CTD 第 2 部
2.4 非臨床試験の概括評価
用語及び略語一覧
ADME absorption, distribution, metabolism and
excretion 吸収、分布、代謝及び排泄
ALP alkaline phosphatase アルカリフォスファターゼ
ALT alanine aminotransferase アラニンアミノトランスフェラーゼ
ASV asunaprevir, BMS-650032 アスナプレビル
AUC area under the plasma concentration-time
curve 血漿中濃度曲線下面積
BCRP breast cancer resistance protein 乳癌耐性蛋白
BDC bile duct-cannulated 胆管カニューレ挿入
BID twice daily 1 日 2 回
BMS-650032 NS3 protease inhibitor, asunaprevir, ASV NS3 阻害薬、アスナプレビル
BMS-790052 NS5A inhibitor, daclatasvir, DCV NS5A 阻害薬、ダクラタスビル
BSEP bile salt export pump 胆汁酸塩輸送ポンプ
BVDV bovine viral diarrhea virus ウシウイルス性下痢ウイルス
CC50 50% cytotoxic concentration 50%細胞毒性濃度
cDNA complementary DNA 相補的デオキシリボ核酸
CHO Chinese hamster ovary チャイニーズハムスター卵巣
Cmax highest observed plasma concentration 最高血漿中濃度
CYP cytochrome P450 チトクロームP450
DCV daclatasvir, BMS-790052 ダクラタスビル
EC50 50% effective concentration 50%有効濃度
FDA Food and Drug Administration 米国食品医薬品局
F0 founder generation 親世代
F1 first generation 第1 世代
GBV GB virus GB ウイルス
GGT gamma glutamyltransferase γ-グルタミルトランスフェラーゼ
GLP Good laboratory practice 医薬品の安全性に関する非臨床試験の
実施の基準
GSH glutathione グルタチオン
HCoV Human corona virus ヒトコロナウイルス
HCV hepatitis C virus C 型肝炎ウイルス
hERG human ether-a-go-go-related gene ヒトether-a-go-go 関連遺伝子
HIV human immunodeficiency virus ヒト免疫不全ウイルス
HRV human rhinovirus ヒトライノウイルス
IC50 concentration that causes 50% inhibition 50%阻害濃度
ICH International conference on harmonization 日米EU 医薬品規制調和国際会議
IFN-α interferon-alfa インターフェロンα
IKr rapidly activating, delayed rectifier cardiac
potassium current 急速活性化遅延整流カリウム電流
LC-MS/MS liquid chromatography with tandem mass
MCH mean corpuscular hemoglobin 平均赤血球ヘモグロビン量
MCHC mean corpuscular hemoglobin concentration 平均赤血球ヘモグロビン濃度
MCV mean corpuscular volume 平均赤血球容積
MRP multidrug resistance protein 多剤耐性蛋白
NOAEL no observed adverse effect level 無毒性量
NS3 nonstructural protein 3 of HCV HCV 非構造蛋白 3
NS5A nonstructural protein 5A of HCV HCV 非構造蛋白 5A
NS5B nonstructural protein 5B of HCV HCV 非構造蛋白 5B
OAT organic anion transporter 有機アニオントランスポーター
OATP organic anion transporter polypeptide 有機アニオン輸送ポリペプチド
OCT organic cation transporter 有機カチオントランスポーター
P-gp p-glycoprotein P 糖蛋白
PEG polyethylene glycol ポリエチレングリコール
QD once daily 1 日 1 回
QT beginning of Q wave to the end of T wave
in the ECG 心電図における点まで Q 波の始点から T 波の終
QTc QT interval corrected for heart rate 心拍数補正したQT 間隔
RDW red cell distribution width 赤血球分布幅
RNA ribonucleic acid リボ核酸
SCN5A sodium channel ナトリウムチャネル
TPGS d-α-tocopheryl polyethylene glycol 1000
succinate d-α-トコフェリルポリエチレングリコール1000 コハク酸エステル
TK toxicokinetics トキシコキネティクス
UGT uridine diphosphate glucuronosyl
目次
1 非臨床試験計画概略 ... 6 2 薬理試験 ... 10 2.1 酵素活性及び特異性 ... 10 2.2 抗HCV 活性 ... 10 2.3 細胞毒性 ... 11 2.4 作用機序 ... 11 2.5 耐性 ... 11 2.6 併用試験 ... 13 2.7 安全性薬理試験 ... 13 3 薬物動態試験 ... 17 3.1 吸収及びバイオアベイラビリティ ... 17 3.2 分布 ... 18 3.3 代謝 ... 18 3.4 排泄 ... 19 3.5 蛋白結合 ... 20 3.6 薬物動態学的薬物相互作用 ... 20 3.7 トキシコキネティクス ... 21 4 毒性試験 ... 24 4.1 単回投与毒性試験 ... 24 4.2 反復投与毒性試験 ... 25 4.3 併用投与毒性試験 ... 30 4.4 遺伝毒性試験 ... 31 4.5 がん原性試験 ... 31 4.6 生殖発生毒性試験 ... 32 4.7 局所刺激性試験 ... 33 4.8 その他の毒性試験 ... 33 5 総括及び結論 ... 34 6 参考文献 ... 39表一覧
表1-1 アスナプレビルの毒性試験 ... 8 表3.7-1 主要な反復経口投与毒性試験におけるアスナプレビルの投与量、AUC 値 及びヒトAUC に対する動物 AUC の比 ... 22 表4-1 アスナプレビルの無毒性量及び主な毒性発現用量における曝露量と ヒト曝露量との比 ... 24 表4.2.3.1-1 反復投与毒性試験における肝臓の所見及びアスナプレビルの曝露量... 291 非臨床試験計画概略 アスナプレビル(BMS-650032)は、C 型肝炎ウイルス(HCV)の非構造蛋白 3(NS3)プロテ アーゼに対する低分子阻害薬(直接作用型抗ウイルス薬)である。本薬にはヒトにおける機能あ るいは代謝に関して重要な標的は知られていないことから、過剰な薬理作用により副作用が発現 する可能性は低いことが期待される。アスナプレビルはPI’末端にアシルスルホンアミド基を有す るトリペプチドであり、PI’末端にケトアミド基を有するテトラペプチドであるテラプレビル(既 承認の NS3/4A プロテアーゼ阻害剤)とは構造が大きく異なることから、テラプレビルとの毒性 学的プロファイルの比較から、あまり有益な情報は得られないと考えられる。 アスナプレビルは他の直接作用型抗ウイルス薬との併用で相加又は相乗作用を示し、現在、C 型慢性肝炎患者の治療を目的として、HCV NS5A 複製複合体阻害薬であるダクラタスビル塩酸塩 (BMS-790052、以下、ダクラタスビル)との併用で開発が進められている。本概括評価では、ア スナプレビルの非臨床試験計画並びにアスナプレビル単剤及びダクラタスビルとの併用(ASV + DCV)による薬理、薬物動態及び毒性試験結果の概略を記載する。 アスナプレビルの臨床第2/3 相用量は、軟カプセル 100 mg 1 日 2 回投与又は錠剤 200 mg 錠剤 の1 日 2 回投与である。第 3 相試験では軟カプセルが用いられたが、初期の臨床試験(第 2 相試 験を含む)で用いた錠剤と同様の曝露量であることが確認されている。第 2/3 相用量における定 常状態でのアスナプレビルの曝露量(以下、ヒト曝露量)は、最高血漿中濃度(Cmax)0.419 μg/mL、 血漿中濃度曲線下面積(AUC)3.69 μg•h/mL であった。アスナプレビルはヒト及び動物のいずれ においても高い蛋白結合率を示した。In vitro における蛋白結合率は、毒性試験に用いた各種動物 の血清で同程度(マウス99.2%、ラット 98.8%、イヌ 98.5%、サル 97.2%)であり、ヒト血漿では 99.7%であった。動物におけるアスナプレビルの曝露量のヒト曝露量との比(動物の曝露量 ÷ ヒ トの曝露量)は、総血漿AUC を基に第 2/3 相用量におけるヒトの AUC との比を算出し、アスナ プレビルの遊離体としての補正は行わなかった。 アスナプレビルの主な薬理学評価(効力、細胞毒性及び耐性)については、HCV レプリコンアッ セイを用いた。HCV レプリコンの複製及び阻害に対するアスナプレビルの単独効果及びダクラタ スビルを含む他の抗HCV薬との併用効果の評価には、複数の検出系(Luciferase, FRET及びTaqman) を用いた。心血管系、中枢神経系及び呼吸系に及ぼす影響については、一連のin vitro 及び in vivo における心血管系安全性薬理評価を安全性薬理試験で実施し、また、in vivo における心血管系、 中枢神経系及び呼吸系の安全性薬理評価を主要な毒性試験の一部として実施した。すべての薬理 試験は適切な試験計画に基づき、日米 EU 医薬品規制調和国際会議(ICH)ガイドラインに準拠 して実施した。 アスナプレビルとその代謝物の吸収、分布、代謝及び排泄(ADME)を動物及びヒトを用いて 検討した。医薬品の安全性に関する非臨床試験の実施の基準(GLP)適用下で実施したトキシコ キネティクス(TK)試験でアスナプレビルの血漿中濃度を測定するため、高感度かつ特異的な液 体クロマトグラフィー・タンデム質量分析(LC-MS/MS)法を開発し、その妥当性を検証した。 アスナプレビルの肝臓中又は胆汁中濃度は、科学的に妥当性が検証された GLP 非適用の LC-MS/MS 法を用いて測定した。有色ラット及びアルビノラットを用いたアスナプレビルの組織
内分布は、定量的オートラジオグラフィー法を用いて検討した。 アスナプレビルは、経口投与により吸収され全身循環し、体内に広範に分布した。代謝物プロ ファイルはすべての種で質的に類似しており、ヒトに特有な又は主要な代謝物は検出されなかっ た。アスナプレビルはヒト血漿中の主化合物であった。多様な血中代謝物が存在したが、いずれ の動物試験においてもアスナプレビルの曝露量の 6%未満であった。ヒトにアスナプレビルを反 復投与後、代謝物の曝露量は増加したが、いずれの代謝物もアスナプレビルのAUC 値の 20%未 満又はアスナプレビルとその代謝物の総曝露量の10%未満であった。代謝物の解析結果に基づく と、ヒトにアスナプレビル200 mg を 1 日 2 回、10 日間反復経口投与後の代謝物曝露量に比べて、 動物にアスナプレビル[100 mg/kg(マウス)、80 mg/kg(ラット)又は 50 mg/kg(イヌ)]を単回 経口投与後の代謝物曝露量の方が大きく、ヒトのAUC(0-12 h)値に対する動物の AUC(0-8h)値の比 は2.2~46.5 であった。このことから、代謝物を用いた試験は別途実施しなかった。 ヒト肝ミクロソーム及びヒト相補的デオキシリボ核酸(cDNA)発現酵素を用いた in vitro 試験 により、アスナプレビルの代謝物の生成に関与する酵素を検討した。人工膜透過性測定法及び Caco-2 細胞モデルを用いて、アスナプレビルの膜透過性を検討した。トランスポーター発現細胞 株及びP 糖蛋白(P-gp)ノックアウトマウスを用いて、P-gp 及びその他のトランスポーターとの 相互作用を検討した。赤血球への分布、複数の種(マウス、ラット、イヌ、サル、ヒト)由来の 血清蛋白との結合、ラット肝細胞への取込みのメカニズムを検討した。 アスナプレビルの毒性を評価するための非臨床毒性試験として、単回投与毒性試験(マウス、 ラット、イヌ)、反復投与毒性試験(ラット最長6 ヵ月間、イヌ最長 9 ヵ月間)、併用投与(ASV
+DCV)毒性試験(ラット 1 ヵ月間、サル最長 3 ヵ月間)、遺伝毒性試験(in vitro 及び in vivo)、
がん原性試験[Tg-rasH2 マウス、ラット(実施中)]、生殖発生毒性試験(ラット、マウス、ウサ
ギ)、光毒性試験(in vitro 及び in vivo)を実施した。重要な毒性試験はいずれも GLP 適合下で ICH ガイドラインに準拠して実施した。 肝臓が治療の標的器官であることから、一部の反復投与毒性試験においてアスナプレビルの肝 臓中及び胆汁中濃度を測定した。胆汁中濃度は非臨床試験に用いた動物種及びヒトで測定したが、 GLP 適合下で実施していない。これらの測定以外に特殊な検査は実施しなかった。 アスナプレビルは波長290~700 nm の領域の光線を吸収するため、アスナプレビルの光毒性に ついてマウス線維芽細胞及びLong-Evans 有色ラットを用いて評価した。 アスナプレビル単剤の毒性試験に用いる主要な動物種として、ラット(げっ歯類)及びイヌ(非 げっ歯類)を選択した。ラットは毒性試験に用いる標準的動物種で背景データが豊富であること から選択した。イヌは、経口バイオアベイラビリティ(42%以上)がサル(10%)より高く、高 い全身曝露量が得られることから選択した。胚・胎児発生に関する試験では、構造が類似した先 行化合物の試験でマウスはラットより高い感受性を示したため、げっ歯類動物種としてマウスを 選択した。マウス、ラット、イヌ及びヒトにおけるアスナプレビルの代謝プロファイルは質的に 類似していた。併用投与(ASV + DCV)毒性試験における動物種は、単剤投与の毒性に基づいて 選択した。ダクラタスビルの毒性試験では、イヌと比較してサルのダクラタスビルの代謝プロファ イルがよりヒトに類似しているため、非げっ歯類としてサルを選択した。アスナプレビルの曝露
量はイヌがサルより高かったが、サルにおいても十分な曝露量が得られ、代謝プロファイルは類 似していることから、併用投与毒性試験における非げっ歯類としてサルを選択した。 アスナプレビル単剤の重要な試験の用量選択は、十分に高い用量で評価するため、先行して実 施した試験に基づき設定した。併用投与毒性試験の用量選択に関しては、明らかな毒性が発現す る用量よりも、臨床におけるそれぞれの薬剤のヒト曝露量範囲での影響を評価する AUC が得ら れる用量を設定した。併用投与毒性試験の投与期間については、ICH M3(R2)ガイダンス及び FDA
のHCV ガイダンス案(Chronic Hepatitis C Virus Infection: Developing Direct-Acting Antiviral Agents for Treatment, CDER, September 2010)に基づき、単剤の高用量による長期投与毒性試験の方が臨
床曝露量に関連した AUC が得られる用量で実施した併用投与毒性試験よりも安全性の評価に有 用と考えられることから、最長3 ヵ月間とした。毒性試験の投与経路は臨床投与経路と同様の経 口投与とし、媒体[60% ポリエチレングリコール 400(PEG-400)及び 40%ビタミン E d-α-トコ フェリルポリエチレングリコール1000 コハク酸エステル(TPGS)]に溶解して投与した。 実施したアスナプレビルの毒性試験を表1-1 に示す。 表1-1 アスナプレビルの毒性試験 試験の種類及び投与期間 投与経路 試験系 単回投与毒性 経口 マウス、ラット、イヌ 反復投与毒性 1 週間投与毒性試験 経口 サル 2 週間投与毒性試験 経口 ラット 1 ヵ月間投与毒性試験 経口 ラット、イヌ 6 ヵ月間投与毒性試験 経口 ラット 9 ヵ月間投与毒性試験 経口 イヌ 併用投与毒性(ASV + DCV) 1 ヵ月間投与毒性試験 経口 ラット、サル 3 ヵ月間投与毒性試験 経口 サル 遺伝毒性
復帰突然変異試験 In vitro S. typhimurium, E. coli 染色体異常試験 In vitro CHO 細胞 小核試験 経口 ラット がん原性 26 週間投与がん原性試験 経口 Tg-rasH2 マウス 2 年間投与がん原性試験 経口 ラット(実施中) 生殖発生毒性 受胎能及び着床までの初期胚発生に関する試験 経口 ラット 胚・胎児発生に関する試験 経口 マウス、ウサギ 出生前及び出生後の発生並びに母体の機能に関する試験 経口 ラット その他の毒性 光毒性試験 In vitro Balb/c 3T3 マウス線維芽細胞 単回投与光毒性試験 経口 ラット
以上の非臨床薬理試験、薬物動態試験及び毒性試験の成績は、C 型慢性肝炎患者におけるアス ナプレビルのダクラタスビルとの併用療法の有効性及び安全性を裏付けるものと考えられる。
2 薬理試験 HCV NS3 プロテアーゼは NS3 の N 末端ドメインにコードされる 181 のアミノ酸から成るセリ ンプロテアーゼである1)。NS3 プロテアーゼは酵素活性の発現に必要な補因子である NS4A 蛋白 とヘテロ二量体酵素を形成している。アスナプレビルの主な薬理学評価(効力、細胞毒性及び耐 性)には、HCV レプリコンアッセイを用いた2)。また、本薬のNS3 プロテアーゼに対する選択性 及び結合性を検討した。心血管系、中枢神経系及び呼吸系に及ぼす影響を評価するため、一連の
in vitro 及び in vivo における心血管系安全性薬理評価を安全性薬理試験で実施し、また、in vivo に
おける心血管系、中枢神経系及び呼吸系の安全性薬理評価を主要な毒性試験の一部として実施し た。 2.1 酵素活性及び特異性 NS3/4A プロテアーゼ複合体に対するペプチド類似阻害薬の構造は、プロテアーゼが基質ペプ チドから放出されるC 末端側産物ではなく、N 末端側産物によるフィードバック阻害を受けやす いという知見によって決定された。アスナプレビルは、効力と選択性のいずれも最大化できるよ うに、最適化したトリペプチド阻害薬の中心部を利用している。アスナプレビルのジェノタイプ スペクトラムを評価するため、HCV の 6 種の主要なジェノタイプに対する IC50値を測定した。そ の結果、IC50値は0.3~320 nM の範囲であった。アスナプレビルの in vitro 活性をテラプレビル3) を対照薬として評価した。アスナプレビルのin vitro 活性は、ジェノタイプ 2b 以外の試験したす べてのジェノタイプに対しテラプレビルよりも優れていた。 プロテアーゼの基質特異性は、一般的に切断部位に隣接する側鎖によって決定される 4)。HCV NS3/4 プロテアーゼ複合体によりトランス切断を受ける天然基質は、切断される結合部位の P1 位 に、システイン残基があることが必須である。この特殊な必要条件により、高い選択性を有する HCV プロテアーゼ阻害薬を開発することが可能となる。体内には多くのプロテアーゼが存在する ため、治療薬としてのプロテアーゼ阻害薬には選択性が重要である。アスナプレビルは、HCV と 近縁関係にあるGBV-B の NS3 プロテアーゼ及びヒトのセリン又はシステインプロテアーゼに対 し、ほとんどあるいは全く活性を示さなかったことから、本薬がHCV NS3 セリンプロテアーゼ に対して選択的であることが示された(選択指数は21200 倍以上)。In vitro におけるアスナプレ ビルの選択性は、BMS-697372(テラプレビルの P1 プロピル官能基位光学異性体の混合物)より も優れていた。 2.2 抗HCV活性 アスナプレビルのHCV RNA の複製に及ぼす影響を評価するため、HCV レプリコンシステム2) を用いた。本薬のジェノタイプ1a, 1b, 2a の HCV レプリコン及びジェノタイプ 2b, 3a, 4a の NS3 プロテアーゼ配列をコードしたハイブリッドレプリコンに対する活性を検討した。レプリコンに おける抗HCV 活性及び算出された治療係数について、アスナプレビルはジェノタイプ 1a, 1b 及 び 4a のレプリコンに対しテラプレビルよりも強力な作用を示した。一方、ジェノタイプ 2b, 3a
のハイブリッドレプリコンに対するアスナプレビルの効果は、テラプレビルと同等又はそれより 弱かった。 アスナプレビルの感染性HCV ジェノタイプ 2a(JFH-1)ウイルス5) に対する抗ウイルス活性を 検討した。その結果、アスナプレビルのウイルス産生に対する50%有効濃度(EC50)は39 nM で あり、テラプレビルで認められた116 nM よりも約 3 倍強力な阻害作用を示した。 アスナプレビルのウイルス特異性を評価するため、様々なウイルスに対する検討を行った。ア スナプレビルは細胞培養試験において、ウシウイルス性下痢ウイルス(BVDV)(EC50 = 12 μM)、 ヒト免疫不全ウイルス(HIV-1)(EC50 = 14 μM)、ヒトライノウイルス(HRV)(EC50 > 100 μM) 及びヒトコロナウイルス(HCoV)(EC50 > 100 μM)に対して、ほとんどあるいは全く活性を示さ なかった。 抗ウイルス薬の臨床効果は血清蛋白との結合によりしばしば減弱する。そこで、アスナプレビ ルの抗ウイルス活性に及ぼす血清蛋白結合の影響を調べるため、40%ヒト血清を含む培養液中に おいてレプリコンアッセイを実施した。その結果、アスナプレビルのジェノタイプ 1b(Con 1) レプリコンに対する EC50値が、40%ヒト血清を含まない標準的なアッセイの場合と比較して 6.5 倍に増大したことから、本薬の抗ウイルス活性はヒト血清により軽度にしか変化しないことが示 唆された。BMS-697372 の抗ウイルス活性も、アスナプレビルの場合と類似した変化であった (EC50値が6.8 倍に増大)。 2.3 細胞毒性 アスナプレビルの細胞毒性を解剖学的に異なる起源のヒト細胞株を用いて検討した。4~6 日間 培養後に50%細胞毒性濃度(CC50)を測定した。アスナプレビルの細胞毒性はBMS-697372 と同 程度であったが、HCV ジェノタイプ 1a(H77)レプリコンに対する EC50値との比から算出した 治療係数は、アスナプレビルの方が著明に高かった(治療係数2750 以上)。 2.4 作用機序 アスナプレビルの作用機序について、組換え酵素アッセイを用いて複数のHCV ジェノタイプ 1 分離株の NS3/4A プロテアーゼ複合体で検討した。純粋な組換え NS3/4A プロテアーゼ複合体に 対し、アスナプレビルは競合的阻害薬の速度論的パラメータの特徴を示した。これは、最大酵素 代謝回転率が一定の間、基質に対するみかけのMichaelis-Menten 定数が増加した Lineweaver-Burke プロットにより確定された。試験したウイルス株により、阻害定数(Ki)は0.24~1.0 nM の範囲 を示した。この競合的阻害様式は、その後に実施した X 線結晶構造解析によって裏付けられた。 アスナプレビルはNS3 プロテアーゼドメインの活性部位である S4 から S1’結合部位6) を占有し ていた。これらのサブサイトは、通常HCV の基質によって占有されると考えられる。 2.5 耐性 アスナプレビルの耐性機序を把握するため、HCV サブゲノムレプリコンシステムを用いて臨床
で発現する耐性置換の特定及びその特徴を明らかにした。また、テラプレビル及びboceprevir(NS3 プロテアーゼ阻害剤、国内未承認)といったNS3 プロテアーゼ阻害薬の臨床での耐性変異をジェ ノタイプ1a 及び 1b のレプリコンに発現させ、アスナプレビルの阻害作用に対する感受性を評価 した。 表現型解析の結果、ウイルス学的ブレイクスルーを起こしたジェノタイプ1a レプリコンの感受 性は、対照とした野生型細胞の約1/49~1/11 であった。耐性レプリコンの NS3 及び NS4A 蛋白の 遺伝子型解析の結果、NS3 プロテアーゼドメイン内に多数の非保存的変化がみられた。これらの
置換は、T40A, Q41E, V51A, R62K, D79E, T95A, I114V, R123G, R155K, D168G, I170T, N174Y, L175P
及びG176E であった。表現型解析の結果、ウイルス学的ブレイクスルーを起こしたジェノタイプ
1b レプリコンの感受性は、対照とした野生型細胞の約 1/400~1/170 に低下した。耐性レプリコン
群のNS3 及び NS4A 蛋白の遺伝子型解析の結果、NS3 プロテアーゼドメイン内に非保存的変化が
みられた。これらの置換は、Q41R, Q80R, Q86R, P89L, Y105C, D168A/G/H/V/Y, E173G 及び E176G
であった。発現した置換の多くは、本薬の結合部位周辺に存在することがX 線結晶構造解析試験 によって確認された。 アスナプレビル耐性レプリコンは、NS5A 阻害薬のような HCV の複製サイクルにおける他のウ イルス蛋白を標的とする HCV 阻害薬に対する感受性を維持している。このことは、複数のウイ ルス蛋白を標的とする薬剤と組み合わせることにより、アスナプレビルによる治療中に発現し得 る耐性ウイルスに対しても治療が期待できることを示唆している。 HCV NS3 プロテアーゼに結合したアスナプレビルのコンピューターモデルを用いた解析の結 果、Q80, R155, D168 及び I170 のアミノ酸における置換が酵素活性部位の近傍において優先的に 認められた。これらの置換は、いずれも種々のNS3 プロテアーゼ阻害薬の耐性に関連することが 以前に報告されている7)8)9)。D168 は切り出された NS3/4A プロテアーゼ複合体とアスナプレビル のX 線結晶構造において、酵素活性部位の S2 及び S4 ポケットの間に存在している。D168 のア ラニン、グリシン、バリン又はチロシンのようなアミノ酸残基への置換は、アスナプレビルのP2 イソキノリン部分との相互作用に重要な R155 との塩橋の形成を妨害することが予測された。構 造解析の結果、I170 のアミノ酸がアスナプレビル結合時の R155 と D168 の塩橋関係周辺のポケッ ト形成部分であるR155 の頭部基と相互作用を有することが示された10)。I170 がより極性の高い スレオニンへ置換されることにより、R155 の周辺環境を変化させると考えられ、その結果、アス ナプレビルの P2 イソキノリン部分との相互作用が軽度に減少した。変異を有するレプリコンの 作製により、アスナプレビル耐性におけるこれらの置換の役割が裏付けられた。 アスナプレビルに対するレプリコンの耐性プロファイルは、テラプレビルや boceprevir につい て報告されているものとわずかに異なる 7)8)。テラプレビルや boceprevir の耐性レプリコンでは、 V36 及び A156 のアミノ酸残基に変化がみられた。概して、アスナプレビルはテラプレビルに比 べて良好な耐性プロファイルを示した。一方、A156 のアミノ酸残基に変異を有するレプリコンに 対するテラプレビルの効力は、アスナプレビルに比べて著明に低下した。
2.6 併用試験 テラプレビルの臨床試験において、C 型慢性肝炎患者にテラプレビルを単剤投与した場合には、 高い頻度で耐性が発現した 8)。HIV 治療と同様に、C 型慢性肝炎治療においても併用療法により 耐性発現を効果的に防御できると考えられる。アスナプレビルが他の抗 HCV 薬と併用投与が可 能か否かHCV レプリコンシステムを用いて検討した。 HCV ジェノタイプ 1b のレプリコン細胞をアスナプレビルとインターフェロン α(IFNα)又は リバビリン、あるいはダクラタスビルや と併用 した場合、相加又は相乗効果が認められた。 HCV ジェノタイプ 1a のレプリコン細胞にアスナプレビルと IFNα を併用した場合、HCV ジェ ノタイプ1b レプリコンの場合と同様に、相乗効果又は相加効果を示す結果が得られた。検討した すべての併用試験において相加又は相乗効果が認められ、抗ウイルス活性の拮抗は認められな かった。これらの結果から、アスナプレビルは他の抗 HCV 薬との併用により、C 型慢性肝炎に 対する有効な治療薬となる可能性が示唆された。 ジェノタイプ1a-NS3 の野生型レプリコン及びジェノタイプ 1a-NS3-R155K の NS3 プロテアー ゼ阻害薬耐性レプリコンに、アスナプレビルとダクラタスビル及びIFNα を 25 日間併用すると、 いずれのレプリコンの複製も同等に抑制された。これらの結果は、アスナプレビルとダクラタス ビル及びIFNα の投与レジメンが、テラプレビルや boceprevir を含む治療法が無効であった C 型 慢性肝炎患者において、HCV RNA の複製を抑制できる可能性を示唆している。 2.7 安全性薬理試験 ICH S7A ガイドラインで推奨される心血管系、中枢神経系及び呼吸系の各指標に及ぼすアスナ プレビルの影響について、一連のin vitro 及び in vivo 安全性薬理試験を実施し、また、単回投与 毒性及び反復投与毒性試験の一部として評価した。In vitro 試験では受容体及びイオンチャネルの リガンド結合並びに酵素活性の相対的阻害を評価した。心血管系については、ヒト ether-a-go-go 関連遺伝子(hERG/IKr)電流及び心筋イオンチャネル、ウサギプルキンエ線維の活動電位に及ぼ す影響並びに摘出ウサギ心臓を用いた試験をin vitro で評価し、また、麻酔下ウサギを用いた単回 投与試験及びイヌを用いた単回投与テレメトリー試験により、心血管系パラメータに及ぼす影響 をin vivo で評価した。アスナプレビルの曝露量はイヌの方がサルより高かったため、安全性薬理 試験にはイヌを用いた。アスナプレビルの代謝プロファイルが安全性薬理試験に用いた動物種及 びヒトにおいて質的に類似しており、また、ヒトに特有の代謝物も検出されなかったことから、 代謝物の安全性薬理試験は実施しなかった。In vitro 安全性薬理試験では本質的に蛋白を含まない
ことから、in vitro 試験における曝露量比は、in vitro 濃度 ÷ 臨床用量での遊離体 Cmax(0.005 μg/mL) として算出した。
2.7.1 受容体/イオンチャネル結合及び酵素アッセイ
(受容体、酵素、イオンチャネル、セカンドメッセンジャー及び取込み部位)に対し、明らかな 結合や阻害を示さなかった(50%以上の変化を明らかな作用と定義した)。アスナプレビル 10 μM (7.5 μg/mL)の濃度は、アスナプレビルの臨床用量での遊離体 Cmax の 1500 倍であることから、 本薬がヒトにおいて有害作用を引き起こす可能性は低いことが示唆された。 2.7.2 心血管系 アスナプレビルの心筋カリウムチャネル(hERG/IKr)、ナトリウムチャネル(SCN5A)及び L 型カルシウムチャネル電流、並びにウサギプルキンエ線維の活動電位に及ぼす影響をin vitro 試験 で検討した。 hERG 及びナトリウムチャネルアッセイでは、アスナプレビルの 10 及び 30 μM(7.5 及び 22.5 μg/mL)を試験した。アスナプレビルのナトリウムチャネルに及ぼす頻度依存性作用を、1 及 び4 ヘルツ(Hz)の 2 つの刺激頻度を用いて評価した。L 型カルシウムチャネルアッセイでは、 アスナプレビルの30 μM(22.5 μg/mL)を試験した。再分極に及ぼす影響を検討するため、アス ナプレビル3, 10 及び 30 μM(2.2, 7.5 及び 22.5 μg/mL)の摘出ウサギプルキンエ線維の活動電位 に及ぼす影響を検討した。 アスナプレビルは10 及び 30 μM で、hERG カリウム電流をそれぞれ 8.2%及び 20.6%阻害し、IC50 値は30 μM 超であった。アスナプレビルは 10 及び 30 μM で、心筋ナトリウム電流を 1 Hz 刺激頻 度で26.6%及び 65.9%、また、4 Hz 刺激頻度で 29.8%及び 71.6%阻害した。更に、アスナプレビル はL 型カルシウム電流を 30 μM で 18.3%阻害した。アスナプレビルは 30 μM 以下の濃度ではウサ ギプルキンエ線維の活動電位持続時間及び他の活動電位パラメータに影響を及ぼさなかった。 以上より、アスナプレビルは10 μM で hERG/IKr、ナトリウム及び L 型カルシウム電流を軽度 に阻害し、また、30 μM でプルキンエ線維活動電位パラメータには影響を及ぼさなかった。アス ナプレビルのヒト遊離体 Cmax の 1500 倍高い濃度においても軽度なイオンチャネル阻害作用 (hERG)が認められのみであり、また、同様に 4500 倍でも活動電位持続時間に影響を及ぼさな かったことから、本薬がヒトの心電図に影響を及ぼす可能性は低いことが示唆された。 摘出ウサギ心臓における電気生理学的試験では、アスナプレビル10 μM(7.5 μg/mL)を大動脈 の逆行性灌流により注入して20 分又は 60 分間の試験を、また、右心房を 3.5, 4, 4.5 及び 5 Hz で それぞれ30 秒間刺激した。心房電位図、心電図及び冠灌流量を継続的に記録した。洞結節回復時 間及び心拍数の測定により洞房結節機能を評価した。 その結果、摘出ウサギ心臓において、アスナプレビル10 μM(7.5 μg/mL)を 60 分以下で灌流 した場合の心房及び心室のアスナプレビル濃度はそれぞれ93.4 μM(69.9 μg/mL)及び 151.5 μM (113.4 μg/mL)であったが、洞房結節機能や冠動脈灌流量に本薬に関連した変化は認められな かった。これら心房内及び心臓内のアスナプレビル濃度は、ヒト遊離体 Cmax のそれぞれ 13980 倍及び22680 倍という高濃度であったことから、上記の結果は、本薬がヒトに心血管系の作用を 引き起こす可能性が低いことを示唆するものである。 麻酔下ウサギを用いた in vivo 心臓電気生理学的試験において、アスナプレビルを 3, 10 及び 30 mg/kg の用量で静脈内に漸増投与し、アスナプレビルの心血管系への作用を更に評価した。ア
スナプレビルの平均血漿中濃度は15~180 μg/mL(ヒト Cmax の 36 倍~430 倍)の範囲であり、 用量依存性が認められた。アスナプレビルの血漿に対する心臓の平均濃度比が 1.2 であったこと から、本薬の心組織への蓄積は少ないことが示された。アスナプレビルは30 mg/kg 以下の用量(血 漿中濃度は180 μg/mL 以下)では、心電図パラメータに影響を及ぼさなかったが、軽微で一過性 の用量依存的な血圧上昇が認められた(3, 10 及び 30 mg/kg でそれぞれ投与前値に比して 10%、 21%及び 24%の上昇)。 単回経口投与の心血管系テレメトリー試験において、アスナプレビルの100 mg/kg をイヌに投 与した結果、心拍数、全身動脈血圧、左心室圧、心電図の各種波形間隔(QTc 間隔を含む)、身体 活動及び深部体温にアスナプレビルの投与に関連した変化はみられなかった。 本試験における投与4 時間後のアスナプレビルの平均血漿中濃度(57 μg/mL)は、イヌの 9 ヵ 月間投与毒性試験における100 mg/kg/day の Cmax(50.3 μg/mL)と同等であった。テレメトリー 試験における投与4 時間後の血漿中濃度は、ほぼ定常状態の Cmax であことから、イヌの心血管 系テレメトリー試験における無作用量でのCmax は、ヒト曝露量 Cmax の 120 倍であった。 イヌの反復投与毒性試験においてもアスナプレビルの心血管系に及ぼす影響を検討した。その 結果、イヌの 1 ヵ月間反復経口投与試験で 300 mg/kg/day まで、9 ヵ月間反復経口投与試験で 100 mg/kg/day までの用量を投与しても心拍数及び心電図に本薬投与に関連した変化は認められ なかった。100 mg/kg/day 投与時の Cmax 及び AUC は、それぞれヒト曝露量の 120 倍及び 82 倍で あった。 アスナプレビルとダクラタスビルの併用投与による最長3 ヵ月間のサル反復経口投与毒性試験 においても、本薬投与に関連した心拍数及び心電図への影響は認められなかった。併用投与毒性 試験におけるアスナプレビルのAUC は、ヒト曝露量 AUC の 18 倍以下であった。 健康被験者への単回投与(1200 mg 以下の用量、AI447001 試験)及び反復投与(600 mg 以下の 用量の1 日 2 回を 14 日間、AI447003 試験)並びに C 型慢性肝炎被験者に単回投与(600 mg 以下 の用量、AI447002 試験)及び反復投与(600 mg 以下の用量を 3 日間、AI447004 試験)した臨床 試験においても、バイタルサインや心電図に一貫した変化あるいは臨床的に関連した傾向は認め られなかった。また、健康被験者を対象としたthorough QT 試験(AI447025 試験)において、ア スナプレビル軟カプセル300 mg 1 日 2 回の 10 日間反復投与では QTc 間隔に臨床上懸念される変 化はみられなかった。 以上、アスナプレビルが心血管系に影響を及ぼす可能性は低いことが示された。唯一認められ た心血管系の作用は、ヒト遊離体Cmax の 1500 倍の濃度で認められた軽度な hERG 阻害作用及び ヒト曝露量Cmax の 36 倍以上の濃度で認められた麻酔下ウサギの血圧上昇であった。また、本薬 は高曝露量でも、プルキンエ線維活動電位持続時間(ヒト遊離体Cmax の 4500 倍)、ウサギ摘出 心臓試験での心電図(ヒト遊離体Cmax の最大 22680 倍)、イヌの単回経口投与毒性試験(ヒト曝 露量Cmax の 120 倍)、最長 9 ヵ月間の反復経口投与毒性試験(ヒト曝露量 AUC の最大 82 倍)、 及びサルにアスナプレビルとダクラタスビルを最長3 ヵ月間投与した併用投与毒性試験(ヒト曝 露量AUC の最大 18 倍)における心血管系パラメータのいずれに対しても影響を及ぼさなかった。
2.7.3 中枢神経系
アスナプレビルの中枢神経系に及ぼす影響を評価するための独立した安全性薬理試験は実施し なかった。有色Long-Evans ラット及び白色 Sprague Dawley ラットに[14C]アスナプレビルを投与 した試験において、本薬の脳組織内濃度は定量下限未満であった。本結果と一致し、マウス、ラッ ト及びイヌの毒性試験において、神経学的な臨床症状や神経系の組織学的所見にアスナプレビル 投与に関連した変化は認められなかった。
アスナプレビルは、イヌの 1 ヵ月間反復経口投与毒性試験(300 mg/kg/day 以下、ヒト曝露量
AUC の最大 375 倍)及び 9 ヵ月間反復経口投与毒性試験(100 mg/kg/day 以下、ヒト曝露量 AUC
の最大82 倍)において、行動及び運動、末梢及び中枢神経機能に影響を及ぼさなかった。 また、アスナプレビルの反復併用投与毒性試験において、本薬とダクラタスビルをラット(ヒ ト曝露量AUC の最大 11 倍)又はサル(ヒト曝露量 AUC の最大 18 倍)に投与した場合でも、本 薬投与に関連した中枢神経系への影響は認められなかった。 以上より、アスナプレビルがヒトの中枢神経系に影響を及ぼす可能性は低いと考えられた。 2.7.4 呼吸系 アスナプレビルの呼吸系に及ぼす影響を評価するための独立した安全性薬理試験は実施しな かった。マウス、ラット及びイヌの毒性試験において、呼吸系の臨床症状にアスナプレビル投与 に関連した変化は認められなかった。 アスナプレビルは、イヌの9 ヵ月間までの反復経口投与毒性試験において 100 mg/kg/day 以下 の用量(臨床用量でのAUC の 82 倍以下)では、呼吸数及び肺音(胸部聴診による判断)、ある いは動脈血酸素飽和度(パルス酸素濃度計により測定)に影響を及ぼさなかった。 また、アスナプレビルをダクラタスビルと併用してラット(臨床用量での AUC の 11 倍以下) 又はサル(臨床用量でのAUC の 18 倍以下)に反復投与した場合でも、本薬投与に関連した呼吸 系への影響は認められなかった。 以上より、アスナプレビルがヒトの呼吸系に影響を及ぼす可能性は低いと考えられた。
安全性薬理評価の結果、ヒトでの有害事象を懸念させるin vitro 試験での標的以外の作用、in vivo 試験での心血管系、中枢神経系及び呼吸系への影響は認められなかった。
3 薬物動態試験 一連のin vitro 試験及びマウス、ラット、ウサギ、イヌ及びサルを用いた in vivo 試験において、 アスナプレビルの非臨床薬物動態を評価した。マウス、ラット、ウサギ、イヌ及びサル由来の各 種生体試料中のアスナプレビルは、LC-MS/MS 法により分析した。使用した分析法は、高感度で、 精度良く、正確であった。 アスナプレビルは、経口投与により吸収され全身循環し、体内に広範に分布した。代謝物プロ ファイルはすべての種で質的に類似し、ヒトに特有の代謝物は検出されなかった。多様な血中代 謝物が存在したが、動物におけるアスナプレビルの曝露量の 6%未満であった。ヒトにアスナプ レビルを反復投与した結果、代謝物の曝露量は増加したが、いずれの代謝物もアスナプレビルの AUC 値の 20%未満又はアスナプレビルとその代謝物の総曝露量の 10%未満であった。代謝物の 解析結果に基づくと、代謝物の曝露量はヒトよりも動物の方が大きかった。アスナプレビルは主 としてチトクロームP450(CYP)3A4 及び CYP3A5 により代謝された。動物におけるアスナプレ ビルの消失には、胆汁クリアランス、代謝クリアランス及び腸内分泌などの複数の経路が関与し、 アスナプレビルとその代謝物は主に糞便中に排泄された。腎クリアランスは、アスナプレビルの 主要な消失経路ではなかった。アスナプレビルは多様な酵素及びトランスポーターの基質、阻害 剤及び誘導剤であり、CYP3A、CYP2D6、有機アニオン輸送ポリペプチド(OATP)及び P-gp を 介した薬物相互作用が臨床試験で確認された。しかしながら、ダクラタスビルとアスナプレビル の臨床薬物動態学的相互作用は顕著ではなかった。 3.1 吸収及びバイオアベイラビリティ アスナプレビルの膜透過性は高く、アスナプレビルを溶液として経口投与したとき吸収は速や かで、最高血中濃度到達時間(Tmax)は、試験した動物種で 1.3~9.6 時間であった。アスナプレ ビルはP-gp の基質であることから、P-gp がアスナプレビルの経口吸収を抑制する可能性がある。 ラットにアスナプレビルを静脈内及び門脈内投与したときの曝露量は同程度であったことから、 初回通過効果がバイオアベイラビリティを抑制するとは考えられない。マウス、ラット及びサル における絶対バイオアベイラビリティは1%~28%の範囲であったが、イヌでは 42%以上と高値で あった。ラット及びイヌにおけるバイオアベイラビリティは用量に依存し、ラットでは 10~ 30 mg/kg の用量範囲で 4~8 倍増加し、イヌでは 4~10 mg/kg の用量範囲で約 6 倍増加した。イヌ に錠剤 400 mg を経口投与したとき、食事の影響(吸収の増加)が認められた。これは、脂質の 存在下で緩やかな消化管運動による溶解性及び溶解度の増加が原因であると考えられた。 イヌ及びサルにアスナプレビルを静脈内投与すると、アスナプレビルは速やかに消失し、半減 期はそれぞれ1 時間以下及び 1.3 時間であった。アスナプレビルの消失半減期はマウス及びラッ トの方が長く、それぞれ4.6 時間及び 4.2~8.1 時間であった。平均滞留時間も同様の傾向を呈し た。しかしながら、マウスの血清クリアランス(57.3 mL/min/kg)及びラットの総血漿クリアラン ス(38.4~38.9 mL/min/kg)は、イヌ及びサルの総血漿クリアランス(15.4~18.7 mL/min/kg)より も高い値を呈した。これは、イヌ及びサルの定常状態分布容積が高値であることが原因と考えら れた。
3.2 分布 マウス、ラット、イヌ及びサルの定常状態分布容積は、報告されている全身水分量11) と同程度 又はそれ以上であったことから、アスナプレビルの血管外分布が示唆された。 アスナプレビル1 μM(0.748 μg/mL)の血漿中濃度に対する血液中濃度の比は、ヒトで 0.55、 マウス、ラット、イヌ及びサルで0.34~0.82 の範囲であったことから、血液中のアスナプレビル は主として血漿中に分布することが示唆された。 [14C]アスナプレビルを用いた組織分布試験において、放射能は体内に広範に分布し、反復経口投 与後の蓄積は認められなかった。動物において、薬理作用の標的臓器である肝臓への取込みは顕 著であり、血漿中アスナプレビルに対する肝臓中アスナプレビルのAUC 比は 40~257 以上であっ た。In vitro 試験結果から、受動拡散とエネルギー依存性の能動的取込み過程の両方が肝取込みに 関与することが示唆された。アスナプレビルの肝取込みは速やかで、飽和性があることから、ト ランスポーターが肝取込みに関与し、アスナプレビルがOATP1B1 及び OATP2B1 の基質であるこ とが示唆された。これらトランスポーターは肝取込みに関与することから、肝臓中にアスナプレ ビルを濃縮する役割を担う可能性がある。 ラットに[14C]アスナプレビルを経口投与したとき、[14C]アスナプレビル関連物質は血液脳関門 を通過しなかったことから、アスナプレビルが中枢神経系に影響を及ぼすとは考えられない。妊 娠ラットに[14C]アスナプレビルを投与すると放射能は胎盤を通過し、また、授乳中のラットに[14C] アスナプレビルを投与すると放射能が乳汁中に検出されたことから、アスナプレビルの投与を受 けている女性の胎児及び乳児は、アスナプレビルとその代謝物に曝露される可能性のあることが 示唆された。 3.3 代謝 ヒト肝ミクロソームを用いた反応表現型解析試験から、アスナプレビルは主としてCYP3A4 及
びCYP3A5 により代謝され、CYP2A6、CYP2B6、CYP2C9、CYP2C19 及び CYP2D6 もわずかな がらアスナプレビルの代謝に寄与することが示された。 様々な種の肝ミクロソーム、肝S9 画分及び肝細胞において、13 種類の代謝物が生成した。[14C] アスナプレビルの主要な代謝経路として、一及び二酸化、N-脱アルキル化、イソキノリン環の脱 離、O-脱メチル化及びアミド加水分解が挙げられる。ヒトに特異的な代謝物は検出されなかった。 アスナプレビルの酸化物のグルタチオン(GSH)付加体が、試験したすべての種で検出された。 [14C]アスナプレビルとラット、イヌ、サル及びヒトのミクロソームをインキュベートすると、放 射性物質とミクロソーム蛋白との不可逆的結合が起こり、この結合はGSH 存在下で減少した。こ の試験結果は反応性代謝物の生成を示すものであるが、アスナプレビルを 200 mg/kg/day までの 用量で6 ヵ月間投与したラット(AUC 値はヒトにアスナプレビル 200 mg を 1 日 2 回投与したと きのAUC 値の最大 136 倍)及び 100 mg/kg/day までの用量で 9 ヵ月間投与したイヌ(AUC 値はヒ トにアスナプレビル200 mg を 1 日 2 回投与したときの AUC 値の最大 82 倍)において、肝毒性
ないことが示唆された。 [14C]アスナプレビルを経口投与したときの in vivo 代謝プロファイルは、すべての種で質的に類 似しており、ヒトに特有な代謝物は検出されなかった。アスナプレビルは主に酸化物へと代謝さ れ、排泄物中に検出された代謝物は、ウサギの10 種類からマウス、ラット及びサルの 16 種類に 及んだ。ヒトの排泄物中では、15 種類の代謝物が検出された。動物の排泄物中(主に糞便中)で 確認された最も量の多い代謝物は、マウス及びラットで BMS-558364(脱イソキノリン環体、そ れぞれ投与量の8.3%及び 7.0%)、ウサギで M7(一酸化物、投与量の 3.1%)、イヌでは BMS-558364 及びM3(一酸化物)(両代謝物とも投与量の 2.3%)であった。ヒトの主要な代謝物は、M8(ア ミド加水分解生成物)及びM12(脱イソキノリン環体)で、それぞれ投与量の 14.6%及び 8.3%に 相当した。以上より、いずれの種でも投与量の15%以上に相当する代謝物はなかった。いずれの ヒト代謝物も、少なくとも1 種類の動物(マウス、ラット、イヌ又はサル)において生成された。 アスナプレビルは、動物及びヒトの血清中又は血漿中の主化合物であった。動物血液中に様々 な代謝物が存在したが、いずれの代謝物もアスナプレビル曝露量の 6%未満であった。ヒトにア スナプレビルを反復投与すると、代謝物の曝露量は増加したが、アスナプレビルの曝露量の20% 未満又はアスナプレビルとその代謝物の総曝露量の 10%未満で、最も量の多い血中代謝物は M3 であった。 アスナプレビルの開発において、慢性毒性試験のげっ歯類及び非げっ歯類動物として、ラット 及びイヌを使用した。動物及びヒトで忍容性がある投与量において、ヒトの代謝物曝露量は動物 のそれよりも小さかった。このことから、毒性試験でのこれら代謝物の曝露量は、安全性を評価 する上で十分な量であったことが示唆された。ヒトと動物のアスナプレビル曝露量のより詳細な 比較検討は、本概括評価のトキシコキネティクスの項(3.7)に記載した。 3.4 排泄 動物におけるアスナプレビルの消失には、胆汁クリアランス、代謝クリアランス及び腸内分泌 などの複数の経路が関与し、アスナプレビルとその代謝物は主に糞便中に排泄された。 動物で投与量の18%~54%が代謝により消失し、ヒトではもっと多い量(投与量の 76.4%)が 代謝により消失した。ヒトにおけるアスナプレビルの消失が代謝クリアランスに依存することは、 肝障害患者でアスナプレビルの曝露量が増加することと合致する(AI447012 試験)。胆管カニュー レ挿入(BDC)ラットに[14C]アスナプレビルを静脈内投与したときの放射能回収率から、アスナ プレビル関連物質の直接的な腸内分泌が示唆された。また、アスナプレビルの腸内分泌も生じる 可能性はあるものの、証明されていない。BDC ラット、BDC サル及びヒト(投与後 3~8 時間に 胆汁を採取)に[14C]アスナプレビルを経口投与した結果、放射能はラット及びサルでそれぞれ投 与放射能の25.5%及び 32.9%、ヒトでは投与放射能の 8.14%が胆汁中で回収された。アスナプレビ ルがラット、サル及びヒトの胆汁中に検出されたことから、胆汁中排泄がこれら種におけるアス ナプレビルとその代謝物の消失経路であることが示唆された。 マスバランス試験において、マウス、ラット、ウサギ、イヌ及びヒトに[14C]アスナプレビルを 単回経口投与した結果、投与放射能の 77%~88%が糞便中で回収された。投与放射能の 0.2%~
1.4%が尿中で回収されたことから、尿中排泄がアスナプレビルの主要な消失経路ではないことが 示唆された。動物において、糞便中の未変化体は投与量の35%~66%を占めた。なお、胆汁中排 泄、腸内分泌、糞便中に検出された未変化体のうちで体内に吸収されなかった未変化体の割合は 不明である。ヒトにおいて、投与量の7.5%が糞便中で未変化体として検出されたことから、アス ナプレビルは動物よりも多く体内に吸収されたか又は胆汁中排泄や腸内分泌が少なかったことが 示唆された。 3.5 蛋白結合 アスナプレビルのin vitro 血清蛋白結合は高値を呈し、すべての動物種において同程度で、濃度 10 μM(7.48 μg/mL)で 97.2%~99.2%の範囲であった。また、ヒト血漿蛋白結合率は濃度に依存 せず、濃度1 μM(0.748 μg/mL)で 99.7%であった。ヒト血漿中のアスナプレビル(平均血漿中濃 度0.050~1.588 μg/mL)の ex vivo 蛋白結合率は約 99.8%であった(AI447004 試験)。したがって、 動物及びヒトのアスナプレビル曝露量を比較検討するとき、アスナプレビルの遊離形分率を補正 しなかった。 3.6 薬物動態学的薬物相互作用 ヒトにおいてアスナプレビルは主に CYP3A を介した代謝により消失するため、アスナプレビ ルと CYP3A の阻害剤又は誘導剤が併用投与された場合、薬物相互作用が起こる可能性がある。 このことは、薬物相互作用試験(AI447014 試験)において、CYP3A 及び P-gp の阻害剤であるケ トコナゾールを併用投与した健康被験者のアスナプレビル曝露量が増加したことから裏付けられ る。しかしながら、AI447018 試験でアスナプレビルとリファンピシン(CYP3A の誘導剤)を反 復併用投与しても、アスナプレビルの曝露量は顕著に減少しなかった。これは、アスナプレビル がOATP の基質であるのに対し、リファンピシンは OATP の阻害剤であることが原因であると考 えられる。すなわち、リファンピシンによるOATP の阻害はアスナプレビルの肝取込みを減少さ せ、リファンピシンによる CYP3A 誘導の影響を減弱した可能性があり、結果としてアスナプレ ビルの血漿中濃度に対し、臨床的に意義のある変化がみられなかったものと考えられた。 アスナプレビルはOATP1B1 及び OATP2B1 の基質である。したがって、これらトランスポーター の活性を変化させる薬物の併用投与は、アスナプレビルの曝露量に影響を及ぼす可能性がある。 FDA の薬物相互作用に関するガイダンス(案)に説明されているベーシックモデルを用いて
in vitro データを解析した結果、アスナプレビルは CYP3A4 の誘導剤であるが、CYP1A2 又は
CYP2B6 の誘導剤ではなかった。アスナプレビルは CYP3A の時間依存的阻害剤であるため、ア スナプレビルを反復投与すると、CYP3A の誘導と阻害が同時に起こり得る。アスナプレビル (AI447007 試験:600 mg を 1 日 2 回 7 日間投与、AI447020 試験:200 mg を 1 日 2 回 10 日間投 与)とミダゾラム(CYP3A の基質)を併用投与したとき、ミダゾラムの曝露量が 29%~44%減少 した。これは、アスナプレビルが CYP3A を誘導したことを示すものである。また、アスナプレ ビルはCYP2D6 の時間依存的阻害剤であることが示唆され、AI447020 試験において、アスナプレ ビルとデキストロメトルファン(CYP2D6 の基質)との間で薬物相互作用が観察された。したがっ
て、アスナプレビルの投与がCYP3A 及び CYP2D6 の基質との薬物相互作用をもたらす可能性が 示唆された。 アスナプレビルは、in vitro でウリジン二リン酸グルクロン酸転移酵素(UGT)1A1 を阻害した。 しかしながら、AI447011 試験において、アスナプレビルによる UGT1A1 阻害に起因した、総ビ リルビン(UGT1A1 の基質)の増加は認められなかったため、アスナプレビルが UGT1A1 を介し たグルクロン酸抱合に明らかな影響を及ぼす可能性は低いと考えられた。 OATP1B1、OATP2B1 及び OATP1B3 などの多様なトランスポーターがアスナプレビルにより阻 害されることが示された[IC50 = 0.3~3.0 μM(0.22~2.2 μg/mL)]。AI447016 試験で被験者にアス ナプレビル200 mg を 1 日 2 回投与したときの Cmax 値 0.56 μM(0.42 μg/mL)から、OATP1B1 の 基質との薬物相互作用の発現が示唆された。AI447015 試験において、アスナプレビルとロスバス タチン(OATP1B1 及び OATP1B3 の基質)との薬物相互作用が観察され、アスナプレビルの投与 によりロスバスタチン曝露量が増加した。更に、P-gp 阻害に対する IC50値が50.6 μM(37.9 μg/mL) と高いにもかかわらず、AI447021 試験で健康被験者にアスナプレビルを 10 日間投与したとき、 ジゴキシン(P-gp の基質)の曝露量の増加がみられた。これは、吸収相で高濃度のアスナプレビ ルが存在する腸管でP-gp が阻害され、結果として腸管内腔へのジゴキシンの排泄が減少し、吸収 が増加したためと考えられた。 アスナプレビルは、ダクラタスビルと併用投与される。ダクラタスビルの相当量がCYP3A4 を 介した代謝により消失し、ダクラタスビルはP-gp の基質、CYP3A の誘導剤かつ阻害剤、また P-gp 及びOATP の阻害剤である。したがって、アスナプレビルとダクラタスビルとの間で薬物相互作 用が起こる可能性がある。しかしながら、健康被験者を対象としたAI447009 試験及び C 型慢性 肝炎患者を対象とした AI447011 試験において、ダクラタスビルとアスナプレビルの併用投与に おいて臨床的意義のある薬物相互作用は認められなかった。 3.7 トキシコキネティクス 主要な毒性試験で用いたラット及びイヌの血漿中アスナプレビル濃度は、特に高用量、更にイ ヌで変動したが、嘔吐がアスナプレビルの曝露量を抑える一因になったと考えられた。ラットに アスナプレビルを反復投与したときのAUC 値は、30~200 mg/kg/day の用量範囲では概して用量 比以上の増加を示したものの、より高い用量では用量比未満の増加を示した。ラットにおけるア スナプレビルのAUC 値は、30~100 mg/kg/day の用量範囲では概して雌雄で同程度であったもの の、100 mg/kg/day を上回る用量では雄性よりも雌性の方が約 2 倍大きかった。イヌにおける反復 投与毒性試験では、概して15~300 mg/kg/day の用量範囲ではアスナプレビルの AUC 値は用量比 以上に増加し、雄性よりも雌性の方が約2 倍大きかった。また、アスナプレビルの反復投与後の 蓄積はみられなかった。ラット及びサルにアスナプレビルとダクラタスビルを併用投与したとき、 アスナプレビルの AUC 値に増加傾向が認められたが、アスナプレビルの曝露量は個体間変動が 大きいためトキシコキネティクスの相互作用の可能性については明らかでなく、DCV + ASV 併用 療法の臨床試験では薬物相互作用は報告されていない(AI447009 試験、CTD 2.7.2.2.8)。数値の 変動や血漿中濃度の性差がおおむね 2 倍以下であったことを考慮して、TK パラメータ値及び曝
露量比は、雌雄を合わせた平均値として記載した。血漿中代謝物量はアスナプレビルの AUC 値
の 6%未満であったことから、非臨床毒性試験においてアスナプレビルの代謝物を測定しなかっ
た。
主要な毒性試験におけるアスナプレビルの投与量、AUC 値及びヒト AUC に対する動物 AUC
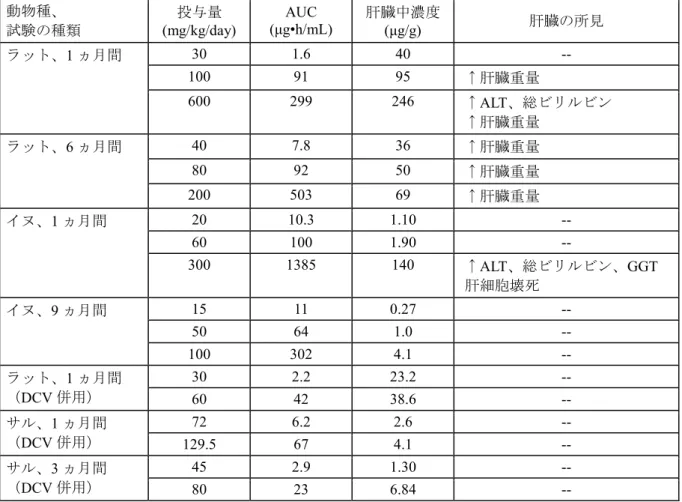
の比を表 3.7-1 に要約した。アスナプレビルの毒性の包括的評価を行うため、また、ヒトでのリ スクの可能性を判断するため、適切な投与量が毒性試験で用いられたことが AUC 比から示され た。 肝臓が治療の標的器官であり、ラットのADME 試験で肝臓中の[14C]アスナプレビルが高濃度で あったことから、いくつかの反復投与毒性試験でアスナプレビルの肝臓中濃度を測定した(表 4.2.3.1-1)。 表3.7-1 主要な反復経口投与毒性試験におけるアスナプレビルの投与量、AUC値及び ヒトAUCに対する動物AUCの比 動物種 試験 (試料採取時点) 投与量 (mg/kg/day) AUC (μg•h/mL)a AUC 比b 雄 雌 雄 雌 マウス 6 ヵ月間反復投与毒性 試験(26 週目) 25 14 14 3.8× 3.8× 100 256 417 69× 113× 200 983 1600 266× 434× ラット 1 ヵ月間反復投与毒性 試験(28 日目) 30 1.9 1.3 0.5× 0.4× 100 83 98 22× 27× 600 227 371 62× 101× ラット 1 ヵ月間併用投与毒性 試験(28 日目) 30 2.2 2.1 0.6× 0.6× 60 28 55 7.6× 15× ラット 6 ヵ月間反復投与毒性 試験(26 週目) 40 4.0 12 1.1× 3.3× 80 39 144 11× 39× 200 321 684 87× 185× イヌ 1 ヵ月間反復投与毒性 試験(28 日目) 20 4.6 16 1.2× 4.3× 60 102 99 28× 27× 300 1410 1360 382× 369× イヌ 9 ヵ月間反復投与毒性 試験(39 週目) 15 5.5 16 1.5× 4.3× 50 47 81 13× 22× 100 223 380 60× 103× サル 1 ヵ月間併用投与毒性 試験(28 日目) 72 4.6 7.8 1.2× 2.1× 129.5 74 60 20× 16× サル 3 ヵ月間併用投与毒性 試験(13 週目) 45 2.8 2.9 0.8× 0.8× 80 32 14 8.7× 3.8× ラット 受胎能及び着床まで の初期胚発生に関す る試験(14 日目) 50 53 30 14× 8.1× 200 454 545 123× 148× 600 386 373 105× 101×
妊娠マウス 胚・胎児発生に関する 試験(妊娠15 日) 10 - 0.9 - 0.2× 50 - 19 - 5.1× 250(母動物) - 737 - 200× 500(胎児) - 1740 - 472× 妊娠ウサギ 胚・胎児発生に関する 試験(妊娠19 日) 50 - 1.2 - 0.3× 100 - 2.8 - 0.8× 200 - 4.4 - 1.2× 授乳ラット (母動物曝露量) 出生前及び出生後の 発生に関する試験 (哺育4 日) 40 - 27 - 7.3× 125 - 282 - 76× 400 - 711 - 193× 下線を施した投与量は無毒性量(NOAEL)を示す。 a 投与後0 時間から最終測定可能時間(4~24 時間)までの血漿中濃度曲線下面積 b ヒトに臨床推奨用量を投与したときのアスナプレビルのAUC 値は 3.69 μg•h/mL である。
4 毒性試験 アスナプレビルの毒性について、ICH ガイドラインに準拠して実施した非臨床毒性試験(表 1-1) により評価した。重要な試験はGLP 適合下で実施した。ヒトにおける安全性を評価するため、総 合的な毒性評価を担保する高い曝露量が得られる適切な投与量を設定した。 重要な試験で得られた動物の曝露量と臨床用量における定常状態のヒトの曝露量との比(AUC 又はCmax)を表 4-1 に示す。毒性が発現した曝露量についてもヒト曝露量との比を算出した。 表4-1 アスナプレビルの無毒性量及び主な毒性発現用量における曝露量と ヒト曝露量との比 動物種、試験の種類 無毒性量(mg/kg/day) AUC (μg•h/mL) 又は Cmax (μg/mL) ヒト曝露量との比a ラット1 ヵ月間 100 AUC: 91 25 ラット6 ヵ月間 200 AUC: 503 136 イヌ1 ヵ月間 60 AUC: 100 27 (心電図に関する無毒性量)b 300 Cmax: 110 263 イヌ9 ヵ月間 100 AUC: 302 82 (心電図に関する無毒性量)b 100 Cmax: 50 120 マウスがん原性 200 AUC: 1292 350 ラット生殖能 600 AUC: 380 103 マウス胚・胎児発生 母動物:250 胎児:500 AUC: 737 1740 200 472 ウサギ胚・胎児発生 母動物・胎児:200 AUC: 4.4 1.2 ラット出生前及び出生後発生 F0 母動物:40 F1 新生児:125 AUC: 26.8 282 7.3 76 毒性所見 動物種、 試験の種類 毒性発現用量 (mg/kg/day) AUC (μg•h/mL) ヒト曝露量との比a 消化管(腸細胞肥大) ラット1 ヵ月間 600 299 81 肝臓(血液生化学的変化) ラット1 ヵ月間 600 299 81 肝臓(血液生化学的変化を伴 う肝細胞壊死) イヌ1 ヵ月間 300 1385 375
a 臨床推奨用量100 mg BID におけるヒトの Cmax 0.419 μg/mL 及び AUC 3.69 μg•h/mL に基づき算出
(動物のAUC/Cmax ÷ ヒトの AUC/Cmax)、AUC 及び Cmax は雌雄合計平均値(生殖発生毒性試験を除く)
b 最終心電図測定時(1 ヵ月間試験:投与 28 日、9 ヵ月間試験:投与 39 週)
4.1 単回投与毒性試験
マウス及びラットに200~2000 mg/kg、イヌに 30~300 mg/kg の用量でアスナプレビルを単回経 口投与した。マウス及びラットにおいて600 mg/kg まで忍容性が認められたが、2000 mg/kg では マウスで死亡例(剖検及び病理組織学的検査で胃腸管毒性を示唆する変化)がみられ、ラットで