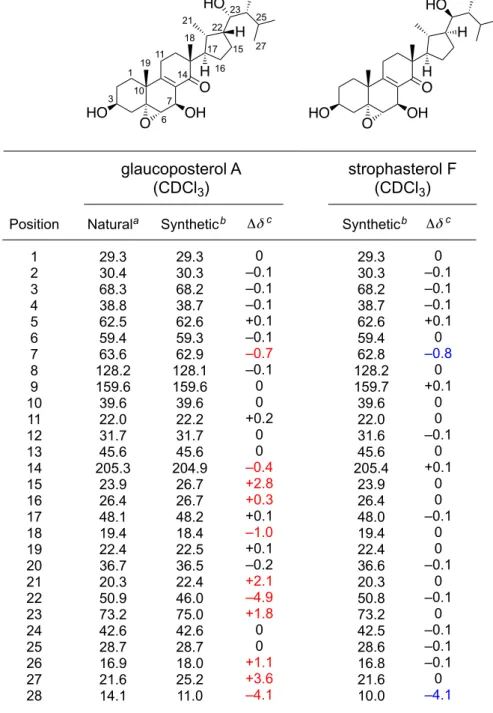
abeo-エルゴスタン型骨格を有するストロファステ
ロール類の合成研究
著者
佐藤 俊太郎
学位授与機関
Tohoku University
学位授与番号
11301甲第19326号
URL
http://hdl.handle.net/10097/00127871
abeo-エルゴスタン型⾻格を有する
ストロファステロール類の合成研究
東北⼤学⼤学院農学研究科
⽣物産業創成科学専攻
⽣物有機化学分野
佐藤 俊太郎
指導教員 桑原 重⽂ 教授
⽬次
緒⾔……… 1 本論 第1章 研究の背景と⽬的………. 4 1.1 転位型ステロイド化合物………. 4 1.2 Strophasterol 類について………..………. 6 1.3 研究⽬的………..………..……. 8 第2章 Strophasterol A及びBの合成研究..………..……..………. 10 2.1 Strophasterol 類の予想⽣合成経路….………..……….……. 10 2.2 Heretsch らによる strophasterol A の合成………..………..……. 11 2.3 Strophasterol A 及び B の合成計画………..………..………. 12 2.4 a-ヒドロキシケトンの合成と D 環の酸化開裂………..…..…..…………. 13 2.5 亜鉛を⽤いた新規変換法の発⾒と D 環の酸化開裂.……..……….……... 16 2.6 D 環開裂の新戦略.……..………..……….…………... 17 2.7 ⼆重結合の異性化の検討とラジカル環化反応による D’環の構築………….. 18 2.8 Strophasterol A 及び B に対応する側鎖の⽴体選択的な構築…...………... 20 2.9 B 環の官能基修飾……….………...………...…... 23 2.10 Strophasterol A の合成……….………...………..…...…... 27 2.11 Strophasterol B の合成……….………...………..…....…... 29 2.12 第 2 章の⼩括………..…………...………....…... 30 第3章 Strophasterol C, E, Fの合成研究.…………....……….... 32 3.1 側鎖への酸素官能基の導⼊の試み……...………..…...………... 32 3.2 ニトリルオキシドの 1,3-双極⼦付加環化を⽤いる D’環及び 23 位ヒドロキシ基の構築………..…...………... 34 3.3 D’環上のケトン基の還元的除去の検討…….…………...…...………... 38 3.4 B 環部エポキシアルコール構造の導⼊…………..……..…...…………... 393.5 Strophasterol C 及び F の合成………..………….…...………... 43 3.6 Strophasterol E の合成………..………….…...………... 44 3.7 第 3 章の⼩括………..………….…...………... 47 第4章 Strophasterol D及びglaucoposterol Aの合成研究……….48 4.1 ジアステレオ選択的な 1,3-双極⼦付加環化反応の検討…... 48 4.2 側鎖の⽴体選択的な構築………..………..…...………... 50 4.3 Glaucoposterol A の提唱構造の合成と⽴体化学の改訂.…………... 53 4.4 Strophasterol D の合成と⽴体化学の決定………..…….……... 56 4.5 第4章の⼩括…………..…...………... 58 総括………...………..…...………... 60 引⽤⽂献..…...………... 61 実験の部.………..………... 65 結⾔..………... 128 謝辞..……….………... 129
略語表
Ac acetyl ADDP 1,1’-(azadicarbonyl)dipiperidine AIBN 2,2’-azobis(isobutyronitrile) Am amyl aq aqueous AZADOL 2-hydroxy-2-azaadamantane 9-BBN 9-borabicyclo[3.3.1]nonane BSTFA N,O-bis(trimethylsilyl)trifluoroacetamide Bu butylCSA 10-camphorsulfonic acid
DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DDH 1,3-dibromo-5,5-dimethylhydantoin DIAD diisopropyl azodicarboxylate DIBAL diisobutylaluminum hydride DIPEA N,N-diisopropylethylamine DMF N,N-dimethylformate DMSO dimethyl sulfoxide
dpm 2,2,6,6-tetramethyl-3,5-heptanedionato dr diastereomeric ratio ee enantiomeric excess eq equivalent Et ethyl HMDS 1,1,1,3,3,3-hexamethyldisilazane IBX 2-iodoxybenzoic acid
Im imidazole
KHMDS potassium hexamethyldisilazide
mCPBA m-chloroperoxybenzoic acid
Me methyl
MMPP magnesium monoperoxyphthalate
MOM methoxymethyl
MRSA methicillin-resistant staphylococcus aureus
MS molecular sieves Ms methanesulfonyl NBP N-bromophthalimide NBS N-bromosuccinimide NBSa N-bromosaccharin NCS N-chlorosuccinimide NIS N-iodosuccinimide
NMO N-methylmorpholine N-oxide NMR nuclear magnetic resonance NOE nuclear Overhauser effect
NOESY nuclear Overhauser effect spectroscopy NPSP N-phenylselenenylphthalimide Ph phenyl PPTS pyridinium p-toluenesulfonate py pyridine quant quantitative rt room temperature
TBAF tetrabutylammonium fluoride TBDPS tert-butyldiphenylsilyl TBHP tert-butyl hydroperoxide TBS tert-butyldimethylsilyl TES triethylsilyl
Tf trifluoromethanesulfonyl TFA trifluoroacetic acid
THF tetrahydrofuran TMS trimethylsilyl Ts p-toluenesulfonyl
V-70 2,2’-azobis(4-methoxy-2,4-dimethylvaleronitrile) VO(acac)2 vanadyl acetylacetonate
緒⾔
幼い頃にアリの⾏列を観察したことがある⼈は多いだろう。当時はこれを不思議な ことだとは思わず眺めていたが、後に集団⾏動をしているアリは視覚が発達しておら ず、触覚によってある化学物質を感知し、それに応答することで⾏列を成すというこ とを知る。これが「道標フェロモン」である。働きアリは餌を⾒つけると、それぞれ の個体が道標フェロモンと呼ばれる特定の化学物質を分泌し、後続のアリがそのフェ ロモンを感知することで、あとに続くと考えられている。アリだけではなく、他の動 物や植物、菌類など、様々な⽣物の⽣命現象にもこのような化学物質が関与している。 天然物化学の分野ではこのような⽣理活性物質の存在や機能を明らかにし、⽣命現象 を解明する研究が⾏われてきた。しかしながら、機能が解明された⽣命現象はごく⼀ 部であり、多くは未解明のままである。アリの道標フェロモンもアリの種によって化 合物の構造は異なり、現在もなお、道標フェロモンの同定に関する研究は続いている。 さて、歴史を遡ると世界で初めて発⾒されたフェロモンは、1959 年にドイツの科学 者である Butenandt によって単離・同定されたボンビコールである。 ボンビコールは雌のカイコガが分泌する性フェロモンであり、その構造は E-Z に共 役した⼆重結合を含む炭素数 16 個の第⼀級アルコールから成っている。このように 単純な構造を有するボンビコールであるが、他の⽣物種には作⽤することなく、カイ コガの雄に特異的に作⽤する。この単純有機化合物をカイコガの雄が如何にして選択 的に受容しているかは多くの科学者の興味を引き、昆⾍の⽣態や⽣活環を物質レベル で解明する研究へと発展していった。⼀⽅で、このような⽣命現象の仕組みを解明で きれば我々の⽣活に応⽤することも可能である。例えば近年、フェロモンの性質を逆 ⼿に取った害⾍防除剤の開発が⾏われている。その仕組みは、農業において害となる 昆⾍の雌が放出する性フェロモンを⼈為的に散布することで雌雄の交信を錯乱し、繁 OH bombykol殖を防ぐというものである。性フェロモンは特定の害⾍にのみ低濃度で強⼒に作⽤す ることから、効果的で低毒性の農薬として普及しつつある。 しかしながらフェロモンをはじめとする天然有機化合物は、⽣物体内ではごく微量 しか⽣産されず、天然からの単離量だけでは詳細な⽣物学的研究やその後の応⽤展開 のためには不⼗分である。そのような研究材料の供給不⾜を補うことは有機合成化学 の役割の⼀つであり、今⽇までに合成化学の技術を駆使した、天然物の標品供給、さ らには実⽤的農薬・医薬品の開発が数多く実施されてきた。農薬や医薬品開発におけ る有機合成化学の⼤きな貢献とは裏腹に、その意義がしばしば疑問視されることもあ るが、合成化学には以下のような⼤きな役割があると考える。 1つ⽬として、新たな反応の開発が挙げられる。⼀⼝に新たな反応と⾔ってもその ⽬指すところは様々であり、新規⾻格の構築や収率・選択性の向上を志向した反応、 操作の簡便性やコストの軽減に繋がる反応、環境⾯に配慮したクリーンな反応など多 岐に渡る。そのどれもが精密な実験操作や条件検討といった試⾏錯誤によってのみ実 現可能であり、AI が発達した現代においても変わることのない有機合成化学の技と ⾔えよう。 2つ⽬として化合物の構造決定がある。単離された天然物の構造は、その構造が複 雑になればなるほど NMR 的な⼿法で決定することが難しくなり、X 線による構造決 定も適⽤できない場合、最後の砦となるのが化学合成による構造決定である。構造決 定の技術が進歩した昨今においても、合成研究による構造改訂の報告は絶えないこと から、有機合成化学が構造決定の分野で依然として重要な位置を占めていることが分 かる。 3つ⽬として天然物の誘導化や部分構造の合成を可能にする点が挙げられる。⽣理 活性物質の活性発現に重要な部分構造(ファーマコフォア)を特定することは化合物 の構造簡略化に繋がり、新たな薬剤をデザインするための⼀歩となる。この部分構造 を特定するための様々な構造修飾や複雑天然物の部分合成は、有機合成化学によって のみ成し得る専売特許である。 以上のように、有機合成化学は有⽤な⽣理作⽤を持つ微量天然物を供給することで ⽣命現象の理解に寄与するとともに、天然物に勝る機能物質を⽣み出すことのできる
重要な研究領域であると⾔えよう。 本博⼠論⽂研究では、キノコ⼦実体形成誘導活性を⽰し、前例のない転位型ステロ イド⾻格を持つ strophasterol A とそのエピマーである strophasterol B、及びそれらの酸 化型類縁体である strophasterol C-F と glaucoposterol A を対象として、有機合成化学の 進捗と⽣物学的研究の進展に寄与することを⽬指し、以下の合成化学的研究を実施し た。
本論
第1章 研究の背景と⽬的 1.1 転位型ステロイド化合物 ステロイド化合物は⻑い研究の歴史を持ち、動物や植物など様々な⽣物のホルモン として働いていることが明らかにされている。1953 年に単離されたアルドステロン は、我々の腎臓や腸の電解質を調整する役割を担う鉱質コルチコイドの⼀種として知 られている(Figure 1)。また、植物の伸⻑促進や発芽促進の役割を担うカスタステロン や昆⾍の変態を促すエクジソンの発⾒により、哺乳動物以外の⽣物の⽣理現象におい てもステロイドホルモンが関与していることが明らかになった。また、世界で初めて 臨床試験が⾏われたステロイド医薬品であるコルチゾンが関節リウマチの治療に⼤ きく貢献したことを⽪切りに、ステロイド化合物は医薬品分野で広く⽤いられるよう になった。これら4つの化合物を含め、多くのステロイド化合物は、A, B, C, D と略 記される4つの環から成る基本⾻格を有している。ところが、⽣合成の過程で炭素鎖 の開裂や炭素⾻格の転位反応が起きることで、そのような基本⾻格から逸脱した天然 ステロイド型化合物の単離・合成研究が近年数多く報告されてる1)。 Figure 1. 4 環性の基本⾻格(ABCD 環)を有するステロイド化合物 A B C D H H O O OH O H Cortisonemedicine for rheumatoid arthritis Castasterone
plant growth-promoting hormone
HO HO H H OH OH OH H O Ecdysone insect molting hormone
H H O H OH HO O OH O Aldosterone mineralocorticoid hormone HO HO H H H O OH HO H H
その代表例として、seco-ステロイドと abeo-ステロイドがある。seco-ステロイドは⽣ 合成の過程で環の切断が起きたステロイド化合物の総称であり、その代表例としてビ タミン D 類が挙げられる(Scheme 1)。ビタミン D 類の⼀種であるビタミン D3の⽣合 成経路については、コレステロールが酸化されて 7-デヒドロコレステロールとなった 後に、紫外線による B 環の開環が起こり、(6Z)-tacalciol となる。その後、⼆重結合の 異性化が進⾏することでビタミン D3が⽣合成されている。即ち、ビタミン D3はステ ロイド⾻格の 9–10 位の結合が開裂した構造を有しており、9,10-secosteroid の⼀つで ある。 Scheme 1. Vitamin D3の⽣合成経路 ⼀⽅、abeo-ステロイドは⽣合成の過程で炭素⾻格の転位反応が起きたステロイド化 合物の総称であり、近年多種多様な⾻格を持つ abeo-ステロイドの単離報告が相次い でいる。例えば 2017 年に単離・構造決定がなされた pleurocin A2)と ganotheaecolin A3) はともに abeo-ステロイド型の⾻格を有しており(Scheme 2)、天然のステロイド化合物 であるエルゴステロールから⽣合成されると考えられている。Pleurocin A については、 エルゴステロールが酸化を受けて 1 となった後、ケトンの⽣成に伴って C9–11 結合が 開裂して 2 となり、最後に 11 位が新たに 7 位と結合形成するという予想⽣合成経路 が提唱されている。このように 11 位の炭素が 9 位から 7 位に転位したエルゴスタン 型の化合物は 11(9→7)abeo-エルゴスタンと呼ばれている。同様に ganotheaecolin A は エルゴステロールの C5–10 の結合が開裂して 3 となった後、11 位が 9 位から 10 位へ HO H H H HO H H H H HO H H H H cholesterol 7-dehydrocholesterol
(6Z)-tacalciol (9, 10-secosteroid)vitamin D3 9 10 7 6 UV light isomerization
転位した 4 を経て、最後に 9 位が 4 位と新たに結合形成しているため、9(11→4), 11(9 →10)diabeo-5,10-seco エルゴスタンに属する。このような abeo-ステロイド型の⾻格を 有する天然物は、ステロイド研究の⻑い歴史の中でも発⾒されることのなかった⾻格 であり、キノコからの単離が多いことも興味深い(pleurocin A と ganotheaecolin A は それぞれ、マツタケ(Tricholoma matsutake)とマンネンタケ(Ganoderma theaecolum)から 単離されている)。
Scheme 2. pleurocin A 及び ganotheaecolin A の予想⽣合成経路
1.2 Strophasterol 類について
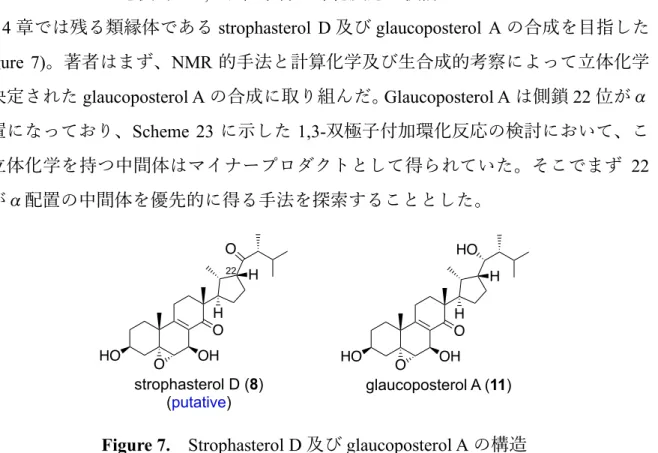
2012 年に静岡⼤学の河岸らは abeo-エルゴスタン型の天然物である strophasterol A–D (5–11)を⾷⽤のキノコのサケツバタケ(stropharia rugosoannulata)から単離した(Figure
2)4)。このうち、5 及び 6 は対応する bis(p-bromo)benzoate 体の X 線結晶構造解析によ
り絶対⽴体配置が決定されたが、7 及び 8 に関しては平⾯構造のみの決定にとどまっ た。その後 2017 年に Vidari らによってアオアシフウセンタケ(Cortinarius glaucopus)か ら 7 及び glaucoposterol A (11)が単離され、NOESY を含む NMR 的⼿法と計算化学に よって両者の⽴体化学がアサインされた5)。さらに 2019 年、7 の 23 位ケトン基の還 pleurocin A 11(9→7)abeo-ergostane O H OH HO HO H H HO R H O HO O 11 9 7 O HO H OH H ganotheaecolin A 9(11→4), 11(9→10)diabeo-5,10-secoergostane O H R O H O R H H O 11 10 5 9 9 4 O O HO O R H H ergosterol 7 11 9 1 2 3 4 [O] [O]
元体である strophasterol E (9)及び F (10)が菊池らによってエリンギ(Pleurotus eryngii)か ら単離され、それらの tris(p-bromo)benzoate 体の X 線結晶構造解析によって絶対⽴体 配置が決定された6)。本化合物群は全て、通常のステロイド⾻格を有するエルゴステ ロールの D 環の C14–15 結合が開裂して、新たに側鎖 22 位と D’環を形成した 15(14 →22)abeo-エルゴスタン構造を有しており、本構造を持つ化合物群は Vidari らによ り”strophastane”と名付けられた。 Figure 2. Strophasterol 類 本化合物群の天然からの単離量はそれぞれ微量である(サケツバタケの乾燥重量 20 kg から 5: 11 mg、6: 1.1 mg、7: 3.7 mg、8: 0.9 mg。エリンギの乾燥重量 11 kg から 9: 1.1 mg、10: 7.2 mg。アオアシフウセンタケの乾燥重量 3.7 kg から 11: 1.6 mg)。そのた め 2–7 は詳細な活性評価を⾏うために⼗分な量を確保することができず、⽣物活性は 未だ不明である。⼀⽅、河岸らにより唯⼀活性評価が⾏われた 5 は⼩胞体ストレスへ の抵抗活性や抗 MRSA 活性に加えて、数種のキノコに対して⼦実体形成誘導活性を ⽰すことが明らかになった7)。さらに、5 は分類学的に異なる 82 種のキノコに、7 は 52 種のキノコに内在していることも確認されたことから、河岸らは本化合物群が未 だ発⾒の報告がないキノコのホルモンではないかという仮説を⽴てている。もしそう であれば、キノコホルモンの⽣理学・⽣化学・化学という新しい研究領域に発展する HO O H OH O C22β: strophasterol A (5) C22α: strophasterol B (6) A B C D' HO O H OH O strophasterol C (7) HO O H OH O strophasterol D (8) (putative) O O HO O H OH O glaucoposterol A (11) HO H HO O H OH O strophasterol E (9) HO HO O H OH O strophasterol F (10) HO H H H H HO H H 22 15 14 15(14→22)abeo-ergostane “strophastane”
可能性を秘めている。 1.3 研究⽬的 キノコは、遡ること古代ローマの時代から⼈々の⾷卓にあったとされている。⽇本 では縄⽂時代から⾷べられてきたという説もあり、昔からキノコは⼈々の⾷⽣活に⽋ かせないものであった。多くの⼈に好まれるが故に、⼈々は早くからキノコの⼈⼯栽 培に取り組み、現在では多くの種類のキノコが⾷卓に並ぶようになった。そもそもキ ノコの⽣活環は以下に⽰すように、まず⼦実体(⾷⽤に供されるいわゆるキノコ)か ら放出された胞⼦が発芽して、核を1つ持つ⼀核菌⽷、それが融合した⼆核菌⽷とな る(Figure 3)。その後⼦実体原基が形成され、それが成⻑することで普段私たちが⽬に する⼦実体となる。ところが、各段階における形態変化は現在までのところ、物質レ ベルでは全く解明されていない。 Figure 3. キノコの⽣活環 河岸らは strophasterol 類がキノコの⽣活環の中で、⼆核菌⽷から⼦実体原基の形成、 もしくは⼦実体原基から⼦実体への⽣⻑のいずれかの段階で作⽤していると考えて いる。もし本化合物群がキノコの⼦実体形成を促す因⼦であるならば、⼈⼯栽培が難 しいマツタケやトリュフなど(外⽣菌根菌)の⼈⼯栽培に道を開くとともに、キノコ Fruiting body Spore Mononuclear myceria Dinuclear myceria Primordium Germination Fusion Primordia formation Growth
の⽣活環を分⼦レベルで解明する端緒にもなりうると考えられる。 Figure 1 でも⽰したように、他の⽣物界においてはステロイドがホルモンとして使わ れていること、キノコには元来、エルゴステロールをはじめとするステロイド類が多 く含まれており、それらを利⽤してホルモンを⽣合成していたとしても不思議ではな いと思われることから、著者も strophasterol 類がキノコの⽣活環を制御するキノコ界 初のホルモンである可能性はあると考えている。また、構造化学的に⾒た場合、 strophasterol 類は、通常のステロイド⾻格の D 環が開裂した後、側鎖との間で新たな 5 員環を形成した、これまでに全く類例のない転位型ステロイド⾻格を有する点で、 ⽣合成及び有機合成の観点からも注⽬に値する天然物であると⾔える。 以上の背景の下、本博⼠論⽂研究では、基礎及び応⽤の両⾯における興味深い活性 に加え、ステロイド研究の⻑い歴史の中でも⾒つからなかった特異な新規構造を有す る strophasterol A-F 及び glaucoposterol A に関して、以下の研究を実施した。
(1)全合成の達成
(2)詳細な⽣物活性評価のための標品供給
(3)NMR で構造決定された strophasterol C 及び glaucoposterol A の⽴体化学の検証 (4)Strophasterol D の⽴体化学の決定
第2章 Strophasterol A及び Bの合成研究 2.1 Strophasterol 類の予想⽣合成経路 Strophasterol 類はステロイド⾻格の C 環と、本化合物群に特徴的な5員環(D’環)が炭 素−炭素単結合により連結した、これまでのステロイド化合物には類を⾒ない特異な ⾻格を有しているが、河岸らは、2011 年にサケツバタケ(Stropharia rugosoannulata)か ら、B 環に strophasterol 類と同⼀のエポキシアルコール構造を有する 12 も単離してい る(Scheme 3)8)。この事実をもとにして、河岸らは本化合物群の⽣合成経路を以下のよ うに推定している3)。 Scheme 3. Strophasterol 類の予想⽣合成経路 即ち、まずキノコに多く含まれるステロイド化合物であるエルゴステロールが酸化 を受けることで 12 となる。次に 12 の D 環がラジカル的に開裂して 13 となった後に、 側鎖の⼆重結合と環化反応を起こし、新たな5員環(D’環)を形成した中間体 15 とな る。環化によって⽣じた炭素ラジカルが⽔素化されると strophasterol A 及び B が⽣成 HO O OH OH H HO O OH O H HO O OH H - H• O HO O OH O HO O OH O O HO O OH O + H• strophasterols A (5) and B (6) strophasterols C (7) and D (8) 12 13 14 15 HO H ergosterol H H [O] [O] H H H D’
し、酸化されると strophasterol C 及び D が⽣成するという経路である。
2.2 Heretsch らによる strophasterol A の合成
河岸らが提唱した予想⽣合成経路に則り、エルゴステロールを出発原料とした strophasterol A の合成が、2016 年に Heretsch らによって報告された(Scheme 4)9)。
Scheme 4. Heretsch らによる strophasterol A の合成
Heretsch らはエルゴステロールを出発原料とし、3⼯程を経て既知の 16 へと導いた。 その後、⼆酸化セレンによるアリル位の酸化と Burgess 試薬を⽤いた脱⽔により、D 環に⼆重結合を導⼊した。続くモノペルオキシフタル酸マグネシウム(MMPP)を⽤い たエポキシ化と PCC によるa-ヒドロキシケトンの形成を経て、17 を合成した。17 を ⽔酸化カリウムで処理すると、⾚の⽮印で⽰した⾻格転位が進⾏して 18 となった後、 19 に⽰すように D 環の開裂に伴い塩素が脱離し、カルボン酸 20 が得られることを⾒ 出した。20 から5⼯程を経てヨウ化物 21 へ変換した後、ラジカル環化反応を⽤いる HO O O OH H HO H ergosterol H strophasterol A (5) R H 16 (known) H O 3 steps R H 17 O O 1) SeO2 2) Burgess 3) MMPP 4) PCC ClO H OH 18 O Cl O O R Cl O R O OH O KOH 20 O O R OH O H O I H AcO 19 AcO O 22 H 4 steps 5 steps Et3B, O2 Bu3SnH 21 A B C D D’ R H H 22
ことで D’環を構築し、22 を単⼀の⽴体異性体として得た。最後に4⼯程で B 環の官 能基修飾を⾏い、strophasterol A の合成を達成した。 しかしながら Heretsch らの合成経路は、21 から 22 に⾄るラジカル環化反応におい て、22b型の⽴体化学を持つ 22 しか得ることができず、22a型の⽴体化学を持つ strophasterol B や glaucoposterol A の合成には適⽤できないという難点があった。 2.3 Strophasterol A 及び B の合成計画 著者はまず、X 線結晶構造解析でその構造が決定され、側鎖に酸素官能基を持たな い Strophasterol A (5)及び B (6)の合成に取り組んだ。合成計画の策定に関しては、 Heretsch らの合成における課題を踏まえ、両者を作り分けることのできる合成経路を ⽴案した(Scheme 5)。 Scheme 5. Strophasterol A 及び B の合成計画 著者はまず 5 及び 6 の D’環及び側鎖を合成し、その後 B 環に官能基を導⼊するとい う順序で合成することとした。出発原料としては、河岸らの予想⽣合成経路に基づき RO OH O OH RO O O RO O radical cyclization oxidative cleavage HO O O OH 25 26 27 strophasterol A (5) stereodivergent formation of D’-ring H H H H H H H H H H H H D' B-ring functionalization RO H Rubottom oxidation 24 H H H HO O O OH H RO OH H H H 22-epi-27 strophasterol B (6) B-ring functionalization 22 HO H ergosterol O OH 23 (known) O H H H H 6 steps
エルゴステロールを選択した。市販のエルゴステロールから6⼯程を経て既知のケト ン 23 へ導いた後、ルボトム酸化反応に付すことでa-ヒドロキシケトン 24 が得られる と考えた。24 の D 環を酸化開裂することで、ケトカルボン酸 25 に変換できるはずで ある。25 のカルボキシル基を活性化してアシルラジカルを発⽣できれば、側鎖⼆重結 合との 5-exo-tirg 型のラジカル環化反応を経て新規⾻格の特徴を成す D’環が合成でき ると考えた。次に D’環上のケトンを利⽤し、これを除去する過程で⼆種の異性体 27 及び 22-epi-27 を作り分け、27 に対し B 環に官能基修飾を施すことで 5 の合成が達成 できると考えた。また同様の変換により、22-epi-20 からは 6 の合成が達成できると考 えた。 2.4 a-ヒドロキシケトンの合成と D 環の酸化開裂 実際の合成について以下に記す。市販のエルゴステロールを出発原料とし、(1)の条 件で塩酸を⽤いて⼆重結合を異性化し、(2)の条件で A 環のヒドロキシ基を Ac 基で保 護して 28 とした(Scheme 6)。28 に対し Jones 試薬を作⽤させ、g-ヒドロキシ-a,b-不飽 和エノン 29 へと導いた。その後酸性条件下で亜鉛を⽤いて三級ヒドロキシ基を還元 的に除去して 30 を得た後、(1)の条件で Ac 基を除去し、(2)の条件でバーチ還元によ るエノンの 1,4-還元を⾏い、既知のケトン 31 を合成した10)。31 に対し、(1)の条件で シリルエノールエーテルへと変換し、(2)の条件でルボトム酸化を⾏うことで D 環に a-ヒドロキシケトン構造を導⼊することができた。⽣じたヒドロキシ基の⽴体化学は 決定していないが、32 は単⼀の⽴体異性体として得られた。その後 A 環のヒドロキ シ基を TBDPS 基で保護し、酸化開裂前駆体 33 の調製が完了した。
Scheme 6. a-ヒドロキシケトン 33 の合成 次に、33 の D 環の開裂を試みた(Table 1)。⼀⾒すると本反応は単純な酸化開裂に⾒ えるかもしれないが、ステロイド D 環の選択的な開裂はこれまで⾮常に例が乏し く、本反応の検討も困難を極めた。entry 1–3 に⽰すように、過ヨウ素酸ナトリウム 11)や四酢酸鉛12)、塩化ルテニウム13)を⽤いた⼀般的な条件では反応は全く進⾏しな かった。種々検討を重ねた結果、entry 4 に⽰すように、Jones 試薬を作⽤させた時の み良好な収率で D 環が開裂し、ケトカルボン酸 34 が得られることを⾒出した14)。 HO H ergosterol AcO H O OH AcO H AcO H O HO H O 1) HCl, MeOH CHCl3, 70 ºC 2) AcCl, pyridine rt, 2 steps 90% Jones reagent acetone, rt, 44% Zn, H2SO4 MeOH/CH2Cl2 rt, 86% 1) K2CO3, MeOH H2O, 60 ºC, 73% 2) Li, liq. NH3 THF, –78 ºC, 80% H H H 28 29 30 31 (known) H H H H H H 1) TMSCl, HMDS LiI, CH2Cl2, rt 2) mCPBA, NaHCO3 CH2Cl2, rt 2 steps 54% RO H O OH 32 (R = H) 33 (R = TBDPS) H H H TBDPSCl, Im. CH2Cl2, rt A B C D
Table 1. D 環の酸化開裂の検討① 以上のように、ステロイド⾻格 D 環の開裂法を⾒出すことはできたが、本合成ルー トは出発原料のエルゴステロールから 34 まで 10 ⼯程と⻑く、総収率も 7.8%と満⾜ のいくものではなかった。また、Jones 試薬を⽤いてg-ヒドロキシ-a,b-不飽和エノンを 合成する反応(28→29)が⼤量系においては収率が低下する(5 g 以上のスケールで ⾏うと収率が 20%まで低下)という問題もあり、研究⽬的の⼀つである標品供給を考 えると難の残るものであった。そこで、28 の D 環の⼆重結合を選択的に酸化開裂で きれば、これらの問題を解決することができると考え、検討を⾏った(Table 2)。 Table 2. D 環の酸化開裂の検討② TBDPSO H O OH 33 H H H TBDPSO O OH O H H H H 34 entry 1 2 3 4 conditions NaIO4, acetone, 60 ºC Pb(OAc)4, AcOH, 60 ºC
NaIO4, RuCl3, CCl4, MeCN, H2O, 50 ºC Jones reagent, acetone, rt
results no reaction no reaction no reaction 79% (2 steps) AcO H 28 H Table AcO O OH O H H 35 entry 1 2 3 4 5 conditions
OsO4, py., NaIO4, 1,4-dioxane, H2O, rt NaIO4, RuCl3, CCl4, MeCN, H2O, rt NaIO4, KMnO4, THF, H2O, 0 to 50 ºC OsO4, Pb(OAc)4, 1,4-dioxane, H2O, 60 ºC O3, CH2Cl2, Me2S, –78 ºC results no reaction no reaction no reaction no reaction many products Table
しかしながら entry 1–4 に⽰す条件では反応が全く進⾏せず、entry 5 に⽰すように、 オゾン分解を⽤いたときには反応点を制御できず、多くの⽣成物を与える結果となっ た。以上の検討結果から、本合成経路では短⼯程化や収率改善が困難であると判断し、 新たな合成ルートを探索することとした。 2.5 亜鉛を⽤いた新規変換法の発⾒と D 環の酸化開裂 新規合成経路を探索している中で著者は、興味深い反応を発⾒した。先に⽰した合 成経路の中で、g-ヒドロキシ-a,b-不飽和エノン 29 に対し、亜鉛を⽤いてヒドロキシ 基を除去し、30 を得ていた。ところがこの反応において亜鉛を⼤過剰量⽤いたと き、ヒドロキシ基が還元的に除去されただけの 30 が消失し、D 環に⼆重結合を有す る 38 が得られることを⾒出した(Scheme 7)。 Scheme 7. 亜鉛を⽤いた新規変換法の発⾒ 38 の⽣成は次のような反応が連続的に起こった結果であると考えられる。即ち、29 のヒドロキシ基が還元的に除去されて 30 となった後に、エノンの 1,4–還元が起こる ことでケトン 36 となる。さらにケトンの還元が進⾏して 37 となり、⽣じたヒドロ キシ基が酸により脱⽔することで D 環に⼆重結合が導⼊された 38 が⽣成したと考え た。亜鉛を⽤いたこのような変換反応はこれまでに類例がなく、ステロイド⾻格に おける新たな変換反応を⾒出すことができた15)。 AcO H O OH AcO R H O Zn (60 eq.) H2SO4 MeOH/CH2Cl2 rt, 63% H H 29 30 H AcO R H O H 36 H H H AcO R H OH H H H H AcO H H H H 1,4-reduction of enone reduction of ketone 37 38 R
得られた 38 は D 環に⼆重結合を有しており、これを利⽤することで D 環の酸化開 裂が可能であると考えた。まず、38 に対しモノペルオキシフタル酸マグネシウム (MMPP)を⽤いて D 環の⼆重結合をエポキシ化した(Scheme 8)。⽣じたエポキシドの ⽴体化学は決定していないが、39 は単⼀の⽴体異性体として得られた。39 に対し、 Jones 試薬を作⽤させると、a-ヒドロキシケトンの時と同様に速やかに D 環が開裂 し、良好な収率でケトカルボン酸 35 を得ることができた。 Scheme 8. D 環の酸化開裂 以上により原料のエルゴステロールから6⼯程で 35 を合成することができたが、 38 の原料である 29 を得るための Jones 酸化が最⾼でも 44%と低収率であったため、 さらなる合成経路の改善が必要と判断した。 Scheme 8 の結果から、38 を合成することができれば良好な収率で 35 に導くことが できるという知⾒が得られたため、著者は 38 をより効率的に得る⽅法を探索するこ ととした。 2.6 D 環開裂の新戦略 著者はこれまでの検討の中でエルゴステロールを塩酸で処理することで、⼆重結合 が C 環及び D 環に異性化した 40 を得ていた(Scheme 6)16)。⼀⽅ Corey らは、エルゴ ステロールをバーチ還元の条件に付すことによって、B 環の⼆重結合の⼀⽅が還元 AcO R H H H H 38 AcO R H H H H 39 AcO O OH O H H H H 35 O MMPP EtOH CH2Cl2, rt Jones reagent acetone, rt 2 steps 61% R =
された 41 が合成できることを報告している(Scheme 9)17)。これらの知⾒をもとに著 者は、41 に対し酸を作⽤させれば⼆重結合が D 環まで異性化して 38 が得られるの でないかと期待した。その後 Scheme 8 で確⽴した⼿法を⽤いて D 環の⼆重結合を選 択的に酸化開裂することで 35 へ導こうと考えた。 Scheme 9. D 環開裂の新戦略 2.7 ⼆重結合の異性化の検討とラジカル環化反応による D’環の構築 市販のエルゴステロールに対し、Corey らの条件でバーチ還元を⾏い、A 環のヒドロ キシ基を Ac 化することで 42 へと導いた(Scheme 10)。42 に対し、種々の酸を⽤いて ⼆重結合の異性化を検討した。Table の entry 1, 2 ではトルエン溶媒でカンファースル ホン酸もしくはトシル酸を⽤いたが、⼆重結合が C 環に異性化した 43 のみが得られ る結果となった。そこで entry 3 ではキシレンを溶媒として、より⾼温条件で反応を⾏ った。すると、マイナープロダクトではあるが、⼆重結合が D 環まで異性化した 38(約 20%)が得られることが分かった。しかし、⾼温条件ではこれ以上の収率の向上が困難 であったため、entry 4‒6 ではより酸性度の強い、濃塩酸やトリフルオロメタンスルホ ン酸を⽤いてより低温で反応を⾏った。しかしこの場合も同様に、⽬的とする 38 が マイナープロダクトとして得られるのみであった。その後も種々の酸を⽤いて検討を ⾏った結果、entry 7 に⽰すように、クロロホルム溶媒中、‒20 ºC で塩化⽔素ガスを作 HO R H 40 H HO R H H H H HCl/MeOH CHCl3, 60 ºC (Scheme 6) Li, liq. NH3 t-AmOH THF –78 ºC 41 isomerization ? AcO R H 38 H H H HO H ergosterol H AcO O OH O H 35 H H H H H R
⽤させることにより、38 の⽣成⽐を増加させることができた。最終的に entry 8 に⽰ すように、ジクロロメタン溶媒中、‒78 ºC で塩化⽔素ガスを作⽤させることにより 38 と 43 の⽣成⽐が逆転することを⾒出し、望みの 38 を 71%の収率で得ることができた 18)。また 38 と 43 は硝酸銀含有シリカゲルカラムクロマトグラフィーにより分離可能 であり、43 に対し再度塩化⽔素ガスを作⽤させることで 38 への異性化が可能である ことも分かった。 Scheme 10. ⼆重結合の異性化 望みの 38 が得られたので、続いて Scheme 8 で確⽴した⼿法を⽤いて D 環の酸化開 裂を⾏い、カルボン酸 35 を合成した(Scheme 11)。これによりエルゴステロールから 5⼯程、通算収率 42%で共通中間体 35 の合成を達成し、⼯程数及び収率の問題を解 決することができた。また各反応は 10 g 以上のスケールで⾏っても収率を損なうこ となく進⾏したため、以降の合成経路を進めるための基盤が整った。 HO H ergosterol AcO H
1) liq. NH3, Li, t-AmOH THF, -78 ºC, 99% 2) AcCl, pyridine rt, 98% 42 H H H H Table AcO H 38 H H H AcO H 43 H H
+
entry 1 2 3 4 5 6 7 8 conditions CSA, toluene, 110 ºC TsOH•H2O, toluene, 110 ºC TsOH•H2O, xylene, 140 ºC conc. HCl, CHCl3, 60 ºC conc. HCl, CHCl3, –20 ºC TfOH, CHCl3, –20 ºC HCl gas, CHCl3, MgSO4, –20 ºC HCl gas, CH2Cl2, –78 ºC results 43 (~90%) 43 (~90%) 38 (~20%) + 43 (~70%) 38 (~15%) + 43 (~70%) 38 (~20%) + 43 (~70%) 38 (~15%) + 43 (~70%) 38 (~40%) + 43 (~50%) 38 (71%) + 43 (26%) Table separable HCl gas, CH2Cl2, –78 ºC, 50%⼤量供給が可能となった 35 を対応するセレノエステル 4419)とした後にラジカル条 件に付すと、⽣じたアシルラジカルと側鎖⼆重結合との間で 5-exo-trig 型のラジカル 環化反応が円滑に進⾏し、Dʼ環を有する 45 を単⼀の⽴体異性体として得ることがで きた 20)。45 の⽴体化学については、点線の枠内に⽰した NOE 相関により決定した。 本環化反応は、側鎖が擬エクアトリアルとなる配座で進⾏するために単⼀の⽴体異性 体を与えたと考えられる。以上より、strophasterol 類の炭素⾻格の構築が完了したの で、次に strophasterol A 及び B に対応する側鎖の⽴体選択的な構築法の検討に移った。 Scheme 11. ラジカル環化反応による D’環の構築 2.8 Strophasterol A 及び B に対応する側鎖の⽴体選択的な構築 Scheme 11 で得られたジケトン 45 の D’環上のケトンを除去する過程で側鎖の⽴体化 学を作り分ける検討を開始した(Scheme 12)。45 に対し、まず(A)に⽰したように室温 でチオアセタール化を⾏うと、Dʼ環上のケトン基選択的なチオアセタール化と同時に 22 位のエピマー化も進⾏し、46 と 22-epi-46 が 1.9:1 の⽐率で得られた。これらを分 AcO H 38 H H H MMPP EtOH/CH2Cl2 rt, 82% AcO H H O Jones reagent acetone rt, 77% H H 39 AcO O OH O H PhSeCl Bu3P, Et3N THF, rt, 93% AcO O H O Bu3SnH, AIBN benzene 80 ºC, 66% 35 H H H H H H 45 single diastereomer D D' oxadative cleavage of D-ring O SePh O H H 44 AcO H H R H H3C H O H H H NOE H H radical cyclization
離後、それぞれに対し Raney-Ni を⽤いた脱硫反応を⾏い、strophasterol A の⽴体化学 を有する 47 と B の⽴体化学を有する 22-epi-47 を得た。47 は X 線結晶構造解析によ り構造決定を⾏った。⼀⽅ 45 に対し、(B)に⽰したように低温でチオアセタール化を ⾏うと 22 位のエピマー化が進⾏しないことを⾒出し、46 を単⼀異性体として得るこ とができた。これにより strophasterol A と同じ 22b型の⽴体化学を有する 47 を⽴体選 択的に合成することができた。 Scheme 12. 側鎖の⽴体選択的構築①
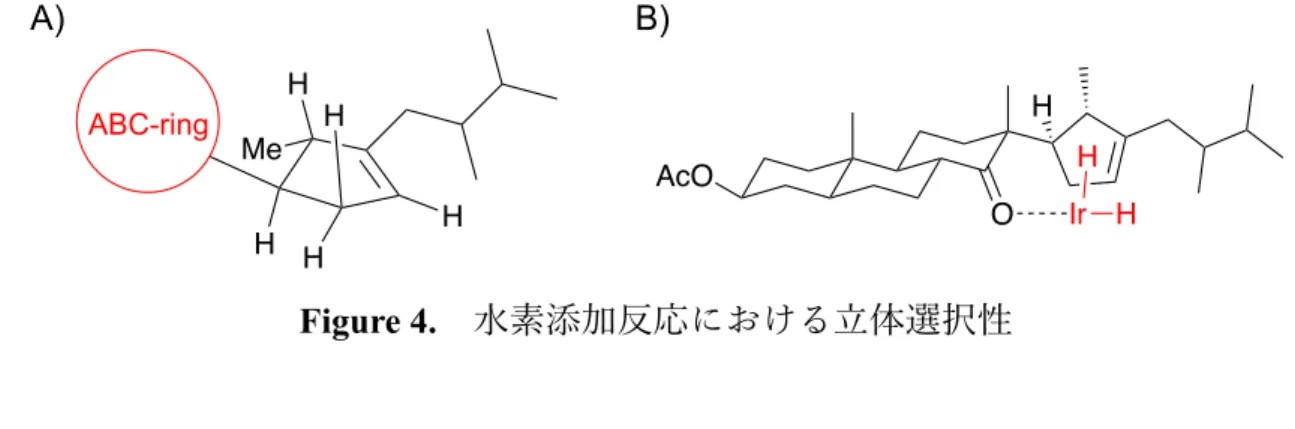
続いて strophasterol B と同じ 22a型の⽴体化学を持つ 22-epi-47 の⽴体選択的な合成 を⽬指した(Scheme 13)。まず 45 に対し、(1)の条件で Dʼ環上のケトンを位置選択的に ビニルスルフィド21)へと変換した後に、(2)の条件で Raney-Ni を⽤いて脱硫し、48 へ と導いた。当初、48 に対する⽔素添加反応は、20 位のメチル基の⽴体障害により紙 ⾯⼿前側から進⾏し、22-epi-47 を主⽣成物として与えるだろうと期待した。しかしな がら Table の entry 1–3 に⽰すように、パラジウム炭素を⽤いた⽔素添加反応はいずれ の溶媒を⽤いたときも、期待に反して 47 を単⼀もしくはほぼ単⼀の⽣成物として与 えた。これは 20 位のメチル基よりも ABC 環部位の⽴体障害の寄与が⼤きいことを意 味している(Figure 4A)。entry 4–7 ではジイミド還元や Wikinson 触媒を⽤いた⽔素添加
AcO O O 45 H H H H AcO O 46 H H H H S S AcO O H H H H S S AcO O 47 (X-ray) H H H H AcO O 22-epi-47 H H H H HS SH HS SH A) BF3•OEt2, CH2Cl2 rt, 2 h, 95% 46/22-epi-46 = 1.9:1 B) BF3•OEt2, CH2Cl2 –40 to –20 ºC, 95% 5 h, 46 only H 2, Raney-Ni EtOH, 50 ºC 8 h, 66% H2, Raney-Ni EtOH, 50 ºC 8 h, 65% 22-epi-46 22 + + D’ (separable) ≣
反応を試みたが、反応は進⾏しなかった。entry 8では Shenvi らによって報告された ⼀電⼦的な⽔素化反応 22)を⽤いたが、やはり 47 を主⽣成物として与える結果となっ た。そこで C 環上のケトン基に着⽬し、酸素官能基に対して配位能を有する Crabtree 触媒を⽤いれば⽴体選択性が逆転するのではないかと期待した(Figure 4B)。実際に反 応を⾏ったところ entry 9, 10 に⽰すように、Crabtree 触媒を 0.12 当量⽤いると 47 と 22-epi-47 の⽐率が 1:1 に変化し、さらに 0.07 当量まで触媒量を減らすと両者の⽐率が 逆転し、1:1.7 となることが判明した。幸いにも、47 と 22-epi-47 はシリカゲルカラム クロマトグラフィーで分離可能であり、strophasterol B 型の⽴体化学を有する 22-epi-47 についても、優先的に得る⽅法を⾒出すことができた。なお、触媒の量を 0.025 当 量まで減じると、反応の収率そのものが低下した(entry 11)。 Scheme 13. 側鎖の⽴体選択的構築② AcO O H H entry 1 2 3 4 5 6 7 8 9 10 11 conditions H2, Pd/C, AcOEt, rt H2, Pd/C, EtOH, rt H2, Pd/C, hexane, rt
H4N2•H2O, AcOH, EtOH, O2, rt to reflux
H4N2•H2O, H2O2, EtOH, rt to reflux
H2, Wilkinson’s cat., CH2Cl2, rt
H2, Wilkinson’s cat., EtOH, rt to 50 ºC
Mn(dpm)3, PhSiH3, TBHP, i-PrOH, rt
H2, Crabtree’s cat. (0.12 eq.), CH2Cl2, rt
H2, Crabtree’s cat. (0.07 eq.), CH2Cl2, rt
H2, Crabtree’s cat. (0.025 eq.), CH2Cl2, rt
results 47/22-epi-47 = 21:1 47/22-epi-47 = 20:1 47 only (92%) no reaction no reaction no reaction no reaction 47/22-epi-47 = 3:1 47/22-epi-47 = 1:1 47/22-epi-47 = 1:1.7 (95%) low yield Table AcO O H H 47 22-epi-47 H H H
+
H AcO O O 45 H H H H AcO O 48 H H H H 1) EtSH, TMSCl CH2Cl2, rt 88% 2) H2, Raney-Ni EtOH, 50 ºC 83% 20 D’ TableFigure 4. ⽔素添加反応における⽴体選択性 2.9 B 環の官能基修飾 D’環及び側鎖の⽴体選択的構築が完了したので、strophasterol A 及び B の合成の達成 まで、残るは B 環の官能基修飾のみとなった。まず、strophasterol A の⽴体化学を有 する 47 を対応するシリルエノールエーテル 49 に変換した後に、B 環に⼆重結合を導 ⼊する経路(49→50)を検討した(Scheme 14)。 Scheme 14. C 環ケトンに対するシリルエノールエーテル化の検討 しかしながら entry 1–4 に⽰すように、47 に対するシリルエノールエーテル化は進⾏ しなかった。これは 10 位及び 13 位に存在する2つのメチル基の⽴体障害により、ケ トンα位の⽔素が引き抜かれにくかったことに加え、⽣成物である 42 のシリルオキ シ基と D’環部分の⽴体反発が⼤きいことが原因と考えられた。そこで次に 47 のエノ ン 50 への直接的な酸化を試みた(Table 3) H H Me H H H ABC-ring O AcO Ir H H H A) B) AcO O 47 H H H entry 1 2 3 4 conditions TMSOTf, Et3N, THF, 0 ºC to rt HMDS, LiI, TMSCl, CH2Cl2, reflux KHMDS, TMSCl, THF, 0 ºC to rt BSTFA, Et3N, THF, 0 ºC to rt results no reaction no reaction no reaction no reaction Table AcO OTMS 49 H H H Table F3C OTMS N OTMS BSTFA 10 13 AcO O 50 H H
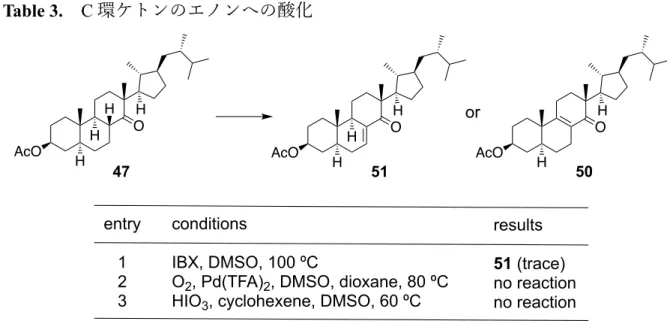
Table 3. C 環ケトンのエノンへの酸化 entry 1 では⼀般的な IBX 酸化の条件に付した。この時、痕跡量ではあったが 51 を 得ることができた。そこで entry 2 では、IBX 酸化の別法として知られるパラジウム触 媒と酸素を⽤いた⼿法 23)、entry 3 ではヨウ素酸を⽤いた⼿法 24)を試みた。しかしな がらいずれの条件でも反応は進⾏せず、⽬的物は得られなかった。これらの検討結果 より、C 環ケトンの直接的なエノンへの酸化は困難であると判断し、次にケトンα位 の臭素化の検討を⾏った(Table 4)。 Table 4. C 環ケトンの臭素化 AcO O 47 H H H H entry 1 2 3 conditions IBX, DMSO, 100 ºC
O2, Pd(TFA)2, DMSO, dioxane, 80 ºC HIO3, cyclohexene, DMSO, 60 ºC
results 51 (trace) no reaction no reaction AcO O 51 H H H AcO O 50 H H or AcO O 47 H H H H entry 1 2 3 conditions NBS, p-TsOH•H2O, CH2Cl2, reflux Py•Br3, AcOH, 50 ºC
PhNMe3•Br3, THF then DBU, 50 ºC
results no reaction no reaction 50 (89%) AcO O 52 H H H Br AcO O 50 H H
entry 1, 2 に⽰すように、NBS や Py•Br3を臭素化剤として⽤いたときは反応が全く進 ⾏しなかった。これに対し、entry 3 の PhNMe3•Br3を⽤いたときには速やかにケトン α位が臭素化されることを発⾒した25)。そこで、ワンポットで DBU を加えたところ、 臭素の脱離により⼆重結合が導⼊され、エノン 50 を 89%の収率で得ることができた。 続いて B 環アリル位への官能基導⼊に取り組んだ(Scheme 15)。まず、エノン 50 を酸 化クロムを⽤いたアリル位の酸化反応に付したところ、望みの B 環アリル位ではな く、C 環のアリル位にケトン基が導⼊された 53 が得られるのみであった。そこで次 に Wohl–Ziegler 臭素化の条件を⽤いて、アリル位のラジカル的な臭素化を試みた。ま ずは entry 1 に⽰すように、NBS と AIBN を⽤いた⼀般的な条件で臭素化を⾏ったと ころ、所望の位置での臭素化が進⾏し、54 を 42%の収率で得ることができた 26)。こ の時副⽣成物として⼆箇所のアリル位が共に臭素化されたジブロモ体 55 が得られて いることが分かった。そこで分解点がより低いラジカル開始剤である V-70 を⽤い、 より温和な条件での臭素化を試みたところ、収率が 50%まで向上した(entry 2)。さら に臭素化剤の検討を重ねたところ、entry 5 に⽰すように、DDH を⽤いると収率を 61% まで改善できることを⾒出した。
Scheme 15. B 環アリル位の官能基化 次に得られたアリルブロミド 54 の臭素の脱離による⼆重結合の導⼊に取り組んだ (Scheme 16)。まず、54 に対し DBU を作⽤させて脱臭素化し、ジエン 56 を得ようと 試みたが、実際に反応を⾏うと、主⽣成物として得られたのは望みの 56 ではなく、B 環から⽔素が引き抜かれ、縦に⼆重結合が導⼊された 57 であった。また、57 に対し、 酸や塩基の添加や求核種の付加による 56 への異性化を試みたが、反応は進⾏しなか った。そこで、54 の臭素をフェニルセレネニルアニオンで求核置換して、アリルセレ ニド中間体 58 とした後に、セレノキシドへの酸化と脱離をワンポットで⾏ったとこ ろ、望みのジエン 56 を単⼀⽣成物として得ることができた。 AcO O H Br H byproduct (55) Br entry 1 2 3 4 5 conditions NBS, AIBN, CCl4, 80 ºC NBS, V-70, CCl4, 40 ºC NBP, V-70, CCl4, 40 ºC NBSa, V-70, CCl4, 40 ºC DDH, V-70, CCl4, 40 ºC results 42% 50% 45% 53% 61% Table N Br O O N N O O Br Br N S O Br O O NBP DDH NBSa N N NC CN MeO OMe V-70 AcO O H H 53 AcO O H H 50 AcO O H H 54 Br Table O CrO3 CH2Cl2 rt
Scheme 16. ジエン構造の構築 2.10 Strophasterol A の合成 最後に、ジエン部分を⾜がかりとして strophasterol 類に特徴的な構造である B 環の エポキシアルコール部位の構築を⽬指した(Scheme 17)。まず、56 に対しジオール化を 試みたが、種々条件を検討しても反応は進⾏せず、望む 59 は得られなかった。次に ブロモアセトキシ化を⾏ったところ、1.7 : 1 の⽐率で望むジアステレオマー60 とその bis-epi 体 61 が得られた27)。この⽴体選択性は 10 位のメチル基と臭素との⽴体反発に よって⽣じていると思われ、より原⼦半径が⼤きいヨウ素を⽤いることで⽴体選択性 を向上できると考えた。そこでヨードヒドロキシ化と⽣じたヒドロキシ基の TMS 保 護を⾏ったところ、予想通りに⽴体選択性は向上し、ヨードヒドリン TMS 保護体 62 と 63 を 5.5 : 1 の⽐率で得ることができた。本反応は、ヨウ素が 10 位のメチル基との ⽴体反発を避けるように紙⾯奥側から接近し、⽣成した三員環型陽イオン中間体に⽔ がアリル位側から攻撃することで進⾏したと解釈できる。そのような三員環型陽イオ ンの開環は⼀般的には不利であるため(Fürst-Plattner rule)28)、反応はアリルカチオニッ クな化学種を経ているものと推測している。 AcO O H H 56 AcO O H H 54 Br PhSeSePh NaBH4 THF/EtOH rt AcO O H H DBU, THF rt, 80% 56/57 = 1 : 4 56 AcO O H H 57
+
isomerization AcO O H H SePh then H2O2 rt, 74% 58Scheme 17. ジエンに対する官能基修飾 望みの⽴体化学を有するヨードヒドリン TMS 保護体 62 が得られたので、5–6 位に ⼆重結合を導⼊するべく、ヨウ素をヒドロキシ基に変換することを試みた(Scheme 18)。 まず mCPBA を⽤いたヒドロキシ基への変換を試みた。即ち、mCPBA によりヨウ素 が酸化を受けてヨードソ化合物 64 となった後に、系中の⽔による求核置換が進⾏す ることによって 65 へ変換できると考えた29)。ところが実際に反応を⾏うと、予想し ていた 65 は得られず、驚くべきことに stropahsteol 類のエポキシアルコール構造を有 する 67 が単⼀の⽴体異性体として⼀挙に得られることが分かった。これは、ヨウ素 が mCPBA による酸化を受けて 64 となった後に、次亜ヨウ素酸の syn 脱離に伴い⼆ 重結合が⽣成して 66 となり、最後に過剰に⼊っていた mCPBA により⽴体的に空い AcO O H H 56 K2OsO4•2H2O NMO t-BuOH, H2O rt to 60 ºC AcO O H HOH OH 59 O 1) NIS, H2O acetone, 0 ºC 2) TMSOTf 2,6-lut., rt AcO O H AcO O H OTMS I I OTMS H H 62 (2 steps 49%) 63 (2 steps 9%) OH OH TS 62 TS 63 R I AcO R AcO I δ δ O 10
+
NBS, AgOAc AcOH, rt 60/61 = 1.7 : 1 AcO O H AcO O H OAc Br Br OAc H H 60 61+
10ている紙⾯奥側からエポキシ化が進⾏したと考えれば説明できる30)。この予想外の反 応の進⾏により、strophasterol 類に特徴的なエポキシアルコール構造を効率的に合成 する⼿法を確⽴することができた。最後に⽔酸化カリウムを⽤いて Ac 基と TMS 基 を⼀挙に除去することで strophasterol A (5)の合成を達成した。合成品の各種スペクト ルデータ及び⽐旋光度は天然物と良い⼀致を⽰した4)。 Scheme 18. Strophasterol A の合成 2.11 Strophasterol B の合成 次に strophasterol A (5)の 22 位に関するエピマーである strophasterol B (6)の合成を⽬ 指した。Crabtree 触媒を⽤いた⽔素添加反応(Scheme 13)の際に主⽣成物として得られ た 22-epi-47 に対し、strophasterol A の合成と同様の⼿法を⽤いて合成を⾏った(Scheme 19)。即ち、epi-47 の C 環ケトンα位の臭素化と DBU による脱離によりエノン 22-epi-50 へと変換した後、続くアリル位の臭素化により 22-epi-54 とした。その後セレネ ニル基の付加とセレノキシドの脱離により⼆重結合を導⼊し 22-epi-56 を得、ヨード AcO O H OTMS I H O 64 AcO O H OTMS 66 IOH AcO O H OTMS O HO O H OH O KOH MeOH, rt 82% 67 strophasterol A (5) mCPBA NaHCO3 CH2Cl2 rt, 78% Oxidation of iodide syn-Elimination of hypoiodous acid Stereoselective epoxidation AcO O H OTMS I H 62 AcO O H OTMS OH H 65 mCPBA AcO OH OTMS I H 64 O H2O 5 6 lit. [α]22 D = +2.3 (c 0.93, MeOH) [α]20 D = +2.3 (c 0.26 MeOH)
ヒドロキシ化と TMS 保護により 22-epi-62 を得た。先に⾒出したエポキシ環構築法は 本基質においても良好な収率で進⾏し、エポキシド 22-epi-67 を得ることができた。 最後に2つの保護基を⼀挙に除去し、strophasterol B (6)の初の合成を達成した。合成 品の各種スペクトルデータ及び⽐旋光度は天然物と良い⼀致を⽰した4)。 Scheme 19. Strophasterol B の合成 2.12 第 2 章の⼩括 第2節では、キノコ⼦実体形成誘導活性を⽰す新規ステロイド誘導体 strophasterol A とその 22 位エピ体である strophasterol B の合成に取り組んだ(Scheme 20)。市販のエル ゴステロールを出発原料とし、塩化⽔素ガスによる B 環から D 環への⼆重結合の異 性化反応を⾒出すとともに、これまで⾮常に例が乏しかったステロイド D 環の酸化 開裂を Jones 試薬を⽤いることにより達成した。これによりエルゴステロールから5 ⼯程 42%で効率的に seco-ステロイド型カルボン酸 28 を調製することができた。その AcO O R H OTMS O HO O H OH O KOH MeOH, rt 82% 22-epi-67 strophasterol B (6) AcO O R H OTMS I H 22-epi-62 AcO O H H 22-epi-47 H H AcO O R H H 22-epi-56 1) NIS, H2O acetone, 0 ºC 2) TMSOTf 2,6-lut., rt 2 steps 47% mCPBA NaHCO3 CH2Cl2, rt 81% PhNMe3•Br3 THF, rt then DBU 50 ºC, 85% AcO O R H H 22-epi-50 AcO O R H H 22-epi-54 Br DDH, V-70 CCl4, 40 ºC 51% PhSeSePh NaBH4 THF/EtOH then H2O2, rt 71% lit. [α]22 D = –15 (c 0.08, MeOH) [α]20 D = –16.1 (c 0.20 MeOH) 22 R
後ラジカル環化反応により strophastane ⾻格の特徴を成す D’環を備えた 38 を単⼀の ⽴体異性体として得た。⽣じた D’環上のケトン基を除去する過程でパラジウムまた は Crabtree 触媒を⽤いることで、2 種の異性体 40 と 22-epi-40 を作り分けた。最後に ヨードヒドリン保護体を経由した効率的な B 環へのエポキシアルコール構造の導⼊ に成功し、strophasterol A の合成を原料のエルゴステロールから 17 ⼯程、通算収率 2.3%、strophasterol B の合成を 17 ⼯程、通算収率 1.3%で達成した。 Scheme 20. 第 2 章の⼩括 AcO O H 35 H H H HO H ergosterol H AcO O H O H H H 45 HO O H OH O isomerization of double bond radical cyclization HO O H OH O strophasterol A (5) strophasterol B (6) D’ D B-ring functionalization stereodivergent reduction OH O AcO O H H H 47 AcO O H H H H B-ring functionalization 22-epi-47 22 oxidative cleavage Pd/C Crabtree’s cat. Total 17 steps 2.2% yield Total 17 steps 1.3% yield B
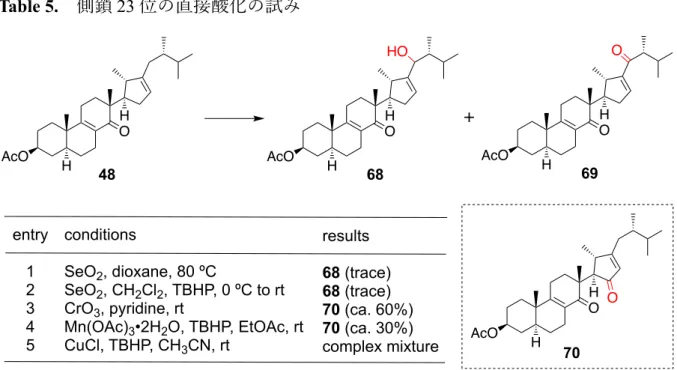
第3章 Strophasterol C, E, Fの合成研究 3.1 側鎖への酸素官能基の導⼊の試み 第 3 章では strophasterol A (5)と同様の 22b配置を持つ類縁体である、strophasterol C (7), E (9), F (10)の合成を⽬指した(Figure 5)。これらは側鎖 23 位に酸素官能基(ヒド ロキシ基もしくはケトン基)を有しており、これらを如何にして導⼊するかが合成 の鍵である。著者は、5 の合成中間体を⽤いることで、本化合物群を効率的かつ網羅 的に合成することができると考え、合成研究に取り組んだ。 Figure 5. Strophasterol C, E, F の構造 まず、先に合成した中間体 48 (Scheme 13)のアリル位を直接酸化することで側鎖に酸 素官能基を導⼊しようと考えた(Table 5)。Table の entry 1, 2 では⼆酸化セレンを⽤い たアリル位の酸化を⾏ったが、⽬的とする 68 は痕跡量しか得られなかった。次に entry 3 に⽰す酸化クロムを⽤いた酸化や、entry 4 に⽰す Mn(OAc)3•2H2O を⽤いた酸化 31) を試みたが、共に D’環上のアリル位が酸化された 70 が得られるのみであった。また entry 5 に⽰す、塩化銅と TBHP を⽤いた酸化条件 32)では反応系中が複雑化する結果 となった。以上より、48 に対するアリル位の酸化は困難であることが分かった。 HO O H OH O strophasterol C (7) O HO O H OH O strophasterol E (9) HO HO O H OH O strophasterol F (10) HO H H H 22 23
Table 5. 側鎖 23 位の直接酸化の試み 次に、D’環を形成する環化反応の際に、側鎖 23 位に官能基を導⼊する取り組みを⾏ った(Scheme 21)。まず、strophasterol A 及び B を合成した際の中間体であるカルボン 酸 35 をチオエステル 71 とした後、PhNMe3•Br3を⽤いて C 環ケトンのα位を臭素化 し、その後 DBU で処理することにより、エノン 72 へと変換した。72 を福⼭還元反 応に付すことでアルデヒド 73 とした。まず著者はこのアルデヒドに対し、23 位への ハロゲンの導⼊を伴う分⼦内プリンス環化反応を⾏おうと考えた。即ち、ルイス酸と して四塩化スズを⽤いて、プリンス環化反応による D’環の構築と側鎖への塩素原⼦ の導⼊を試みた 33)。その結果、⽬的とする環化体 74 をジアステレオマー混合物とし て得ることができたものの、低収率にとどまった。反応条件を精査しても収率の向上 が⾒込めなかったため、環化⽅法を変更することにした。アルデヒド 73 を還元して アルコール 75 へと導き、アッペル反応を⾏うことでヨウ化物 76 を得た。76 をラジカ ル条件に付すことで、原⼦移動を伴う 5-exo-trig 型のラジカル環化反応を⾏い、77 を 合成しようと考えた。(A)の条件ではビストリブチルスズと AIBN34)、(B)の条件では酸 素とトリエチルボラン35)を⽤いたラジカル反応を試みたが、両条件とも原料は消失し たものの、⽬的物は得られなかった。副⽣成物として、通常のラジカル環化反応が進 ⾏した、側鎖に官能基を持たない化合物(47, Scheme 12)が得られるのみであった。以 上より、プリンス環化反応やラジカル環化反応では収率よく側鎖に官能基を導⼊する AcO O H H 70 entry 1 2 3 4 5 conditions SeO2, dioxane, 80 ºC SeO2, CH2Cl2, TBHP, 0 ºC to rt CrO3, pyridine, rt Mn(OAc)3•2H2O, TBHP, EtOAc, rt CuCl, TBHP, CH3CN, rt results 68 (trace) 68 (trace) 70 (ca. 60%) 70 (ca. 30%) complex mixture AcO O H H 48 AcO O H H 68 AcO O H H 69 O HO O +
ことが困難であることが分かったので、新たな合成経路を探索することとした。 Scheme 21. D’環の構築と 23 位のハロゲン化の試み 3.2 ニトリルオキシドの 1,3-双極⼦付加環化を⽤いる D’環及び 23 位ヒドロキシ基の 構築 これまでの検討結果を受けて、次に、ニトリルオキシドの 1,3-双極⼦付加環化反応を ⽤い、D’環の構築と 23 位ヒドロキシ基の導⼊を同時に⾏う⽅法について検討を⾏う こととした(Scheme 22)。即ち、先に合成したアルデヒド 73 (Scheme 21)をまずオキシ ム 78 へ変換する。その後、オキシムの酸化によって⽣じるニトリルオキシドと側鎖 ⼆重結合との 1,3-双極⼦環化付加反応を利⽤することで、Dʼ環の構築と側鎖への酸素 官能基の導⼊を⼀挙に⾏うという計画である。その後、⽣じた 79 のイソオキサゾリ ン環の還元的開裂により、23 位にヒドロキシ基が導⼊された 2 種の異性体 80 及び 81 に導く。 AcO O OH O H 35 H H H EtSH, EDC DMAP CH2Cl2 rt, 92% AcO O R H 73 H H AcO O SEt O H 71 H Pd/C Et3SiH acetone, rt 2 steps 58% R AcO O SEt O H 72 H R H H Me3NPhBr3 THF, rt then DBU 50 ºC O AcO O R H 75 H OH AcO O 74 H Cl OH AcO O R H 76 H I AcO O 77 H I SnCl4 CH2Cl2, 0 ºC low yield NaBH4, MeOH 0 ºC, 84% I2, Im. PPh3 Et2O, rt 74% A ) (Bu3Sn)2 AIBN PhH, 80 ºC B) O2, Et3B PhH, 0 ºC Intramolecular Prince Cyclization Atom Transfer Radical Cyclization R
Scheme 22. ニトリルオキシドの 1,3-双極⼦付加環化を⽤いる D’環及び 23 位ヒドロキシ基の構築 実際の合成について以下に⽰す(Scheme 23)。35 から 3 ⼯程を経て導いたアルデヒド 73 と、ヒドロキシルアミンの縮合により対応するオキシム 78 を調製した。オキシム 78 に対し、酸化によるニトリルオキシドの⽣成とその 1,3-双極⼦付加環化反応の検討 を⾏った。Table の entry 1 では酸化剤として NCS を⽤いた36)。その結果、1,3-双極⼦ 付加環化反応が速やかに進⾏し、79A と 79B の混合物が 2.7:1 の⽐率で得られること が分かった(収率 51%)。79A の優先的⽣成は、20 位のメチル基が擬エクアトリアル位 に位置する遷移状態 TS 79A が擬アキシアル位に位置する TS71B よりも有利な遷移状 態であると考えると説明できる(Figure 6)。なお、79A と 79B は分離が困難であったた め、化合物の⽐率は1H NMR スペクトルの積分⽐から算出している。続いて、entry 2‒ 8 では酸化剤として次亜塩素酸ナトリウム⽔溶液もしくは次亜塩素酸ナトリウム五⽔ 和物を⽤いて検討を⾏った。このとき entry 2, 3 や entry 5, 6 に⽰すように、環化の⽴ 体選択性は反応温度に影響を受け、低温条件で選択性がやや向上する結果となった。 これに加え、本環化反応は反応溶媒によって⼤きく影響を受けることがわかった。即 ち、entry 7 のように THF を⽤いたとき系中は複雑化し、低収率に留まったものの、 entry 8 のように塩化メチレンを⽤いたときは収率、選択性ともに向上した。最終的に AcO O AcO O 1,3-dipolar cycloaddition 79 81 H H H H D' N isoxazoline ringopening
O AcO O 80 H H HO H H HO O O AcO H O OH 73 H H H AcO N OH 78 H OH oxime formation B
entry 5 に⽰すように、トルエンを⽤いたときに最も良い結果を与えることを⾒出した。 以上より、収率・選択性共に最も良かった entry 5 を 1,3-双極⼦付加環化の最適条件 として選択した。 Scheme 23. ニトリルオキシドの 1,3-双極⼦付加環化を⽤いる D’環の構築と 23 位酸素官能基の導⼊ Figure 6. 1,3-双極⼦付加環化反応の⽴体選択性 AcO O H 78 H N OH NH2OH•HCl pyridine EtOH, rt quant. R N H O H H H 1,3-dipolar cycloaddition AcO O H O H 73 H nitrile oxide Table entry 1 2 3 4 5 6 7 8 reagents NCS, Et3N NaOCl aq. NaOCl aq. NaOCl aq. NaOCl•5H2O NaOCl•5H2O NaOCl•5H2O NaOCl•5H2O results (79A : 79B) 2.7 : 1 (51%) 3.8 : 1 (86%) 3.0 : 1 (80%) 5.3 : 1 6.4 : 1 (90%) 5.0 : 1 many spot 2.8 : 1 Table AcO O H H 79A N O D’ AcO O H H 79B N O H H
+
temp. 0 ºC to rt 0 ºC rt 0 ºC to rt 0 ºC to rt 80 ºC –78 ºC to rt –78 ºC to rt solv. CH2Cl2 CH2Cl2 CH2Cl2 toluene toluene toluene THF CH2Cl2 strophasterol C, E, F strophasterol D glaucoposterol A 35 3 steps 23 O H N O H H AcO O N O H AcO H H TS 79A TS 79B 20次に得られた 79A 及び 79B のイソオキサゾリン環の還元的加⽔分解によるアルドー ル型化合物 80 及び 81 の調製について検討を⾏った(Table 6)。2種の⽴体異性体 79A 及び 79B は分離困難であったため、混合物のまま開環反応に付した。まず entry 1 に ⽰すようにモリブデンヘキサカルボニルを⽤いた環開裂37)を試みたところ、複雑な混 合物を与える結果となった。entry 2, 3 では⽔素及び Pd/C と弱酸(酢酸もしくはホウ 酸)による開裂38)を試みたが、いずれの場合においても⽬的物を得ることはできなか った。次に entry 4, 5 に⽰すようにラネーニッケルを⽤いた加⽔素分解に付した39)。 この時、酸として酢酸を加えると系中が複雑化したが、ホウ酸を加えると収率良く⽬ 的物が得られることが分かった。得られた 80 及び 81 はシリカゲルカラムクロマトグ ラフィーで分離可能であり、両異性体の⽴体化学は図に⾚で⽰した NOE 相関によっ て決定した。 Table 6. イソオキサゾリン環の開裂反応 entry 1 2 3 4 5 conditions Mo(CO)6, CH3CN, H2O, rt to 80 ºC H2, Pd/C, AcOH, MeOH, H2O, rt H2, Pd/C, H3BO3, THF, MeOH, H2O, rt to 50 ºC H2, Raney-Ni, AcOH, MeOH, H2O, rt
H2, Raney-Ni, H3BO3, MeOH, H2O, rt results many products many products no reaction many products 80 (81%) + 81 (13%) AcO O H H 79A N O AcO O H H 79B N O H H
+
AcO O 81 H H AcO O 80 H H HO H H HO O O+
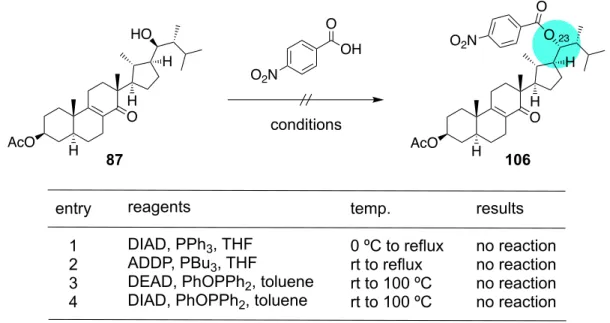
inseparable separable H H H NOE3.3 D’環上のケトン基の還元的除去の検討 次に strophasterol C, E, F の⽴体化学を持つ 80 の Dʼ環上のケトン基を還元的に除去す る検討を⾏った(Scheme 24)。まず(A)に⽰す条件でチオアセタール化を試みたところ ⽬的の 82 は得られず、側鎖のヒドロキシ基が脱離した 83 とそこからさらにチオアセ タール化が進⾏した 84 が得られる結果となった。また、過塩素酸リチウムを⽤いた より温和な(B)の条件では反応が全く進⾏しなかった 40)。そこで⼀旦側鎖のヒドロキ シ基を保護しようと Table に⽰す 3 つの条件を検討したが、⽬的の 85 は得られず、全 て側鎖のヒドロキシ基が脱離し、83 が⽣成する結果となった。以上の結果より、側鎖 のケトン基存在下では 23 位のヒドロキシ基が容易に脱離してしまうことが分かった。 Scheme 24. D’環上のケトン基の還元的除去の試み そこで、Dʼ環上のケトンをアルコールに還元してから除去しようと考えた(Scheme
25)。還元剤を種々検討した結果、Table の entry 1, 2 に⽰すように NaBH4や DIBAL を
AcO O H H 80 O H HO AcO O H H 82 HO S S H AcO O H H 85 (R = TBS or MOM) O H RO entry 1 2 3 conditions TBSCl, Im., DMF, 60 ºC TBSOTf, 2,6-lut., CH2Cl2, –60 ºC MOMCl, DIPEA, CH2Cl2, rt results 83 83 83 Table AcO O H H 84 S S AcO O H H 83 O A) BF3•OEt2, CH2Cl2 –40 ºC to rt B) LiClO4, Et2O –40 ºC to rt Table HS SH HS SH 23