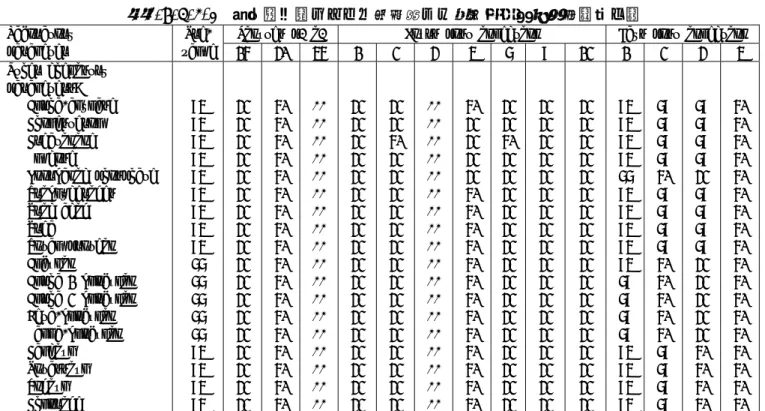
2.7.4.3 臨床検査値の評価 統合安全性解析(ISS)では,第Ⅱ相及び第Ⅲ相臨床試験の全臨床検査値は標準的な国際単位で表 示されているが,一方,一部の試験報告書では米国単位が用いられている. 報告書 A04410(特別な集団での試験),報告書 BA13(日本での第Ⅱ相試験),及び報告書 A05868(日本での第Ⅲ相試験)は検査施設が異なるため,別に評価された.これら以外の第Ⅱ相及 び第Ⅲ相臨床試験は,すべて同一の検査施設で測定され,まとめて評価された. 2.7.4.3.1 方法 臨床検査値結果は,第Ⅰ相,第Ⅱ相及び第Ⅲ相臨床試験から収集された. 第Ⅰ相臨床試験の臨床検査結果は,報告書 A336,A337,A954,A08607 に記載されており,次項 の 2.7.4.3.2 健康人(第Ⅰ相)にまとめた.第Ⅱ相及び第Ⅲ相臨床試験結果は ISS に含まれており, 2.7.4.3.3 患者(第Ⅱ相及び第Ⅲ相臨床試験)の項にまとめた. 臨床検査項目としては,生化学検査(特に肝機能,膵機能,腎機能についての項目),血液学的 検査,凝固系,電解質,及び尿検査について検討された. 第Ⅰ相臨床試験の各臨床検査項目の正常範囲は,個々の報告書に記載されている.第Ⅱ相及び第 Ⅲ相臨床試験における各臨床検査項目の基準値は,各治験実施施設で測定された日本での第Ⅱ相及 び第Ⅲ相試験を除き,2.7.4.7 付録 表 24~26 に示す. 2.7.4.3.2 健康被験者(第Ⅰ相臨床試験) SH L569B を投与され,臨床検査が実施された全被験者数を測定項目別に表 2.7.4.3-1 に示す.
表 2.7.4.3-1 第Ⅰ相試験における各時点での臨床検査測定例数
Minutes p.i. Hours post injection Days post injection
Laboratory parameter
Pre-value 15 30 45 1 2 3 4 6 8 12 1 2 3 4
Liver function tests: Alkaline phosphatase Gamma-GT GOT GPT LDH LAP Cholinesterase Bilirubin total Bilirubin direct Bilirubin indirect 84 84 84 84 84 32 32 84 84 84 32 32 32 32 32 32 32 32 32 32 50 50 50 50 50 32 32 50 50 32 32 32 32 32 32 32 32 32 32 32 50 50 50 50 50 32 32 50 50 50 50 50 50 50 50 32 32 50 50 50 32 32 32 32 32 32 32 32 32 32 68 68 68 68 68 32 32 68 68 68 50 50 50 50 50 32 32 50 50 50 50 50 50 50 50 32 32 50 50 50 50 50 50 50 50 32 32 50 50 50 84 84 84 84 84 32 32 84 84 84 68 68 68 68 68 32 32 68 68 68 50 50 50 50 50 32 32 50 50 50 50 50 50 50 50 32 32 50 50 50 Blood coagulation factors:
Quick time Fibrinogen PTT Thrombin time 66 66 66 66 32 32 32 32 48 48 48 48 32 32 32 32 32 32 32 32 48 48 48 48 32 32 32 32 30 50 48 47 48 48 48 48 32 32 32 32 48 48 48 48 66 66 66 66 48 48 48 48 32 32 32 32 48 48 48 48 Iron related parameters:
Iron
Iron-binding
Capacity (free and total) Ferritin 84 68 84 -- -- -- 50 50 50 32 32 32 50 50 50 50 50 50 32 32 32 68 68 68 50 50 50 50 50 50 50 50 50 84 68 84 68 68 68 50 50 50 50 50 50 Reference Documentation: Report nos. A336, A337, A954 and A08607
表 2.7.4.3-1 第Ⅰ相試験における各時点での臨床検査測定例数(続き)
Minutes p.i. Hours post injection Days post injection
Laboratory parameter
Pre-value 15 30 45 1 2 3 4 6 8 12 1 2 3 4
Hematology
White blood cells Red blood cells Hemoglobin Hematocrit MCV MCH Platelets Leucocytes, neutrophil Leucocytes, eosinophil Leucocytes, basophil Lymphocytes Monocytes Reticulocytes 84 84 84 84 84 84 84 84 84 84 84 84 6 32 32 32 32 32 32 32 32 32 32 32 32 --50 50 50 50 50 50 50 50 50 50 50 50 --32 32 32 32 32 32 32 32 32 32 32 32 --50 50 50 50 50 50 50 50 50 50 50 50 --50 50 50 50 50 50 50 50 50 50 50 50 -- 32 32 32 32 32 32 32 32 32 32 32 32 -- 68 68 68 68 68 68 68 68 68 68 68 68 6 50 50 50 50 50 50 50 50 50 50 50 50 -- 32 32 32 32 32 32 32 32 32 32 32 32 -- 50 50 50 50 50 50 50 50 50 50 50 50 --84 84 84 84 84 84 84 84 84 84 84 84 6 68 68 68 68 68 68 68 68 68 68 68 68 6 50 50 50 50 50 50 50 50 50 50 50 50 6 50 50 50 50 50 50 50 50 50 50 50 50 6 Urinalysis U-specific density Urobilinogen U-erythrocytes U-hemoglobin U-protein U-beta-NAG U-albumin U-glucose U-creatinine Creatinine,clearance per test point Creatinine,clearance per 24 hrs. 68 84 84 66 84 50 50 84 50 50 50 --50 50 50 50 50 50 50 50 50 50 50 -- -- -- -- -- -- -- -- -- -- -- 68 50 50 50 32 50 32 32 50 50 50 50 50 50 50 50 50 50 50 50 50 50 -- -- -- -- -- -- -- -- -- -- -- 50 50 50 50 50 50 50 50 50 50 50 68 84 84 66 84 50 50 84 50 50 50 68 68 68 50 68 50 50 68 50 --50 68 68 68 50 68 50 50 68 50 -- 50 50 50 50 50 32 50 50 50 32 32 32 Reference Documentation: Report nos. A336, A337, A954 and A08607
表 2.7.4.3-1 第Ⅰ相試験における各時点での臨床検査評価例数(続き)
Minutes p. i. Hours post injection Days post injection
Laboratory parameter Pre-value 15 30 45 1 2 3 4 6 8 12 1 2 3 4 Other chemistry parameters: Alpha-amylase Cholesterol Creatinine Glucose Inorganic phosphate Triglycerides Uric acid Urea Total protein Albumin Alpha 1 globulin Alpha 2 globulin Beta-globulin Gamma-globulin Calcium Potassium Sodium Chloride 84 84 84 84 84 84 84 84 84 66 66 66 66 66 84 84 84 84 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 --32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 50 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 --50 32 32 32 32 50 50 50 50 50 50 50 50 50 50 50 50 50 32 32 50 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 32 84 84 84 84 66 84 84 84 84 84 68 68 68 68 84 84 84 84 68 68 68 68 50 68 68 68 68 50 50 50 50 50 68 68 68 68 68 68 68 68 32 68 68 68 68 32 32 32 32 32 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 50 Reference Documentation: Report nos. A336, A337, A954 and A08607
血液学的検査,生化学検査,凝固系検査,尿検査項目は,概して,投与した SH L569B の用量に依 存した,又はプラセボ投与群と比較して臨床上有意な変化や傾向を示さなかった.臨床検査値は最 終時点の検査でほぼ個体の変動範囲内であり,個々の被験者における正常範囲からのわずかな逸脱 は,臨床上有意ではなく,問題とは考えられなかった. 第Ⅰ相臨床試験の 2 試験では,血清鉄値又は遊離鉄結合能(fIBC)に関して二つの現象が注目さ れた. 報告書 A336 本剤投与前の血清鉄値が正常範囲内又は下限未満であった 2 例(100μmol/kg 群)の被験者にお いて,投与後 4 又は 6 時間に正常範囲の上限をわずかに超える値が延べ 3 回認められた.Ferrocine complexion 法で測定した血清鉄の値の解析では,本剤投与後 4~8 時間に,プラセボ投与群と比較 して,投与に関連した血清鉄の平均値の上昇が認められた.しかし,血清鉄平均値は正常範囲内で あり,さらに,上記の 3 回の測定値以外では個々の血清鉄値もすべて正常範囲内又は正常範囲下限 未満であった.血清鉄濃度は,造血骨髄への移行と網内系組織からの放出のバランスによる動的な 平衡関係を一時点で見ているものであり,その 1 日の交代率は極めて大きく,日内変動があること も知られている.血清鉄濃度が上昇又は低下しても,生体からの損失又は過剰蓄積を必ずしも意味 するわけではない.また,わずかに異なる時点の検体で誘導結合プラズマ発光分析(ICP)を用いて 測定した場合には,血清鉄値に影響が見られなかったことから測定系への影響も示唆される.本剤 投与後に見られた,一時的な血清鉄の上昇又は低下は臨床上問題のないものと判断している.遊離 鉄結合能(fIBC)では,投与後 4~12 時間に正常範囲下限未満の測定値が多く見られたが,投与量 に応じた変動はなく,また,各時点の平均値はプラセボ投与群と差がなかった. 報告書 A954(高用量 CT 試験) 本剤投与後の最初の 1 時間以内に血清鉄と総鉄結合能(tIBC)の一時的な減少が認められ,投与 4~8 時間後には,血清鉄の一時的な増加とそれに伴う遊離鉄結合能(fIBC)の低下が用量に依存し て見られた.
2.7.4.3.3 患者(第Ⅱ相及び第Ⅲ相臨床試験) SH L569B の単回投与を受け,臨床検査データが得られた全患者が,安全性データベースに含まれ た.表 2.7.4.3-2 に,以下に示す各時点で臨床検査値の評価が行われた第Ⅱ相及び第Ⅲ相臨床試験 の患者数の概要を示す. • 投与 4 時間後(2-4 時間後) • 投与 24 時間後(20-28 時間後) • 投与 24-72 時間後(報告書 BA13 の第Ⅱ相臨床試験のみ) • 投与 48 時間後(44-52 時間後) • 投与 72 時間後(68-76 時間後) • 投与 144 時間後(140-148 時間後)(報告書 A04410 の第Ⅲ相臨床試験のみ)
*新薬承認情報提供時に置き換えた。
表 2.7.4.3-2 第Ⅱ,Ⅲ相試験における各測定時点での臨床検査評価例数
Hours post injection Laboratory
parameters
Pre-value
2-4 20-28 24-72∗ 44-52 68-76 140-148
Liver function tests: Alkaline phosphatase Gamma-GT GOT GPT LDH Cholinesterase Bilirubin total Bilirubin direct 1688 1691 1698 1698 1658 1198 1698 508 1191 1197 1198 1198 1161 1198 1198 146 1507 1510 1513 1513 1475 1194 1513 326 181 181 185 185 183 160 185 182 119 119 119 119 111 119 119 16 611 613 615 615 609 438 616 226 54 54 54 54 54 54 54 7 Blood coagulation factors:
PTT
Prothrombin time (%) Prothrombin time (sec)
700 697 67 541 555 42 700 697 67 -- -- -- -- -- -- 492 483 18 39 -- 45 Iron related parameters:
Iron
Iron-binding
Capacity (free and total) Ferritin 1682 952 949 1195 835 834 1512 952 949 170 -- -- 119 -- -- 613 438 437 54 54 54 Hematology: WBC (other cells) Hemoglobin Hematocrit MCV Erythrocytes Platelets Leucocytes Leucocytes, neutrophil Leucocytes, eosinophil Leucocytes, basophil Lymphocytes Monocytes Stab cells Segmented neutrophils 225 1603 1603 720 1603 1599 1603 1557 1556 1553 1557 1556 1127 1127 225 1113 1113 340 1113 1112 1113 1127 1127 1127 1127 1127 1127 1127 221 1418 1418 535 1418 1417 1418 1383 1383 1380 1383 1383 1110 1110 -- 185 185 185 185 182 185 174 173 173 174 173 -- -- -- 7 7 7 7 6 7 -- -- -- -- -- -- -- 218 579 579 177 579 579 579 576 576 572 576 576 433 433 -- 54 54 -- 54 54 54 54 54 54 54 54 47 47 Urinalysis: U-specific density Urobilinogen U-erythrocytes U-casts U-protein U-leucocytes U-albumin U-glucose U-creatinine U-pH Osmolality 49 115 1246 589 1243 1249 1148 163 363 49 491 49 -- 12 -- -- 14 48 49 -- 49 49 48 115 1246 589 1243 1249 1247 163 363 48 491 -- 20 -- -- 20 -- -- 20 -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- 112 441 441 440 441 441 112 -- -- 440 48 -- 8 -- -- 12 44 48 -- 48 44 Reference Documentation: ISS Table L.1.4, WBC =White blood cells,
*新薬承認情報提供時に置き換えた。
表 2.7.4.3-2 第Ⅱ,Ⅲ相試験における各測定時点での臨床検査評価例数(続き)
Hours post injection Laboratory parameters Pre-value 2-4 20-28 24-72∗ 44-52 68-76 140-148 Other chemistry parameters: Alpha-amylase Cholesterol Creatinine Glucose Inorganic phosphate Triglycerides Uric acid Urea Total protein Albumin Alpha 1 globulin Calcium Potassium Sodium Chloride 1686 952 1697 951 951 952 952 1695 1697 1697 363 1288 1665 1695 1311 1198 835 1198 833 834 835 835 1198 1198 1198 -- 836 1166 1196 832 1512 952 1512 951 951 952 952 1512 1512 1513 363 1109 1481 1511 1127 174 -- 185 -- -- -- -- 183 185 184 -- 179 184 184 184 119 -- 119 -- -- -- -- 119 119 119 -- -- 111 118 -- 614 436 615 434 435 436 438 613 613 616 -- 597 614 615 613 54 54 54 54 54 54 54 54 54 54 -- 54 54 54 54 Reference Documentation: ISS Table L.1.4
∗The time point 24-72 hours refers only to Study no. H*356, Report no. BA13
これらの臨床検査データを解析した結果,試験の全経過を通じて個々の血液検査並びに尿検査項 目の平均値には注目するべき変動は認められなかった.減少及び増加は,全測定項目において均一 に分布していた.個々の項目の変動については,そのほとんどが正常範囲内にあり,特に肝機能及 び/又は腎機能に関連する項目の変化に伴って,その他の検査項目が変動することはなかった.ほ とんどの患者に対して投与された臨床投与量は 25μmol/kg である.欧米で実施された第Ⅱ相及び第 Ⅲ相試験(報告書 A04410 を除く)において,同一の測定施設で測定された 25μmol/kg 投与後の平 均値並びに中央値を 2.7.4.7 付録 表 27 に示す. 報告書 A04410 では,臨床検査値の評価対象となったいずれの項目においても,一貫した又は投 与群に特有な経時的変化は認められなかった.平均値並びに中央値を 2.7.4.7 付録 表 28 に示す. 報告書 BA13 及び報告書 A05868(日本での第Ⅱ相及び第Ⅲ相試験)において,個々の臨床検査項 目に顕著な変化(増加及び/又は減少)は認められなかった.これらの試験には治験実施施設が 22 (BA13)又は 15(A05868)施設含まれ,血液サンプル及び尿サンプルは異なる正常範囲を持った各 施設の検査室で評価された.ISS では臨床検査値の平均値,または中央値(報告書 A05868)は,施 設ごとに算出した. 2.7.4.3.4 治験責任医師が評価した臨床上有意な臨床検査値異常 臨床検査データの解析の結果,それぞれの検査値に個々の変動が存在することは明らかであるが, すべての臨床検査項目並びに尿検査項目は,全体としてだけでなく,肝臓又は腎臓の機能障害を有 する患者などの特定のリスク集団においても安定していた(2.7.4.3.5 臨床検査項目の部分集団解 析 参照).個々の検査項目の変動は,これらの患者の基礎疾患又は併用薬に起因する可能性が非 常に高く,SH L569B の安全性プロフィールに関する問題とはなり得ないと考えられた.
健康被験者(第Ⅰ相臨床試験) 血液学的検査,生化学検査,凝固系検査,尿検査項目は,様々な SH L569B の投与量で,プラセボ 投与群と比較しても臨床上有意な変化や傾向を一切示さなかった. 患者(第Ⅱ相及び第Ⅲ相臨床試験) 報告書 AH34 ベースラインと比べ本剤投与後の臨床検査項目に臨床的に有意な変化が認められた症例として, 12.5,25μmol/kg 投与群の各 1 例,及び 50μmol/kg 投与群の 3 例の計 5 例が治験責任医師から報 告された.これらの患者に見られた変化は,治験責任医師により,3 例では,それぞれ「検体(尿) 不良」,「化学療法」及び「溶血血清」の結果の可能性があると評価された.他の 2 例のうち 1 例では, 白血球尿と診断され,もう 1 例では基準値範囲の上限をわずかに上回る総ビリルビン増加(投与前 12.0μmol/L,24 時間後 18.8μmol/L;正常範囲上限 17μmol/L)が観察された.
報告書 AI94 臨床検査項目でベースラインからの臨床上有意な変化を示した症例として,プラセボ投与群の 1 例が治験責任医師より報告されたが(項目:ALP・ヘモグロビンの低下,AST/GOT・ALT/GPT・アミ ラーゼ・白血球数の上昇),SH L569B 投与群の患者については報告されなかった. 報告書 AI83(高用量 CT 試験) 臨床検査項目においてベースラインからの臨床上有意な変化を示した症例として,治験責任医師 より 4 例が報告された. これら全例が本剤 350μmol/kg 投与群であった.2 例は,本剤投与前の血 清鉄の値が基準値範囲未満で,20-28 時間後に基準値範囲内の値まで増加した.他の 2 例では, AST/GOT 及び ALT/GPT が,本剤投与前に比べ 20-28 時間後に基準値範囲を超える上昇を示した.さ らに,このうちの 1 例ではアミラーゼ値,別の 1 例では LDH 値が 20-28 時間後に基準値範囲外の値 に上昇した. 報告書 BA13(日本) 日本で実施した第Ⅱ相試験においては,投与後 24-72 時間に採血,採尿を行い各施設で測定した. それらの測定値に関して,臨床的有意性に関わらず,投与前と比較した異常変動の有無,及び治験 薬との関連性を評価した.臨床検査値の異常変動は 84 例(12.5,25,50μmol/kg 投与群でそれぞ れ 25,32,27 例)で認められた.そのうち「関連性あり」と判断された変動は 36 例であり,12.5, 25,50μmol/kg 群でそれぞれ 6,16,14 例(発現率はそれぞれ 9.8,25.8,22.2%)であった.用 量別の変動が最も多かった項目は血清鉄であったが,上昇及び低下の両方が認められ,一定の傾向 は見られなかった.総ビリルビンが投与後に 2.0mg/dL を越える変動が 25,50μmol/kg 群で各 1 例 ずつ認められたが,投与前より正常範囲を超える値からの変動で,変動幅は小さかった.これらの 変動は患者の個体内変動範囲とも考えられたが,治験責任医師は「関連の可能性あり」と判断した. 報告書 A00518 本剤投与前後の臨床検査項目の中で臨床上有意な変化を示した症例が,治験責任医師から 2 例報 告された.1 例では血液希釈(24 時間後の採血が,術中になされた)に起因する血液学的検査項目
(ヘモグロビン,ヘマトクリット)の変化が認められ,もう 1 例では肝臓外科手術後のビリルビン 上昇であった. 報告書 A03779 本剤投与前後に認められた臨床上有意な臨床検査項目の変化(項目:カリウム,白血球,総たん 白,無機リン,好中球,分葉核球,総ビリルビン,グルコース,ナトリウム,尿中アルブミン)は 7 例で報告され,治験責任医師により全例で治験薬と「関連性なし」と評価された.これらの変化 は主に併用薬,感染症,基礎疾患,若しくは採血前の食物摂取によるものであった. 報告書 A05742 臨床上有意な臨床検査値の変化を示した症例として,5 例が治験責任医師により報告された(ビ リルビン上昇 4 例,血清鉄上昇 1 例)が,一定の傾向や一過性でない変動は認められなかった. 報告書 A01908 臨床検査値に関して臨床上有意な変化を示した症例として,14 例が治験責任医師より報告された. このうち 6 例では,一つ又は複数の尿検査パラメータに臨床上有意な変化が発現した.個々の変動 は,これらの患者の基礎疾患や併用薬に起因する可能性が高かった.4 例では,採尿又は採血の失 敗により試験結果が異常になった可能性があった.他に原因が説明できなかった検査値異常は,尿 中赤血球の一過性増加 1 件と,24 時間後の軽度のアミラーゼ上昇 1 件であった. 報告書 A04410 特別な集団での試験として,軽度から重度の肝障害(肝硬変 Child-Pugh 分類 A-C),中等度腎機 能障害,末期腎不全,及び中等度の肝・腎障害を合併する患者及び対照の健康被験者を対象として 検討した.臨床的に有意な臨床検査値の変動は,いずれの項目でもいずれの時点でも報告されな かった. 報告書 A05868(日本) 臨床検査値における臨床上有意な異常変動は,11 例において 13 件報告された.2 件以上の変動が 報告された項目は,白血球数 3 件(増加1,減少 2 件),尿中ブドウ糖 2 件,血清鉄(上昇,低下 各 1 件)であり,他の項目は 1 件ずつであった(BUN 上昇,総ビリルビン上昇,アミラーゼ上昇, 血小板数減少,APTT[秒]延長及び PT[%]の活性低下).13 件のうち,5 件は投与後 24 時間の測定 において異常が認められたもので,3 日後の測定では異常変動は認められなかった.薬剤との関連 性では,8 件は「関連なし」又は「関連性は考えにくい」,5 件は「関連の可能性あり」であった. 試験番号 305654 臨床検査値における臨床上有意な異常変動は,SH L569B 群の 1 例(総ビリルビンの増加)及び Gd-BOPTA 群の 2 例(LDH の増加,及び尿中の白血球数・赤血球数・総たん白・アルブミンの増加) で報告された.治験責任医師は,SH L569B 群で見られた総ビリルビンの増加は肝硬変によるもので, 過去にも総ビリルビンの変動が認められたとした.Gd-BOPTA 投与後にみられた尿検査項目の異常は, 尿検体が尿道カテーテル抜去直後に採取されたことにより説明できるとされ,また,LDH の増加は 検体の溶血によるものとされた.
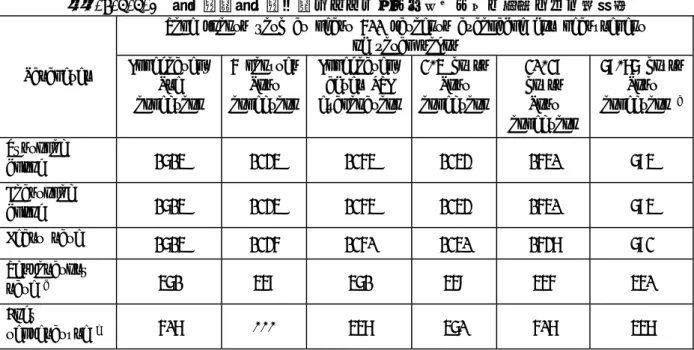
2.7.4.3.5 臨床検査項目の部分集団解析 第Ⅱ相及び第Ⅲ相臨床試験すべての全安全性評価対象集団,肝酵素又は血清クレアチニンが高値 の部分集団,肝硬変の有無別の部分集団,及び Child-Pugh 分類スコア別の部分集団のそれぞれにお いて臨床検査値を評価したが,血液学的検査,血液生化学検査,尿検査項目において,SH L569B 投 与との因果関係を示唆する所見はなかった.唯一の特徴は,肝硬変患者における肝機能検査項目の ベースライン値が,全安全性評価対象集団に比較して,コリンエステラーゼ,PT(%)は低く, AST/GOT,ALT/GPT,γ-GTP,LDH,総ビリルビン,APTT(秒)では高かった.この他に,抗痙攣薬と 抗不整脈の併用の有無による部分集団解析を行った.これらの薬は肝臓で代謝される代表的な併用 薬であること,また,本剤との相互作用の可能性を検討する時,抗不整脈併用患者は循環器系疾患 を合併するリスク患者であることから解析を実施した. クレアチニン値別 投与前血清クレアチニン値が正常範囲内,正常範囲を超えるが 265.2μmol/L(=3.0mg/dL)未満, 265.2μmol/L 以上の 3 部分集団に分類して解析した.同一の測定施設で測定された欧米での第Ⅱ相 及び第Ⅲ相試験における 25μmol/kg 投与(報告書 A04410 を除く),及び報告書 A04410(特別な 集団での試験)におけるクレアチニン及び BUN の測定値の記述統計量を部分集団ごとに 2.7.4.7 付録 表 29 に示す.また,本剤の全投与量を投与後の「正常範囲未満,正常範囲内,正常範囲を超 える」の推移を 2.7.4.7 付録 表 30 に示す.クレアチニン値が 265.2μmol/L 以上の患者は,9 例 認められた(報告書 A04410 における末期腎障害患者 6 名を含む).報告書 A04410 における末期腎 不全 6 例のクレアチニン及び BUN の平均値及び中央値は,個体内変動が大きく,投与後 2-4,20-28,68-76 時間のいずれの時点でも投与前より低下した. 日本で実施した第Ⅲ相試験において,同様の部分集団解析を行った結果は報告書 A05868 に示さ れる.投与前クレアチニン値が 3.0mg/dL 以上の患者は認められず,正常範囲を超えるが 3.0mg/dL 以下の患者が 8 例認められた.これらの患者でクレアチニン,BUN の変動は,本剤投与後認められ なかった. 肝機能別 投与前臨床検査値において,AST/GOT,ALT/GPT,又はγ-GTP が正常範囲上限の 2 倍を超えている か,又は総ビリルビン値が 1.5mg/dL 以上を示した患者を肝機能異常ありととらえ,これらの異常値 がない患者部分集団とともに解析した.同一の測定施設で測定された欧米での第Ⅱ相及び第Ⅲ相試 験における 25μmol/kg 投与(報告書 A04410 を除く),及び報告書 A04410(特別な集団での試 験)における肝機能に関連した測定値の記述統計量を,部分集団ごとに 2.7.4.7 付録 表 31,32 に示す.なお,日本での第Ⅲ相試験において同様の部分集団解析を行い評価したが,投与前値に差 が見られた以外には,肝機能異常群での投与後の臨床検査値変動を示唆する結果は認められなかっ た(報告書 A05868).
肝硬変の有無別
肝硬変の有無別部分集団について,同一の測定施設で測定された欧米での第Ⅱ相及び第Ⅲ相試験 における 25μmol/kg 投与(報告書 A04410 を除く),及び報告書 A04410(特別な集団での試験) における肝機能に関連した項目の測定値の記述統計量を 2.7.4.7 付録 表 33,34 に示す. 日本での第Ⅱ相,第Ⅲ相試験において,同様の部分集団解析を行ったが,投与前値に差が見られ る以外には,肝硬変群での投与後の変動を示唆する結果は認められなかった(報告書 BA13, A05868). Child-Pugh 分類スコア別 肝硬変について,Child-Pugh 分類スコアに従い,不明,A,B,C に分類し部分集団解析を行った. 同一の測定施設で測定された欧米での第Ⅲ相試験における 25μmol/kg 投与(報告書 A04410 を除 く),及び報告書 A04410(特別な集団での試験)における肝機能に関連した測定値の記述統計量を, 部分集団ごとに 2.7.4.7 付録 表 35,36 に示す.これらの部分集団解析において,スコア C の患 者は 17 例認められた(報告書 A04410 での 6 例を含む). 日本での第Ⅱ相,第Ⅲ相試験において,同様の部分集団解析を行ったが,投与前値に差が見られ る以外には,スコア A,B 群で異なる変動を示唆する結果は認められなかった(報告書 BA13, A05868).スコア C の患者は報告書 BA13 で 3 例,報告書 A05868 において 1 例であった.
抗痙攣薬併用の有無別 抗痙攣薬を併用していた患者は,第Ⅱ相及び第Ⅲ相試験の 1755 例中に 15 例認められた.併用の 有無別の部分集団解析を行ったが,併用群に一定の変動傾向は認められなかった. 抗不整脈薬併用の有無別 抗不整脈薬を併用していた患者は,第Ⅱ相及び第Ⅲ相試験の 1755 例中に 265 例認められた.同一 の測定施設で測定された欧米での第Ⅱ相及び第Ⅲ相試験における 25μmol/kg 投与(報告書 A04410 を除く)後の,全検査項目の測定値の記述統計量を,2.7.4.7 付録 表 37 に示す. 病変のタイプ別(SOR 又は最終診断名により,悪性肝病変,良性肝病変,及び両方を有する患者 に分類) SOR(Standard of reference)における診断名又は最終診断名より,悪性肝病変のみ,良性肝病 変のみ,悪性,良性肝病変の両方を有する,不明,病変なしに分類して部分集団解析を実施した. 同一の測定施設で測定された欧米での第Ⅱ相及び第Ⅲ相試験における 25μmol/kg 投与(報告書 A04410 を除く)後の,全血液臨床検査項目の測定値の記述統計量を部分集団ごとに 2.7.4.7 付録 表 38 に示す.
2.7.4.4 バイタルサイン,理学所見及び安全性に関連する他の観察項目 2.7.4.4.1 バイタルサイン 健康被験者(第Ⅰ相臨床試験) 収縮期血圧,拡張期血圧,心拍数を含むバイタルサインは,第Ⅰ相臨床試験 4 試験のうち 3 試験 において評価した(第Ⅰ相臨床試験の報告書 A337 においては,投与前及び投与 24 時間後のみ測定さ れたため,ここには含めない).1 時点の測定が欠測であった 3 例を除く全被験者が,すべての時 点でバイタルサインの測定を受けた. 血圧及び心拍数において個々の症例での変動は認められたが,治験責任医師が臨床上有意である 又は SH L569B と関連があると考えた変化はなかった. 血行動態学的項目は,SH L569B の投与量を増量してもその影響を受けなかった.血行動態学的反 応をおこし得るすべての可能性を考慮しても,認められた変化は,検査環境によるストレスに起因 するものと考えられた. 患者(第Ⅱ,Ⅲ相臨床試験) 第Ⅱ相及び第Ⅲ相の全臨床試験の多く,又はいずれかの試験で,以下の時点においてバイタルサ イン(収縮期血圧,拡張期血圧,心拍数)が測定された: ベースライン(投与前 24 時間以内),SH L569B 投与直前,投与 5 分後,10 分後,15 分後,20 分 後,30 分後,45 分後,60 分後,80-90 分後,120 分後,MRI 検査終了直後*,フォローアップ時 (2-4 時間後,6 時間後,8 時間後,20-28 時間後,44-52 時間後,68-76 時間後,92-100 時間後, 116-124 時間後,140-148 時間後). *A04410 を除く第Ⅲ相試験においては,投与 20 分後からの撮像が終了した後の測定.第Ⅱ相試験では, 投与 45 分後(ヨーロッパでの第Ⅱ相試験;報告書 AH34, AI94)からの,又は投与 60~120 分後(日本で の第Ⅱ相試験;報告書 BA13)の間の撮像が終了後の測定. 呼吸数が測定された時点:SH L569B 投与直前,投与直後,5 分後,フォローアップ時(2-4 時間 後,20-28 時間後,68-76 時間後).(報告書 A05742,A01908 のみで測定された) 体温が測定された時点:SH L569B 投与直前,投与直後,フォローアップ時(2-4 時間後,20-28 時間後,68-76 時間後,140-148 時間後)に測定された.(報告書 A05742,A01908 のみで測定され た) 収縮期血圧,拡張期血圧,心拍数,呼吸数,体温については,ベースラインから投与直前までの 平均値に変化はほとんどなかったので,特定の変化を算出するための参照値として,投与直前値を 用いた. 表 2.7.4.4-1 に,収縮期血圧,拡張期血圧,及び心拍数が少なくとも 500 例以上で測定された投 与後の特定の時点での,全用量での患者数を示す.呼吸数及び体温が測定された患者数もあわせて 示した.
表 2.7.4.4-1 第Ⅱ相,第Ⅲ相試験においてバイタルサインが測定された患者数 Time points with at least 500 patients available for measurement
of vital signs Parameter Immediately Pre injection 5 minutes Post injection Immediately after MRI examination 2-4 hours Post injection 20-28 hours Post injection 68-76 hours Post injection a Systolic blood 1614 1234 1254 1243 1540 684 Diastolic blood 1614 1234 1254 1243 1540 684 Heart rate 1614 1235 1250 1240 1537 682 Respiratory rate b 461 458 461 459 455 450 Body temperature c 507 --- 457 460 507 447
a Evaluations for this time point were not done in 4 phase III studies(A05742, A01908, A04410, A05868).
b and c performed in Report A05742 and A01908.
Reference Documentation: ISS Tables VS 1.1.2 overall, VS 1.2.2 overall, VS 1.3.2 overall, VS 1.4.2 overall, VS 1.5 overall. 最も投与された患者数の多い投与量である 25μmol/kg とすべての用量について,それぞれの患者 群での収縮期血圧,拡張期血圧,心拍数,呼吸数,体温の平均値並びに変化量の平均値を表 2.7.4.4-2 に示す.MRI 検査直後でも,投与 2-4 時間後の時点でも,SH L569B 投与後に有意な変化 は生じなかった.それ以外の時点で測定されたバイタルサイン,呼吸数,体温の平均値と平均変化 量にも同じように SH L569B 投与に関連した有意な変化は生じなかった.全用量群の記述統計量を 2.7.4.7 付録 表 39~43 に示す.
表 2.7.4.4-2 MRI 検査終了直後及び投与 2-4 時間後の収縮期/拡張期血圧,心拍数,呼吸数,及び体温の平均値 Parameter Treatment (25µmol/kg)/ and Overall* N Pre injection Mean±SD Immediately after MRI Mean±SD Difference Mean±SD N Pre injection Mean±SD 2-4 hours post injection Mean±SD Difference Mean±SD Systolic BP (mm Hg) 25 µmol/kg 1151 133.3±19.52 133.6±19.34 0.4±13.00 961 134.3±19.30 132.0±17.07 -2.3±13.13 Overall 1254 133.4±19.25 133.8±19.30 0.4±12.85 1243 134.2±19.04 132.5±17.27 -1.7±12.51 Diastolic BP (mm Hg) 25 µmol/kg 1151 77.8±11.12 77.3±10.90 -0.7±8.64 961 77.8±11.31 76.5±10.17 -1.3±8.88 Overall 1254 77.9±11.09 77.6±10.89 -0.6±8.52 1243 77.5±11.52 76.6±10.31 -0.9±8.88 Heart Rate (bpm) 25 µmol/kg 1147 73.6±12.74 72.4±12.12 -1.2±8.11 958 73.9±12.16 74.9±11.70 1.0±9.23 Overall 1250 74.0±12.62 72.9±12.06 -1.1±8.00 1240 74.8±12.06 75.6±11.52 0.8±8.85 Respiration rate (breaths/min) 25 µmol/kg 461 16.4±3.35 16.6±3.41 0.2±2.50 459 16.4±3.34 16.6±3.20 0.3±2.63 Overall 461 16.4±3.35 16.6±3.41 0.2±2.50 459 16.4±3.34 16.6±3.20 0.3±2.63 Body temperature (oC) 25 µmol/kg 457 36.62±0.425 36.58±0.469 -0.04±0.370 460 36.62±0.423 36.63±0.466 0.01±0.375 Overall 457 36.62±0.425 36.58±0.469 -0.04±0.370 460 36.62±0.423 36.63±0.466 0.01±0.375
*Overall includes all dose groups of Gd-EOB-DTPA from 3 to 500μmol/kg BW
BP = blood pressure; bpm = beats per minute; N = number of patients; SD = standard deviation. Reference Documentation: ISS Tables VS 1.1.2, VS 1.2.2, VS 1.3.2, VS 1.4.2, VS 1.5
バイタルサイン(血圧,心拍数)において「特定した変動」が認められた患者数の概要を,25μ mol/kg 投与群について測定された全時点毎に 2.7.4.7 付録 表 44~46 に示す.第Ⅱ相及び第Ⅲ相 臨床試験のほとんどの患者において,バイタルサインは安定しており,SH L569B 投与後,収縮期血 圧については±20mmHg,拡張期血圧については±15mmHg,心拍数については±15bpm の特定した範 囲内の変動にとどまった.さらに「特定した変動」についての,投与直前に対し各測定時点で見られ た収縮期血圧,拡張期血圧,心拍数の個々の変化は,最も多くの患者が組み入れられた 25μmol/kg 投与群で最も明らかであり,以下に記載する. 収縮期血圧については,投与直前から MRI 検査直後に測定値が>20~40mmHg 上昇した患者は, 1151 例中 42 例(3.6%)であったが,投与 2-4 時間後には 961 例中 26 例(2.7%)に減少した.> 20~40mmHg 低下した患者数は,MRI 直後に 1151 例中 54 例(4.7%)であり,投与 2-4 時間後には 961 例中 60 例(6.2%)に増加した.投与 20-28 時間後の時点で,1259 例中 120 例(9.5%)で>20 ~40mmHg までの低下が観察された.投与 2-4 時間後に 40mmHg を超える低下が生じたのは 961 例中 12 例(1.2%),40mmHg を超える上昇が生じたのは 961 例中 3 例(0.3%)であった. 拡張期血圧については,投与直前から MRI 検査直後に測定値が>15~30mmHg 上昇した患者は, 1151 例中 35 例(3.0%)であったが,投与 2-4 時間後の時点でもほぼ同じ数であった(961 例中 30 例,3.1%).測定値が>15~30mmHg 低下した患者数は,MRI 直後には 1151 例中 43 例(3.7%)で あったが,投与 2-4 時間後では 961 例中 40 例(4.2%)に増加した.投与 20-28 時間後には,1259 例中 73 例(5.8%)で>15~30mmHg までの低下が観察された.投与 2-4 時間後に 30mmHg を超える 低下が生じたのは 961 例中 8 例(0.8%),30mmHg を超える上昇は 961 例中 1 例(0.1%)で認めら れた. 心拍数については,測定値が>15~30bpm 増加した患者は,MRI 検査直後には 1147 例中 20 例 (1.7%)であった.この数は,投与 2-4 時間後には 958 例中 53 例(5.5%)に増加した.投与 20-28 時間後には 1256 例中 76 例(6.1%)で>15~30bpm までの増加が観察された.>15~30bpm まで の減少が観察された患者数は,MRI 直後では 1147 例中 52 例(4.5%),投与 2-4 時間後には 958 例 中 32 例(3.3%)であった.投与 20-28 時間後には,1256 例中 68 例(5.4%)で>15~30bpm まで の減少が観察された. 25μmol/kg 以外のすべての投与量群の患者においても,わずかな変動が上記と同程度か,それ未 満見られた. 20mmHg を超える収縮期血圧の変化と 15mmHg を超える拡張期血圧の変化は,各試験報告書に個々 の変化の詳細が記載されている(日本での第Ⅱ相試験を除く).概して,各変化は,患者の既往歴, 体位の変化(立ち上がる/横になる),検査による神経の高ぶりにより説明された.欧米での第Ⅱ 相及び第Ⅲ相試験では,これらの患者の中で,治験責任医師が「臨床上問題となる」,又は/かつ「治 験薬と関連あり」と判断した変化は認められなかった.国内での第Ⅲ相試験において,規定した時間 のバイタルサイン測定で異常変動が認められた症例が,1 例見られた.患者番号 でベースラ インの血圧 146/80 に対し,投与前 50 分の血圧が 196/95 に上昇していた.本例の血圧上昇は,有害 事象として報告された.また,国内での第Ⅱ相試験のバイタルサイン測定では,MRI 検査後に 12.5, 25, 50μmol/kg 投与投与群においてそれぞれ 1,2,1 例の血圧変動が認められた.血圧上昇 3 件, 低下 1 件であった.このうち収縮期血圧が 30mmHg 以上増加した 50μmol/kg 投与群の 1 例は有害事 象として報告されている(投与前 122/84,検査後 160/84mmHg).他の変動は,臨床的に有意なもの ではなかった. 呼吸数と体温については,観察された変化は小さく,臨床上有意なものはなかった.
バイタルサインの部分集団解析 性別,人種別,年齢別,国別,体重別,病変のタイプ別にバイタルサイン,呼吸数,体温の記述 統計量を算出したが,全評価対象集団と同様,これらの部分集団においても,本剤に関連した有意 な又は特異的な問題は見られなかった. 2.7.4.4.2 理学所見 健康被験者(第Ⅰ相臨床試験) 第Ⅰ相臨床試験全 4 試験において,理学所見に臨床上有意な変化は観察されなかった. 患者(第Ⅱ,Ⅲ相臨床試験) 日本の第Ⅱ相臨床試験,報告書 BA13 の 186 例を除く,すべての第Ⅱ相及び第Ⅲ相臨床試験にお いて,個々の患者に対して種々の臓器を含む全身の理学検査が実施された.したがって,SH L569B 投与を受けた 1558 例で理学所見がとられた.
今回提出する申請資料に含まれる欧米での第Ⅱ相臨床試験(報告書 AH34, AI94, AI83)及び第Ⅲ 相臨床試験(報告書 A00518, A03779, A05742, A01908, A04410,試験番号 305654)においては, ベースライン検査に合わせ,フォローアップのための理学所見が MRI 検査 24 時間後にとられた.欧 米で実施された二つの第Ⅲ相試験(報告書 A05742, A01908)では,これに加えて 2-4 時間後及び 72 時間後にも理学所見がとられた.日本での第Ⅲ相試験では,投与後 72 時間に理学所見をとった. 報告書 A04410 では,7 日後にも理学所見がとられたが,ISS では投与後 72 時間までの理学所見デー タのみを対象とした. 表 2.7.4.4-3 は,治験責任医師が評価した理学所見において,正常,異常,及び臨床上有意な所 見が認められた患者数を示す.1588 例中総計 24 例(1.5%)が,治験責任医師により SH L569B 投 与後の理学所見に臨床上有意な変化があったと評価された.そのうち 22 例は 25μmol/kg,1 例は 50μmol/kg,1 例は 350μmol/kg の SH L569B 投与であった.
*新薬承認情報提供時に置き換えた。 表 2.7.4.4-3 治験責任医師により臨床的に有意と判断された理学所見が見られた患者数 Physical examination Normal Abnormal (no cl.s. change) Abnormal (cl.s. change) Overall Treatment (SH L569B) (μmol/kg BW) N % N % N % N % 3 4 11.4 31 88.6 0 35 100.0 6 6 18.2 27 81.8 0 33 100.0 12.5 29 26.6 80 73.4 0 109 100.0 25 632 49.5 622 48.7 22 1.7 1276 100.0 50 9 11.5 68 87.2 1 1.3 78 100.0 200 0 5 100.0 0 5 100.0 350 1 8.3 10 83.3 1 8.3 12 100.0 500 2 20.0 8 80.0 0 10 100.0 Overall 683 43.8 851 54.6 24 1.5 1558 100.0 Placebo 8 22.9 27 77.1 0 35 100.0 Gd-BOPTA 95 63.8 54 36.2 0 149 100.0 cl.s. = clinically significant
Note: The ISS database includes only the time points 2 to 4 hours p.i., 24 hrs p.i. and 72 hrs p.i. Reference Documentation: ISS Table PE.1
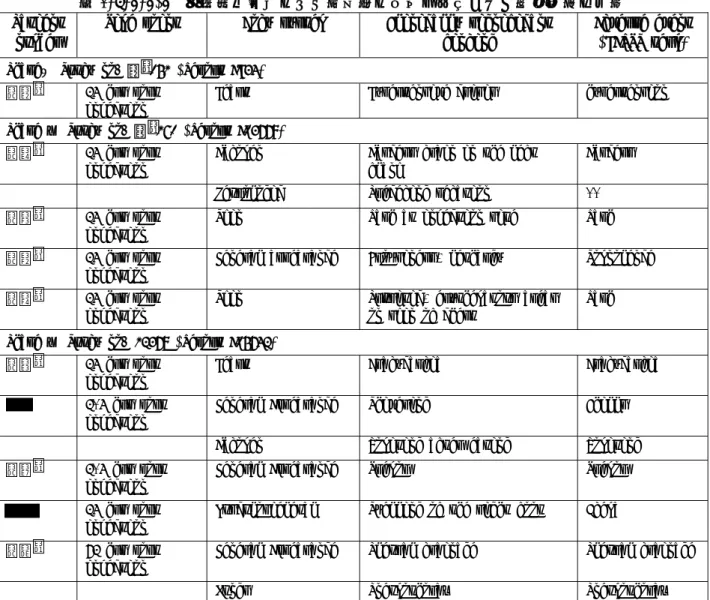
理学所見における臨床的に有意な変化は,SH L569B 投与 2-4 時間後,24 時間後,及び 72 時間後 に認められた.表 2.7.4.4-4 にその一覧を示す.これらの所見のほとんどは有害事象として記録さ れた. 報告書 A04410 では,第 3 群(重度の肝障害)の患者(番号AN*)において,腹部にのみ臨床上 有意な変化が見られた.この患者の腹水はベースライン時には相当な量であったが,退院時(投与 7 日後)には極めて少なくなっていた.
*新薬承認情報提供時に置き換えた。
表 2.7.4.4-4 治験責任医師により臨床的に有意と判断された理学所見の一覧 Patient
number
Time point Body system Clinically significant finding
Adverse event (HARTS term)
PhaseⅡ Study no. G*051 (Report AH34)
AG* 24 hrs post
injection
Heart Hypertensive crisis hypertension
Phase Ⅲ Study no. J*160 (Report A03779)
AI* 24 hrs post
injection
Abdomen Abscess drain in the left
flank
Abscess
Neurologic Preceding sedation --
AX* 24 hrs post
injection
Skin Rash at injection site Rash
AW* 24 hrs post
injection
General appearance Drowsiness, lethargy Somnolence
AY* 24 hrs post
injection
Skin Pruritic, erythematous areas
on skin of chest
Rash
Phase Ⅲ Study no. 12378 (Report A05742)
AH* 24 hrs post
injection
Heart Bradycardia Bradycardia
2-4 hrs post injection
General Appearance Shivering Chills
Abdomen Vomiting after eating Vomiting
AL* 2-4 hrs post
injection
General Appearance Tremor Tremor
24 hrs post injection
Musculoskeletal Swelling of the right foot Edema
AC* 72 hrs post
injection
General Appearance Pleural drainage Pleural drainage
Lungs Pneumothorax Pneumothorax
Note: The ISS database includes only the time points 2 to 4 hours p.i., 24 hrs p.i. and 72 hrs p.i. Reference Documentation: see Report nos. in the table above
*新薬承認情報提供時に置き換えた。
表 2.7.4.4-4 治験責任医師により臨床的に有意と判断された理学所見の一覧(続き) Patient
number
Time point Body system Clinically significant finding
Adverse event (HARTS term)
Phase Ⅲ Study no. 014763 (Report A01908) 24 hr post
injection
Musculoskeletal Back pain, experienced in
past by patients, not related to exam or study drug
Back pain
24 hr post injection
Musculoskeletal Mild right wrist soreness Pain
72 hr post injection
Lymph nodes Submandibular lymph nodes
(2), <1 cm, mobile
Lymphadenopathy 24 hr post
injection
Musculoskeletal Painful area, right
paravertebral medial to scapula, 2-3 cm, soft movable density
Back pain
72 hr post injection
Skin/dermatological Itchy back from day before
(scratch marks)
Pruritus 2 to 4 hr post
injection
Abdomen Mild diffuse abdominal
tenderness when palpated
Abdominal pain 2 to 4 hr post
injection
Skin/dermatological Rash, left thigh Maculopapular
rash
AM* 72 hr post
injection
Mini mental status check
Very drowsy - recent morphine sulfate Somnolence AE* 24 hr post injection, 72 hr post injection
Skin/dermatological Spontaneous serosanguineous
drainage from left buttock wound
Abscess
24 hr post injection
Lungs Right basilar crackles Lung disorder
Phase Ⅲ Study no. 014468 (Report A04410)
AN* 7 days post
injection
Abdomen Ascites ---
Phase Ⅲ Study no. 30082 (Report A05868, Japan) 72 hr post
injection
Skin Eruption Rash
72 hr post injection
Eyes, ears, nose, throat
Stomatitis Stomatitis
Phase Ⅲ Study no. 305654 24 hr post injection
Heart Few extra systolic beats extrasystoles
Note: The ISS database includes only the time points 2 to 4 hours p.i., 24 hrs p.i. and 72 hrs p.i. Reference Documentation: see Report nos. in the table above
*新薬承認情報提供時に置き換えた。 第Ⅲ相臨床試験 2 試験(報告書 A05742,A01908)では,本剤投与 5 分後及び MRI 検査終了直後に, 健康状態につき患者に質問する形で簡単な身体チェックが実施され,後期第Ⅲ相臨床試験(試験番 号 305654)でも MRI 検査終了直後に同様の身体チェックが実施された.ベースラインから投与 5 分 後,MRI 検査直後の患者の状態に関する臨床上有意な変化は,個々の試験報告書に記載されている が,ISS には含まれてない. 質問による簡単な身体チェックの結果,本剤投与 5 分後及び MRI 検査終了後に 6 例で 7 件の臨床 上有意なベースラインからの変化が認められ,これらは有害事象としても記録された(表 2.7.4.4– 5). 表 2.7.4.4–5 簡易身体チェックにおいて治験責任医師により臨床的に有意と判断された所見 Patient number
Time point Body system Clinically significant finding
Adverse event (HARTS
term)
Phase Ⅲ Study no. 012387 (Report A05742)
AL* After completion of
MRI
General appearance Tremor Tremor
Phase Ⅲ Study no. 014763 (Report A01908) After completion of
MRI
General appearance Patient complained of back
pain after getting up from MRI table
Back pain
5 minutes post injection
General appearance Warm feeling that lasted
approximately 30 seconds
Vasodilatatio n
AB* 5 minutes post
injection
General appearance Nausea slightly after
injection that went away just before 5 minute check
Nausea
After completion of MRI
General appearance Back is sore from lying down
(patient’s words)
Back pain
AM* 5 minutes post
injection and after completion of MRI
General appearance Patient became anxious and
asked to stop scan
Anxiety
Phase Ⅲ Study no. 305654
(Gd-BOPTA 投与)
After completion of MRI
General appearance Subcutaneous swelling at the
left parietal cranium
Accidental injury (Gd-BOPTA 投与) After completion of MRI
General appearance Slight pain in the liver
region; slight pain in the back, at the soft tissue tumor; nasal mucosa swollen, and therefore, nasal
respiration hindered Abdominal pain, back pain, rhinitis AK* After completion of MRI
General appearance Claustrophobic attack with
fit of perspiration, tachycardia
Anxiety