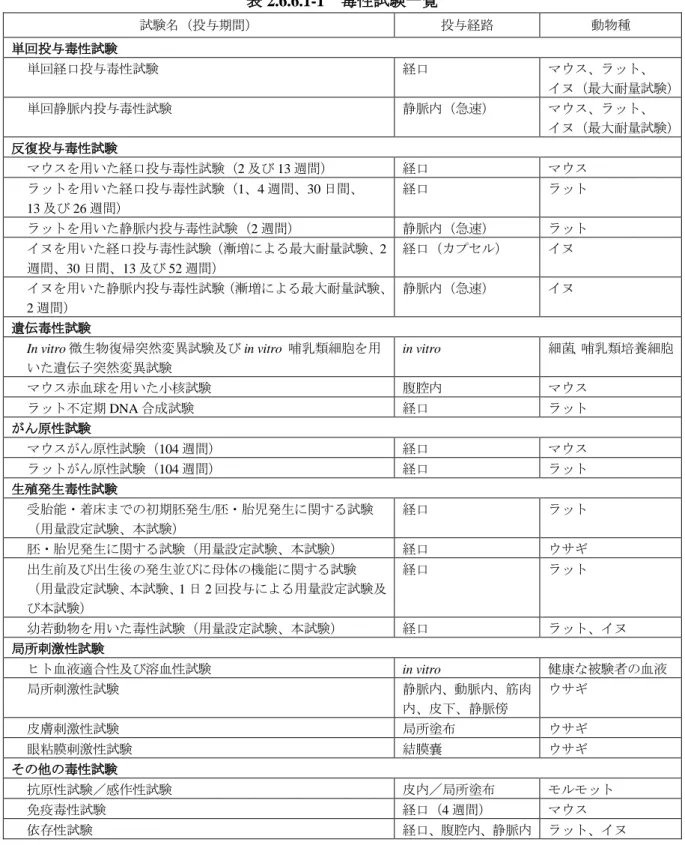
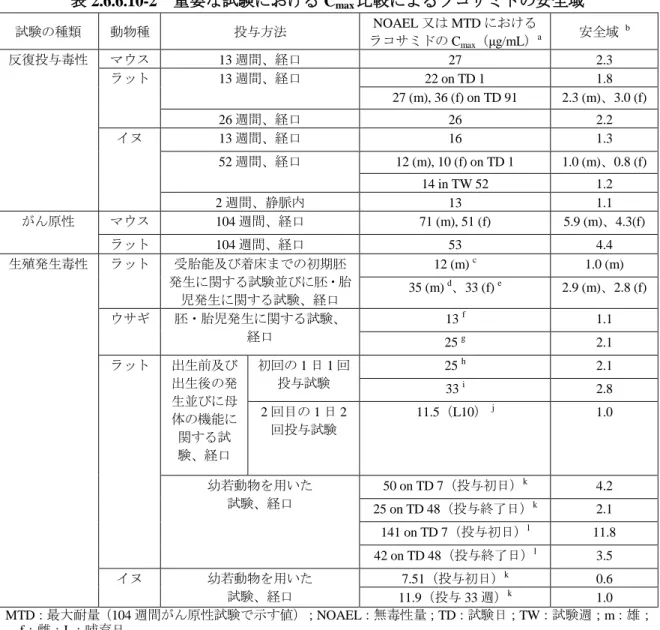
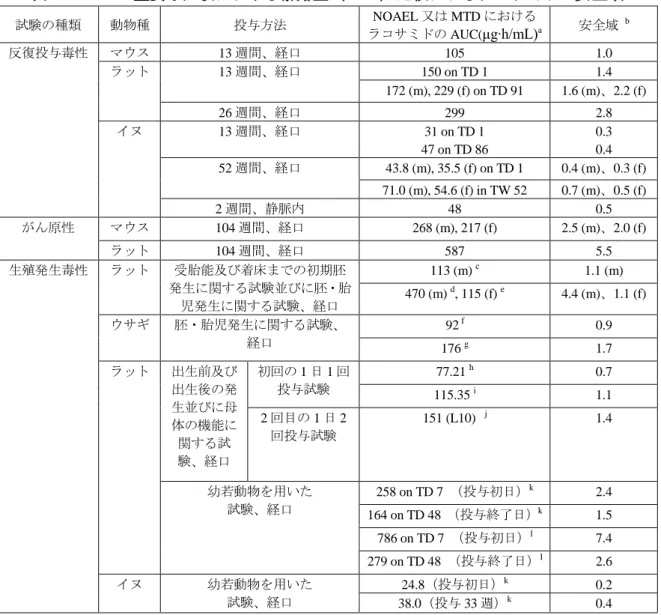
ラコサミド 毒性試験の概要文 Page 毒性試験の概要文 まとめ非臨床毒性試験として 単回投与毒性試験ではマウス ラット及びイヌで 反復投与毒性試験ではマウスで 13 週間まで ラットで 26 週間まで及びイヌで 52 週間まで ラコサミド ( 開発コード
200
0
0
全文
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
図
関連したドキュメント
読書試験の際には何れも陰性であった.而して
実験は,試料金属として融点の比較的低い亜鉛金属(99.99%)を,また不活性ガ
病状は徐々に進行して数年後には,挫傷,捻挫の如き
本実験には,すべて10週齢のWistar系雄性ラ ット(三共ラボラトリ)を用いた.絶食ラットは
毒性の強いC1. tetaniは生物状試験でグルコース 分解陰性となるのがつねであるが,一面グルコース分
試験タイプ: in vitro 染色体異常試験 方法: OECD 試験ガイドライン 473 結果: 陰性.
性状 性状 規格に設定すべき試験項目 確認試験 IR、UV 規格に設定すべき試験項目 含量 定量法 規格に設定すべき試験項目 純度
(b) 肯定的な製品試験結果で認証が見込まれる場合、TRNA は試験試 料を標準試料として顧客のために TRNA
