与が実施された被験者が
1 例あった。本被験者の 1 回目投与は投与液濃度 12 mg/mL で投
与液量
6 mL、2 回目投与(追加投与)は投与液濃度 20 mg/mL で投与液量 3.5 mL であった。
継続投与試験に登録された被験者において累積投与回数が
4 回になった被験者は 1 例で、
累積投与量は
240 mg を超えた。
なお、前期第
II 相臨床試験後の継続提供・治験外提供(参考資料:5.3.5.4-参 01)では、
累積投与回数が
3 回になった被験者は 4 例あり、また、累積投与量が 240 mg を超えた被
験者は
3 例あった。
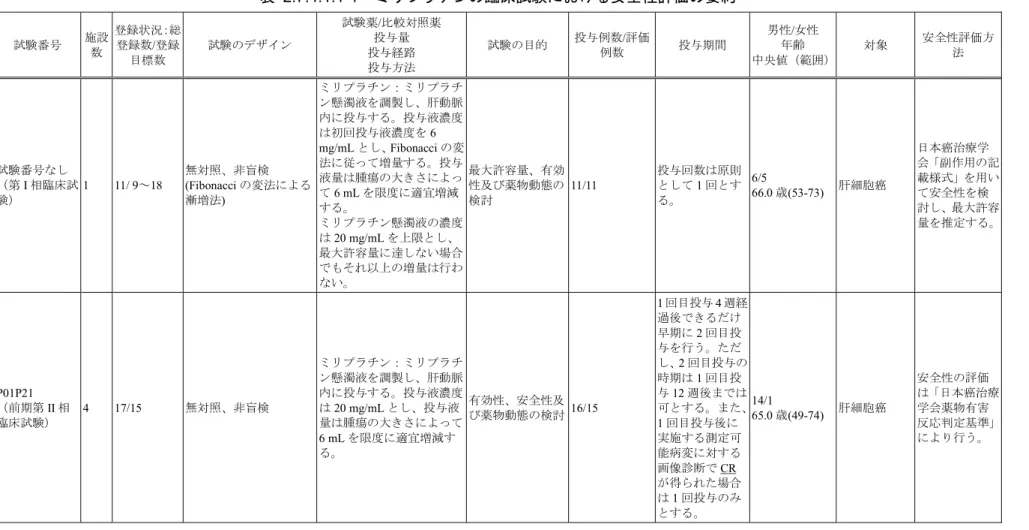
表
2.7.4.1.2-1 ミリプラチンの試験別曝露量
6 12 20
投与回数(回) N 3 3 5
1 3 3 5 11 ( 100 0 % ) 4 ( 26 7 % ) 27 ( 32 5 % )
2 0 0 0 0 ( 0 % ) 11 ( 73 3 % ) 56 ( 67 5 % )
1回目投与液量(mL) N 3 3 5
<1 0 0 0 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % )
1≦ <2 0 0 0 0 ( 0 % ) 0 ( 0 % ) 6 ( 7 2 % )
2≦ <3 0 0 2 2 ( 18 2 % ) 4 ( 26 7 % ) 8 ( 9 6 % )
3≦ <4 0 0 1 1 ( 9 1 % ) 1 ( 6 7 % ) 12 ( 14 5 % )
4≦ <5 0 1 2 3 ( 27 3 % ) 3 ( 20 0 % ) 15 ( 18 1 % )
5≦ <6 0 1 0 1 ( 9 1 % ) 0 ( 0 % ) 5 ( 6 0 % )
6 3 1 0 4 ( 36 4 % ) 7 ( 46 7 % ) 37 ( 44 6 % )
2回目投与液量(mL) N
<1 0 ( 0 % ) 2 ( 3 6 % )
1≦ <2 1 ( 9 1 % ) 2 ( 3 6 % )
2≦ <3 2 ( 18 2 % ) 7 ( 12 5 % )
3≦ <4 2 ( 18 2 % ) 3 ( 5 4 % )
4≦ <5 1 ( 9 1 % ) 4 ( 7 1 % )
5≦ <6 2 ( 18 2 % ) 7 ( 12 5 % )
6 3 ( 27 3 % ) 31 ( 55 4 % )
総投与量(mg) N 3 3 5
<60 3 1 2 6 ( 54 5 % ) 0 ( 0 % ) 9 ( 10 8 % )
60≦ <120 0 2 3 5 ( 45 5 % ) 4 ( 26 7 % ) 21 ( 25 3 % )
120≦ <180 0 0 0 0 ( 0 % ) 7 ( 46 7 % ) 15 ( 18 1 % )
180≦ <240 0 0 0 0 ( 0 % ) 2 ( 13 3 % ) 14 ( 16 9 % )
240 0 0 0 0 ( 0 % ) 2 ( 13 3 % ) 24 ( 28 9 % )
投与間隔(日) N
28≦ <35 0 ( 0 % ) 2 ( 3 6 % )
35≦ <42 0 ( 0 % ) 4 ( 7 1 % )
42≦ <49 0 ( 0 % ) 6 ( 10 7 % )
49≦ <56 2 ( 18 2 % ) 1 ( 1 8 % )
56≦ <63 0 ( 0 % ) 7 ( 12 5 % )
63≦ <70 2 ( 18 2 % ) 5 ( 8 9 % )
70≦ <77 2 ( 18 2 % ) 11 ( 19 6 % )
77≦ <84 5 ( 45 5 % ) 20 ( 35 7 % )
11
11 56
11 56
11 15 83
試験名
投与液濃度(mg/mL)
11 15
第I相臨床試験
全体
前期第II相臨床試験
20
15 83
20
83
後期第II相臨床試験
2.7.4.2.1.1 比較的よく見られる有害事象
2.7.4.2.1.1.1 因果関係別の有害事象
(1) 第 I 相臨床試験
第
I 相臨床試験の因果関係別有害事象発現割合を表 2.7.4.7.2-1<2.7.4.7 付録参照>に
示した。
また、第
I 相臨床試験全体で 2 例以上発現した副作用について抜粋し、表 2.7.4.2.1-2
に示した。
最も頻度の高かった副作用は「発熱」で
11 例全例に発現した。5 例以上で発現した事象
は、
「
CRP 増加」90.9%(10/11)、
「
NAG 増加」88.9%(8/9)、
「嘔吐」
63.6%(7/11)及び「食
欲不振」
54.5%(6/11)であった。
表
2.7.4.2.1-2 第 I 相臨床試験における副作用:2 例以上
N 副作用 N 副作用 N 副作用 N
胃腸障害 嘔吐 3 2 3 2 5 3 11 7 ( 63.6 % )
全身障害および投与局所
様態 発熱 3 3 3 3 5 5 11 11 ( 100 % )
臨床検査 C-反応性蛋白増加 3 3 3 3 5 4 11 10 ( 90.9 % )
β-NアセチルDグルコ
サミニダーゼ増加 3 3 2 2 4 3 9 8 ( 88.9 % )
アスパラギン酸アミノト
ランスフェラーゼ増加 3 1 3 5 3 11 4 ( 36.4 % )
アラニン・アミノトラン
スフェラーゼ増加 3 1 3 5 3 11 4 ( 36.4 % )
血中ブドウ糖増加 3 1 3 5 3 11 4 ( 36.4 % )
好酸球百分率増加 3 1 3 1 5 2 11 4 ( 36.4 % )
血中ビリルビン増加 3 1 3 1 5 2 11 4 ( 36.4 % )
リンパ球百分率減少 3 2 3 5 2 11 4 ( 36.4 % )
血小板数減少 3 1 3 5 2 11 3 ( 27.3 % )
血中カリウム減少 3 1 3 5 2 11 3 ( 27.3 % )
血中乳酸脱水素酵素増加 3 1 3 5 1 11 2 ( 18.2 % )
ヘパプラスチン減少 3 2 3 5 11 2 ( 18.2 % )
代謝および栄養障害 食欲不振 3 2 3 2 5 2 11 6 ( 54.5 % )
基本語
器官別大分類 6 mg/mL 12 mg/mL 20 mg/mL 全体
副作用
表
2.7.4.7.2-1 より 2 例以上発現した副作用を抜粋
(2) 前期第 II 相臨床試験
前期第
II 相臨床試験の因果関係別有害事象発現割合を表 2.7.4.7.2-2<2.7.4.7 付録参照
>に示した。また、全体で
40%以上に認められた有害事象を抜粋して表 2.7.4.2.1-3 に示し
た。
発現割合が
60%を超えた有害事象は「好酸球百分率増加」100%(15/15)、
「発熱」
93.3%
(
14/15)、「CRP 増加」93.3%(14/15)、「NAG 増加」73.3%(11/15)、「リンパ球百分率減
少」
66.7%(10/15)、
「
LDH 増加」66.7%(10/15)、
「尿中クレアチニン減少」
66.7%(10/15)、
「白血球数減少」
66.7%(10/15)、「ロイシンアミノペプチダーゼ上昇」60.0%(9/15)、「γ
-
GTP 増加」60.0%(9/15)、「好中球数減少」60.0%(9/15)であった。
2 回目投与における発現割合が 1 回目投与における発現割合より 20%以上上昇した事象
はなく、
20%以上低下した有害事象は、
「総蛋白減少」
、
「ロイシンアミノペプチダーゼ上昇」
、
「
NAG 増加」、
「
ALP 増加」、
「好酸球百分率増加」
、
「
Na 減少」、
「血中アルブミン減少」
、
「
Ca
減少」及び「
CRP 増加」であった。
表
2.7.4.2.1-3 前期第 II 相臨床試験における有害事象:40%以上
N N N
全身障害および
投与局所様態 発熱 15 13 ( 86 7 % ) 13 ( 86 7 % ) 11 9 ( 81 8 % ) 9 ( 81 8 % ) 15 14 ( 93 3 % ) 14 ( 93 3 % )
投与部位疼痛 15 5 ( 33 3 % ) 5 ( 33 3 % ) 11 5 ( 45 5 % ) 5 ( 45 5 % ) 15 7 ( 46 7 % ) 7 ( 46 7 % )
臨床検査 好酸球百分率増加 15 15 ( 100 % ) 15 ( 100 % ) 11 8 ( 72 7 % ) 8 ( 72 7 % ) 15 15 ( 100 % ) 15 ( 100 % )
C-反応性蛋白増加 15 14 ( 93 3 % ) 14 ( 93 3 % ) 11 8 ( 72 7 % ) 8 ( 72 7 % ) 15 14 ( 93 3 % ) 14 ( 93 3 % )
β-NアセチルDグルコ
サミニダーゼ増加 15 10 ( 66 7 % ) 8 ( 53 3 % ) 11 4 ( 36 4 % ) 3 ( 27 3 % ) 15 11 ( 73 3 % ) 9 ( 60 0 % )
リンパ球百分率減少 15 7 ( 46 7 % ) 6 ( 40 0 % ) 11 6 ( 54 5 % ) 5 ( 45 5 % ) 15 10 ( 66 7 % ) 8 ( 53 3 % )
血中乳酸脱水素酵素増加 15 8 ( 53 3 % ) 8 ( 53 3 % ) 11 4 ( 36 4 % ) 4 ( 36 4 % ) 15 10 ( 66 7 % ) 10 ( 66 7 % )
尿中クレアチニン減少 15 8 ( 53 3 % ) 3 ( 20 0 % ) 10 5 ( 50 0 % ) 2 ( 20 0 % ) 15 10 ( 66 7 % ) 4 ( 26 7 % )
白血球数減少 15 8 ( 53 3 % ) 7 ( 46 7 % ) 11 7 ( 63 6 % ) 5 ( 45 5 % ) 15 10 ( 66 7 % ) 8 ( 53 3 % )
ロイシンアミノペプチ
ダーゼ上昇 15 9 ( 60 0 % ) 8 ( 53 3 % ) 11 3 ( 27 3 % ) 3 ( 27 3 % ) 15 9 ( 60 0 % ) 8 ( 53 3 % )
γ-グルタミルトランス
フェラーゼ増加 15 9 ( 60 0 % ) 9 ( 60 0 % ) 11 5 ( 45 5 % ) 5 ( 45 5 % ) 15 9 ( 60 0 % ) 9 ( 60 0 % )
好中球数減少 15 8 ( 53 3 % ) 8 ( 53 3 % ) 11 6 ( 54 5 % ) 6 ( 54 5 % ) 15 9 ( 60 0 % ) 9 ( 60 0 % )
アスパラギン酸アミノト
ランスフェラーゼ増加 15 6 ( 40 0 % ) 6 ( 40 0 % ) 11 4 ( 36 4 % ) 4 ( 36 4 % ) 15 8 ( 53 3 % ) 8 ( 53 3 % )
血中ブドウ糖増加 15 7 ( 46 7 % ) 3 ( 20 0 % ) 11 5 ( 45 5 % ) 3 ( 27 3 % ) 15 8 ( 53 3 % ) 4 ( 26 7 % )
アラニン・アミノトラン
スフェラーゼ増加 15 6 ( 40 0 % ) 6 ( 40 0 % ) 11 4 ( 36 4 % ) 3 ( 27 3 % ) 15 7 ( 46 7 % ) 7 ( 46 7 % )
血中アルカリホスファ
ターゼ増加 15 7 ( 46 7 % ) 7 ( 46 7 % ) 11 2 ( 18 2 % ) 2 ( 18 2 % ) 15 7 ( 46 7 % ) 7 ( 46 7 % )
単球百分率増加 15 6 ( 40 0 % ) 6 ( 40 0 % ) 11 4 ( 36 4 % ) 4 ( 36 4 % ) 15 7 ( 46 7 % ) 7 ( 46 7 % )
血小板数減少 15 6 ( 40 0 % ) 6 ( 40 0 % ) 11 3 ( 27 3 % ) 3 ( 27 3 % ) 15 6 ( 40 0 % ) 6 ( 40 0 % )
血中アルブミン減少 15 6 ( 40 0 % ) 5 ( 33 3 % ) 11 2 ( 18 2 % ) 15 6 ( 40 0 % ) 5 ( 33 3 % )
血中カルシウム減少 15 6 ( 40 0 % ) 2 ( 13 3 % ) 11 2 ( 18 2 % ) 15 6 ( 40 0 % ) 2 ( 13 3 % )
血中クロール増加 15 6 ( 40 0 % ) 1 ( 6 7 % ) 11 3 ( 27 3 % ) 15 6 ( 40 0 % ) 1 ( 6 7 % )
好塩基球百分率増加 15 4 ( 26 7 % ) 4 ( 26 7 % ) 11 3 ( 27 3 % ) 2 ( 18 2 % ) 15 6 ( 40 0 % ) 5 ( 33 3 % )
全体
有害事象 副作用 有害事象 副作用 有害事象 副作用
器官別大分類 基本語
1回目 2回目
表
2.7.4.7.2-2 より全体における発現割合が 40%以上の有害事象を抜粋
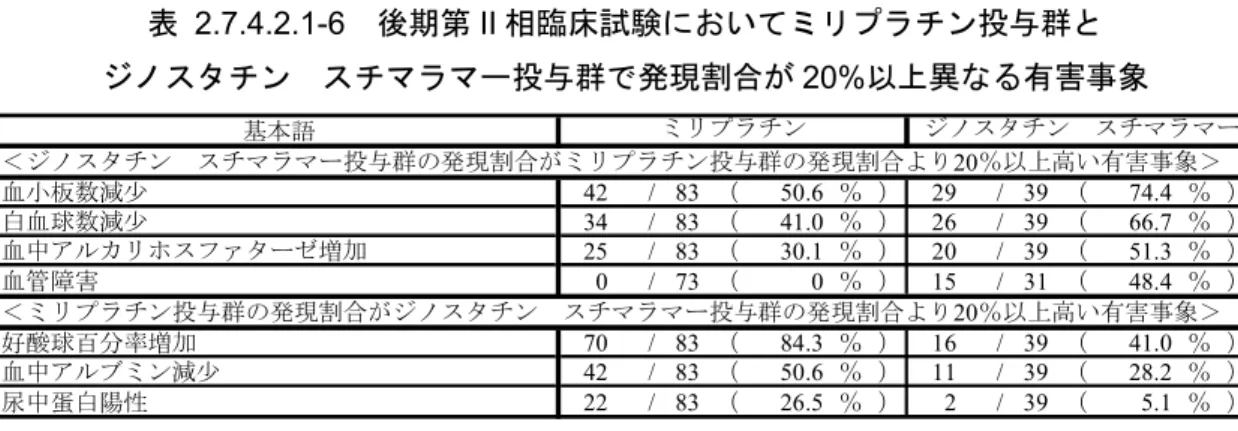
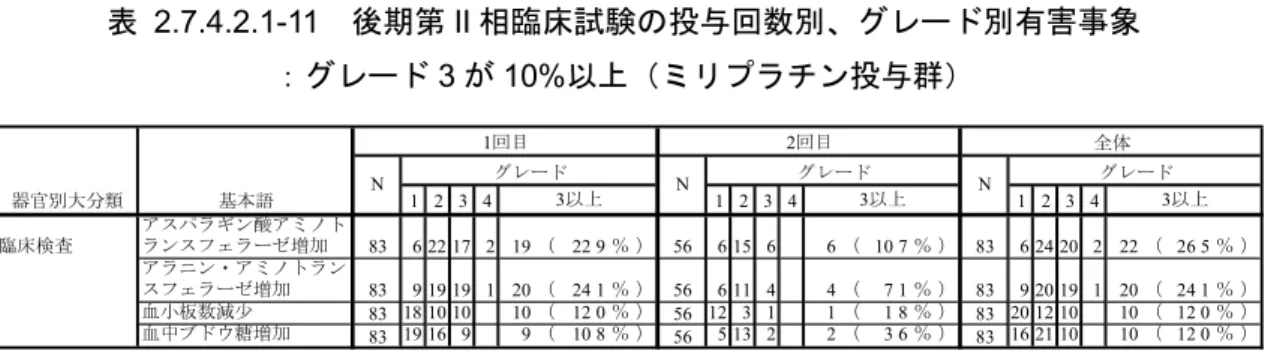
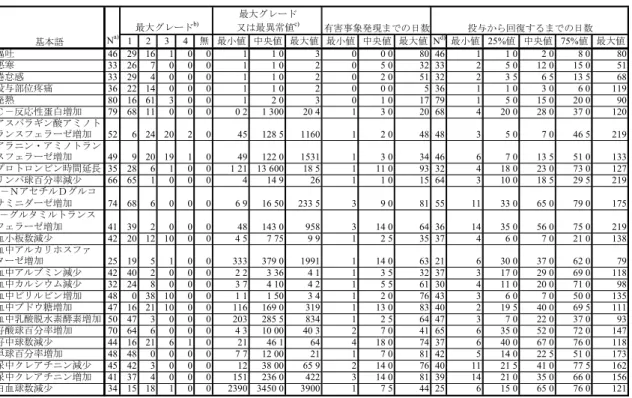
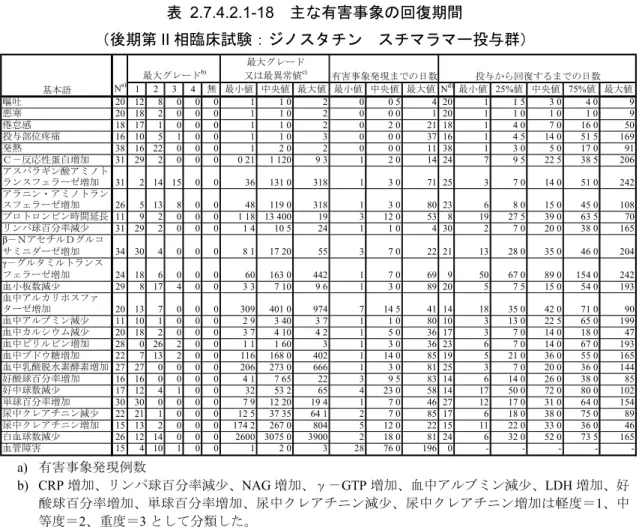
(3) 後期第 II 相臨床試験
後期第
II 相臨床試験における因果関係別有害事象発現割合を表 2.7.4.7.2-3<2.7.4.7 付
録参照>に示した。また、ミリプラチン投与群、ジノスタチン スチマラマー投与群のい
ずれかで全体の有害事象発現割合が
40%以上のものを主な有害事象として抜粋し、表
2.7.4.2.1-4、表 2.7.4.2.1-5 に示した。
ミリプラチン投与群について発現割合が
60%以上であったのは、
「発熱」
96.4%(80/83)、
「
CRP 増加」95.2%(79/83)、
「
NAG 増加」89.2%(74/83)、
「好酸球百分率増加」
84.3%(70/83)、