 event Includes only treatment-emergent adverse events
Preferred terms are sorted by descending order of frequency in the overall denosumab group and coded using MedDRA Version 14.1.
a Includes all adverse events, not only those occurring with ≥ 5% frequency
b Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
表 2.7.4.2-3 全有害事象(基本語): 試験 AMG162-B-J201(Safety Analysis Set, 6-month Cutoff Analysis)
Preferred Term
Denosumab 120 mg Q4W (N = 17)
n (%)
Number of subjects reporting adverse events 16 (94.1)
Injection site reaction 4 (23.5)
Malaise 4 (23.5) Nasopharyngitis 4 (23.5) Dental caries 3 (17.6) Cystitis 2 (11.8) Nausea 2 (11.8) Pyrexia 2 (11.8) Abdominal discomfort 1 (5.9) Back pain 1 (5.9) Burn infection 1 (5.9) Chest discomfort 1 (5.9) Contusion 1 (5.9) Decreased appetite 1 (5.9) Depression 1 (5.9) Dermatitis bullous 1 (5.9) Diarrhoea 1 (5.9) Dizziness postural 1 (5.9) Dysmenorrhoea 1 (5.9) Folliculitis 1 (5.9) Gingival pain 1 (5.9) Headache 1 (5.9) Hordeolum 1 (5.9) Hot flush 1 (5.9) Page 1 of 2 N = Number of subjects who received ≥ 1 dose of investigational product
n = Number of subjects reporting ≥ 1 event Includes only treatment-emergent adverse events Coded using MedDRA version 15.1
Preferred terms are sorted by descending order of frequency.
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
23
表 2.7.4.2-3 全有害事象(基本語): 試験 AMG162-B-J201(Safety Analysis Set, 6-month Cutoff Analysis) Preferred Term Denosumab 120 mg Q4W (N = 17) n (%) Hyperhidrosis 1 (5.9) Hypocalcaemia 1 (5.9) Influenza 1 (5.9) Myalgia 1 (5.9) Nail infection 1 (5.9) Neck pain 1 (5.9) Oral pain 1 (5.9) Otitis media 1 (5.9) Pain 1 (5.9) Paronychia 1 (5.9) Periodontitis 1 (5.9) Pneumonitis 1 (5.9) Pneumothorax 1 (5.9) Rash 1 (5.9) Somnolence 1 (5.9) Temporomandibular syndrome 1 (5.9) Toothache 1 (5.9) Page 2 of 2 N = Number of subjects who received ≥ 1 dose of investigational product
n = Number of subjects reporting ≥ 1 event Includes only treatment-emergent adverse events Coded using MedDRA version 15.1
Preferred terms are sorted by descending order of frequency.
Source: Table 15.3-2.3 of Study AMG162-B-J201 CSR
2.1.2
死亡
GCTB を対象として外国で実施した 2 試験では、投与期間中に 2 名の死亡が報告され、その 内訳は試験 20040215 で 1 名(被験者 : 悪性新生物 neoplasm malignant)、試験 20062004 で 1 名(被験者 : 呼吸不全 respiratory failure)であった(Table tias6-9.2)。 いずれの死亡もデノスマブとの関連性は否定された。これらの叙述は、モジュール 2.7.6 の試 験 20040215 最終解析の項及び試験 20062004 の第 3 回中間解析の項に示す。 試験 AMG162-B-J201 では、6 ヵ月カットオフ解析のデータカットオフ日までに死亡は認め られなかった。
2.1.3
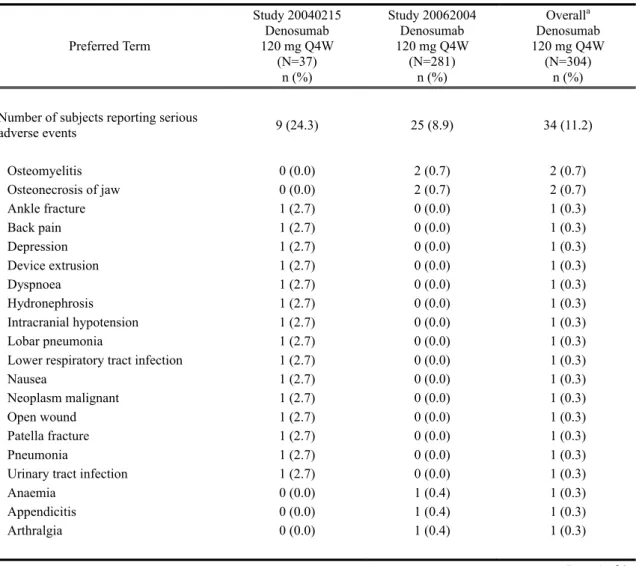
その他の重篤な有害事象
重篤な有害事象は、試験 20040215 では 9 名(24.3%)、試験 20062004 では 25 名(8.9%)に 発現した(表 2.7.4.2-4)。全体で 2 名以上に発現した重篤な有害事象は骨髄炎 osteomyelitis 及 び ONJ のみであり、それぞれ試験 20062004 で 2 名(0.7%)に認められた(第 2.1.4.2.2 項参照)。 試験 20040215 及び 20062004 を通じ、MedDRA の器官別大分類として重篤な有害事象の発 現率が高かった(1.5%以上であった)のは、感染症および寄生虫症 infections and infestations (3.0%)、傷害、中毒および処置合併症 injury, poisoning and procedural complications(3.0%)、097* 105*
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
筋骨格系および結合組織障害 musculoskeletal and connective tissue disorders(2.0%)、良性、悪 性および詳細不明の新生物 neoplasms benign, malignant and unspecified(1.6%)、及び神経系障 害 nervous system disorders(1.6%)であった(Table tias6-10.1)。
治験責任医師により治験薬との関連性があると判定された重篤な有害事象は、試験
20062004 で 3 名(骨髄炎 osteomyelitis と ONJ が各 2 名[0.7%])に認められた(Table tias6-11.2)。 試験 AMG162-B-J201 では、6 ヵ月カットオフ解析のデータカットオフ日までに、重篤な有 害事象として疼痛 pain 及び気胸 pneumothorax が各 1 名報告された(表 2.7.4.2-5)。これらの 有害事象は、治験責任医師により治験薬との関連性は否定された(Table 15.3-2.14)。重篤な有 害事象が認められた被験者の詳細な経過については、モジュール 2.7.6に記載した(6 ヵ月カ ットオフ日以降に発現した事象も含む)。
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
25
表 2.7.4.2-4 重篤な有害事象(基本語): 外国 2 試験併合解析(Safety Subjects, Treatment Analysis Phase, Integrated Analysis of Safety)
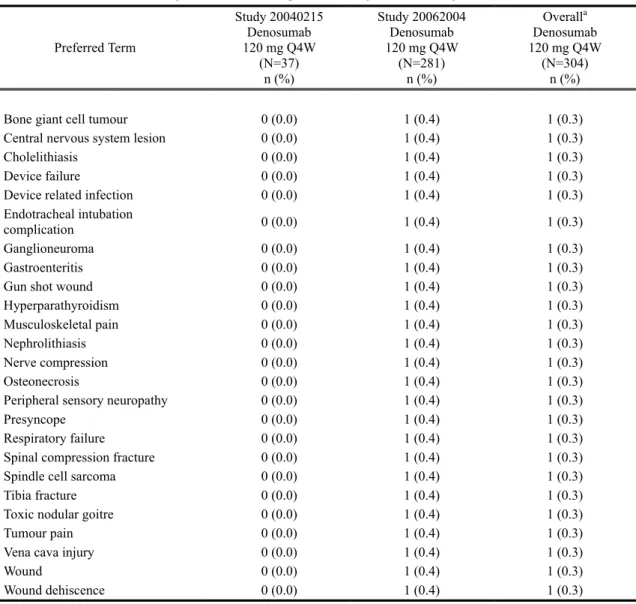
Preferred Term Study 20040215 Denosumab 120 mg Q4W (N=37) n (%) Study 20062004 Denosumab 120 mg Q4W (N=281) n (%) Overalla Denosumab 120 mg Q4W (N=304) n (%)
Number of subjects reporting serious
adverse events 9 (24.3) 25 (8.9) 34 (11.2) Osteomyelitis 0 (0.0) 2 (0.7) 2 (0.7) Osteonecrosis of jaw 0 (0.0) 2 (0.7) 2 (0.7) Ankle fracture 1 (2.7) 0 (0.0) 1 (0.3) Back pain 1 (2.7) 0 (0.0) 1 (0.3) Depression 1 (2.7) 0 (0.0) 1 (0.3) Device extrusion 1 (2.7) 0 (0.0) 1 (0.3) Dyspnoea 1 (2.7) 0 (0.0) 1 (0.3) Hydronephrosis 1 (2.7) 0 (0.0) 1 (0.3) Intracranial hypotension 1 (2.7) 0 (0.0) 1 (0.3) Lobar pneumonia 1 (2.7) 0 (0.0) 1 (0.3)
Lower respiratory tract infection 1 (2.7) 0 (0.0) 1 (0.3)
Nausea 1 (2.7) 0 (0.0) 1 (0.3)
Neoplasm malignant 1 (2.7) 0 (0.0) 1 (0.3)
Open wound 1 (2.7) 0 (0.0) 1 (0.3)
Patella fracture 1 (2.7) 0 (0.0) 1 (0.3)
Pneumonia 1 (2.7) 0 (0.0) 1 (0.3)
Urinary tract infection 1 (2.7) 0 (0.0) 1 (0.3)
Anaemia 0 (0.0) 1 (0.4) 1 (0.3)
Appendicitis 0 (0.0) 1 (0.4) 1 (0.3)
Arthralgia 0 (0.0) 1 (0.4) 1 (0.3)
Page 1 of 2 N = Number of subjects who received ≥ 1 active dose of investigational product
n = Number of subjects reporting ≥ 1 event Includes only treatment-emergent adverse events
Preferred terms are sorted by descending order of frequency in the overall denosumab group and coded using MedDRA Version 14.1.
aSubjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
表 2.7.4.2-4 重篤な有害事象(基本語): 外国 2 試験併合解析(Safety Subjects, Treatment Analysis Phase, Integrated Analysis of Safety)
Preferred Term Study 20040215 Denosumab 120 mg Q4W (N=37) n (%) Study 20062004 Denosumab 120 mg Q4W (N=281) n (%) Overalla Denosumab 120 mg Q4W (N=304) n (%)
Bone giant cell tumour 0 (0.0) 1 (0.4) 1 (0.3)
Central nervous system lesion 0 (0.0) 1 (0.4) 1 (0.3)
Cholelithiasis 0 (0.0) 1 (0.4) 1 (0.3)
Device failure 0 (0.0) 1 (0.4) 1 (0.3)
Device related infection 0 (0.0) 1 (0.4) 1 (0.3)
Endotracheal intubation
complication 0 (0.0) 1 (0.4) 1 (0.3)
Ganglioneuroma 0 (0.0) 1 (0.4) 1 (0.3)
Gastroenteritis 0 (0.0) 1 (0.4) 1 (0.3)
Gun shot wound 0 (0.0) 1 (0.4) 1 (0.3)
Hyperparathyroidism 0 (0.0) 1 (0.4) 1 (0.3)
Musculoskeletal pain 0 (0.0) 1 (0.4) 1 (0.3)
Nephrolithiasis 0 (0.0) 1 (0.4) 1 (0.3)
Nerve compression 0 (0.0) 1 (0.4) 1 (0.3)
Osteonecrosis 0 (0.0) 1 (0.4) 1 (0.3)
Peripheral sensory neuropathy 0 (0.0) 1 (0.4) 1 (0.3)
Presyncope 0 (0.0) 1 (0.4) 1 (0.3)
Respiratory failure 0 (0.0) 1 (0.4) 1 (0.3)
Spinal compression fracture 0 (0.0) 1 (0.4) 1 (0.3)
Spindle cell sarcoma 0 (0.0) 1 (0.4) 1 (0.3)
Tibia fracture 0 (0.0) 1 (0.4) 1 (0.3)
Toxic nodular goitre 0 (0.0) 1 (0.4) 1 (0.3)
Tumour pain 0 (0.0) 1 (0.4) 1 (0.3)
Vena cava injury 0 (0.0) 1 (0.4) 1 (0.3)
Wound 0 (0.0) 1 (0.4) 1 (0.3)
Wound dehiscence 0 (0.0) 1 (0.4) 1 (0.3)
Page 2 of 2 N = Number of subjects who received ≥ 1 active dose of investigational product
n = Number of subjects reporting ≥ 1 event Includes only treatment-emergent adverse events
Preferred terms are sorted by descending order of frequency in the overall denosumab group and coded using MedDRA Version 14.1.
aSubjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
27
表 2.7.4.2-5 重篤な有害事象(基本語): 試験 AMG162-B-J201(Safety Analysis Set, 6-month Cutoff Analysis)
Preferred Term
Denosumab 120 mg Q4W (N = 17)
n (%)
Number of subjects reporting serious adverse events 2 (11.8)
Pain 1 (5.9)
Pneumothorax 1 (5.9)
Page 1 of 1 N = Number of subjects who received ≥ 1 dose of investigational product
n = Number of subjects reporting ≥ 1 event Includes only treatment-emergent adverse events Coded using MedDRA version 15.1
Preferred terms are sorted by descending order of frequency.
Source: Table 15.3-2.10 of Study AMG162-B-J201 CSR
2.1.4
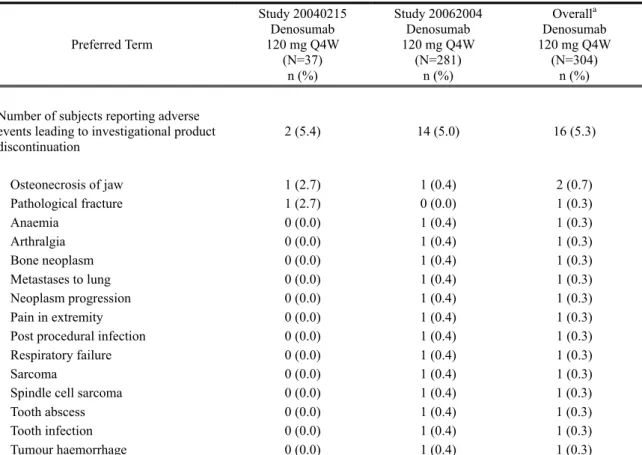
その他の重要な有害事象
2.1.4.1 治験薬の投与中止又は試験中止に至った有害事象 治験薬の投与中止に至った有害事象は、試験 20040215 では 2 名(5.4%)、試験 20062004 で は 14 名(5.0%)で報告された(表 2.7.4.2-6)。両試験を通じて 2 名以上で治験薬の投与中止に 至った有害事象は ONJ のみであった(2 名[0.7%]、試験 20040215 及び 20062004 の各 1 名)。 治験薬との関連性があると判定された有害事象により治験薬の投与を中止した被験者数は 3 名で、その内訳は、試験 20040215 で 1 名(ONJ)、試験 20062004 で 2 名(ONJ 1 件及び関節 痛 arthralgia 1 件)であった(Table tias6-16.2)。有害事象により試験を中止した被験者は、試験 20040215 では 2 名(5.4%)、試験 20062004 では 13 名(4.6%)であった(Table tias6-17.2)。両試験を通じて 2 名以上で試験中止に至った 有害事象は、肺転移 metastases to lung(2 名、試験 20040215 及び 20062004 の各 1 名)及び ONJ
(2 名、試験 20040215 及び 20062004 の各 1 名)のみであった。治験薬との関連性があると判 定された有害事象により試験を中止した被験者は 3 名で、その内訳は、試験 20040215 で 1 名 (ONJ)、試験 20062004 で 2 名(ONJ 1 件、関節痛 arthralgia 1 件)であった(Table tias6-18.2)。 治験薬の投与中止及び試験中止に至った重篤な有害事象は、試験 20062004 で 2 名に認めら れ、その内訳は呼吸不全 respiratory failure(1 名)及び紡錘細胞肉腫 spindle cell sarcoma(1 名) であった(Table tias6-19.2及びTable tias6-21.2)。両事象とも、治験薬との関連性は否定された (Table tias6-20.2及びTable tias6-22.2)。
試験 AMG162-B-J201 では、治験薬の投与中止又は試験中止に至った有害事象は認められな かった(Table 15.3-2.21及びTable 15.3-2.25)。
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
表 2.7.4.2-6 治験薬の投与中止に至った有害事象(基本語): 外国 2 試験併合解析(Safety Subjects, Treatment Analysis Phase, Integrated Analysis of Safety)
Preferred Term Study 20040215 Denosumab 120 mg Q4W (N=37) n (%) Study 20062004 Denosumab 120 mg Q4W (N=281) n (%) Overalla Denosumab 120 mg Q4W (N=304) n (%)
Number of subjects reporting adverse events leading to investigational product discontinuation 2 (5.4) 14 (5.0) 16 (5.3) Osteonecrosis of jaw 1 (2.7) 1 (0.4) 2 (0.7) Pathological fracture 1 (2.7) 0 (0.0) 1 (0.3) Anaemia 0 (0.0) 1 (0.4) 1 (0.3) Arthralgia 0 (0.0) 1 (0.4) 1 (0.3) Bone neoplasm 0 (0.0) 1 (0.4) 1 (0.3) Metastases to lung 0 (0.0) 1 (0.4) 1 (0.3) Neoplasm progression 0 (0.0) 1 (0.4) 1 (0.3) Pain in extremity 0 (0.0) 1 (0.4) 1 (0.3)
Post procedural infection 0 (0.0) 1 (0.4) 1 (0.3)
Respiratory failure 0 (0.0) 1 (0.4) 1 (0.3)
Sarcoma 0 (0.0) 1 (0.4) 1 (0.3)
Spindle cell sarcoma 0 (0.0) 1 (0.4) 1 (0.3)
Tooth abscess 0 (0.0) 1 (0.4) 1 (0.3)
Tooth infection 0 (0.0) 1 (0.4) 1 (0.3)
Tumour haemorrhage 0 (0.0) 1 (0.4) 1 (0.3)
N = Number of subjects who received ≥ 1 active dose of investigational product n = Number of subjects reporting ≥ 1 event
Includes only treatment-emergent adverse events
Preferred terms are sorted by descending order of frequency in the overall denosumab group and coded using MedDRA Version 14.1.
aSubjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
Source: Table tias6-15.2
2.1.4.2 重要な有害事象 低カルシウム血症、ONJ、過敏症と関連する可能性のある有害事象、感染症、悪性新生物、 及び心血管系事象を重要な有害事象として評価した。各事象の評価のために実施した解析を表 2.7.4.1-1に示す。 外国 2 試験併合解析及び 3 試験併合解析でこれらの有害事象の集計に使用した MedDRA(第 14.1 版又は第 15.1 版)用語の一覧を付録 2に示す。
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
29
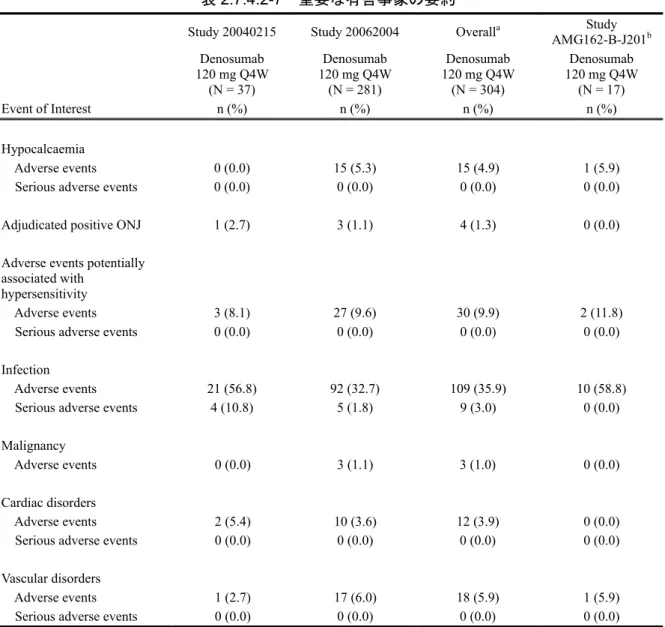
表 2.7.4.2-7 重要な有害事象の要約
Study 20040215 Study 20062004 Overalla AMG162-B-J201Study b Denosumab 120 mg Q4W (N = 37) Denosumab 120 mg Q4W (N = 281) Denosumab 120 mg Q4W (N = 304) Denosumab 120 mg Q4W (N = 17) Event of Interest n (%) n (%) n (%) n (%) Hypocalcaemia Adverse events 0 (0.0) 15 (5.3) 15 (4.9) 1 (5.9)
Serious adverse events 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)
Adjudicated positive ONJ 1 (2.7) 3 (1.1) 4 (1.3) 0 (0.0)
Adverse events potentially associated with
hypersensitivity
Adverse events 3 (8.1) 27 (9.6) 30 (9.9) 2 (11.8)
Serious adverse events 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)
Infection
Adverse events 21 (56.8) 92 (32.7) 109 (35.9) 10 (58.8)
Serious adverse events 4 (10.8) 5 (1.8) 9 (3.0) 0 (0.0)
Malignancy
Adverse events 0 (0.0) 3 (1.1) 3 (1.0) 0 (0.0)
Cardiac disorders
Adverse events 2 (5.4) 10 (3.6) 12 (3.9) 0 (0.0)
Serious adverse events 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)
Vascular disorders
Adverse events 1 (2.7) 17 (6.0) 18 (5.9) 1 (5.9)
Serious adverse events 0 (0.0) 0 (0.0) 0 (0.0) 0 (0.0)
N = Number of subjects who received ≥ 1 dose of investigational product n = Number of subjects reporting ≥ 1 event
Includes only treatment-emergent adverse events and serious adverse events
Coded using MedDRA version 14.1 by preferred term search strategy or standard MedDRA query (SMQ). a
Integrated Analysis (20040215/20062004); Safety Subjects, Treatment Analysis Phase, Integrated Analysis of Safety. Subjects who rolled over from 20040215 to 20062004 or who discontinued 20040215 and re-entered 20062004 are counted only once in the overall column and their analysis period for the overall column will start from study 20040215 and end at study 20062004.
b
From CSR of Study AMG162-B-J201 (6-month Cutoff Analysis)
Source: Table tias6-25.1, Table 1tias4-23.1
2.1.4.2.1 低カルシウム血症
低カルシウム血症は、デノスマブ投与との関連性が知られているリスクであり、現在のラン マークの添付文書では「警告」、「用法・用量に関連する使用上の注意」、「慎重投与」、「重要な 基本的注意」、「重大な副作用」、及び「臨床成績」に記載されている。
2.7.4 臨床的安全性の概要
デノスマブ ランマーク皮下注 120mg
20040215 及び 20062004 のいずれにおいても、カルシウム剤及びビタミン D 製剤の併用投与を 推奨したものの必須とはしなかった。試験 20040215 では、低カルシウム血症の有害事象は認 められなかった。試験 20062004 では、低カルシウム血症の有害事象(カルシウム欠乏 calcium deficiency 及び血中カルシウム減少 blood calcium decreased を含む)が 15 名(5.3%)で報告さ れた。いずれも重篤な有害事象ではなかった(表 2.7.4.2-7)。15 名中 11 名に発現した低カルシ ウム血症は、治験薬との関連性があると判定された(Table tias6-3.2)。重度(治験薬との関連 性なし)は 1 件(被験者 、表 2.7.4.2-8)であり、その他はすべて軽度又は中等度で あった(Table tias6-31.7)。15 名のうち軽度の低カルシウム血症を発現した 1 名(被験者 )が筋痙攣・筋繊維束収縮を併発したが、他に低カルシウム血症に関連して症状を 示した被験者は認められなかった(試験 20062004 治験総括報告書の一覧表 14-6.1.3及び 14-6.2.5)。低カルシウム血症を発現したほとんどの被験者(15 名中 10 名)では報告件数が 1 件であり、多くの事象(25 件中 18 件)は処置の必要がなかった(試験 20062004 治験総括報 告書の一覧表 14-6.2.2)。低カルシウム血症によりデノスマブ投与を中止した被験者は認められ なかった。アルブミン補正血清カルシウム値については第 3.1 項に要約する。 表 2.7.4.2-8 重度の低カルシウム血症の有害事象の要約 Subject ID Age Gender
Brief Description of Subject’s Course on Study
2 years
First dose: Last dose:
On , the subject had a gunshot wound, back and was hospitalized. A computerized tomography scan (CT) showed the bullet in the right anterior axillary line at the chest; liver laceration secondary to gunshot wound with associated subcapsular hematoma. Within this laceration there was an area of increased intensity which may suggest the site of active extravasation. During hospitalization the subject underwent exploratory laparotomy, gastric injury repair, small bowel injury repair, right chest tube thoracostomy placement and appendectomy. The event was resolved on and the subject was discharged from
the hospital on .
Laboratory results of calcium during hospitalization: , 7.8 mg/dL (Grade 2); , 6.5 mg/dL (Grade 3); (76 days from previous dose), 6.8 mg/dL (Grade 3); , 7.6 mg/dL (Grade 2).
Laboratory tests of calcium for study: no Grade 3 or 4. Source: Appendix 11 of CSR, Listing 14-6.2.5 of CSR
試験 AMG162-B-J201 では、高カルシウム血症の場合を除き、併用薬としてビタミン D とカ ルシウムを全被験者に毎日経口投与した。本試験では、非重篤で中等度の低カルシウム血症が 1 名で報告された(表 2.7.4.2-7、Table 15.3-2.34、被験者 )。本事象は、治験薬との関連性 があると判定され(Table 15.3-2.5及びTable 15.3-2.34)、低カルシウム血症に関連した症状は示 day 1* day 393* day 467* 040* day 475* day 476* day 467* day 468* 040* 137* day 469* day 470* 041*