学 位 論 文
真正細菌およびミトコンドリアにおける 翻訳停滞解消機構の解明
Functional analysis of stalled-ribosome rescue factors in bacteria and mitochondria
群馬大学大学院 工学研究科
小暮 裕幸
1
目次
目次... 1
第1章 序 ... 4
翻訳停滞 ... 4
tmRNA・SmpB複合体による翻訳停滞解消機構 ... 5
YaeJタンパク質によるtmRNA非依存的翻訳停滞解消機構の示唆 ... 6
翻訳停滞解消YaeJの機能 ... 7
YaeJのヒトホモログICT1はミトコンドリアの翻訳停滞解消因子として機能する . 8 もう一つのミトコンドリアの翻訳停滞解消因子C12orf65 ... 9
NMRによるICT1の立体構造解析 ... 10
YaeJとリボソームとの共結晶構造 ... 11
本研究の目的および章構成 ... 12
図 ... 13
第2章 C12orf65タンパク質の機能解析 ... 17
序 ... 17
実験方法 ... 17
siRNAを用いた発現抑制 ... 17
細胞培養と遺伝子導入法 ... 18
ウエスタンブロッティング ... 19
定量的PCR(Quantitative PCR,qPCR) ... 20
細胞数測定 ... 21
細胞周期解析 ... 22
Annexin V-FITC / PIアッセイ ... 22
ミトコンドリア 容積と膜電位解析 ... 23
Reactive Oxygen Species(ROS)の測定 ... 24
2
シトクロムcオキシダーゼ活性測定 ... 24
結果 ... 25
siRNAによる発現抑制と細胞増殖の影響 ... 25
細胞周期解析 ... 25
Annexin V-FITC/PIアッセイ ... 26
ミトコンドリアの容積および膜電位 ... 27
シトクロムcオキシターゼ活性 ... 28
ROS活性の測定 ... 28
考察 ... 29
図 ... 31
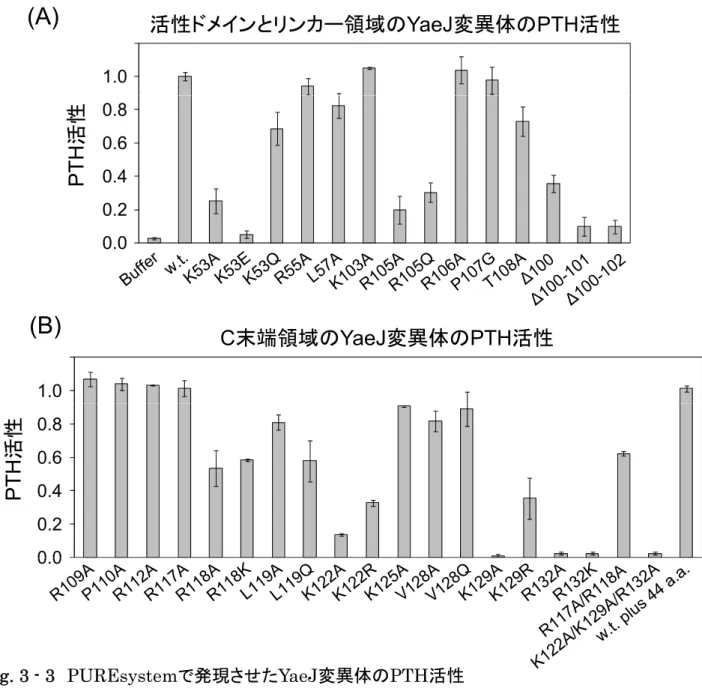
第3章 YaeJタンパク質の変異体解析 ... 37
序 ... 37
実験方法 ... 37
菌体 ... 37
培養 ... 38
DNA操作 ... 39
yaeJ遺伝子の変異導入 ... 40
YaeJとICT1の異生物間でのアミノ酸相同配列比較解析 ... 41
P1形質導入を用いたBL21 yaeJ欠損体(ΔyaeJ)株の作製 ... 42
大腸菌由来PUREsystemを用いたin vitro translationによるPTH活性測定 .. 43
His-tagged YaeJ / ICT1および変異体タンパク質の精製 ... 47
ショ糖密度勾配遠心分離によるリボソーム分画 ... 49
円偏光二色性スペクトル測定 ... 51
結果 ... 52
YaeJタンパク質およびヒトICT1タンパク質のPTH活性の比較 ... 52
3
YaeJおよびICT1の生物種間のアミノ酸配列の相同配列比較解析 ... 53
In vitroにおいて終止コドンを欠損せたmRNAにより滞ったリボソームに対する YaeJ タンパク質のPTH活性 ... 53
考察 ... 62
表と図 ... 65
第4章 総括 ... 76
GGQモチーフをもつ翻訳停滞解消因子のグループ分け ... 76
ミトコンドリアでの二つの翻訳停滞解消因子 ... 77
C12orf65の不完全なタンパク質と様々な疾患... 78
哺乳類のもう一つのミトコンドリアの翻訳停滞解消因子 ... 79
大腸菌における翻訳停滞解消機構~ tmRNA,YaeJ,ArfA ~ ... 79
結語 ... 80
図 ... 82
4
第 1 章 序
翻訳停滞
翻訳は,DNAで構成されている遺伝情報から,タンパク質という実体へ質的 変換させる点で重要かつ複雑な過程である。この過程は,リボソームという巨 大なRNA・タンパク質複合体上で行われ,mRNAの開始コドンから終止コドン までトリプレットをアミノ酸一つずつに変換することで,基本的には一つのタ ンパク質(ポリペプチド)が合成される。この合成過程には,開始,伸長,終 結の3過程があり,それぞれに様々な因子が関わっている。
しかし,細胞内ではこのmRNAの翻訳がスムーズに行われ予定通りのタンパ ク質ができることはまれで,様々な理由により翻訳が滞ることが知られている。
例えば,RNA ポリメラーゼが不完全に転写を終えてしまったり,あるいは mRNAがturnoverされる際にRNaseで切断されてしまったりして終止コドン を失った場合である。この場合,翻訳終結因子(class I peptide release factor,
以下RF)による終止コドンの認識ができず,リボソームでは翻訳終結を起こす
ことができない。結果としてポリソームのまま多くのリボソームをトラップし た状態となる(翻訳停滞;翻訳の滞り)。これに対し,何らかの滞りを解消(rescue)
する仕組みがなければこの異常な状態が続かざるをえない。実際,大腸菌
(Escherichia coli)では250回の翻訳を行われる毎におよそ1回リボソームが 滞ってしまう事が報告されている(Moore and Sauer, 2005)。この頻度でリボソ ームが滞ってしまうと,細胞が一回分裂する間もなく不完全なmRNAによって,
すべてのリボソームが捕われて使えなくなってしまうことになる。
5
tmRNA・SmpB複合体による翻訳停滞解消機構
真正細菌ではこのように頻繁に起こる翻訳の滞りに対してどのように対処し ているのだろうか。これに対し,これまで tRNA と mRNA の両方の機能を有 したtransfer-messenger RNA(tmRNA)という低分子RNAが関わるとされ てきた(Fig. 1-1)(Janssen and Hayes, 2012)。この分子はSmpBという塩基 性タンパク質(160残基)と複合体を形成して初めて機能する。まずtmRNA・ SmpB複合体はtRNAAlaのようにアラニル-tRNA合成酵素によってアラニンが 付加され,そして伸長因子 EF-Tu・GTP と複合体を形成する。そして滞ったリ ボソームのAサイトにtRNAの様に入り伸長中のポリペプチド鎖がtmRNAに 付加されたアラニンに受け渡されtrans-translation(トランス翻訳)を起こす。
その後はmRNAの様にtmRNA内にコードされているORFが翻訳され,その 終止コドン使ってRFが翻訳終結させる。結果として通常の翻訳終結過程を経て 滞ったリボソームは解消される。また合成された未完全なポリペプチド鎖は,
tmRNA内にコードされている短いペプチド(タグペプチド,AANDEYALAA) がC 末端に付加され解消される。このタグペプチドは ClpXP,ClpAP,Lon,
HflB などの ATP 依存性プロテアーゼの標的部位となり速やかに分解される (Moore and Sauer, 2007)。
このように,この翻訳停滞解消機構は,「滞ったリボソームを解消し再利用を 可能とする」だけではなく,「生じてしまった未完全なポリペプチド鎖を速やか に分解させるというタンパク質の品質管理機構」を含む非常に洗練された系で ある。また,最近ではSmpBがトランス翻訳の様々なステップで,重要な役割 を果たしていることが明らかとなってきており(Kurita et al., 2010),本研究で 扱う翻訳停滞解消との類似性については,第3章で考察する。
6
YaeJタンパク質によるtmRNA非依存的翻訳停滞解消機構の示唆
tmRNA による未完全なタンパク質の分解システムを含んだ翻訳停滞解消機
構は,真正細菌では普遍的に保存されていることが配列解析から示されている (Moore and Sauer, 2007)。この普遍性は,細菌にとって翻訳停滞解消機構の必 要性を強く示すものである。しかし,最近になって tmRNA 遺伝子(ssrA)を 欠損させた大腸菌においても,滞ったリボソームが効率よく解消されていると の報告が複数されてきた。例えばS100画分を用いたpoly(Phe)合成システムを 使ったin vitroの実験では,リボソームはtmRNAの助けなしに終止コドンをも たない mRNA から効率よく解消されている(Szaflarski et al., 2008)。また,
tmRNA欠損株を用いたin vivoの実験では,リボソームが滞りやすい配列によ って翻訳が滞っても効率よくリボソームが解消されていた(Kuroha et al., 2009)。
以上の結果から,大腸菌において tmRNA の系とは異なった未知の翻訳停滞 解消機構の存在を示唆していた。この答えの一つが,2010 年の 11 月に本研究 室で初めて報告したYaeJタンパク質である(驚くべきことに,大腸菌にはもう 一つの翻訳停滞解消因子ArfAが存在することが同時期に明らかにされたが,こ のことは第4章で述べる)。
当研究室が全長140残基の小さな塩基性タンパク質YaeJというタンパク質に 注目したのは,RFの活性部位配列を特徴付けるGGQ(Gly-Gly-Gln)モチーフ をもっているからである。RFではこのモチーフのアミノ酸が活性部位を形成し,
ペプチジルtRNAをtRNAとポリペプチド鎖に加水分解する。興味深いことに,
RFと比較するとYaeJタンパク質の配列はより短く,明らかに終止コドンを認 識するドメインが存在しない。この特徴は,YaeJが終止コドン認識を必要とし ない翻訳停滞解消因子と考えるならば,都合がよい。
7 翻訳停滞解消YaeJの機能
本研究室では,YaeJが翻訳の滞ったリボソーム上の,Pサイトに存在するペ プチジルtRNAをpeptidyl-tRNA hydrosis(PTH)活性により,tRNAとポリ ペプチド鎖に加水分解することができるのではないかという作業仮説を立て,
大腸菌由来再構成型無細胞タンパク質合成系(PUREsystem)を用いて検証し た(Handa et al., 2011)。mRNAとしては,crp遺伝子に対して終止コドンを欠 損させたものを使用した(nonstop mRNA)。反応時に蛍光ラベルされたLysを 取り込ませ,中性のSDSゲル電気泳動後,蛍光イメージャーを用いて合成され たタンパク質の量を測定した。YaeJがないと翻訳は滞ってしまうためペプチジ ルtRNAが検出されるが,YaeJを添加することによりペプチジルtRNAは検出 されなくなり,代わりに Crp タンパク質が検出さるようになった。このことか ら,nonstop mRNAに対して,YaeJはPTH活性をもつことが示された。また,
終止コドン上流の24塩基前に4個連続したArgのレアコドン(使用頻度が低い コドン)を挿入したmRNAを用いて作った翻訳の滞りに対しても,YaeJはPTH 活性を示した(この結果は,後述のtmRNAの系との相違点の一つとなる)。一 方,終止コドンがあるmRNAを使った通常の翻訳ではYaeJを加えても翻訳効 率に変化はなかった。
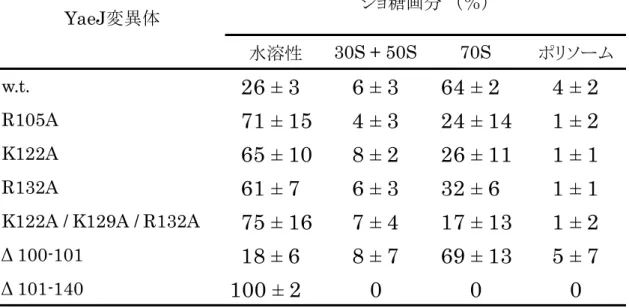
次にショ糖密度勾配法を用いて YaeJ とリボソームとの結合を検証したとこ ろ,YaeJは70Sおよびポリソーム画分に検出された。C末端領域を10残基分 欠損させるとシグナルは低分子画分に検出されるようになることから,YaeJの 塩基性残基に富む C 末端領域は,リボソームとの結合に重要であることが示さ れた。
本研究室の実験は主にin vitroの実験であったが,岡山大の阿保らは,本研究
8
室の研究とは独立に,遺伝学的研究からYaeJが翻訳停滞解消因子であるという 結論に行き着いている。tmRNAと ArfA(後述)を共に欠損させると大腸菌は 致死的であるが,過剰発現により suppress する遺伝子を探索した結果,yaeJ 遺伝子を見いだしたのである(Chadani et al., 2011a)。
以上の結果から,YaeJ が,tmRNAの系とは別の翻訳停滞解消因子であるこ とが明らかとなった。
YaeJのヒトホモログICT1はミトコンドリアの翻訳停滞解消因子として機能す る
YaeJはtmRNAとは異なってグラム陰性菌でしか保存されていないが,YaeJ の研究をさらに興味深くさせているのは,そのオーソログが酵母からヒトに至 るまで真核生物に広く存在し,そこではミトコンドリアの翻訳系の翻訳停滞解 消因子として機能していることが明らかになってきたからである(Handa et al.,
2010)。ミトコンドリアは核とは別に独自のDNA(mtDNA)をもち,それに対
応して独自の翻訳系をもつ(Taylor and Turnbull, 2005)。その翻訳系においても 翻訳停滞は起きているはずであるが,一部の例外を除いてはtmRNA・SmpBは 存在せず,長い間その解消機構の存在が意識されることはなかった。2010年に なって,英国のRichterらのグループにより,YaeJのヒトオーソログと考えら れる ICT1 がミトコンドリアにおける翻訳停滞解消因子であることが明らかに された(Richter et al., 2010)。ICT1は,もともと1995年にヒト結腸腺がん細胞
株(HT29-D4)の分化に伴い発現量が最も抑制された遺伝子の一つとして同定
され,immature colon carcinoma cell transcript 1(ICT1)と名付けられたも のである(van Belzen et al., 1995)。
Richterらの報告によるとICT1は,単離したミトコンドリアリボソームに常 に結合している(そのため彼らは,ミトコンドリアリボソームタンパク質の一
9
つとして考えている)。siRNA法を用いてヒト培養細胞におけるICT1の発現を 抑制すると,細胞増殖は著しく抑えられるが,そこでは mtDNA 内にコードさ れているタンパク質の発現量が著しく減少していた。大腸菌の S30 画分を用い てICT1のPTH活性を調べたところ,nonstop mRNAに対してのみ活性がある ことが示された。以上のことから,ICT1は,ミトコンドリアでの翻訳停滞解消 因子であることが強く示唆された。おそらく,ICT1が機能しなくなると翻訳の 滞りを解消できず,mtDNA上の13種類(哺乳類の場合)のタンパク質をコー ドしている遺伝子から効率よくタンパク質合成することができなくなると考え られる。
当研究室では,Flow cytometry解析を用いてICT1の発現抑制による細胞お よびミトコンドリアへの影響を調べた(Handa et al., 2010)。ヒト培養細胞にお いてRNAi法を用いてICT1の発現を抑制すると,ほとんど増殖できなかった。
また,発現抑制によりアポトーシスが起きており,コントロールに比べると,
正常なミトコンドリアの膜電位と容積をもつ細胞数が著しく減少していた。特 に,ミトコンドリアの膜電位と容積が低下した細胞が増えていた。
もう一つのミトコンドリアの翻訳停滞解消因子C12orf65
ヒトミトコンドリアにはtmRNA・SmpBが存在しないことから,ICT1がミ トコンドリア翻訳系で唯一の翻訳停滞解消因子であると思われたが,同年すぐ に,ICT1同様に GGQモチーフをもつ C12orf65タンパク質が新たに翻訳停滞 解消に関与していることが示唆された(Antonicka et al., 2010)。このタンパク質 の研究の起点は,翻訳停滞解消の研究からではなく,ミトコンドリア脳筋症を 発症している家系の異なる 2 名の患者のゲノムを調べたところにある。そのゲ
ノムではc12orf65遺伝子中に一塩基欠損が起きて終止コドンが生じており(ナ
10
ンセンス変異),その結果,C12orf65 タンパク質が発現されていないことが判 明した。つい最近では,遺伝性痙性対麻痺の日本人の患者(同一家系の 2 名)
からも同様に,c12orf65遺伝子にナンセンス変異が見つかっている(Shimazaki et al., 2012)。Antonickaのグループは,ミトコンドリア脳筋症患者の皮膚線維 芽細胞由来の初代細胞株において,ICT1の発現を抑制した場合と同様に,ミト コンドリアタンパク質の発現量の低下を観察した。興味深いことに,この細胞 に対して ICT1 を過剰発現させると,シトクロム c オキシダーゼ(COX)活性
が約50%回復すると同時に,呼吸鎖複合体の一部の発現量がある程度回復する。
このことは,C12orf65がICT1と同様に翻訳停滞解消因子として機能している ことを示唆している。しかし,C12orf65 に関しては多くのことが未知であり,
またICT1と同等の条件下でその欠損の影響を比べるデータはない。
NMRによるICT1の立体構造解析
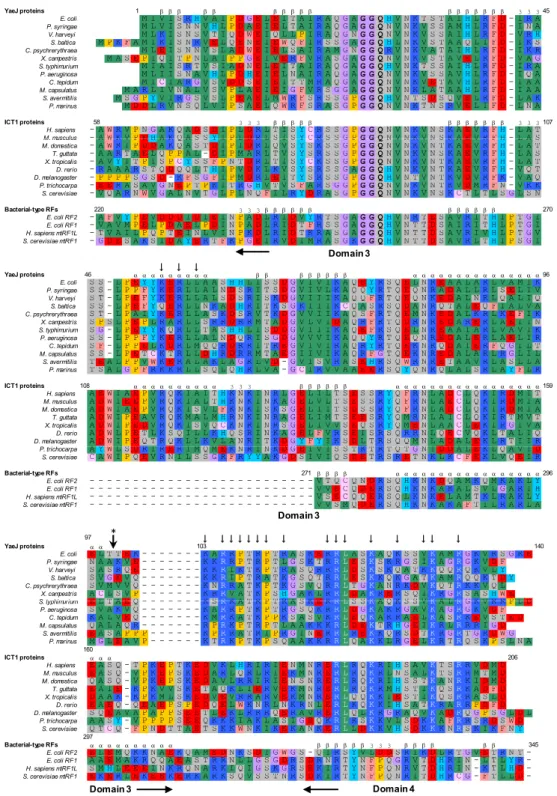
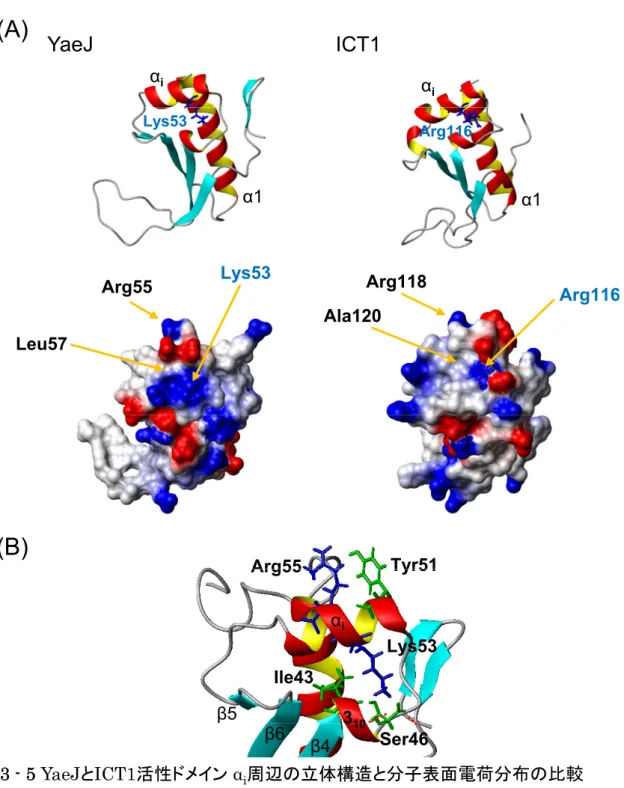
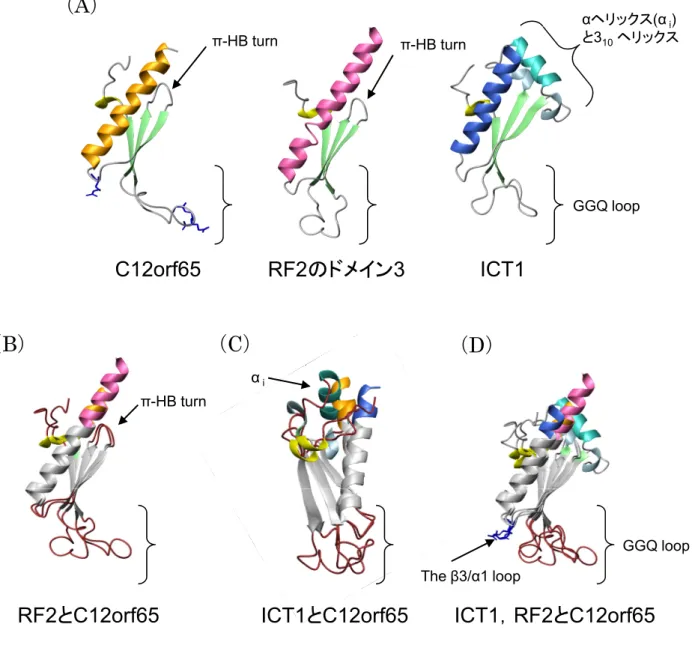
当研究室では,理化学研究所と共同研究よりマウス(Mus musculus)由来の ICT1タンパク質の GGQドメインの構造を決定することに成功した(Handa et al., 2010)(Fig. 1-2)。この構造を用いて構造的相同検索を行った結果,予想通 り真正細菌型のRFのGGQモチーフが含まれるドメイン3と酷似していること がわかった(Frolova et al., 1999; Zavialov et al., 2002)。
真正細菌型(ミトコンドリア由来を含む)のRFは,4つのドメインから構成 されており,ドメイン2と4は終止コドンを認識し30Sサブユニットと結合す る(Fig. 1-3)。ドメイン3 は50S サブユニットのpeptidyltransferase center
(PTC)と結合し,PサイトにあるペプチジルtRNAをPTH活性によりtRNA とポリペプチド鎖に加水分解する事で翻訳を終結される(Klaholz et al., 2003;
Rawat et al., 2003)。ICT1のGGQドメインは,このドメイン3との構造的相
11
同だけでなくGGQモチーフ周辺の活性部位に関わるアミノ酸配列もRFとよく 共通していた。最も異なる点としては,β2とβ3とを結びつける領域において,
RFのドメイン3ではそこが6残基からなるターン(ターンの分類によるとπ-HB turn)を形成するのに対して(Dasgupta and Chakrabarti, 2008),ICT1ではα ヘリックス(αi)が挿入されている。しかし,この挿入によってドメイン3の構 造上のアーキテクチャに変化はない。さらに配列解析の結果をふまえると,こ の決定された構造は,すべての生物種のICT1/YaeJタンパク質で保存されてい ることがわかる。また,ICT1の方が真正細菌由来のYaeJよりN末端が長いの は,ミトコンドリア移行シグナルがあるためである。
以下,このGGQモチーフをもつ活性ドメインのことをGGQドメインとする。
YaeJとリボソームとの共結晶構造
2012 年に,米国の Gagnon らによって,大腸菌由来の YaeJ と高度好熱菌
(Thermus thermophilus)由来のリボソームとの共結晶構造(3.2 Å)が報告 された(Fig. 1-4)(Gagnon et al., 2012)。GGQドメインは,予想通りRFのド メイン3と同様にPサイトにあるペプチジルtRNAのエステル結合(ペプチド とtRNAの間の結合)を分解する位置にあった。注目すべき点は,YaeJの非構 造領域であるC末端領域(113~128)がαヘリックス構造をとって,mRNA entry channel(30S サブユニットにあるmRNAの通る穴)の中に深く入り込んでい た点である。そのαヘリックスとGGQドメインとの間は,flexibleなペプチド 鎖(リンカー領域)で結ばれていて,α ヘリックスが錨のように 30S サブユニ ットに打ち込まれてように見える。ここで興味深いのは,SmpB と YaeJ の C 末端領域がリボソーム上で果たす機能の類似性である(塩基性アミノ酸が多い 以外は配列に相同性はない)。tmRNAと複合体を構成しているSmpBも,Aサ
12
イトに結合時にはその非構造領域であるC末端がαヘリックスとなってmRNA entry channelに結合する(ただし,YaeJとは異なり,それほど深く入り込ま ない)。その結合時には,SmpBあるいはYaeJのどちらの場合でも,16S RNA のG530をsyn型からanti型にフリップさせるが,同じようなフリップは通常 の翻訳におけるコドンとアンチコドンの相互作用のときにも見られるものであ る。
本研究の目的および章構成
本研究の目的は,「真正細菌およびミトコンドリアにおける翻訳停滞解消機構 の解明」である。
そのため,第2章では,ミトコンドリアにおける翻訳停滞解消因子C12orf65 を中心に,ヒト培養細胞においてsiRNAを用いた発現抑制による細胞およびミ トコンドリアへの影響の比較を行った。これにより,ICT1とC12orf65の機能 の相違点を明らかにし,どのような役割分担があるか考察した。
第3章では,YaeJがリボソーム上でどのように機能しているのか,その分子 メカニズムを明らかにするために,YaeJに対し変異体解析を行い,翻訳停滞解 消での PTH 活性に必要な残基を同定した。また,大腸菌リボソームに対して,
ICT1がYaeJとほぼ同様な活性を示すことを明らかにし,このことから真正細 菌由来のYaeJと真核生物由来のICT1との異生物種間でのアライメントに成功 した。以上の結果から,第 4 章では,これらの翻訳停滞解消因子のグループ分 けを提示し,その機能・役割分担,他の翻訳停滞解消因子との関係,進化など 様々な角度から考察する。
Ala
終止 ドンを
アラニル‐tRNA合成酵素
によってアラニンが付加 されたtmRNA
A‐site P‐site
5' 3'
A
滞ったリボソーム 終止コドンを
失ったmRNA tRNA tmRNA
A‐site P‐site
A‐site
5' 3'
5' 3'
5' 3'
A
mRNAの解放
リボソ ムのリサイクル
A
A
tRNAがAサイトに結合
A
リボソームのリサイクル
ClpXP,ClpAP,Lon,HflB
A A
N D E Y
N A L A A
ORF
タンパク質分解酵素 Tag‐peptide
Fig. 1 - 1 tmRNAによるリボソームの翻訳停滞解消機構
終止コドン欠損などにより,リボソームのAサイトにmRNAがなくなるとアラニル-tRNA合成 酵素によってtmRNAにアラニンが付加された状態でAサイトにtmRNAが入る。その後,
tmRNA内にあるタグペプチドに対応するORFを翻訳後,tmRNA内の終止コドンをRFが 認識して翻訳を終わらせる。タグペプチドが付加されたタンパク質はタグペプチド認識プロ テアーゼによって速やかに分解される。tmRNAに結合しているSmpBは省略した。
13
(A) RF2のドメイン3
1
C π-HBturn
N
C
23 N
310
GGQ loop
3
2
1 310
The 3/loop
(B) ICT1
i
N
C
GGQ loop
N
i C
310-2
310-3
2 3
1 310-2
310-1 310-3
GGQ loop
3
2
310-1 1
The 3/loop
Fig. 1 - 2 RFのドメイン3とICT1の活性ドメイン(GGQドメイン)の溶液中の立体構造およびト ポロジー比較
(A)RF2のドメイン3(203-307残基)の立体構造およびトポロジー(PDB code 1MI6)。立体構
GGQ loop
(A)RF2のドメイン3(203 307残基)の立体構造およびトポ ジ (PDB code 1MI6)。立体構 造は3次元立体表示ソフトMOLMOL(Koradi et. al., 1996)を用いて表示した。310ヘリックス,
βストランド, αヘリックス,GGQループはそれぞれ,黄色,黄緑色,桃色,茶色で示した。
(B)本研究室で決定したマウスICT1タンパク質のGGQドメインの立体構造およびトポロジー
(PDB code 1J26) 。31010ヘリックス, βストランド,αiiヘリックス,αヘリックス,GGQQループはそれ ぞれ,黄色,黄緑色,水色,青色,茶色で示した。
14
( A )
ICT1 真核生物(ミトコンドリア)
(365)
(206)
128 224 236 306 322 117
1
3 4
2 ドメイン1
真正細菌(RF1,RF2)
ミトコンドリア(mtRF1a) 365
ICT1 真核生物(ミトコンドリア)
真正細菌
(206)
69 162
YaeJ (140)
1 99
( B )
To the peptidyl- transferase
center (PTC) ドメイン1
ドメイン3
To the decoding center
ドメイン2/4
Fig. 1 - 3 GGQファミリーのドメインの構成とRFの構造
(A) 大腸菌RF2,マウスICT1,大腸菌YaeJのドメイン構成の概略図。数字は残基番号を 示す。
(B) 高度好熱菌における70Sリボソームと結合しているRF2の構造 (PDB code 2WH1)。リ ボソームの構造は書いていない。 ドメイン1,2と4は(A)と同色で書かれている。GGQモ チーフを含むドメイン3はそれぞれαヘリックスとβシートはそれぞれ桃と緑で示した。
15
(B)
(A)
Pサイト
tRNA YaeJ
50Sサブユニット
(B)
(A)
30Sサブユニット 30Sサブユニット
Fig. 1 - 4 大腸菌YaeJと高度好熱菌70SリボソームとのX線結晶構造解析
(A)Nonstop mRNAによる翻訳停滞時の70SリボソームにおけるYaeJの機能(4DH9,
( ) o stop R による翻訳停滞時 70Sリボソ における aeJ 機能( 9, 4DHA)。50Sサブユニット,30Sサブユニット,nonstop mRNA,YaeJタンパク質,P部位 のtRNAをそれぞれ青色,アイボリー,灰色,赤色,緑色で示した。立体構造は3次元立体 表示ソフトUCSF Chimera (Pettersen et al., 2004)を用いて表示した。
(B)30Sリボソームに結合しているYaeJ(4DH9)。YaeJを赤色,PサイトのtRNAを緑色で 示した。
16
17
第 2 章 C12orf65 タンパク質の機能解析
序
本章では,ミトコンドリアにおける翻訳停滞機構の解明を目的とし,翻訳停 滞解消因子ICT1 とC12orf65 を培養細胞においてsiRNAを用いてそれぞれの 発現抑制することにより,細胞およびミトコンドリアにおける影響を調べた。
これによって,2つの因子の影響を調べると共に,詳細な比較を行うことで,機 能的な相違点を明らかにする。方法として,様々な蛍光色素を用いて Flow cytometry(FCM)解析を行い,細胞周期,アポトーシス/ネクローシス解析,
ミトコンドリアの容積と膜電位およびROSの測定を行った。さらにシトクロム cオキシダーゼ活性を測定することで,ミトコンドリア活性の評価も行った。
実験方法
siRNAを用いた発現抑制
本実験では,3 種類の siRNA をミックスした siTrio カクテル(Hokkaido System Science)を用いて,ICT1とC12orf65の発現抑制を行った。
siRNA配列の設計は,コスモバイオ株式会社に委託し,合成はHokkaido System Science社に依頼した。
ただし,c12orf65の発現抑制に関してはミックスした3種類のsiRNAよりも 1種類のみの方が効果的であったために1つを選択的に使用した。
使用したそれぞれのsiRNAの標的配列は以下の通りである。
si-ICT1-A: GCACUGAGUUCAAGAGCAU
18
si-ICT1-B: GCAGAUUGCCUGCAGAAAA si-ICT1-C: CGGGAAAGGCUGAGACAAA si-C12orf65 : CCAGAAGCUCCCAGGGAAU
ネガティブコントロールとして用いたNontarget(NT)siRNAの配列は以下の 通りである。
si-NT-A : AUCCGCGCGAUAGUACGUA si-NT-B : UUACGCGUAGCGUAAUACG si-NT-C : UAUUCGCGCGUAUAGCGGU
細胞培養と遺伝子導入法
本研究にはHeLa細胞を使用した。10% fetal bovine serum(以下,FBS), 100 units/ml ペニシリン,および 100 μg/ml ストレプトマイシンを添加した Dulbecco's Modified Eagle's Medium(以下,DMEM)(Sigma-Aldrich)を使 って培養した。
標的遺伝子の発現抑制は,siRNA を用いてリバーストランスフェクション法 で行った。遺伝子導入条件は,5% FBSのみを含むDMEMを用いて35 mmデ ィッシュ(Corning)にHeLa細胞 [1.0 × 105 cell/well]を播種し,2.5 μlの遺伝 子導入試薬Hily Max(Dojindo)を用い,最終濃度50 nMになるようにsiRNA を導入した。導入後,24時間後に10% FBSを含む新鮮なDMEMに交換して,
数日間 CO2インキュベーターで培養をおこない,以降の実験のサンプルとして 使用した。
以下,非標的なsiRNAを導入した細胞をsi-NT,ict1に対するsiRNAを導入 した細胞を si-ICT1,c12orf65 に対する siRNAを導入した細胞を si-C12orf65 と表記する。
19 ウエスタンブロッティング
サンプル調製
HeLa細胞の培地を捨て,Phosphate Buffered Saline(以下,PBS)で2回 洗浄をした。氷冷PBSを1 ml加えてセルスクレーパーで培養細胞を1.5 ml チ ューブに回収した。遠心分離(3,000 rpm,10分,4℃)をおこない,PBSを除 去した。回収したHeLa細胞にLysis Buffer [7 M urea,2 M thio-urea,1%
Triton X-100,1 mM PMSF,0.5 mM EDTA]を加え,HeLa細胞から全タンパ ク質を抽出した。この操作は常に氷上で操作を行った。細胞塊をよくピペッテ ィングで懸濁し,超音波ホモジナイザー(Taitec)で数分間処理した。その後,
遠心分離(12,000 rpm,30分,4℃)をし,上清を新たなチューブに移し,細 胞抽出液とした。
タンパク質の定量
タンパク質濃度は,ブラッドフォード法に基づくQuick start Bradford Dye Reagent(Bio-Rad)を用いて決定した。標準曲線を得るための標準タンパク質 として,ウシ血清アルブミン(以下, BSA)を用いて,異なる濃度の BSA を 加えたブラッドフォード試薬の波長 595nm における吸光度を分光光度計(GE Healthcare)で測定をした。BSA濃度とブラッドフォード吸光度を基づき,標 準曲線を算出した。これより,濃度未知なタンパク質液の濃度を定量した。
ウエスタンブロット
細胞抽出液50 µg を15% SDS-PAGE で分離し,泳動後のゲルを 200 ml の 転写バッファー [24.8 mM Tris,192 mM Glycine,20% Methanol]に数分間浸 した。あわせて,ゲルと同じサイズにBioTrace NT-Nitrocellulose Membrane φsize 0.2 μm(Pall)とフィルター紙(Advantech)を切り出し,転写バッファ ーに十分に浸しておいた。その後,トランスブロット SD セル(Bio-Rad)を 使用し,SDS-PAGE ゲルからメンブレンにタンパク質を転写した(constant 1
20 mA / cm2,1 hr)。
転写後,メンブレンを1% skim milk / PBS- 0.1% Tween(以下,PBST)で 振とうし(30分,室温),ブロッキングを行った。
ブロッキング終了後,メンブレンを5 mlの抗原抗体反応増強剤can get signal 1(Toyobo)でそれぞれの一次抗体 [ ICT1 MaxPab mouse polyclonal antibody
(1:500),C12orf65 MaxPab mouse monoclonal antibody(1:500)(Abnova)] を希釈し,ハイブリパック(コスモバイオ)に入れ,振とう(一晩,低温室)
し た 。 ま た , 泳 動 コ ン ト ロ ー ル と し て は , ハ ウ ス キ ー ピ ン グ 遺 伝 子 ヒ ト Gamma-tubulinの一次抗体(Cell Signaling Technology)を5 mlに希釈(1%
skim milk/TBST(1:8000))して用いた。
次に,ハイブリパックからメンブレンを取出し,新鮮なPBST で振とう(10 min,室温)し,洗浄した。この洗浄操作を 3 回繰り返し,メンブレンを再び ハイブリパックに入れ,can get signal solution 2で5 mlに希釈した二次抗体 [ Anti-mouse IgG HRP-linked Antibody(1:4000)(Cell Signaling Technology)] で,振とう(70分,室温)した。その後に再び,メンブレンをPBSTで3 回振 とう洗浄(10分,室温)した。
タンパク質の検出は,化学発光試薬ECL Prime Western Blotting Reagents
(GE Healthcare)を使用しX線フィルム(FUJI Medical)に感光し現像した。
定量的PCR(Quantitative PCR,qPCR)
チューブに回収した細胞塊にTriPure Isolation Reagent(Roche)を1 ml加 えて,ボルテックスミキサー(Taitec)で懸濁した。懸濁液にクロロホルムを 200 μl加えて懸濁し,遠心分離(12,000 rpm,15分,4℃)にて,水層と有機 溶媒層に分けた。中間層にある変性したタンパク質を取らないように水層の上
21
清を新しいチューブに回収した。上清に酸性フェノール500 μlとクロロホルム 100 μl加えて,遠心分離(12,000 rpm,15分,4℃)することでDNAとRNA を分離した。上清をチューブに回収し,イソプロパノール沈殿をおこないトー タルRNAを精製した。
精製した100 ng トータルRNAをReverTra Ace qPCR Mix(Toyobo)を用 いて cDNA 化した。合成した 2 ng cDNA と標的遺伝子のプライマーと共に GoTaq qPCR Master Mix(Promega)でMyiQ(Bio-Rad)を用いて付属の説 明書に従った条件でqPCRを行った。解析は,ヒトgapdh遺伝子の発現を内部 標準として,ΔCt法を用いて相対的に行った。
細胞数測定
siRNA導入後 48,72,96 時間に培養している細胞の培養液を取り除き,PBS で洗浄後,Trypsin-EDTA(Sigma-Aldrich)で処理をし,10%FBSを含むDMEM を加えて細胞を回収した。生細胞より膜透過性が高い死細胞を特異的に染色で きるトリパンブルー(Wako)を使用し,生細胞を観察の観察を行った。0.4%ト リパンブルー90 μlと回収した培養細胞を含む懸濁液30 μlを混合し血球計算盤 に入れた。これを位相差顕微鏡で観察し,生細胞数を数えて測定した。
Flow cytometry(FCM)解析
FCMは,一点に集められたレーザー光をフローセルの中心を通過する細胞に 照射し,そこから生じる散乱光と,蛍光を同時に測定する技術または方法のこ とをいう。測定結果は,相関ヒストグラムとして示され,これらのヒストグラ ムを解析することにより統計的にサンプルの特性を明らかにできる。測定機器 は,本学の生体情報ゲノムリソースセンターにある FACSCalibur(Becton
22 Dickinson)を使用した。
なお,FACSCaliburの詰まりや測定のブレを防ぐため,準備した細胞はすべ て測定前にナイロンフィルターを通し,細胞塊を除いた。また,本研究では,
アポトーシスのポジティブコントロールとして,カスパーゼ 3 の活性化による アポトーシス誘導剤のスタウロスポリン(staurosporine)を終濃度50 nMで使 用した(Chae et al., 2003)。
細胞周期解析
培養してあるHeLa細胞をPBS で洗浄後Trypsin-EDTAで処理し,DMEM で再懸濁を行い,遠心(1,000 rpm,3分,室温)して,細胞を回収した。その 後,細胞をPBSで洗浄し,再度遠心(3,000 rpm,10分,4℃)で細胞を回収 し,70%エタノールを 1 ml加えて細胞固定(一晩,4℃)をした。固定した細 胞は遠心分離(3,000 rpm,10分,4℃)をおこないエタノールを除去した後に,
PBSで2回洗浄を行った。その後,1 mlのPBSで再懸濁し,終濃度10 μg/ml のRNase A(Qiagen)と終濃度20 μg/mlのDNA蛍光プローブPropidium iodide
(以下,PI)(Dojindo)を加えることで,擬陽性を示す細胞内のRNAの切断と DNA染色を同時におこった(2時間,4℃)。FCMで 104個の細胞を,PI の蛍 光波長で測定し,サンプルごとの細胞周期の割合を求めた。
Annexin V-FITC / PIアッセイ
細胞のアポトーシス/ネクローシス検出のため Annexin V-FITC Apoptosis Detection Kit I (Mbl)に含まれるPIと,Fluorescein isothiocyanate(以下,
FITC)が結合しているAnnexin Vを使って細胞を染色した(Fadok et al.,1992)。 培養してあるHeLa細胞をPBS で洗浄後Trypsin-EDTAで処理し,DMEM
23
で再懸濁を行い,遠心(1,000 rpm,3分,室温)して,細胞を回収した。回収 した細胞をPBSで洗浄後,遠心分離(1,000 rpm,10分,4℃)をおこない,
500 μlのbinding bufferで再懸濁する。そこに5 μlのAnnexin V-FITCと5 μl のPI を加え,遮光して 15 分間,室温に静置した。その後 FCM解析を行うま で氷上に静置した。この実験も同様に FCM で 104個の細胞に対して FITC と PIの蛍光波長で検出した。
ミトコンドリア 容積と膜電位解析
ミトコンドリアの機能への影響を調べるために,ミトコンドリア標識用の蛍 光プローブMitoTracker Green FM(Life Technologies)とMitoTracker Red Chloromethyl-X-rosamine(CMXRos)(Life Technologies)を用いた。
MitoTracker Green FMは,ミトコンドリアの膜電位非依存的にミトコンド リアに蓄積し,緑色蛍光を示すプローブでミトコンドリア容積を調べることが できる。また,MitoTracker RedCMXRos は,ミトコンドリア膜電位依存的に ミトコンドリアに蓄積し,赤色蛍光を示すプローブでミトコンドリアの膜電位 を調べることができる(Pendergrass et al., 2004)。
これらの蛍光プローブを,siRNAでそれぞれ発現抑制したHeLa細胞に最終 濃度0.1 μMになるように加えて37℃で,1時間CO2ガスインキュベーター内 で培養した。これらの HeLa細胞は PBS で洗浄後,Trypsin-EDTAで処理し,
DMEMで再懸濁を行い,遠心(1,000 rpm,3分,室温)して,細胞を回収し た。回収した細胞を1 mlのPBSで再懸濁し,遠心分離(3,000 rpm,10分,4℃)
をおこない,細胞塊を集めた。回収した細胞に70%冷エタノールを加えて,エ タノール固定(2時間,4℃)を行った。固定した細胞は遠心分離(1,500 rpm, 10分,4℃)をおこない,PBS で 2回洗浄した。この実験も同様に細胞 104個
24
をそれぞれの蛍光色素に適した蛍光波長(MitoTracker Green FM:516 nm, MitoTracker RedCMXRos:599 nm)を測定した。
Reactive Oxygen Species(ROS)の測定
アポトーシスの指標の 1 つである細胞内の活性酸素種(Reactive Oxygen Species( ROS)) を 測 定 す る た め , 細 胞 浸 透 性 蛍 光 プ ロ ー ブ 2’, 7’-Dichlorodihydrofluorescin diacetate(以下,DCFH-DA)(Sigma-Aldrich) を利用した(Bland et al., 2001)。DCFH-DAは,細胞膜内に浸透し,細胞内エス テラーゼにより脱アセチル化し非蛍光型2’, 7’-Dichlorodihydrofluorescin(以下,
DCFH) と な る 。 さ ら に ROS に よ り 素 早 く 酸 化 さ れ 強 く 蛍 光 す る 2’, 7’-Dichlorodihydrofluorescein(以下,DCF)に変化する。したがって,この蛍 光強度は細胞質基質のROS量に比例する。
この蛍光プローブを培養してあるHeLa細胞に最終濃度0.1 μMになるように 加えて 1 時間,37℃で CO2インキュベーター(Espec)内で培養した。HeLa 細胞はPBSで洗浄後,Trypsin-EDTA処理によって細胞をはがし再度1 mlの PBS で再懸濁した。この実験も同様にFCM で 104個の HeLa 細胞の緑色蛍光 波長を測定し,解析を行った。なお,本実験では,ポジティブコントロールと して未処理のHela細胞に蛍光プローブ添加40分後にH2O2(終濃度1 mM)で 刺激を与え,20分間培養することでROSの産出を促したものを用いた。
シトクロムcオキシダーゼ活性測定
シトクロム c オキシダーゼ活性を測定するため,無傷な状態でミトコンドリ アを単離できるMitochondria Isolation Kit(Pierce)を用いてsiRNAによっ て発現抑制した培養細胞からミトコンドリアを抽出した。このミトコンドリア
25
の濃度をQuick Start Bradford Dye Reagent(Bio-Rad)を使用し決定した。
ミトコンドリアを 15 ngに調整した。その後,ミトコンドリア呼吸鎖複合体Ⅳ の酸化反応(下記の反応式)でフェロシトクロムcが酸化されて550 nmの吸光 が弱くなることを利用し,シトクロム c オキシダーゼ活性を測定できる。実験 には,Cytochrome c oxidase activity assay kit(Biochain)を用いて行った。
4Fe ‐ Cytochrome c ferrocytochrome c 4H O
4Fe ‐ Cytochrome c ferricytochrome c H O
結果
siRNAによる発現抑制と細胞増殖の影響
ミトコンドリアでのICT1およびC12orf65の機能を評価するため,siRNAを 用いてHeLa細胞のICT1とC12orf65の発現量を減少させた(ノックダウン)。
qRT-PCRおよびウエスタンブロットの結果,コントロールと比べ,両タンパク
質のmRNAレベルおよびタンパク質レベルで優位に発現量の減少を確認された
(Fig. 2-1)。
siRNAを導入後48,72,96時間で細胞数を測定したところ,72時間で顕著 な差が表れ始めた。96 時間後では si-NT に比べて si-ICT1 は 90%減少し,
si-C12orf65は64%減少した(Fig. 2-1 C)。これより,両タンパク質の発現抑制 は,共に細胞増殖に大きな影響を与えるが,ICT1 の発現抑制の影響の方が,
C12orf65より著しく細胞増殖が低下をすることがわかった。
細胞周期解析
ICT1およびC12orf65の発現抑制された細胞の状態を詳細に解析するために,
96時間後に細胞をPIで染色し,FCMを用いて細胞周期解析を行った。
26
赤色蛍光色素 PI は,DNA にインターカレートすることで蛍光を呈し,この蛍 光強度は核内の総DNA量を表す。PI染色した細胞をFCMで測定することで,
DNA量のヒストグラムのピークが得られる。このヒストグラムピークは,細胞 周期(sub-G1,G1,S,G2/M)にそれぞれ当てはめることができる。これによ
って,sub-G1である細胞がアポトーシスによる断裂した核の状態を検出するこ
とができる。その結果,sub-G1期の割合は,si-NTは6.5%を示したのに対して,
si-ICT1では34.3%,si-C12orf65では13.0%とコントロールと比べて高い割合 を示した(Fig. 2-2)。
したがって,ICT1タンパク質および,C12orf65タンパク質をそれぞれ発現抑 制することでアポトーシスが生じていることが示めされ,また,ICT1を発現抑 制した方が,アポトーシスが強く生じていることがわかった。
Annexin V-FITC/PIアッセイ
アポトーシスによる細胞死をより詳細に分析するため,Annexin VとPIによ る二重染色をし,アポトーシス/ネクローシスの解析を行った。Annexin V は,
アポトーシス初期にみられる細胞膜の構造変化によって細胞内膜から外膜側へ と露出するホスファチジルセリンにカルシウムイオン依存的に結合する。一方,
PI は細胞膜構造が劣化し始めるアポトーシス後期やネクローシスにおいて,細 胞膜を透過してDNAに結合する。この2つの原理を利用して,アポトーシスと ネクローシスを詳細に解析した(Vermes et al., 2000)。
その結果,si-NT は,共に Annexin V および PI の蛍光を示さないため,
Annexin VとPIによる二成分展開による解析では,主に左下に細胞群がみられ,
アポトーシス初期を示す細胞群の割合は 6.6%を示した。それに比べ,si-ICT1 およびsi-C12orf65は,共にAnnexin Vのみの蛍光増加が見られる一方,PIの
27
蛍光増加は見られず,アポトーシス初期を示す左上方向に細胞群がシフトして いた(si-ICT1は24.6%,si-C12orf65は11.9%の割合を示した)(Fig. 2-3)。こ れはアポトーシス誘導をするスタウロスポリン処理した細胞と同様の傾向を示 した。このことから,si-ICT1およびsi-C12orf65はネクローシスによる細胞膜 破壊ではなく,アポトーシス初期が起こっていることが示された。また,細胞 周期解析の結果と同様に,C12orf65の発現抑制した細胞よりICT1の発現抑制 した方が強くアポトーシスが生じていることが示された。
ミトコンドリアの容積および膜電位
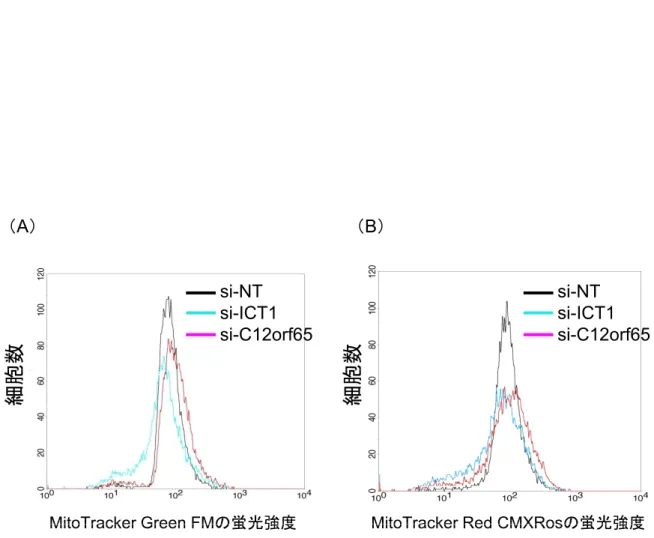
次に,ミトコンドリアへの影響を調べるため,MitoTracker Green FM と MitoTracker Red CMXRos の 2 種類のミトコンドリア蛍光プローブを用いて FCM解析を行った。MitoTracker Green FMはミトコンドリア膜電位とは関係 なく,ミトコンドリア内に選択的に蓄積し,ミトコンドリア容積に比例して蛍 光を示す。対して,MitoTracker Red CMXRosはミトコンドリアの膜電位のみ に依存する。
siRNA で発現抑制した細胞をそれぞれの蛍光プローブで染色し FCM 解析し
たところ,si-ICT1とsi-C12orf65はsi-NTに比べて,全体的にピークの減少が みられた。これは正常なミトコンドリアをもつ細胞が著しく減少したことを示 している(Fig. 2-4)。ピークのシフトに着目すると(シフトは,ある状態を示 すミトコンドリアをもつ細胞集団の変化を示す),si-ICT1 は,si-NT と比べて ピークが左側にシフトした。一方,si-C12orf65は,si-NTと比べて右側にシフ トした。つまり,コントロールと比べ si-ICT1 はミトコンドリアの膜電位や容 積が減少した細胞集団が増加した傾向を示し,si-C12orf65は反対に膜電位や容 積が増加する細胞集団が増加した傾向を示した。
28 シトクロムcオキシターゼ活性
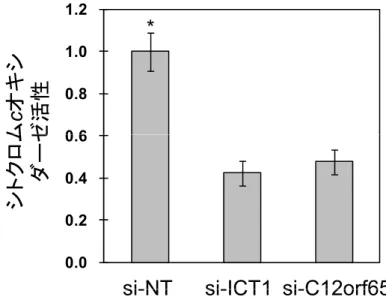
ミトコンドリアにおけるATP生産は,電子伝達系を担うタンパク質複合体Ⅰ
~Ⅴによって生じるプロトン濃度勾配によって合成される。電子伝達系のタン パク質複合体Ⅳであるシトクロム c オキシターゼの活性を測定することでミト コンドリアの活性を測定した。
その結果si-NTと比べ, si-ICT1は約60%のシトクロムcオキシターゼ活性 が低下を示した。また,si-C12orf65は,約50%の活性が低下したことがわかっ た(Fig. 2-5)。
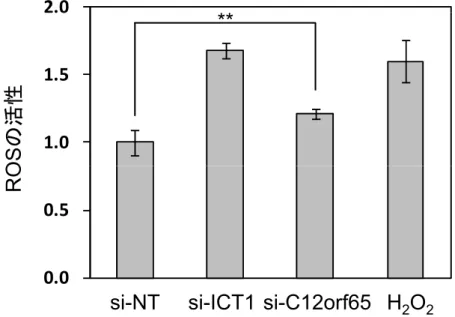
ROS活性の測定
活性酸素種(ROS)とは,酸素分子(O2)に由来する反応性に富む一群の分 子群の総称である。ROSはミトコンドリアの電子伝達系における副産物として,
また窒素酸化物(NOX)などの酵素によって産生される。ミトコンドリアにお いては,電子伝達系で電子を受け取った酸素分子の大部分は水へと変換される が,一部の酸素分子は電子還元されスーパーオキシドになる。通常は産出され たスーパーオキシドは速やかに酸化還元酵素群により水へと代謝される。しか し,細胞死などに伴うミトコンドリア機能が障害される状況では,ROSが過剰 に産出されることが知られている(Liu et al., 2013)。また,mtDNAにコードさ れたタンパク質の合成が完全でない場合は,呼吸鎖複合体の活性における電子 伝 達 中 に 電 子 の 脱 落 が 生 じ る こ と で ,ROS の 産 出 が 増 加 す る (Moreno-Loshuertos et al, 2006)。そこで,ROSの産出量が変化しているかど うか確認するため,FCMを用いてROSを測定した。結果としてsi-NTと比べ,
si-ICT1および si-C12orf65では ROS の産出がそれぞれ約1.6倍と約 1.2倍高 いことがわかった(Fig. 2-6)。si-ICT1のROSの産出量に関しては,ポジティ
29
ブコントロールである過酸化水素(H2O2)処理したものと同程度の非常に高い ROSの産出が示された(Gil et al., 2003; Kim et al., 2010)。
考察
本研究により,細胞およびミトコンドリアにとって,ICT1とC12orf65の両 因子が必須であることが明らかとなった。これらの実験結果をまとめると,翻 訳停滞解消因子の欠損によって細胞にどのようなことが起きているか,以下の ように推測される。これらの因子が一つでもなくなると,mtDNAにコードされ たタンパク質の翻訳の停滞が頻繁に生じることになる。これにより翻訳停滞が 効率よく解消できず,完全な状態で呼吸鎖複合体を合成することができない。
したがってATP生産能の著しい低下となる。これらのミトコンドリア異常によ って,アポトーシスが引き起こされることや,通常より多くのROSが産出され るといったことで,それらの負の連鎖が起き,細胞増殖に大きな影響を与える というモデルが考えられる。
本研究ではさらに,ICT1の抑制のほうがC12orf65の抑制よりも細胞および ミトコンドリアへの影響が大きく,また,両者のミトコンドリアに対する影響 の質が異なっていることを示した。Antonicka らによって,ICT1 と C12orf65 の発現抑制の影響は,共にミトコンドリアDNAにコードされているタンパク質 の合成量が著しく低下することが報告されているが(Antonicka et al., 2010),全 体的にタンパク質合成量が低下しており,ICT1とC12orf65間の差は見いだせ なかった。本研究のFCM解析の結果で,初めて両者の相違点,つまりミトコン ドリアの容積と膜電位のへの影響が有意に異なっていることを示したことにな る。ICT1 の抑制の場合は,ミトコンドリア容積および膜電位が減少しており,
C12orf65の抑制の場合は,ミトコンドリア容積,膜電位が増加を示した。一般
30
的に,ミトコンドリアが異常をきたしATP生産が低下すると,容積を増やして recovery しようとする傾向にあることが報告されている(Auré et al., 2006;
Wredenberg et al., 2002)。これを考慮するとC12orf65の抑制の場合は,その recovery機構が保たれている状態であるが,ICT1の抑制はその機構さえも壊し,
ミトコンドリア・細胞にとって深刻な状態を生じさせると考えられる。これは,
当然ながら,ICT1 の発現抑制の影響の方が,C12orf65 より著しく細胞増殖が 著しく低下するという結果と一致している。
では,両因子でどのような使い分け・役割分担がされているのか。まず,因 子それぞれに停滞解消をする遺伝子が決まっていることが考えられる。これは,
ICT1 とC12orf65 の発現抑制によって,mtDNA由来のタンパク質に差がある ことで示されるが,上記の通り両者に有意な差が見られなかった。しかし,こ の実験ではミトコンドリア量を定量的に揃えることが難しいことも関係してい るので(容量も変化している),この結果をもってこの説は否定しきれない。一 方で,酵母とヒトでは,共通する遺伝子は6個だけで,5つのタンパク質は,酵 母には存在しないことを考慮すると(Gray et al., 1999),遺伝子の種類を通じて,
ICT1とC12orf65の役割を論ずるのは難しい。
本研究室では,ICT1とC12orf65のプロモーター解析を行っており,それに よれば,少なくとも両者で転写因子が明らかに異なっている(未発表データ)。
これは,両者で発現パターンが異なることを示唆しており,両者が機能する時 期が異なっている可能性があると考えている。また,それと関連して,遺伝子 の種類というより,翻訳停滞の種類(終止コドンがない,あるいはmRNAの途 中で止まった場合など)によって使い分けがあるかもしれない。二つの因子の 機能の本質的な相違点を明らかにすることは,今後の重要な課題である。
(A)
0 6 0.8 1.0 1.2 1.4
rf65mRNA
0 6 0.8 1.0 1.2 1.4
T1mRNA
i NT i ICT1 (C) 60
(B)
0.0 0.2 0.4 0.6
C12or
si-NT si-ICT1 si-C12orf65
0.0 0.2 0.4 0.6
si-NT si-ICT1
ICT
si-C12orf65
20 40 60
si-NT si-ICT1 si-C12orf65
胞数(1 ×105)
si-NT si-ICT1 ICT1
γ-tubulin
( )
( )
si-C12orf65 si-NT
Fig. 2 - 1 siRNAの効果と発現抑制による細胞増殖への影響
0 24 48 72 96
0 C12orf65
γ-tubulin
Time [h]
細
(A)siRNAを導入したHeLa細胞の定量的PCR解析。
非標的(NT),ICT1,C12orf65を標的とするsiRNAをHeLa細胞に導入し,96時間後 に回収してトータルRNAを抽出した。その後,ICT1,C12orf65,GAPDHのmRNAの 発現量を解析した。
(B)siRNAを導入したHeLa細胞のウエスタンブロット解析。
NT,ICT1,C12orf65を標的とするsiRNAをHeLa細胞に導入し,96時間に回収して全 タンパク質を抽出した。その後,ICT1,C12orf65とヒトGamma-tubulin抗体を用いてタ ンパク質の発現量を解析した。
(C)siRNAを導入したHeLa細胞の細胞増殖の影響。
それぞれのsiRNAをHeLa細胞に導入後,DMEMで48,72,96時間培養し,細胞数を 測定した。独立した実験を3回行い,それぞれの値の平均値と標準偏差をとった。
31
si-NT Sub-G1:
スタウロスポリン Sub-G1:
6.5±1.5%1 17.4±5.3%1
si-ICT1 Sub-G1: 34.3±5.2%
si-C12orf65 Sub-G1: 13.0±3.2%
細胞数
Fig. 2 - 2 FCMとヨウ化プロピジウム(PI)を用いたDNA量の測定
si-NT,si-ICT1とsi-C12orf65をそれぞれHeLa細胞へ導入後,96時間培養した。そ PI
s ,s C とs C o 65をそれぞれ e a細胞 導入後,96時間培養した。そ の後,FCMを用いてその細胞を解析した。アポトーシスのポジティブコントロールは,
アポトーシス誘導剤であるスタウロスポリンを50 nM添加し,HeLa細胞のアポトーシ スを誘導させた。細胞はPIを用いて染色を行い,FCMによって測定を行った。図内 のバーはsub-G1期を示しており,その割合を右上に示した。独立した実験を3回行 い それぞれの値の平均値と標準偏差をとった
い,それぞれの値の平均値と標準偏差をとった。
32
si-NT スタウロスポリン
6.6±2.6 % 69.5±16.8 %
si-ICT1
Annexin V
24.6±5.9 % 11.9 ±2.3 % si-C12orf65
Fig. 2 - 3 アポトーシス/ ネクローシス細胞の数の散布図
si-NT,si-ICT1とsi-C12orf65のそれぞれのsiRNAをHeLa細胞に導入後,96時間培 PI
s ,s C とs C o 65 それぞれ s R を e a細胞に導入後,96時間培 養した。同時に50 nM スタウロスポリンを測定2時間前より細胞に添加し,培養することで アポトーシス誘導させた。細胞はPIとAnnexin Vで染色しFCMによって解析を行った。
早期アポトーシスの細胞はPIとAnnexin Vを使った散布図の左上に位置し,その割合 を左上の数字で示した。
33
(A) (B)
si-NT si-ICT1 si-C12orf65
si-NT si-ICT1 si-C12orf65
胞数 胞数
MitoTracker Red CMXRosの蛍光強度 MitoTracker Green FMの蛍光強度
細胞 細胞
Fig. 2 - 4 ICT1,C12orf65の発現抑制によるミトコンドリアへの影響
(A)ミトコンドリアの容積に依存的なMitoTracker Green FMによる蛍光強度の比較。
MitoTracker Red CMXRosの蛍光強度 MitoTracker Green FMの蛍光強度
( )ミト ドリア 容積に依存的な to ac e G ee による蛍光強度 比較。
si-NT,si-ICT1とsi-C12orf65をHeLa細胞に導入後,それぞれ96時間培養した。
MitoTracker Green FMで染色後,細胞を固定化しFCM解析を行った。
(B)ミトコンドリア膜電位に依存的なMitoTracker Red CMXRosによる蛍光強度の比較。
(A)と同じ操作を行い,FCM解析を行った。
(A)と同じ操作を行い,FCM解析を行った。
34
0 6 0.8 1.0
1.2 *
ムcオキシ ゼ活性
0.0 0.2 0.4 0.6
si-NT si-ICT1 si-C12orf65
シトクロム ダーゼ
si-NT si-ICT1
Fig. 2 - 5 シトクロムcオキシターゼ活性
si-NT,si-ICT1とsi-C12orf65をHeLa細胞に導入後,それぞれ96時間培養した。それ si-C12orf65
培
ぞれのsiRNAを導入した細胞からミトコンドリアを抽出し,ミトコンドリアのシトクロムcオキ シターゼ活性を測定した。si-NTを導入した細胞のシトクロムcオキシターゼ活性値を基準 として,si-ICT1とsi-C12orf65の活性を相対的に示した。独立した実験を3回行い,それ ぞれの値の平均値と標準偏差)をとった。 * p< 0.01
35
2.0
1.0 1.5 2.0
OSの活性
**
0.0 0.5
si-NT si-ICT1 H2O2
RO
si-C12orf65
Fig. 2 - 6 ICT1,C12orf65発現抑制による細胞内ROS生産量の測定
ROS依存的に蛍光を呈する細胞浸透性蛍光プロー ブDCFH-DAによる蛍光強度の比較。依存的 蛍光 す 細 浸 性蛍光 蛍光強度 較。
si-NT,si-ICT1とsi-C12orf65をHeLa細胞に導入後,それぞれ96時間培養した。細胞を DCFH-DAで染色後, 測定直前に1 mM過酸化水素(H2O2)処理し,ROSを発生させ たものをポジティブコントロールとした。si-NTを導入したHeLa細胞の蛍光強度を基準とし て,si-ICT1とsi-C12orf65の蛍光強度を相対的に示した。独立した実験を3回行い,それ ぞれの値の平均値と標準偏差をとった。** p < 0.05
36
37
第 3 章 YaeJ タンパク質の変異体解析
序
本章では,YaeJがリボソーム上でどのように機能しているのか,その分子メ カニズムを明らかにするために,YaeJに対し変異体解析行った。保存残基に着 目して様々な変異体を作製し,再構成型無細胞タンパク質合成系を用いたPTH 活性測定を行うと共に,YaeJ変異体のリボソームとの相互作用をショ糖密度勾 配遠心分離のウエスタンブロットによって解析をした。また,ヒト ICT1 との PTH活性の比較を行った。これらの結果から,PTH活性およびリボソームとの 結合部位に必要な部位を明らかにすることで,YaeJの機能の分子メカニズムに ついて考察を行った。
実験方法 菌体
使用した大腸菌株とプラスミドDNAはTable 1に示す。大腸菌全遺伝子プラ スミドクローンASKA library (Kitagawa et al., 2005)と網羅的Open-reading frame (ORF)欠損株ライブラリー Keio collection (Baba et al., 2006)は,
National BioResource Project(NIG,Japan)により提供された。また,ヒト ICT1のcDNA(IRAK027I21)はRIKEN BRCより供与された(Ota et al., 2004)。
大腸菌 K-12 BW25113 株を親株として欠損株が作られている Keio collection に関してはP1 形質導入法(本章の実験方法 参照)を用いてBL21株に目的遺 伝子の組み換えを行った。
38 培養
本実験においての培養は,全てLysogeny Broth培地(以下,LB培地) [1%
tryptone,0.5% yeast extract,1% NaCl] で行った。
大腸菌株にプラスミドを用いて形質転換するために,プラスミドの取り込み 効率が上がるように処理してコンピテントセルとした。大腸菌を5 mlのLB培 地で一晩培養(37℃)した。その後,菌溶液を250 μlとり,25 mlのSOB培 地 [(2% Bacto Tryptone,0.5% Yeast extract,10 mM NaCl,25 mM KCl)と
(1 M MgCl2・6H2O,1 M MgSO4・7H2O)を50:1の割合で混合] に植菌した。
このSOB培地を25℃でOD600が0.6 absになるまで培養した。その後,培養 液を氷上で10分間冷却し,遠心分離(3,000 rpm,15分,4℃)し菌体を回収,
上 澄 み を 除 い た 後 ,15 ml の 氷 冷 し た Transformation buffer [10 mM PIPES-KOH (pH 6.7),15 mM CaCl2・2H2O,250 mM KCl, 55 mM MnCl2・ 4H2O ]でタッピングにより懸濁させ10分間氷冷した。培養液を遠心分離(3,000 rpm,15 分,4℃)し菌体を回収,上澄みを除いた後,4 ml の氷冷した Transformation bufferで再懸濁した。また凍結時の細胞へのダメージを軽減さ せるためDMSO を最終濃度が7%になるように添加し,コンピテントセルとし た。
作製したコンピテントセル100 μlに3 μlの目的タンパク質をコードしたプラ スミドDNAを加え氷上で30分間静置した後,ヒートショック(45秒,42℃)
を行った。大腸菌の回復のため100 μlのLB培地を加え37℃,1時間培養し,
抗生物質を含む LB 寒天培地にコンラージ棒で均一に全量植菌し,37℃で一晩 培養した。抗生物質に関しては,アンピシリン,50 μl/ml;カナマイシン,10 μl/ml;クロラムフェニコール,34 μl/mlの最終濃度でそれぞれ使用した。