酵素を検出標的あるいは検出手段として用いたバイ オ分析のための分子システムの創製
登, 貴信
https://doi.org/10.15017/1931882
出版情報:九州大学, 2017, 博士(工学), 課程博士 バージョン:
権利関係:
工学府
材料物性工学専攻 博士論文
酵素を検出標的あるいは検出手段として用いた バイオ分析のための分子システムの創製
平成 30 年 3 月
登 貴信
1-1. 検出ターゲットとしての酵素 ... 1
1-1-1. 創薬・治療標的としての酵素 -プロテインキナーゼ- ... 1
1-1-2. 分子標的薬としてのプロテインキナーゼ阻害剤 ... 3
1-1-3. プロテインキナーゼ阻害剤のハイスループットスクリーニング ... 5
1-1-4. 従来のプロテインキナーゼ活性検出法 ... 5
1-1-5. 蛍光増強を利用したプロテインキナーゼ活性検出システム ... 8
1-1-6. レシオメトリーを用いたプロテインキナーゼ活性検出 ... 10
1-2. 検出ツールとしての酵素 ... 11
1-2-1. バイオマーカーとしての膜タンパク質 ... 11
1-2-2. 膜タンパク質を検出ターゲットとした一細胞解析技術 ... 13
1-2-3. フローサイトメトリーによるマルチカラー解析 ... 13
1-2-4. フローサイトメトリーにおける従来の染色法 -蛍光抗体法-... 15
1-2-5. バイオマーカー検出のための酵素 -酵素反応を利用した免疫染色法- ... 16
1-3. 本小論の構成 ... 17
1-4. 参考文献 ... 19
第 2 章 量子ドットと蛍光修飾基質ペプチドを利用した プ ロ テ イ ン キ ナ ー ゼ 活 性 検 出 シ ス テ ム... 25
2-1. 緒言 ... 25
2-1-1. 融合タンパク質を用いた FRET 型プロテインキナーゼ活性検出 ... 25
2-1-2. 量子ドットを用いた FRET 型プロテインキナーゼ活性検出 ... 26
2-1-3. 錯形成によるリン酸基認識を用いた FRET 型プロテインキナーゼ活性検出 ... 27
2-1-4. 静電相互作用を利用したナノ粒子によるプロテインキナーゼ活性検出 ... 29
2-1-5. 量子ドットと蛍光修飾ペプチドの静電相互作用および錯形成に基づく F R E T 型 プ ロ テ イ ン キ ナ ー ゼ 活 性 検 出 シ ス テ ム の 提 案 ... 31
2-2. 静電相互作用に基づく検出システムについての結果及び考察 ... 34
2-2-1. 蛍光修飾ペプチド (TAMRA-Kemptide) の合成 ... 34
2-2-2. 励起波長の決定と FRET ペアとしての相性 ... 35
2-2-3. 蛍光修飾ペプチドと QD の混合モル比 (Peptide/QD 比) の決定 ... 36
2-2-4. PKA 精製酵素を用いた酵素活性検出 ... 39
2-2-5. PK 阻害剤スクリーニングへの応用検討 ... 40
2-3-3. Phos-tag-QD605 に対する亜鉛イオンの影響 ... 46
2-3-4. 蛍光修飾ペプチドと QD の混合モル比 (Peptide/QD 比) の決定 ... 47
2-3-5. Phos-tag によるリン酸基の認識 ... 49
2-3-6. リン酸化率に対する蛍光強度の変化 ... 50
2-3-7. PKA 精製酵素を用いた酵素活性検出 ... 51
2-4. 結言 ... 52
2-5. 静電相互作用に基づく検出システムについての実験項 ... 55
2-5-1. 試薬 ... 55
2-5-2. 蛍光修飾ペプチド (TAMRA-Kemptide) の合成 ... 56
2-5-3. 蛍光スペクトルと吸収スペクトルの測定 ... 56
2-5-4. QD と蛍光修飾ペプチド間の FRET ... 57
2-5-5. PKA による TAMRA-Kemptide (S) のリン酸化後の蛍光スペクトル測定 ... 57
2-5-6. PK 阻害剤の IC50 値の決定 ... 57
2-6. 錯形成に基づく検出システムについての実験項 ... 58
2-6-1. 試薬 ... 58
2-6-2. 蛍光修飾ペプチド (Cy5-Kemptide) の合成 ... 58
2-6-3. Phos-tag 修飾 QD605 (Phos-tag-QD605) の調製 ... 59
2-6-4. 蛍光スペクトルと吸収スペクトルの測定 ... 59
2-6-5. QD と蛍光修飾ペプチド間の FRET ... 59
2-6-6. PKA による Cy5-Kemptide (S) のリン酸化後の蛍光スペクトル測定 ... 59
2-7. 参考文献 ... 60
第 3 章 蛍光修飾ポリマーを用いた濃度消光に基づく プ ロ テ イ ン キ ナ ー ゼ 活 性 検 出 シ ス テ ム... 65
3-1. 緒言 ... 65
3-1-1. 消光基を利用したプロテインキナーゼ活性検出 ... 65
3-1-2. 集合体形成による濃度消光に基づいたプロテインキナーゼ活性検出 ... 66
3-1-3. 検出シグナルと内部標準シグナルを有する蛍光プローブ ... 69
3-1-4. 内部標準シグナルを有する蛍光プローブを用いた 濃度消光に基づくプ ロ テ イ ン キ ナ ー ゼ 活 性 検 出 法 の 提 案 ... 70
3-2-3. 各蛍光修飾ポリマーの吸収スペクトルの測定 ... 75
3-2-4. PIC の粒子径測定 ... 76
3-2-5. PIC 形成による各蛍光強度の変化 ... 77
3-2-6. PKCα 精製酵素を用いた酵素活性のリアルタイム検出... 78
3-2-7. PKCα 阻害剤スクリーニングへの応用検討 ... 80
3-3. 結言 ... 82
3-4. 実験項 ... 83
3-4-1. 試薬 ... 83
3-4-2. アジ化ペプチドの合成 ... 84
3-4-3. Dex-Alkyne (Compound 1) の合成 ... 85
3-4-4. Dex-Cy5 (Compound 2) の合成 ... 85
3-4-5. Dex-Cy5-Alphatomega (Cationic polymer) の合成 ... 86
3-4-6. pAsp-EDA (Compound 4) の合成 ... 86
3-4-7. pAsp-TAMRA (Anionic polymer) の合成 ... 87
3-4-8. PIC の粒子径測定 ... 87
3-4-9. PIC の蛍光強度測定 ... 87
3-4-10. PKCα によるリン酸化に対する蛍光強度の経時変化... 88
3-4-11. PKCα 阻害剤の IC50 値の決定 ... 88
3-5. 参考文献 ... 89
第 4 章 膜タンパク質検出の高感度化に向けた 膜アンカー型アルカリフォスファターゼ基質による 細胞蛍光染色システム ... 93
4-1. 緒言 ... 93
4-1-1. フローサイトメトリーの現状とその問題点 ... 93
4-1-3. ビオチン修飾抗体と蛍光修飾ストレプトアビジンを用いた染色法 ... 95
4-1-4. 蛍光基内包リポソームを用いた染色法 ... 96
4-1-5. 蛍光ポリマーと抗体の複合体を利用した染色法 ... 97
4-1-6. 酵素による増感を利用した染色法 (Catalyzed reporter deposition: CARD) ... 98
4-1-8. 増感酵素 アルカリフォスファターゼ ... 101
4-2. 結果及び考察 ... 103
4-2-1. 蛍光性膜アンカー型アルカリフォスファターゼ基質の設計と合成 ... 103
4-2-2. Methyl-β-cyclodextrin による基質の分散性向上 ... 105
4-2-3. リポソームを用いた脂質二重膜への分子の分配評価 ... 106
4-2-4. Methyl-β-cyclodextrin の細胞染色能への影響 ... 107
4-2-5. アルキル鎖長の違いによる細胞染色能の変化 ... 109
4-2-6. 阻害剤による内在性アルカリフォスファターゼ活性の抑制 ... 110
4-2-7. 細胞固定による内在性アルカリフォスファターゼ活性の抑制 ... 111
4-2-8. CD20 を標的とした CARP 法による細胞染色とフローサイトメトリー検出... 113
4-2-9. EGFR を標的とした CARP 法による細胞染色とフローサイトメトリー検出 .... 115
4-2-10. CARP 法における細胞染色能の基質濃度依存性 ... 116
4-3. 結言 ... 117
4-4. 実験項 ... 119
4-4-1. 試薬 ... 119
4-4-2. 膜アンカー型アルカリフォスファターゼ基質及び酵素反応後の分子の合成 ... 120
4-4-3. ビオチン化抗体及びビオチン化 AP の調製 ... 121
4-4-4. 蛍光修飾抗体の調製 ... 121
4-4-5. 細胞培養 ... 121
4-4-6. 基質 1-p 及び分子 1 の分散性評価 ... 122
4-4-7. リポソームを用いた脂質二重膜への分子の分配評価 ... 122
4-4-8. 阻害剤による内在性アルカリフォスファターゼ活性の抑制 ... 122
4-4-9. 細胞固定による内在性アルカリフォスファターゼ活性の抑制 ... 123
4-4-10. 膜タンパク質を標的とした CARP 法による細胞染色とその検出 ... 123
4-5. 参考文献 ... 125
第 5 章 低発現膜タンパク質を標的とする一細胞解析に向けた 膜アンカー型 β-ガラクトシダーゼ基質による細胞蛍光染色システム ... 131
5-1. 緒言 ... 131
5-1-1. 増感酵素 β-ガラクトシダーゼ ... 131
5-1-2. Enzyme-Linked ImmunoSorbent Assay における β-ガラクトシダーゼの利用 ... 132
膜アンカー型 β-ガラクトシダーゼ基質による細胞蛍光染色システム ... 137
5-2. 結果及び考察 ... 139
5-2-1. 蛍光性膜アンカー型 β-ガラクトシダーゼ基質の設計と合成 ... 139
5-2-2. Methyl-β-cyclodextrin による基質の分散性向上 ... 141
5-2-3. Methyl-β-cyclodextrin の細胞染色能への影響 ... 144
5-2-4. 染色分子の細胞内局在評価 ... 146
5-2-5. CARP 法による膜タンパク質検出を利用した最適な基質とその濃度の検討 ... 147
5-2-6. CARP 法による膜タンパク質検出を利用した酵素濃度の最適化 ... 150
5-2-7. CARP 法による膜タンパク質検出を利用した染色反応時間の検討 ... 150
5-2-8. CD20 を標的とした CARP 法と従来の染色法の比較 ... 152
5-2-9. CD33 を標的とした CARP 法と従来の染色法の比較 ... 153
5-3. 結言 ... 155
5-4. 実験項 ... 157
5-4-1. 試薬 ... 157
5-4-2. 膜アンカー型 β-ガラクトシダーゼ基質及び酵素反応後の分子の合成... 158
5-4-3. ビオチン化抗体の調製 ... 159
5-4-4. 蛍光修飾抗体の調製 ... 159
5-4-5. 細胞培養 ... 159
5-4-6. 基質及び酵素反応後分子の分散性評価 ... 159
5-4-7. 基質及び酵素反応後分子の細胞染色能評価 ... 160
5-4-8. 膜タンパク質を標的とした蛍光抗体法及び CARP 法による細胞染色 ... 160
5-5. 参考文献 ... 161
第 6 章 結論 ... 167 謝辞 175
1
第 1 章 序論
1-1. 検出ターゲットとしての酵素
我々ヒトを含む全ての生物は、体内における多種多様な化学反応により生命活動の維 持を行っている。その化学反応に対し、特異的触媒作用を示すタンパク質の総称を酵素 という。ゆえに、生命活動の異常状態、つまり疾病と酵素活性は深く関与しており、あ る疾患に関連のある酵素をターゲットとした治療・創薬技術の開発が進んでいる。本節 では、本論文の導入として、創薬研究における酵素反応の利用について概説する。
1-1-1. 創薬・治療標的としての酵素 -プロテインキナーゼ-
我々人間は、外界から受ける大小様々な刺激に対し、無意識の内に適応することがで きる。この驚くべき適応力を可能にしているのは我々の細胞である。人間の体は約 60 兆個の細胞から構成されると言われているが、恒常性を維持するために 1 日に約 3000
~ 4000 億個の細胞が死を遂げ、同時に誕生していると考えられている。また、それぞれ
の細胞は自らが置かれた環境に応じて様々な形態や機能を有する。この多様性と 60 兆 個という膨大な数を活かし、時々刻々と変化する環境に対応し恒常性を維持しているの である。
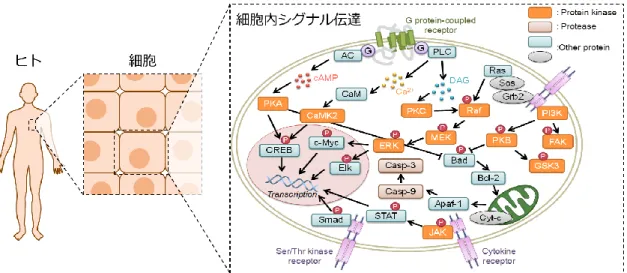
上記のように、ヒトの恒常性を維持しているのは細胞である。これをさらにミクロな 視点から考えてみると、細胞では外界から受けた刺激情報を細胞内部まで正確に伝達す る「細胞内シグナル伝達系」という情報伝達ネットワークが形成されている。これによ り細胞は自身の死や増殖、その他の機能を制御している (Figure 1-1)1-6。
2
Figure 1-1. ヒトにおける細胞内シグナル伝達系の概念図1-6
この細胞内シグナル伝達系において、重要な役割を担っている生体分子の 1 つがタ ンパク質リン酸化酵素 (Protein kinase: PK) である7-12。PK は、基質となるタンパク質 中のセリン、チロシン、スレオニン残基の側鎖にあるヒドロキシル基にアデノシン三リ ン酸 (Adenosine triphosphate: ATP) のリン酸基を転移させる (Figure 1-2)。このリン酸化 反応を手段として、PK は細胞内で情報伝達を行い、細胞を正常な状態に維持している。
ゆえに、PK の働きに異常が生じると、細胞、ひいては我々の健康に支障をきたすとい うことは言うまでもない13。実際に、日本人の死因第 1 位であるがんには PK の異常 が関わっており、その多くは PK の過剰発現とされている3,14,15。そのため、PK は様々 な疾患に対する創薬・治療ターゲットとして利用されている。当研究室においても、こ の PK に応答した遺伝子デリバリーシステムの開発等に成功している16-22。
3
Figure 1-2. プロテインキナーゼによるリン酸化反応
1-1-2. 分子標的薬としてのプロテインキナーゼ阻害剤
多くのがんで見られる過剰な PK 酵素活性を抑制し、細胞の異常を食い止める PK 阻害剤は、そのまま抗がん剤になり得る23。このように、細胞で異常を示すある特定の 分子を標的とし、その機能を制御することで薬理効果を発揮する薬剤を分子標的薬とい
う24-26。分子標的薬は、その名の通り、分子レベルで細胞の異常を認識するため、従来
の治療薬に比べ正常細胞への影響が少なく、副作用の低い治療薬として期待されている。
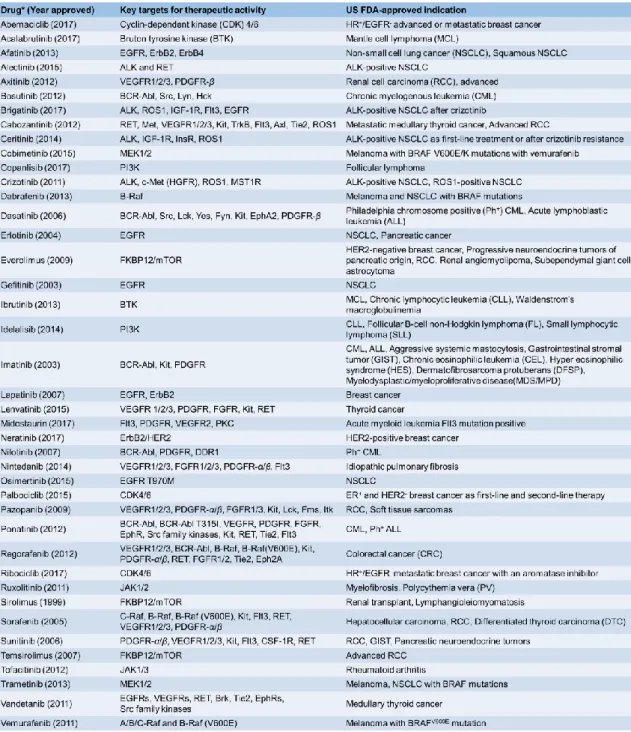
Table 1-1 に示したように、既に PK の一種である上皮細胞増殖因子受容体 (Epidermal growth factor receptor: EGFR)27-31 などをターゲットとした分子標的薬が米国食品医薬品 局 (US Food and Drug Administration: FDA) の認可を受けており、現在では、その他の PK をターゲットとした創薬研究が世界中で盛んに行われている32-34。
4
Table 1-1. FDA の認可を受けている PK 阻害剤一覧32-34
5
1-1-3. プロテインキナーゼ阻害剤のハイスループットスクリーニング
PK 阻害剤が、がんに対する分子標的薬としてどれほどの薬理効果を発揮するかを知 るためには、がん細胞において過剰発現し異常に高くなった PK 活性が、PK 阻害剤に よりいかに抑制されたかを評価しなければならない。ゆえに、この評価を可能にする PK 活性検出法というのは、有効な抗がん剤としての PK 阻害剤を開発する上で必要不 可欠である。特に創薬現場では、数多くの化合物ライブラリーから有効な PK 阻害剤を 効率良く選出するため、一度により多くの化合物ライブラリーを評価できる、所謂ハイ スループットスクリーニング (High-throughput screening: HTS) 可能な PK 活性検出法 が求められている。理想的な HTS に求められる条件として、Bömer らは次の 4 つを 挙げている35。
(1) 酵素反応が均一系である。
(2) 酵素活性依存的に検出シグナルが増加する。
(3) 共役酵素反応 (Coupled Enzyme Assay) ではない。
(4) 夾雑物の影響を受けにくい。
1-1-4. 従来のプロテインキナーゼ活性検出法
現在、この 4 条件を満たし最も広く利用されている PK 活性検出法の 1 つとして Scintillation proximity assay (SPA) がある (Figure 1-3)36,37。これは、ビオチン化した基質 ペプチドとストレプトアビジン (Streptavidin: SA) 修飾をしたシンチレーションビーズ、
さらに放射性同位体である 32P もしくは 33P を標識した ATP を用いる方法である。
SA はビオチンと非共有結合性の強力な相互作用を示す (Kd = 10-15 M) ことで知られて いる38。放射線ラベル化された ATP を用いたリン酸化により基質ペプチドを放射線ラ ベル化した後、ビオチンと SA を結合させると、リン酸化ペプチド上のリンの放射性同 位体から放出された β 線がビーズに伝わりシンチレーターが発光するという測定原 理である。この検出法は基質ペプチドの配列に制限がなく、コストが低いといった利点 を持つが、一方で、放射性物質を利用するため人体への危険性や特殊な設備と技術が求 められ、小型化が難しい等の問題を抱えている35。したがって、近年では放射性物質を 用いない PK 活性検出法の開発が盛んに行われている。
6
Figure 1-3. SPA の検出原理36,37
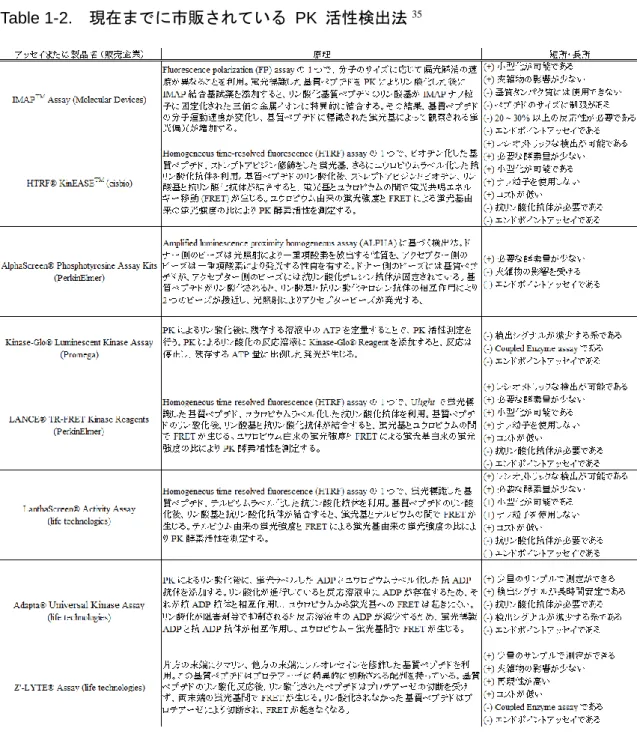
放射性物質を用いない PK 活性検出法は今日までにいくつか市販されている。その
一部を Table 1-2 にまとめた。これからもわかるように、各検出法は放射性物質の使用
はないものの、その他の問題を抱えており、SPA では満たしていた理想的な HTS の 4 条件を満たせていないものも多いのが現状である。したがって、放射性物質を利用せず、
かつ理想的な HTS のための 4 条件を満たす、革新的な PK 活性検出法の開発が期待 されている。そこで、多くの研究者たちが放射性物質の代替として利用しているものが 蛍光である。蛍光を用いた検出系は比較的感度に優れ、放射性物質と異なり特殊な設備 や技術が必要ないため小型化が期待できる。そのため、蛍光を用いた検出系は HTS と 非常に相性が良いとされている。そこで次項からは、最近報告された新規的な PK 活性 検出法について述べる。
7
Table 1-2. 現在までに市販されている PK 活性検出法35
8
1-1-5. 蛍光増強を利用したプロテインキナーゼ活性検出システム
基質のリン酸化に応じて金属錯体を形成することにより、蛍光強度が増加することを 利用した PK 活性検出システムを、いくつかのグループが報告している39-48。
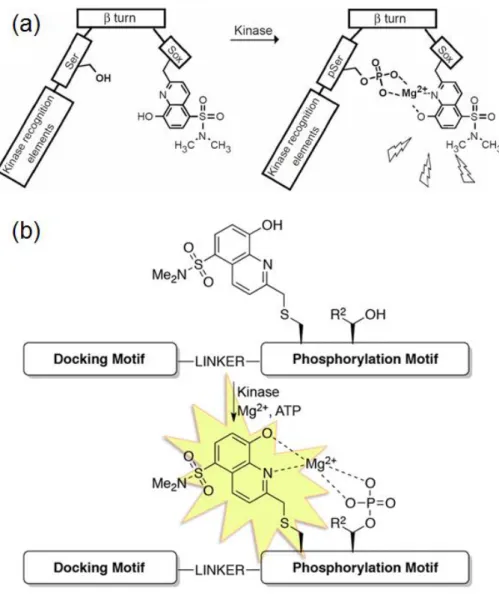
その 1 つとして、Imperiali らは、Mg2+ を中心金属にリン酸化アミノ酸のリン酸基と
Sulfonamido-oxide (Sox) という非天然のアミノ酸が配位することで PK 活性を検出す
る錯体型分子センサーを開発した (Figure 1-4, a)45。Sox とは、キレートにより蛍光強 度を増幅させる性質を有しており、Chelation-enhanced fluorescence (CHEF) の 1 つとし て知られている49。この分子センサーはキナーゼ認識部位を有し、β ターン構造を取る アミノ酸配列を介して Sox と基質ペプチド配列が繋がった構造をしている。検出原理 は、基質ペプチドのリン酸化により、リン酸基と Sox を介して Mg2+ がキレートを形 成することで増強した Sox の蛍光強度を測定するというものである。これにより彼女 らは数種類の PK 活性検出に成功したが、リン酸化されるアミノ酸残基を β ターン構 造の近傍に位置づけなければならないという制限があった。しかし、近年 Imperiali ら は Sox 類似体をシステインの側鎖に組み込んだ CSox を用いることで、β ターン構造 を必要としない新たな分子センサーを報告している (Figure 1-4, b)48。彼女らはこのシ ステムにより細胞ライセート中の PK 活性検出にも成功しているが、β ターン構造を 必要としないとは言え、CSox を基質配列中に組み込む際に、基質ペプチドの特異性の 維持と CSox のキレートによる蛍光増強のバランスを考慮して設計しなければならず、
依然として基質配列に多少の制限が残っている。
また、蛍光増強を用いた PK 活性検出法では 1 つの蛍光波長しか検出しないため、
検出シグナルの強度は酵素活性のみならず、その蛍光プローブの濃度にも依存する。例 えば、生細胞中の酵素活性を検出する際は、酵素活性に加え蛍光プローブの細胞内取り 込み効率も検出シグナルに影響を与える50。このように系内のプローブ濃度が変化して しまう場合、真の酵素活性を正確に測定することは難しい。さらに、生体分子の中には 蛍光性を示すものがあるため、検出の際にその自家蛍光の影響を受けてしまうことが懸
念される51,52。
9
Figure 1-4. Sox (a) 及び CSox (b) を利用した蛍光増強による PK 活性検出44-48 Reprinted by permission from Springer Nature, Nat. Methods, A multiplexed homogeneous fluorescence-based assay for protein kinase activity in cell lysates, Shults, M. D.; Janes, K. A.;
Lauffenburger, D. A.; Imperiali, B., Copyright 2005.
Reprinted with permission from Biochemistry, 2014, 53, 5771–5778. Copyright 2014 American Chemical Society.
10
1-1-6. レシオメトリーを用いたプロテインキナーゼ活性検出
前述したように、蛍光増強を利用した手法では、生細胞のような系での定量性の高い 酵素活性検出は困難である。そこで近年は蛍光増強の他に、2 つのシグナルを検出しそ れらの比を用いて定量を行う、レシオメトリーを用いた PK 活性検出法が数多く報告 されている53。中でも特に盛んに研究・開発が行われている検出系として、蛍光共鳴エ ネルギー移動 (Fluorescence resonance energy transfer または Förester resonance energy
transfer: FRET) を利用したレシオメトリックな PK 活性検出法が挙げられる。その 1
つである Homogeneous time-resolved fluorescence (HTRF) assay は特に広く利用されてお り54、Table 1-2 に示したように、既にいくつか HTRF assay に基づいた検出キットが 販売されている (Figure 1-5)55。HTRF assay では、ビオチン化した基質ペプチド、SA 修 飾をした蛍光基、さらにユウロピウムラベル化した抗リン酸化抗体を用いる。基質ペプ チドのリン酸化後、SA とビオチン、リン酸基と抗リン酸化抗体が結合すると、蛍光基 とユウロピウムの間で FRET が生じる。そのため、ユウロピウム由来の蛍光強度と
FRET による蛍光基由来の蛍光強度の比により PK 酵素活性を測定するのである。こ
の検出法は放射性物質を使わない上に理想的な HTS の 4 条件を満たしており、PK 活性検出法の中では優秀な部類に入る。しかし、ここで用いている抗リン酸化抗体、特 に抗リン酸化セリン抗体は特異性が低いといった問題を抱えている35。
Figure 1-5. HTRF®KinEASETM を用いた PK 活性検出法の測定原理55
11 1-2. 検出ツールとしての酵素
上述してきた通り、PK を代表とする酵素は、創薬研究の検出ターゲットとして扱わ れる。一方で、酵素は温和な条件下で高い触媒能、特異性を示すという優れた特徴から、
生物基礎研究分野における検出ツールや、医療現場における診断用ツールとしても利用 されている。そこで本節では、前節までの検出ターゲットとしての酵素ではなく、生体 分子、中でも膜タンパク質の一細胞解析における、検出ツールとしての酵素について説 明する。
1-2-1. バイオマーカーとしての膜タンパク質
PK 阻害剤のような分子標的薬の目覚ましい発展の裏には、多くの疾患の原因がゲノ ム・分子レベルで解明されたという重要な背景がある。この疾患の原因や病態に関連す る分子をバイオマーカーといい、疾患の予測や進行度を判断する客観的な指標として用 いられている56-60。代表的なバイオマーカーには、前述した EGFR 等の PK を含む膜 タンパク質が挙げられる61-67。1-1-1 の項でシグナル伝達について述べたが、このシグ ナル伝達には、リン酸化あるいは脱リン酸化による系の他に、サイトカインなどの水溶 性リガンドが膜タンパク質、つまり膜受容体に結合することで生じる系も存在する68,69。 そのため、膜タンパク質の検出も PK 活性検出と同様、生物基礎研究・創薬研究・医療 診断分野において必要不可欠となっている70,71。
実際に、Table 1-3 に分子標的薬の中でも特に抗体医薬を示したが、これらの中でも、
HER272、PD-173,74, 75、PD-L175,76、EGFR27,28,30、その他の CD 抗原25,77など、多くの膜タ ンパク質が治療ターゲットとして頻用されていることから、その検出標的としての意義 深さが窺える。
12
Table 1-3. FDA の認可を受けているタンパク質を標的とした分子標的薬一覧34
13
1-2-2. 膜タンパク質を検出ターゲットとした一細胞解析技術
創薬・診断・生物基礎研究分野、特に細胞生物学分野において、重要な検出標的とな る膜タンパク質であるが、この検出において欠かせないのが一細胞解析技術である78-81。
1-1-1 でも述べたように、細胞は自らの置かれた環境や発生段階に応じて不均一な形態
や機能を有し、それは遺伝子やタンパク質の発現量によって制御されている78,79。ゆえ に、一細胞レベルでの遺伝子やタンパク質の検出は、細胞の構造や機能における重要な 知見の獲得に繋がり、さらに薬剤に対する応答等を検出すれば、創薬・診断分野での有 用な情報を得ることが可能となる80。
代表的な一細胞解析技術として、顕微鏡とフローサイトメトリー (Flow cytometry:
FCM) が挙げられる。前者は、顕微鏡の中でも特に共焦点顕微鏡によって、単一細胞の
詳細な画像解析が可能であり、染色分子や細胞小器官の局在等の構造や形態における情 報を得られるが、大量解析には向いていない。一方、FCM は、細胞の高速・大量解析 やセルソーターによる形態学的に限界付近の細胞の分類・分取も可能であり、遺伝子や タンパク質の発現量を定量的に解析できるが、単一細胞の詳細な画像解析は困難である
81。このように一長一短ある一細胞解析技術であるが、本小論においては、膜タンパク 質を検出ターゲットとするため、その発現量の定量的解析が可能な FCM に着目してい く。
1-2-3. フローサイトメトリーによるマルチカラー解析
FCM とは、細胞懸濁液を細く流すことで細胞を一列に並べ、その一つひとつの細胞 にレーザー光を照射することで、個々の細胞の情報を光学的に解析する技術である
(Figure 1-6)82,83。1 秒間に数千から数万個の細胞を測定できるという優れたハイスルー
プット性78-83と、一細胞レベルでの解析が可能78-81といった利点に加え、最近ではフロ
ーサイトメーターの小型化が進んでいるため、細胞生物学分野における基礎研究のみな らず、実際の医療現場におけるヒト免疫不全ウイルス (Human immunodeficiency virus:
HIV) 感染症の病態把握や血液疾患の簡易検査などにも利用されている82,84-86。
14
Figure 1-6. フローサイトメトリーの基本原理
前述の通り、FCM は HIV 感染症などの免疫疾患や白血病などの血液疾患の診断、
移植治療などに利用されており、具体的には、リンパ球サブセット解析、CD34 陽性細 胞数の測定、白血病・悪性リンパ腫タイピングの 3 つの分析法が挙げられる。この中 でも、例えば白血病の簡易検査である白血病タイピングでは、白血病に関連する複数の 膜タンパク質の発現量を調べ、その結果から「どの細胞」が「どの段階」でがん化した のかを総合的に判定することで、その後の治療方針が構築される (Table 1-4)87。このよ うに、一度に複数の標的を検出できるマルチカラー解析は、上記のような臨床検査だけ でなく、創薬研究現場においても威力を発揮しており、その重要性が見てとれる 88,89。
Table 1-4. 白血病タイピング87
15
1-2-4. フローサイトメトリーにおける従来の染色法 -蛍光抗体法-
Figure 1-6 にも示したように、FCM においては、細胞を光学的に解析するために、
ターゲットとなる膜タンパク質を有した標的細胞を、事前に蛍光標識するのが一般的で ある。これにより、蛍光強度から膜タンパク質の発現の有無やその発現量を定量的に評 価できる。この染色技術の中でも従来用いられている染色システムが蛍光抗体法である
90。Fiugre 1-7 に示したように、蛍光基を修飾した一次抗体または二次抗体によって、
標的抗原となる膜タンパク質を蛍光標識するという非常に簡単な染色システムである。
一次抗体・二次抗体による染色をそれぞれ直接蛍光抗体法・間接蛍光抗体法といい、共 通した特徴としては、蛍光基の種類を変えるだけでマルチカラー解析に応用できるとい う点が挙げられる。しかし、この蛍光抗体法による染色では、FCM 解析において十分 な感度が得らず、特に発現量の少ない膜タンパク質の検出には利用できないといった問 題を抱えている91-95。これについては第 4 章で詳しく述べていく。
Figure 1-7. 蛍光抗体法による蛍光免疫染色の基本原理
16
1-2-5. バイオマーカー検出のための酵素 -酵素反応を利用した免疫染色法-
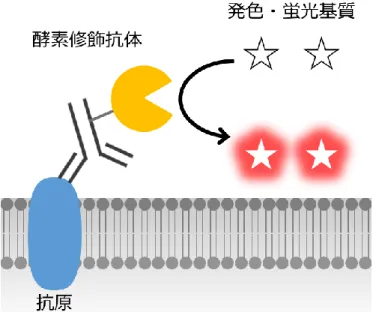
FCM に限らず、免疫組織染色等の蛍光免疫染色全般での利用において、上記の蛍光 抗体法の染色能に関する問題を改善すべく開発された染色システムが、酵素抗体法であ る (Figure 1-8)96-101。これはある酵素を修飾した抗体により膜タンパク質を標識し、そ こに基質を添加し、酵素反応によって生じた発色・発光性生成物によって染色を行うと いうシステムである。代表的な酵素として、アルカリフォスファターゼ (Alkaline phosphatase: AP) や西洋ワサビペルオキシダーゼ (Horseradish peroxidase: HRP) が用い られ、過剰量の基質を加えることで多量の生成物を産生できることから、蛍光抗体法に 比べ一般的に染色能が強く、検出シグナルの増感が可能である。特に FCM では、HRP を用いた、Catalyzed reporter deposition (CARD) 法が開発されている98-101。しかし、従来 の蛍光抗体法で可能であった FCM によるマルチカラー解析が、この CARD 法では困 難であるという問題を抱えており、さらに、内在性の酵素活性による非特異的な染色が 生じる場合があるため利用には注意が必要である。これについては、第 4 章で詳細を 述べる。
Figure 1-8. 酵素抗体法による蛍光免疫染色の基本原理
17 1-3. 本小論の構成
以上の通り、酵素は我々ヒト体内において疾患と深く関わっているため、創薬研究に おける検出ターゲットとなることが多く、また、その優れた特性から創薬・診断・生物 基礎研究分野における検出ツールとしても利用されている。そこで本論文では、検出タ ーゲットとして酵素を扱う『酵素を標的とした分子システム』、検出ツールとして酵素 を扱う『酵素を手段とした分子システム』と、大きく分けて 2 つのバイオ分析のため の分子システムの創製を目指した。
まず『酵素を標的とした分子システム』について、ハイスループットな PK 活性検出 法の開発は、創薬研究での薬剤スクリーニングにおいて必要不可欠であるが、現在まで に研究・開発・販売されている検出法はどれも一長一短であるため、それらの問題点を 1 つでも多く克服した革新的な検出システムの構築が求められている。理想的な HTS のための 4 条件を満たす検出法が数多く開発されている現在において、新たな検出法 を考える上でさらに考慮すべき条件としては、「放射性物質を使用しないこと」、「レシ オメトリーによりバックグラウンドノイズを低減させること」、「抗リン酸化抗体を使用 しないこと」が挙げられる。
そこで第 2 章では、上記を満たす、より理想に近い PK 活性検出システムとして、
抗リン酸化抗体を用いず FRET を利用した新規 PK 活性検出システムの確立を試み た。ここでは、近年バイオセンシングやバイオイメージングの分野で活発に使用されつ つある量子ドット (Quantum dot: QD) と蛍光修飾ペプチドを利用した 2 つの検出シス テムの構築を検討した。
第 3 章では、FRET ではなく濃度消光を利用したレシオメトリックな検出システム の開発を試みた。蛍光基を修飾したカチオン性ポリマーとアニオン性ポリマーが静電相 互作用により複合体を形成することで、一方の蛍光基は濃度消光を起こし、他方の蛍光 基は濃度消光を起こさないよう分子設計を行った。これにより、前者の蛍光基を PK 応 答性シグナル、後者の蛍光基を PK 非応答性の内部標準シグナルとして有する新規高 分子複合体型キナーゼプローブの開発及びそれを用いた PK 活性検出システムの構築 を検討した。
次に『酵素を手段とした分子システム』について、一細胞解析技術の 1 つである FCM を用いた膜タンパク質検出は、従来の蛍光抗体法による細胞染色では染色能が不十分で
18
ある一方で、増感法である酵素抗体法ではマルチカラー解析が困難であるといった、ト レードオフの問題を抱えている。
これを踏まえ第 4 章では、感度向上を目指した新たな酵素抗体法を開発し、それに よって染色された標的細胞の FCM による検出を試みた。標的細胞の認識には膜タンパ ク質を抗原として利用し、さらにここでは、既に免疫染色等のバイオ分析で利用されて いる AP を増感酵素として選択した。また、基質としては蛍光基を修飾した膜アンカー 型基質を設計・開発した。
第 5 章では、哺乳類細胞膜上に発現していないことで知られる β—ガラクトシダーゼ
(β-gal) を用いて、第 4 章と同様の酵素抗体法の開発を試みた。第 4 章で用いた AP は
内在性の酵素活性が存在するため、それを不活化する必要がある上、それによるバック グラウンドシグナルが懸念された。そこで、細胞膜上に内在性酵素活性がない β-gal を 増感酵素として用いることで、より簡便かつ細胞に優しい条件での細胞染色が可能にな ると考えた。さらに、酵素抗体法を構築する中で、基質の設計や、基質・酵素濃度、染 色時間等の染色反応における重要なパラメータの影響を評価することで、低発現膜タン パク質を標的とした細胞染色に本染色システムが応用可能かどうかを検討した。
第 6 章では、本論文中で行った検討を総括し結論を述べるとともに、今後の課題と 展望について記す。
19 1-4. 参考文献
(1) Nishizuka, Y. Nature 1984, 308, 693–697.
(2) Hunter, T. Cell 2000, 100, 113–127.
(3) Cohen, P. Eur. J. Biochem. 2001, 268, 5001–5010.
(4) Manning, G.; Whyte, D. B.; Martinez, R.; Hunter, T.; Sudarsanam, S. Science 2002, 298, 1912–
1934.
(5) Pearce, L. R.; Komander, D.; Alessi, D. R. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22.
(6) Taylor, S. S.; Kornev, A. P. Trends Biochem. Sci. 2011, 36, 65–77.
(7) Karin, M. Curr. Opin. Cell Biol. 1994, 6, 415–424.
(8) Davis, R. J. Cell 2000, 103, 239–252.
(9) Adams, J. A. Chem. Rev. 2001, 101, 2271–2290.
(10) Li, Q.; Verma, I. M. Nat. Rev. Immunol. 2002, 2, 725–734.
(11) Lieser, S. A.; Aubol, B. E.; Wong, L.; Jennings, P. A.; Adams, J. A. Biochim. Biophys. Acta 2005, 1754, 191–199.
(12) Ubersax, J. A.; Ferrell Jr., J. E. Nat. Rev. Mol. Cell Biol. 2007, 8. 530–541.
(13) Flajolet, M.; He, G.; Heiman, M.; Lin, A.; Nairn, A. C.; Greengard, P. PNAS 2007, 104, 4159–
4164.
(14) Critchfield, J. W.; Coligan, J. E.; Folks, T. M.; Butera, S. T.; PNAS 1997, 6110–6115.
(15) Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O’Meara, S.; Smith, R.; Parker, A.; Barthorpe, A.; Blow, M.; Brackenbury, L.; Butler, A.; Clarke, O.; Cole, J.; Dicks, E.; Dike, A.; Drozd, A.; Edwards, K.; Forbes, S.; Foster, R.; Gray, K.; Greenman, C.;
Halliday, K.; Hills, K.; Kosmidou, V.; Lugg, R.; Menzies, A.; Perry, J.; Petty, R.; Raine, K.;
Ratford, L.; Shepherd, R.; Small, A.; Stephens, Y.; Tofts, C.; Varian, J.; West, S.; Widaa, S.; Yates, A.; Brasseur, F.; Cooper, C. S.; Flanagan, A. M.; Knowles, M.; Leung, S. Y.; Louis, D. N.;
Looijenga, L. H. J.; Malkowicz, B.; Pierotti, M. A.; Teh, B.; Chenevix-Trench, G.; Weber, B. L.;
Yuen, S. T.; Harris, G.; Goldstraw, P.; Nicholson, A. G.; Futreal, P. A.; Wooster, R.; Stratton, M.
R. Nature 2004, 431, 525–526.
(16) Katayama, Y.; Fujii, K.; Ito, E.; Sakakihara, S.; Sonoda, T.; Murata, M.; Maeda, M.
Biomacromolecules 2002, 3, 905–909.
20
(17) Oishi, J.; Kawamura, K.; Kang, J.-H.; Kodama, K.; Sonoda, T.; Murata, M.; Niidome, T.;
Katayama, Y. J. Control. Release 2006, 110, 431–436.
(18) Kang, J.-H.; Asai, D.; Kim, J.-H.; Mori, T.; Toita, R.; Tomiyama, T.; Asami, Y.; Oishi, J.; Sato, Y.
T.; Niidome, T.; Jun, B.; Nakashima, H.; Katayama, Y. J. Am. Chem. Soc. 2008, 130, 14906–
14907.
(19) Toita, R.; Kang, J.-H.; Tomiyama, T.; Kim, C. W.; Shiosaki, S.; Niidome, T.; Mori, T.; Katayama, Y. J. Am. Chem. Soc. 2012, 134, 15410–15417.
(20) Kim, C. W.; Toita, R.; Kang, J.-H.; Li, K.; Lee, E. K.; Zhao, G. X.; Funamoto, D.; Nobori, T.;
Nakamura, Y.; Mori, T.; Niidome, T.; Katayama, Y. J. Control. Release 2013, 170, 469–476.
(21) Kushio, S.; Tsuchiya, A.; Nakamura, Y.; Nobori, T.; Kim, C. W.; Zhao, G. X.; Funamoto, T.; Lee, E. K.; Niidome, T.; Mori, T.; Katayama, Y. Biomed. Eng. Appl. Basis Commun. 2013, 25, 1340005.
(22) Nakamura, Y.; Kim, C. W.; Tsuchiya, A.; Kushio, S.; Nobori, T.; Li, K.; Lee, E. K.; Zhao, G. X.;
Funamoto, D.; Niidome, T.; Mori, T.; Katayama, Y. J. Biomater. Sci. Polym. Ed. 2013, 24, 1858–
1868.
(23) Zhang, J.; Yang, P. L.; Gray, N. S. Nat. Rev. 2009, Cancer 9, 28–39.
(24) Glassman, P. M.; Balthasar, J. P. Cancer Biol. Med. 2014, 11, 20–33.
(25) Ujjani, C. Oncology 2015, 29, 760–768.
(26) Santos, R.; Ursu, O.; Gaulton, A.; Bento, A. P.; Donadi, R. S.; Bologa, C. G.; Karlsson, A.; Al- Lazikani, B.; Hersey, A.; Oprea, T. I.; Overington, J. P. Nat. Rev. Drug Discov. 2017, 16, 19–34.
(27) Ciardiello, F.; Tortora, G. Clin. Cancer Res. 2001, 7, 2958–2970.
(28) Grünwald, V.; Hidalgo, M. J. Natl. Cancer Inst. 2003, 95, 851–867.
(29) Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M. R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D. S. Gene 2006, 366, 2–16.
(30) Sharma, S. V; Bell, D. W.; Settleman, J.; Haber, D. Nat. Rev. Cancer 2007, 7, 169–181.
(31) De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M. R.; Aldinucci, D.; Pinto, A.;
Normanno, N. J. Cell. Physiol. 2008, 214, 559–567.
(32) Knight, Z. A.; Lin, H.; Shokat, K. M. Nat. Rev. Cancer 2010, 10, 130–137.
(33) FDA-approved protein kinase inhibitors compiled by Robert Roskoski Jr.
http://www.brimr.org/PKI/PKIs.htm
21
(34) Targeted Cancer Therapies Fact Sheet - National Cancer Institute
https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact- sheet
(35) von Ahsen, O.; Bömer, U. ChemBioChem 2005, 6, 481–490.
(36) Park, Y.-W.; Cummings, R. T.; Wu, L.; Zheng, S.; Cameron, P. M.; Woods, A.; Zaller, D. M.;
Marcy, A. I.; Hermes, J. D. Anal. Biochem. 1999, 269, 94–104.
(37) Wu, J. J. Methods Mol. Biol. 2002, 190, 65–85.
(38) Weber, P. C.; Ohlendorf, D. H.; Wendoloski, J.J.; Salemme, F. R. Science 1989, 243, 85–88.
(39) Chen, C.-A.; Yeh, R.-H.; Lawrence, D. S. J. Am. Chem. Soc. 2002, 124, 3840–3841.
(40) Yeh, R.-H.; Yan, X.; Cammer, M.; Bresnick, A. R.; Lawrence, D. S. J. Biol. Chem. 2002, 277, 11527–11532.
(41) Wang, Q.; Lawrence, D. S. J. Am. Chem. Soc. 2005, 127, 7684–7685.
(42) Sharma, V.; Wang, Q.; Lawrence, D. S. Biochim. Biophys. Acta 2008, 1784, 94–99.
(43) Shell, J. R.; Lawrence, D. S. Biochim. Biophys. Acta 2013, 1834, 1359–1363.
(44) Shults, M. D.; Imperiali, B. J. Am. Chem. Soc. 2003, 125, 14248–14249.
(45) Shults, M. D.; Janes, K. A.; Lauffenburger, D. A.; Imperiali, B. Nat. Methods 2005, 2, 277–284.
(46) Shults, M. D.; Carrico-Moniz, D.; Imperiali, B. Anal. Biochem. 2006, 352, 198–207.
(47) Luković, E.; Taylor, E. V.; Imperiali, B. Angew. Chem. Int. Ed. 2009, 48, 6828–6831.
(48) Peterson, L. B.; Yaffe, M. B.; Imperiali, B. Biochemistry 2014, 53, 5771–5778.
(49) Minta, A.; Kao, J. P. Y.; Tsien, R. Y. J. Biol. Chem. 1989, 264, 8171–8178.
(50) Liu, X.; Zhang, N.; Bing, T.; Shangguan, D. Anal. Chem. 2014, 86, 2289–2296.
(51) Boumaza, S.; Arribas, S. M.; Osborne-Pellegrin, M.; McGrath, J. C.; Laurent, S.; Lacolley, P.;
Challande, P. Hypertension 2001, 37, 1101–1107.
(52) Lee, W. K.; Bell, J.; Kilpatrick, E.; Hayes, M.; Lindop, G. B. M.; Dominiczak, M. H.
Atherosclerosis 1993, 98, 219–227.
(53) Kikuchi, K.; Hashimoto, S.; Mizukami, S.; Nagano, T. Org. Lett. 2009, 11, 2732–2735.
(54) Mathis, G. Clin. Chem. 1995, 41, 1391–1397.
(55) Harbert, C.; Marshall, J.; Soh, S.; Steger, K. Curr. Chem. Genomics 2008, 1, 20–26.
(56) Loukopoulos, P.; Thornton, J. R.; Robinson, W. F. Vet. Pathol. 2003, 40, 237–248.
22
(57) Loukopoulos, P.; Mungall, B. A.; Straw, R. C.; Thornton, J. R.; Robinson, W. F. Vet. Pathol. 2003, 40, 382–394.
(58) Liu, R.; Wang, X.; Aihara, K.; Chen, L. Med. Res. Rev. 2014, 34, 455–478.
(59) Dienstmann, R.; Jang, I. S.; Bot, B.; Friend, S.; Guinney, J. Cancer Discov. 2015, 5, 118–123.
(60) Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; Wu, E.; Dakin, K.; Petzold, M.; Blennow, K.; Zetterberg, H. Lancet.
Neurol. 2016, 15, 673–684.
(61) Filipe, MI.; Osborn, M.; Linehan, J.; Sanidas, E.; Brito, MJ.; Jankowski, J. Br. J. Cancer 1995, 71, 30–36.
(62) Pirker, R.; Pereira, J. R.; von Pawel, J.; Krzakowski, M.; Ramlau, R.; Park, K.; de Marinis, F.;
Eberhardt, W. E. E.; Paz-Ares, L.; Störkel, S.; Schumacher, K.-M.; von Heydebreck, A.; Celik, I.; O’Byrne, K. J. Lancet. Oncol. 2012, 13, 33–42.
(63) Licitra, L.; Störkel, S.; Kerr, K. M.; Cutsem, E. V.; Pirker, R.; Hirsch, F. R.; Vermorken, J. B.;
von Heydebreck, A.; Esser, R.; Celik, I.; Ciardiello, F. Eur. J. Cancer 2013, 49, 1161–1168.
(64) Yoshie, O.; Fujisawa, R.; Nakayama, T.; Harasawa, H.; Tago, H.; Izawa, D.; Hieshima, K.;
Tatsumi, Y.; Matsushima, K.; Hasegawa, H.; Kanamaru, A.; Kamihira, S.; Yamada, Y. Blood, 2002, 99, 1505–1511.
(65) Ishida, T.; Utsunomiya, A.; Iida, S.; Inagaki, H.; Takatsuka, Y.; Kusumoto, S.; Takeuchi, G.;
Shimizu, S.; Ito, M.; Komatsu, H.; Wakita, A.; Eimoto, T.; Matsushima, K.; Ueda, R. Clin.
Cancer Res. 2003, 9, 3625–3634.
(66) Ishida, T.; Ishii, T.; Inagaki, A.; Yano, H.; Komatsu, H.; Iida, S.; Inagaki, H.; Ueda, R. Cancer Res. 2006, 66, 5716–5722.
(67) Cornish, A. L.; Freeman, S.; Forbes, G.; Ni, J.; Zhang, M.; Cepeda, M.; Gentz, R.; Augustus, M.;
Carter, K. C.; Crocker, P. R. Blood, 1998, 92, 2123–2132.
(68) Miyajima, A.; Kitamura, T.; Harada, N.; Yokota, T.; Arai, K. Annu. Rev. Immunol. 1992, 10, 295–
331.
(69) Kishimoto, T.; Taga, T.; Akira, S. Cell 1994, 76, 253–262.
(70) Henel, G.; Schmitz, J. L. Lab. Med. 2007, 38, 428–436.
(71) Jaye, D. L.; Bray, R. A.; Gebel, H. M.; Harris, W. A. C.; Waller, E. K. J. Immunol. 2012, 188, 4715−4719.
23
(72) Cho, H.-S.; Mason, K.; Ramyar, K. X.; Stanley, A. M.; Gabelli, S. B.; Denney Jr, D. W.; Leahy, D. J. Nature 2003, 421, 756–760.
(73) Topalian, S. L.; Hodi, F. S; Brahmer, J. R.; Gettinger, S. N.; Smith, D. C.; McDermott, D. F.;
Powderly, J. D.; Carvajal, R. D.; Sosman, J. A.; Atkins, M. B., Leming, P. D.; Spigel, D. R.;
Antonia, S. J.; Horn, L.; Drake, C. G.; Pardoll, D. M.; Chen, L.; Sharfman, W. H.; Anders, R. A.;
Taube, J. M.; McMiller, T. L.; Xu, H.; Korman, A. J.; Jure-Kunkel, M.; Agrawal, S.; McDonald, D.; Kollia, G. D.; Gupta, A.; Wigginton, J. M.; Sznol, M. N. Engl. J. Med. 2012, 366, 2443–2454.
(74) Garon, E. B.; Rizvi, N. A.; Hui, R.; Leighl, N.; Balmanoukian, A. S.; Eder, J. P.; Patnaik, A.;
Aggarwal, C.; Gubens, M.; Horn, L.; Carcereny, E.; Ahn, M.-J.; Felip, E.; Lee, J.-S.; Hellmann, M. D.; Hamid, O.; Goldman, J. W.; Soria, J.-C.; Dolled-Filhart, M.; Rutledge, R. Z.; Zhang, J.;
Lunceford, J. K.; Rangwala, R.; Lubiniecki, G. M.; Roach, C.; Emancipator, K.; Gandhi, L. N.
Engl. J. Med. 2015, 372, 2018–2028.
(75) Naidoo, J.; Page, D. B.; Li, B. T.; Connell, L. C.; Schindler, K.; Lacouture, M. E.; Postow, M. A.;
Wolchok, J. D. Ann. Oncol. 2015, 26, 2375–2391.
(76) Brahmer, J. R.; Tykodi, S. S.; Chow, L. Q. M.; Hwu, W.-J.; Topalian, S. L.; Hwu, P.; Drake, C.
G.; Camacho, L. H.; Kauh, J.; Odunsi, K.; Pitot, H. C.; Hamid, O.; Bhatia, S.; Martins, R.; Eaton, K.; Chen, S.; Salay, T. M.; Alaparthy, S.; Grosso, J. F.; Korman, A. J.; Parker, S. M.; Agrawal, S.;
Goldberg, S. M.; Pardoll, D. M.; Gupta, A.; Wigginton, J. M. N. Engl. J. Med. 2012, 366, 2455–
2465.
(77) Glassman, P. M.; Balthasar, J. P. Cancer Biol. Med. 2014, 11, 20–33.
(78) Klepárník, K.; Foret, F. Anal. Chim. Acta 2013, 800, 12–21.
(79) Pedreira, C. E.; Costa, E. S.; Lecrevisse, Q.; van Dongen, J. J. M.; Orfao A. Trends Biotechnol.
2013, 31, 415–425.
(80) Heath, J. R.; Ribas, A.; Mischel, P. S. Nat. Rev. Drug Discov. 2016, 15, 204–216.
(81) 山村 昌平 生物工学会誌 2013, 91, 583.
(82) Brown, M.; Wittwer, C. Clin. Chem. 2000, 46, 1221–1229.
(83) 金山 直樹 生物工学会誌 2012, 90, 785–789.
(84) Landay, A.; Ohlsson-Wilhelm, B.; Giorgi, J. V. AIDS 1990, 4, 479–497.
(85) Campana, D.; Coustan-Smith, E. Cytometry A 1999, 38, 139–152.
24
(86) Coustan-Smith, E.; Sancho, J.; Hancock, M. L.; Boyett, J. M.; Behm, F. G.; Raimondi, S. C.;
Sandlund, J. T.; Rivera, G. K.; Rubnitz, J. E.; Ribeiro, R. C.; Pui, C.-H.; Campana, D. Blood 2000, 96, 2691–2696.
(87) Ⅶ. フローサイトメトリーの臨床検査応用 検査室のための FCM 検査の基礎 サイト
メトリードットコム https://www.bc-cytometry.com/FCM/FCM/fcm_07.html
(88) Jaso, J. M.; Wang, S. A.; Jorgensen, J. L.; Lin, P. Bone Marrow Transplant. 2014, 49, 1129–1138.
(89) Rühle, P. F.; Fietkau, R.; Gaipl, U. S.; Frey, B. Int. J. Mol. Sci. 2016, 17.
(90) Schmid, I.; Krall, W. J.; Uittenbogaart, C. H.; Braun, J.; Giorgi, J. V. Cytometry A 1992, 13, 204–
208.
(91) Zhang, L.; Zhao, W.; Liu, X.; Wang, G.; Wang, Y.; Li, D.; Xie, L.; Gao, Y.; Deng, H.; Gao, W.
Biomaterials 2015, 64, 2−9.
(92) Truneh, A.; Machy, P.; Horan, P. K. J. Immunol. Methods 1987, 100, 59−71.
(93) Scheffold, A.; Miltenyi, S.; Radbruch, A. Immunotechnology 1995, 1, 127−137.
(94) Cohen, J. H. M.; Aubry, J. P.; Jouvin, M. H.; Wijdenes, J.; Bancherau, J.; Kazatchkine, M.;
Revillard, J. P. J. Immunol. Methods 1987, 99, 53−58.
(95) Zola, H.; Flego, L.; Sheldon, A. Immunobiology 1992, 185, 350−365.
(96) Bates, D. L. Trends Biotechnol. 1987, 5, 204−209.
(97) Yamada, H.; Mori, S.; Ueda, S.; Kawata, M.; Sano, Y. Acta Histochem. Cytochem. 1987, 20, 629−637.
(98) Clutter, M. R.; Heffner, G. C.; Krutzik, P. O.; Sachen, K. L.; Nolan, G. P. Cytometry A 2010, 77A, 1020−1031.
(99) Kaplan, D.; Smith, D. Cytometry A 2000, 40, 81−85.
(100) Kaplan, D.; Smith, D.; Meyerson, H.; Pecora, N.; Lewandowska, K. PNAS 2001, 98, 13850−13853.
(101) Kaplan, D.; Meyerson, H.; Lewandowska, K. Am. J. Clin. Pathol. 2001, 116, 429−436.
25
第 2 章 量子ドットと蛍光修飾基質ペプチドを利用した プ ロ テ イ ン キ ナ ー ゼ 活 性 検 出 シ ス テ ム
2-1. 緒言
本章では、『酵素を標的とした分子システム』として、「放射性物質を使用しないこと」、
「レシオメトリーによりバックグラウンドノイズを低減させること」、「抗リン酸化抗体 を使用しないこと」という条件を満たした 2 つの新規プロテインキナーゼ (Protein
kinase: PK) 活性検出システムの確立を試みた。本節では、まず近年報告されている抗リ
ン酸化抗体を用いない蛍光共鳴エネルギー移動 (Fluorescence resonance energy transfer または Förester resonance energy transfer: FRET) 型 PK 活性検出法及びナノ粒子を用い た PK 検出法をいくつか概説する。その後、本研究の戦略と目的について示す。
2-1-1. 融合タンパク質を用いた FRET 型プロテインキナーゼ活性検出
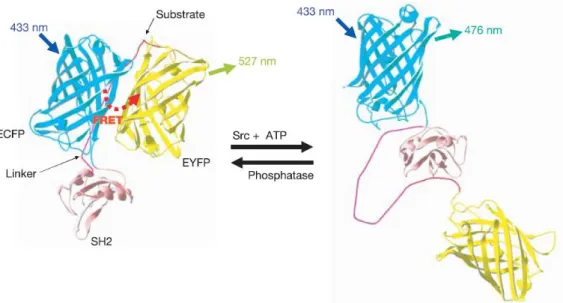
2 つの蛍光基の代わりに、2 つの蛍光タンパク質を含む融合タンパク質を利用し、基 質のリン酸化に応じてその融合タンパク質の立体構造が大きく変化することにより、
FRET 効率が変化することを利用した PK 活性検出システムが現在までにいくつか報
告されている 1-8。ここではその中の 1 つである Chien らの開発した PK 活性検出法 を取り上げる (Figure 2-1)9。彼らは、リン酸化認識部位である SH2 ドメイン10とペプ チドリンカー、基質ペプチドを介して、ECFP1と EYFP1という 2 つの蛍光タンパク質 を繋げた融合タンパク質を作製した。基質ペプチドのリン酸化前では、2 つの蛍光タン パク質が近傍に存在するため、ECFP を励起した場合、ECFP から EYFP への FRET が
生じ、EYFP 由来の蛍光が現れる。一方リン酸化が起きると、SH2 ドメインがリン酸化
された基質ペプチドと相互作用するため、融合タンパク質のコンフォメーションが大き く変化し FRET が解消される。その結果、ECFP を励起すると ECFP 由来の蛍光が見 られる。この FRET 効率の変化に伴う ECFP 由来の蛍光/EYFP 由来の蛍光の蛍光強度 比の変化からレシオメトリックな PK 活性検出行う。
融合タンパク質を用いた PK 活性検出法は、生細胞中での PK 活性検出が可能であ るという大きなメリットがあるが、その一方で、融合タンパク質を生産させる細胞株を 作製するのが困難であることや、蛍光の変化が小さいといった問題がある。
26
Figure 2-1. 融合タンパク質を利用した FRET 型 PK 活性検出法の測定原理9 Reprinted by permission from Springer Nature, Nature, Visualizing the mechanical activation of Src, Wang, Y.; Botvinick, E. L.; Zhao, Y.; Berns, M. W.; Usami, S.; Tsien, R. Y.; Chien, S., Copyright 2005.
2-1-2. 量子ドットを用いた FRET 型プロテインキナーゼ活性検出
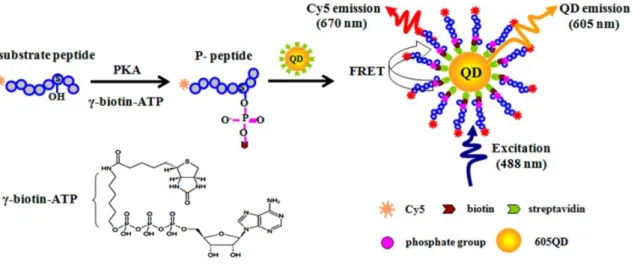
蛍光性を示すものとして、蛍光基や蛍光タンパク質の他にナノ粒子が挙げられる。量 子ドット (Quantum dot: QD)11-13 はその中の 1 つで、モル吸光係数や量子収率、光安定 性が高いなどの優れた光学特性から、近年ではバイオテクノロジーの分野においても活 発に利用されつつある14-21。キナーゼ活性検出への応用は 2010 年頃に報告され始めた 研究分野であり、比較的新しい分野と言えよう22-29。その 1 つとして、Zhang らは QD と蛍光修飾した基質ペプチド、さらに -biotin-ATP を用いた FRET 型 PK 活性検出法 を報告している (Figure 2-2)29。彼らは粒子表面にストレプトアビジン (SA) が修飾さ れた QD を用いており、リン酸化前は FRET は生じないため、QD 由来の蛍光が検出 される。一方で -biotin-ATP 存在下でリン酸化を行うと、蛍光修飾基質ペプチドのリ ン酸化部位に -biotin-ATP 由来のビオチンが修飾される。SA はビオチンと非共有結合 性の強力な相互作用を示す (Kd = 10-15 M)30 ため、この biotin-SA の相互作用により基 質ペプチドは QD 表面に吸着し、QD から蛍光基への FRET が生じ、QD 由来の蛍光 に加えて蛍光基由来の蛍光が現れる。この手法を用いて彼らはリン酸化の検出に成功し ている。
27
Figure 2-2. QD を用いた FRET 型 PK 活性検出法の測定原理29
Reprinted with permission from Anal. Chem., 2015, 87, 4696–4703. Copyright 2015 American Chemical Society.
2-1-3. 錯形成によるリン酸基認識を用いた FRET 型プロテインキナーゼ活性検出
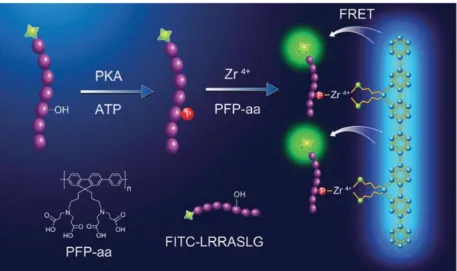
上記の報告においては、抗リン酸化アミノ酸抗体に代わるリン酸化部位の認識に SH2 ドメインや biotin-SA 相互作用を用いていた。その他の認識方法として知られて いるのが第 1 章の 1-1-5 でも紹介した錯形成である 31-40。実際に、リン酸化タンパク 質解析における二次元電気泳動やアフィニティークロマトグラフィーでは古くから利 用されているが 41-45、Li らはこの錯形成によるリン酸化部位の認識を利用した FRET による PK 活性検出法を報告している (Figure 2-3)46。彼らは、poly[(9,9-bis(3’-N,N- bis(2’’-carboxyethyl) amino)propyl)fluorenylene phenylene] (PFPaa) という、リン酸基と共同 して錯形成を行う蛍光性ポリマーを用いてリン酸基の認識を行っている。PK によって リン酸化を受けた蛍光修飾基質ペプチドのリン酸基が PFPaa と Zr4+ との錯形成によ り PFPaa に捕捉されるため、PFPaa から基質ペプチドに修飾された蛍光基への FRET が生じる。これを検出することで彼らは PK 活性検出に成功している。
28
Figure 2-3. 錯形成によるリン酸化部位認識を用いた FRET 型 PK 活性検出法46 Reproduced from Chem. Commun. 2013, 49, 3887–3889 with permission of The Royal Society of Chemistry.
さらに、彼らは特有の希土類元素に、リン酸化基質ペプチド上のリン酸基を特異的に 捕捉する能力があるという非常に興味深い発見をした47。この特性を利用し、磁性マイ クロビーズ上に修飾した希土類元素イオン Dy3+ によるリン酸基の認識を行い、リン酸 化された蛍光修飾基質ペプチドの蛍光を検出することで、一細胞レベルでの PK 活性 検出に成功している (Figure 2-4)48。
Figure 2-4. 希土類元素イオンによるリン酸化部位認識を用いた PK 活性検出法48
From Angew. Chem. Int. Ed., 54, 1–6 (2015). Copyright © 2015 by John Wiley Sons, Inc.
Reprinted by permission of John Wiley & Sons, Inc.
29
2-1-4. 静電相互作用を利用したナノ粒子によるプロテインキナーゼ活性検出
もう 1 つリン酸化の認識方法として忘れてはならないのが静電相互作用である49-54。
Katayama らは、金ナノ粒子 (Gold nanoparticle: GNP) が基質ペプチドにより凝集するこ
とを利用して PK 活性を検出している (Figure 2-5)49。アニオン性の表面電荷を有する GNP にカチオン性の基質ペプチドを添加すると、静電相互作用により GNP の周囲に 基質ペプチドが吸着し電荷が相殺され GNP は凝集する。その結果、GNP 溶液が赤か ら青へと変化する様子が肉眼で見て取れるほど、GNP の吸収スペクトルに大きな変化 が生じる。しかし一方で、リン酸化された基質ペプチドは正味の電荷がリン酸化前から -2 だけ変化するため、リン酸化前に比べ凝集能が下がり、吸収スペクトルの変化が小 さくなる。この違いを利用し PK 活性検出に成功している。
Figure 2-5. GNP の凝集を利用した PK 活性検出法の原理49
From ChemBioChem, 8, 875–879 (2007). Copyright © 2007 by John Wiley Sons, Inc. Reprinted by permission of John Wiley & Sons, Inc.
30
また Yao らも、QD を用いて Katayama らと同様の、凝集による PK 活性検出シス テムを報告している (Figure 2-6)54。彼らは凝集により生じる QD の蛍光スペクトル変 化を利用した。
Figure 2-6. QD の凝集を利用した PK 活性検出法の原理54
Reprinted with permission from Anal. Chem., 2011, 83, 52–59. Copyright 2011 American Chemical Society.
31
2-1-5. 量子ドットと蛍光修飾ペプチドの静電相互作用および錯形成に基づく F R E T 型 プ ロ テ イ ン キ ナ ー ゼ 活 性 検 出 シ ス テ ム の 提 案
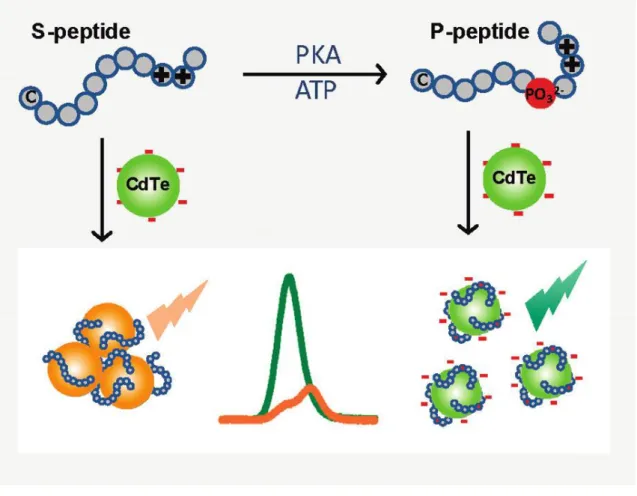
以上のように、PK 活性検出システムにはリン酸化を認識するために様々な分子認 識・相互作用が利用されている。そこで本章では、QD と基質ペプチドに修飾された蛍 光基の間で生じる FRET を利用した PK 活性検出システムを 2 つ提案する。まず 1 つ目の検出システムでは、アニオン性の表面電荷を有する QD とカチオン性の基質ペ プチドを用いることで、Katayama らや Yao らの報告と同様、ナノ粒子と基質ペプチド 間で生じる静電相互作用を利用した (Figure 2-7)55。この作用により QD と基質ペプチ ド上の蛍光基が近づくと FRET が生じる。一方でリン酸化ペプチドの場合、リン酸化 により基質ペプチドは正味の電荷が -2 だけ変化するため、QD との静電相互作用が弱 まり、QD と蛍光基間の距離がリン酸化前に比べ広がると考えられる。そうすると、
FRET 効率は 2 つの蛍光基間の距離に依存する56ため、リン酸化後は FRET が抑制さ
れる。ゆえに、この蛍光基由来の蛍光強度と QD 由来の蛍光強度の比からレシオメト リックな検出が可能であると考えられる。
Figure 2-7. 本章で提案する静電相互作用に基づく FRET 型 PK 活性検出システム55 Reproduced from Ref. 55 with permission from The Royal Society of Chemistry.
32
2 つ目のシステムでは、phos-tag というリン酸基認識分子を用いた (Figure 2-8)57-66。 これはアルカリフォスファターゼのリン酸基認識部位を模倣して開発された機能性低 分子であり、亜鉛二核錯体を形成することでリン酸基を認識・捕捉する (Figure 2-8, a)。
リン酸基に対するその親和性と特異性の高さから、リン酸化タンパク質・ペプチドの解 析・検出等において広く利用されている。ここで提案する検出システムでは、biotin-SA の相互作用により phos-tag が表面修飾された QD と、蛍光修飾基質ペプチドを用いる
ことで、phos-tag がリン酸化基質ペプチド上のリン酸基を捕捉することを利用した。こ
の作用により QD とペプチドに修飾された蛍光基が近づき FRET が生じる。一方で非 リン酸化ペプチドの場合、リン酸基を持たないため phos-tag による基質ペプチドの QD 表面への捕捉は生じず、QD と蛍光基間の距離がリン酸化基質ペプチドの場合に比 べ広がり、FRET が抑制される。ゆえに、1 つ目のシステム同様、蛍光基由来の蛍光強 度と QD 由来の蛍光強度の比からレシオメトリックな検出が可能であると考えられる。
Figure 2-8. (a) 用いたビオチン修飾 phos-tag の構造とリン酸基捕捉の原理57-66 (b) 本章で提案する錯形成に基づく FRET 型 PK 活性検出システム
33
上記の 2 つの検出システムは「放射性物質を使用しないこと」、「レシオメトリーに よりバックグラウンドノイズを低減させること」、「抗リン酸化抗体を使用しないこと」
という条件を満たしており、理想的な HTS として期待できる。
検 出 タ ー ゲ ッ ト の PK と し て は cAMP 依 存 性 プ ロ テ イ ン キ ナ ー ゼ (cAMP- dependent protein kinase: PKA) を採用した。この理由の 1 つとしては、PK 活性検出系 において、PKA をターゲットとした研究は数多く報告されており 26,28,29,35-38,46-50,53,54,67,68
比較対象が多いため、本システムのコンセプトの証明や他のシステムとの比較を示しや すいと考えたからである。また、PKA の基質ペプチドとして広く知られている
Kemptide69は生理条件下における正味の電荷が +2 であるため、リン酸化を受けると電
荷が完全に相殺される。これにより、リン酸化を受けても電荷を有するような基質ペプ チドに比べ、QD 等との非特異的な相互作用を抑制できると考えられ、より理想的な挙 動を示すことが期待される。以上の理由から、本研究においても Kemptide を用いた PKA 活性検出システムとすることで、リン酸化後の基質ペプチドに電荷を残さないよ うにした。
34
2-2. 静電相互作用に基づく検出システムについての結果及び考察 2-2-1. 蛍光修飾ペプチド (TAMRA-Kemptide) の合成
ペプチドの配列には、PKA により特異的にリン酸化される基質ペプチド配列
Kemptide を用いた。この Kemptide の N 末端にスペーサーとして miniPEG を修飾し、
さらに FRET アクセプターである蛍光基としてカルボキシテトラメチルローダミン (5/6-carboxy-tetramethyl-rhodamine: TAMRA) を修 飾 し た 。 ス ペ ー サ ーを 介 し た の は
TAMRA が Kemptide のリン酸化へ影響することを抑えるためである。Kemptide の合
成は Fmoc 固相合成法に基づいて行い、ペプチド N 末端への TAMRA の修飾は N-ヒ ドロキシスクシンイミド (N-hydroxysuccinimide: NHS) 活性化型の TAMRA (TAMRA SE) を用いた。また、活性検出用の蛍光修飾ペプチドに加え、ポジティブコントロール ペプチドとして、リン酸化部位であるセリン残基をリン酸化セリンに置き換えたものも 合成した。以後、活性検出用のペプチドを TAMRA-Kemptide (S)、ポジティブコントロ ールペプチドを TAMRA-Kemptide (pS) とする。各ペプチドの配列、総電荷、分子量を
Table 2-1 に示す。合成した各ペプチドの確認は、逆相高速液体クロマトグラフィー
(High performance liquid chromatography: HPLC) による精製後、マトリックス支援レーザ ー脱離イオン化飛行時間型質量分析 (Matrix assisted laser desorption/ionization-time of flight mass spectrometry: MALDI-TOF MS) によって行った (Figure 2-9)。Table 2-1 及び Figure 2-9 より、TAMRA-Kemptide (S)、TAMRA-Kemptide (pS) のいずれも合成できて いることが確認できた。精製した各ペプチドの濃度は、ペプチドに修飾された TAMRA の吸光度測定によって決定した。この時、モル吸光係数には ε552 = 80,000 M-1cm-1 を利 用した。
Table 2-1. 各ペプチドの配列
ペプチド名 配列 総電荷 分子量
TAMRA-Kemptide (S) TAMRA-miniPEG-LRRASLG-NH2 +2 1328.52 TAMRA-Kemptide (pS) TAMRA-miniPEG-LRRApSLG-NH2 0 1408.50
※ 総電荷はペプチド鎖の側鎖に由来する電荷の総和であり、蛍光基由来のものは含んでいない。
35
Figure 2-9. TAMRA-Kemptide (S) (a) と TAMRA-Kemptide (pS) (b) の質量分析
2-2-2. 励起波長の決定と FRET ペアとしての相性
本研究では、FRET ドナーとして QD545-COOH (以降は QD545 と略す) を用いた。
この QD545 は粒子表面にカルボキシル基を持つため、アニオン性の表面電荷を帯びて
いる。FRET アクセプターには、前述したように、蛍光基 TAMRA を用いた。Figure 2-
10 に QD545 の蛍光スペクトルと吸収スペクトル、TAMRA-Kemptide (S)、TAMRA-
Kemptide (pS) の吸収スペクトルを示した。この図から、まず QD545 は 500 nm より
も短波長側に広く吸収を有することがわかる。また、蛍光修飾ペプチドはどちらも 450
~ 600 nm の波長に加え、およそ 380 nm 以下の波長でも吸収を示した。したがって、
QD545 は吸収を示すが、蛍光修飾ペプチドはほとんど吸収を示さない 400 nm を励起
波長として選択した。さらに、QD545 の蛍光スペクトルとペプチドに修飾された
TAMRA の吸収スペクトルが十分重なっていることから、これらの間で FRET が生じ
ると期待された。