博士論文
題目『納豆発酵過程の分子生理学的研究』
株式会社ミツカングループ本社 中央研究所
加田茂樹
目次 頁 序論---4 第1章 納豆の曳糸性に重要な役割を果たす菌体外プロテアーゼの同定 1-1 背景と目的---10 1-2 材料と方法---13 1-3 結果---21 (1)納豆菌における菌体外プロテアーゼ遺伝子セット (2)各遺伝子破壊株の菌体外プロテアーゼ活性とg-PGA 生産性 (3)プロテアーゼ遺伝子 6 重破壊株及びaprE 破壊株の遊離アミノ酸濃度
(4)Glu の添加によるaprE 破壊株のg-PGA 生産性に対する効果
(5)菌体外ペプチダーゼに関する解析 1-4 考察---26 (1)菌体外プロテアーゼ遺伝子セットとプロテアーゼ活性 (2)g-PGA 生産に材料を供給するプロテアーゼ AprE (3)CSF 成熟化因子としての AprE のg-PGA 生産への関与の可能性 (4) AprE 以外のプロテアーゼの納豆発酵における役割 第2章 納豆菌の 2 種のグルタミン酸ラセマーゼに関する生理学的解析 2-1 背景と目的---31 2-2 材料と方法---34 2-3 結果---41 (1)r22 株における 2 種のグルタミン酸ラセマーゼ遺伝子の配列 (2)破壊株の取得 (3)各破壊株を用いた表現型解析 (4)各遺伝子の転写解析
(5)グルタミン酸ラセマーゼの分子系統樹解析 2-4 考察---46 (1)グルタミン酸ラセマーゼ Glr の必須性 (2)g-PGA 生産にも関わるグルタミン酸ラセマーゼ Glr (3)推定される YrpC の起源と生理学的役割 第3章 納豆発酵におけるアンモニア生成の主要経路の同定 3-1 背景と目的---51 3-2 材料と方法---55 3-3 結果---60 (1)アルカリプロテアーゼ遺伝子aprE 破壊株のアンモニア生成 (2)納豆菌のアミノ酸資化性と二次発酵中のアミノ酸の消長 (3)グルタミン酸脱水素酵素遺伝子破壊がアンモニア生成に与える影響 (4)二次発酵における尿素の生成と分解 (5)グルタミン酸脱水素酵素とウレアーゼの組み合わせによる効果 3-4 考察---65 (1)二次発酵におけるグルタミン酸脱水素酵素の生理学的役割 (2)二次発酵におけるウレアーゼの生理学的役割 (3)アンモニア臭を低減した納豆への応用 参考文献---69 謝辞---90 表---91 図---101 要旨---116
序論 納豆は、特有の香りと粘質物による糸引きを大きな特徴とする、日本の伝統 的な発酵食品の一つであり、一般的に納豆菌と呼ばれる Bacillus subtilis に 分類される一群の有胞子性グラム陽性細菌の胞子を、蒸煮した大豆に散布する ことで製造される。その製造は古くは鎌倉時代に遡ると言われており、古来よ り日本人になじみのある食品となっている。平安時代の京の庶民の生活を描い た『新猿楽記』(11 世紀ごろ成立)には「うす塩辛納豆」が、室町時代の『精進 魚類物語』(15 世紀ごろ成立)中には登場人物として「納豆太郎糸重」の記載が ある(藤井 1993)。従って平安期から室町期にかけて、納豆は日本人に一般的な 食品となっていたことが推測される。元来納豆は、藁苞と呼ばれる藁に蒸煮し た大豆を納め、藁に付着している天然の納豆菌により発酵させて仕上げるもの である。しかし、藁に付着している納豆菌は一定していないため糸を引かない など、発酵にばらつきが出るといった製造上の理由から、安定した製造を達成 するために、工業的には培養した納豆菌胞子を蒸煮した大豆に散布し、ポリス チレン製の容器に入れて製造されることが多い。このような工業ラインの発達 により、納豆の年間生産量は国内で約 22 万 t に達すると推定されており、近年 の健康志向の高まりとあいまって日本人の食卓に欠くことのできない食品とな っている。 納豆の発酵の品質は一般的に官能検査によって、すなわち、「色」、「香り」、「糸 引き」、「柔らかさ」、「呈味」によって決定される。納豆試験法(納豆試験法研究 会著 平成 2 年)には納豆の官能検査法が記載されており、①納豆菌の被り、② 溶菌状態、③割れ、つぶれ、皮むけ、④豆の色、⑤香り、⑥硬さ、⑦味、⑧糸 引きの各項目が評価の基準となっている。納豆菌の被りでは、ムラなく一定の
厚さの菌が大豆の表面を覆っているものが良く、斑になっていたり菌に覆われ ていない素豆のものがある場合は悪いと判定される。溶菌状態は、菌が溶けて べたべたしたものが悪いと判定される。割れ、つぶれ、皮むけはないものが良 く、また豆の色は薄茶色をしているものほどよく、こげ茶や黒っぽいものほど 悪い。香りについては、アンモニア臭やこげ臭、酸臭などの異臭がしないもの が良いと判定される。硬さは近年特に柔らかいものが好まれる傾向にある。味 については、アミノ酸などの旨味、苦味、甘味などから適切な味をしているも のが良好と判定される。また、糸引きについては、粘りが強く糸引きが良いも のが良く、粘りが少なく糸引きが弱いものは悪いと判定される。これらのうち、 特にアンモニア臭と糸引きは納豆の品質を決定づける重要な要素となっており、 品質の高い納豆は糸引きが強く、また官能的にアンモニア臭がしない。納豆は 通常アンモニア臭がしないが、発酵が進み過ぎたり、保存状態が悪いと再度発 酵してアンモニアを生成させてしまう。これにより納豆の品質は著しく劣化す る。また、何らかの原因で納豆発酵がうまく進行しない場合には、納豆の糸引 きが不良となることで最も顕著に現われる。加えて、納豆産業においてはしば しば納豆菌ファージによる汚染を受けることがあり、この場合には糸引きが失 われて製品として成立できなくなってしまう。 これまで良質な納豆を製造すべく、発酵条件、すなわち、温度、湿度、発酵 むろの形状、換気システム、容器の素材や形状など様々な改良が行われてきた。 納豆製造に使用される大豆についても、国産及び外国産の大豆で、大粒、中粒、 小粒、極小粒、ひきわりなどが開発されてきており、様々な製品が店頭に並ぶ ようになってきている。一方納豆菌については、ほとんどの場合、希釈するこ とにでそのまま製造に使用できる状態となった納豆菌胞子液(種菌と呼ぶ)がそ のまま使用されており、品質や風味の改善対象とされてこなかった。これは酒
の製造に用いられる麹菌や酒精酵母、グルタミン酸ソーダの製造に用いられる コリネ菌について盛んに育種・選抜・改良が行われているのと比較して対照的 である。
納豆菌は、明治 38 年に帝国大学農科大学(現在の東京大学農学部)の沢村真博 士によって納豆から発見、Bacillus subtilis Sawamura と命名されて以来、い
くつかの系統が分離されて現在に伝承されている。納豆菌の種菌としては、「宮 城野菌」、「高橋菌」、「成瀬菌」の 3 種が主要なものであるが、これらは共通の 株に由来すると伝えられている。納豆菌のゲノム上には、可動性の因子である 挿入配列(IS)が存在していることが報告されている(Nagai et al.2000)が、そ のうちの1つ IS4Bsu1 をプローブにしてサザン解析を行うと、3 つの種菌は全て IS4Bsu1 を保持していることが確認されており、特に宮城野菌と成瀬菌ではいず れも 6 コピーの IS4Bsu1 を保持しており、そのパターンも完全に一致している ことがわかっている。高橋菌では 11 コピーの IS4Bsu1 を保持しているが、その うちの 6 コピーは宮城野菌と成瀬菌と共通である(伊藤&稲津 2000)。これらの 解析結果も、これら 3 種の種菌が共通の株から派生したことを支持している。 従って、これらのいずれを使用したとしても納豆の品質に大きな差異はなく、 種菌の選択が品質の改善に与える効果は限定的と考えられる。 納豆菌が納豆品質の改善の対象とされてこなかった原因の 1 つは、遺伝学的操 作の困難さにあると考えられる。納豆菌と同一種とされるB. subtilis Marburg 168 株は、高い形質転換能を有しており、その取り扱いの容易さから大腸菌と共 に最も分子遺伝学的な解析が進んだ細菌となっている。胞子形成、蛋白質分泌 などの独自の生物学的プロセスの解明に加えて、1996 年のゲノム解読後、シグ ナル伝達、転写制御、蛋白質間相互作用などの網羅的な解析が急速に進展した。 ただし、Marburg 168 株では納豆は製造できない。一方、納豆菌ではその形質転
換能の低さのために、168 株で開発されてきた解析手法を直接適用できないまま となっており、分子生物学的解析が遅れてきた。しかし、立教大学の Ashikaga らによって納豆菌の低形質転換能の原因が解明された(Ashikaga et al. 2000) ことを契機に、納豆菌の逆遺伝学的解析が進展し始めた。特に B. subtilis Marburg 168 株が生産しないため解析されてこなかった、納豆の糸の主成分であ るポリ-g-グルタミン酸(g-PGA)の生産(Ashiuchi et al. 1999; 2001; 2004; Urushibata et al. 2002A; Stanley&Lazazzera 2005; Ohsawa et al. 2009; Kimura et al. 2009)、 バイ オフィ ルム形 成(Branda et al. 2001;Connelly et al. 2004;Chu et al. 2008;Nagórska et al.2010) な ど の 詳 細 が 納 豆 菌 を 含 む undomesticated strain と称される株を用いて解析されている。
納豆の糸の主成分であるg-PGA は、納豆菌を含むバチルス属細菌のほか、古細
菌(Hazayen et al. 2001)やヒドラ(Szczepanek at al. 2001)などによって生産
されるポリアミノ酸であり、L または D 体の Glu のa位のアミノ基とg位のカルボ
キシル基がアミド結合により重合した構造となっている。 納豆菌の生産する g-PGA にはD-Glu とL-Glu が含まれる(Kubota et al. 1993; Kunioka 1994; Tanaka
et al. 1997)が、炭そ病の原因菌である B.anthracis のg-PGA には D-Glu のみ
(Makino et al. 1989)が、好アルカリ性菌である B.halodurans のg-PGA にはL-Glu
のみ(Aono 1987)が含まれる。その他、B.licheniformis(Pérez-Camero et al. 1999) 、 B.megaterium(Torii 1956) 、 B.amyloliquefaciens(Meerak et al. 2007;2008)等もg-PGA を生産することが知られている。g-PGA の合成酵素は古く
は内在性のプラスミドにコードされる(Hara et al. 1982;1992)とされていたが、
内在性プラスミドをアクリジンオレンジでキュアリングした納豆菌が単離され、
その株がg-PGA 生産性を持っていた(Nagai at al. 1997)ことから、合成遺伝子
子の単離及び反応メカニズムの解明が精力的に進められてきてはいるものの、 未だ異論の多い分野となっている。細胞膜に存在する PgsB (YwsC または CapB とも呼ばれる)が、触媒反応を担う活性中心を持つ蛋白質であり、合成に必須で あることはいずれの研究者も合意している(Ashiuchi et al.2001; Urushibata et al.2002a; Kimura et al. 2009)。PgsB 蛋白質はアミドリガーゼスーパーフ ァミリーに属する蛋白質と相同性があり、このファミリーに属する蛋白質はア ミノ酸をアミド結合によりリボソーム非依存的に重合し、ポリアミノ酸を生成 する(Eveland et al. 1997; Berg et al.2000)。pgsB は、下流に存在する pgsC(ywtA
またはcapC とも呼ばれる)、pgsA(pgsAA、ywtB、capA とも呼ばれる)、ywtC(pgsE
とも呼ばれる)と共転写されることが報告されており(Urushibata et al. 2002b)、
これらの遺伝子産物が PgsB 蛋白質と複合体を形成し、g-PGA 生産にも必須とす
る研究者もいる。高知大の Ashiuchi らは、韓国の大豆発酵食品である戦国醤か
ら納豆菌に近縁なB.subtilis chunkookjang を単離し、そこから PgsBCA 蛋白質
を精製して、この複合体にg-PGA 合成活性があることを報告している(Ashiuchi et al.2004)。静岡大の Urushibata らは、納豆菌 IFO16449 株を用いて ywsC を
破壊しそこにywsC を発現させたところ、同じ ORF から大きさの異なる 2 種の蛋
白質(44 kDa 及び 33kDa)が生成し、これら 2 種の蛋白質がg-PGA 合成に必須であ
ることを報告している(Urushibata et al. 2002a)。また、g-PGA 合成酵素は基
質としてL-Glu もD-Glu の両方を利用するが、D-Glu を好むとする報告(Ashiuchi
et al.2004)も、L-Glu を優先的に利用するという報告もある(Urushibata et
al.2002; Kimura et al. 2009)。これらの研究の流れとは別に、undomesticated strain と呼ばれるg-PGA 生産性 B.subtilis を用いてバイオフィルムの研究も進
展している。ここでも、g-PGA がバイオフィルムの主成分であるという報告
(Branda et al. 2006;Chaugneau&Saier 2004)もあり、結論は出されていない。 いずれにしても、これらの解析も液体培養や固体培地を用いてのケースが主で あり、納豆発酵に焦点を当てて解析された例はほとんどない。 納豆発酵の複雑な過程はこれまでの研究から、①蒸煮した大豆上における納豆 菌の発芽・生育、②大豆蛋白質を分解する菌体外プロテアーゼの分泌、③アミ ノ酸やオリゴペプチドの取り込みと代謝、④菌体内でのグルタミン酸のラセミ 化と合成、⑤高分子g-PGA の生産と分泌の各過程から構成されていると考えられ
る(Itaya & Matsui 1999)。本論文では、納豆菌に分子生物学的手法と生化学的 手法を適用し、納豆発酵の過程を分子生理学的に解析することで、納豆菌の育 種及び改良において鍵となる遺伝子を同定することを目的とした。第 1 章では、 納豆菌の分泌するプロテアーゼが納豆発酵に果たす役割について解析し、7種 の菌体外プロテアーゼのうち、アルカリセリンプロテアーゼ AprE が納豆発酵に おける“糸引き”すなわちg-PGA 生産に不可欠であることを明らかにした。第 2 章では、グルタミン酸のラセミ化に関わるグルタミン酸ラセマーゼホモログの 機能を分子生物学的に解析し、2 種のグルタミン酸ラセマーゼのうち、Glr が主 要なグルタミン酸ラセマーゼであり、g-PGA 生産にも細胞壁合成にもD-Glu を供 給することを見出した。第 3 章では、納豆発酵においてしばしば問題となるア ンモニアが、二次発酵過程において rocG 及び gudB にコードされるグルタミン 酸脱水素酵素と、ureABC にコードされるウレアーゼによって生じていることを 逆遺伝学的手法と生化学的手法を組み合わせることによって明らかにした。
第 1 章 納豆の曳糸性に重要な役割を果たす菌体外プロテアーゼの同定 1-1 背景と目的 納豆発酵に使用される納豆菌は、B. subtilis に分類されるグラム陽性細菌で あり、旺盛に菌体外酵素を分泌することが知られている。B. subtilis は、デン プン糖化用アミラーゼ、洗剤用プロテアーゼ、セルラーゼなどの生産菌として 工業的にも利用されている(表 1-1)。これまで、納豆菌に極めて近縁な B. subtilis Marburg168 株とその派生株、及び変異株を用いて、菌体外酵素に関す る研究が精力的に行われてきた。菌体外酵素のうち、最も詳細に解析されてき たものの 1 つがプロテアーゼであり、168 株のゲノム解析により、8 種の菌体外 プロテアーゼ遺伝子(aprE,bpr,epr,mpr,nprB,nprE,vpr,wprA)が存在すること が明らかとなった(Kunst et al. 1997)。これらのプロテアーゼは、セリンプロ テアーゼ(aprE,bpr,epr,vpr,wprA)または金属プロテアーゼ(mpr,nprB,nprE)を コードしており、wprA のみが細胞壁に結合するが、その他の 7 種は培地上に遊 離した形で存在すると考えられる。ゲノム解読後に行われた菌体外蛋白質のプ ロテオーム解析により、LB 液体培地中には aprE,bpr,mpr,nprE,vpr の各遺伝子
産物が(Märder et al. 2002)、液体最少培地中には wprA 遺伝子産物がそれぞれ 検出されている(Hirose et al. 2000)。さらに、液体最少培地の炭素源をグル コースからマルトースや可溶性のデンプンに変更した場合には、vpr 遺伝子産物 が検出されるようになることも報告されており(Hirose et al. 2000)、栄養源 によりプロテアーゼの分泌パターンが変化することが確認されている。また、 油田から単離されたg-PGA 生産性の B. subtilis B-1 株では、静置培養により浮 遊性バイオフィルム形成させた場合における主要な膜蛋白質として Vpr 前駆体
が検出されることが報告されており、振とう培養でバイオフィルムを形成させ なかった場合とは異なる挙動を示すことが報告されている(Morikawa et al. 2006)。このことは、栄養条件に加えて、培養条件もプロテアーゼの分泌に影響 を及ぼす可能性を示唆している。 これまで、菌体外プロテアーゼは細菌が外界の蛋白質を栄養源として獲得す る、「分解酵素」としての機能が着目され、その詳細な酵素反応メカニズムが解 析されてきた。しかし、近年、栄養源の獲得以外の広範囲な生物学的プロセス に関与することが報告されている。B. subtilis は Competence and sporulation factor(CSF)と呼ばれるペプチド性のシグナル分子を菌体外に放出し、菌体密度 を感知する機構を発達させている(Lazazzera et al.1997)。CSF は未熟なプロ体 として菌体外に放出され、AprE、Epr、Vpr によって成熟体に変換されて機能す ることが報告されている(Lanigen-Gerdes et al. 2007)。また、分泌性抗菌ペ プチドであるズブチリンも CSF と同様に前駆体として菌体外に放出されるが、 AprE、WprA、Vpr によって成熟体に変換されることが報告されている(Corvey et al. 2003)。これらの現象では、外界の環境に適応していくために自ら生産する 菌体外プロテアーゼが欠かせないことを示している。さらに、168 株では Epr(Dixit et al.2002)が、undomesticated strain である A164 株では NprB を 除く 7 種の菌体外プロテアーゼ全て(AprE,Bpr,Epr,Mpr,NprE,Vpr,WprA)が、そ れぞれ swarming molitily(軟寒天培地上における運動性)に重要な役割を果たす ことが報告されており、A164 株では 7 種の菌体外プロテアーゼ遺伝子を破壊す ると swarming molitily を喪失してしまう(Connelly et al. 2004)。A164 株の 7 種の菌体外プロテアーゼ遺伝子破壊株では swarming molitily に加えて、バイ オフィルムの形成が喪失する現象も観察されており(Connelly et al. 2004)、 菌体外プロテアーゼが様々な生物学的現象に深く関与していると考えられる。
しかし、これらの解析はいずれも納豆菌ではない B. subtilis を用いて解析 されており、また、液体培養や寒天培地上での解析結果であり、納豆発酵にお けるプロテアーゼの機能を反映したものではない可能性がある。既に述べたよ うに、プロテアーゼの分泌パターンは栄養条件や培養条件によって変化するが、 納豆発酵においていずれのプロテアーゼ種が重要な役割を果たしているのか解 析された例はほとんどない。本章では、納豆の「糸」の主成分であり、発酵の 品質を決定づけるg-PGA 生産性を指標に、納豆菌ゲノムに存在する 7 種の菌体外 プロテアーゼ遺伝子のうち、納豆発酵に重要な役割を果たす菌体外プロテアー ゼを同定することを目的とした。
1-2 材料と方法 使用菌株と培地 本章で使用した菌株を表 1-2 に示した。 納豆菌の各遺伝子破壊株はB. subtilis r22 株を親株として用いた。r22 株は、 市販納豆から分離した 0-2 株に N-メチル-N’-ニトロ-N-ニトロソグアニジンを 用いて変異原処理することによって得られた株であり、O-2 株と異なり遺伝子組 換えが可能な変異株である(竹村ら 2000)。r22 株は、納豆製造適性があり、官 能的に親株の O-2 株と同様の糸引きを示すことが確認されている。遺伝子破壊 用プラスミドの構築などの遺伝子操作には大腸菌 JM109 株を用いた。 納豆菌の通常の培養には Nutrient broth(NB)培地(1 % ポリペプトン(日本製 薬)、0.5 % NaCl (シグマアルドリッチ)、0.5 % 肉エキス (OXOID))を用いた。 胞子作製には、胞子形成培地(0.0068 % KH2PO4(関東化学)、0.0535 % NH4Cl(シグ マアルドリッチ)、0.0106 % Na2SO4(関東化学)、0.00965 % NH4NO3(和光純薬工業)、
0.000006 % FeCl2・7H2O(和光純薬工業)、0.00126 % MnCl2・4H2O(和光純薬工業)、
0.934 % MgSO4・7H2O(シグマアルドリッチ)、0.238 % グルタミン酸ナトリウム(シ
グマアルドリッチ)、0.152 % 酵母エキス(Becton, Dickinson and Company))を 用いた。納豆菌の形質転換には、Spizizen の最少培地(0.25 % グルコース(関東 化学)、5 mM MgSO4・7H2O(シグマアルドリッチ)、0.6 % KH2PO4(関東化学)、1.4 % K2HPO4(関東化学)、0.1 % クエン酸ナトリウム(シグマアルドリッチ)、0.1 mg/mL ビオチン))を用いた。g-PGA の生産性を検討する大豆粉培地には、納豆発酵に用 いる大豆をミルで挽き、得られた大豆粉を 10 %含む 2 %寒天培地を用いた。大 豆粉培地にグルタミン酸を添加する場合には、L-グルタミン酸ナトリウムを添加 した。
大腸菌 JM109 株の培養には、LB 培地を用い(1 % Bacto Tryptone(Becton, Dickinson and Company)、0.5 % Bacto yeast extract(Becton, Dickinson and
Company)、1 % NaCl(シグマアルドリッチ))、必要に応じて 50 mg/mL アンピシ リン硫酸塩(シグマアルドリッチ)となるように添加した。 菌体外プロテアーゼ遺伝子の検出 菌体外プロテアーゼ遺伝子は PCR を用いて検出した。PCR に用いたプライマー を表 1-3 に示した。これらのプライマーは、B. subtilis 168 株の配列を基に構 築した。PCR は Ex Taq(TaKaRa)を用いて行い、95 ℃ 3 分の変性後、95 ℃ 1 分、 55 ℃ 1 分、72 ℃ 4 分で 30 サイクル行った。得られた PCR 産物を 1 % TAE ア ガロースを用いて電気泳動することで検出した。 nprB 遺伝子座については、表 1-3 に記載した yitL-N 及び yitS-C プライマー を用いて同様の PCR を行い、得られた PCR 産物を pT7Blue に TA クローニングし、
Applied Biosystems 3130xl Genetic Analyzer を用いてシーケンスを行った。
遺伝子破壊株の作製 遺伝子破壊は、破壊したい遺伝子の上流及び下流を PCR で増幅し、薬剤耐性 遺伝子の両端に挟んだコンストラクションにより目的遺伝子を置換する方法か、 または破壊した遺伝子の内部に薬剤耐性遺伝子を挿入する方法のいずれかで行 った。遺伝子破壊用プラスミドの構築に使用したプライマーは表 1-3 に示した。 r22 株への形質転換は、Marburg168 株のコンピテンスセル作製方法と同様に r22 株のコンピテンスセルを作製し、目的遺伝子ごとに構築した破壊用プラスミド を鋳型に PCR で増幅した供与核酸を作製して混合することで行った。 アルカリプロテアーゼ遺伝子aprE の遺伝子破壊は、aprE コード領域をエリス
ロマイシン耐性遺伝子ermC で置換する方法で行った。表 1-3 に示したプライマ
ーaprE-F1 及び aprE-F2 を用いてaprE の開始メチオニンから上流 2 kb を増幅し、
その 5’端を SphI で、3’端を XbaI で消化して、pUC19 の SphI/XbaI サイトに
クローニングした。次に、aprE の終止コドンから下流 2 kb をプライマーaprE-B1
及び aprE-B2 を用いて増幅し、その 5’側をBamHI で、3’端を KpnI で消化して、
上述のaprE 上流 2 kb をクローニングした pUC19 の BamHI/KpnI サイトにクロー
ニングした。このように aprE の上流及び下流をクローニングしたベクターの
XbaI/BamHI サイトに ermC をさらにクローニングして aprE 破壊用ベクターを得 た。ermC 耐性遺伝子は pE194 上のプロモーター領域を含む ermC をプライマー
ermC-F 及び ermC-B を用いて増幅し、両端を XbaI/BamHI で消化して使用した。
破壊用ベクターを鋳型にして、プライマーaprE-F1 及び aprE-B2 を用いて、aprE
の上流 2 kb、 ermC 遺伝子 0.8 kb 及び aprE の下流 2 kb を含む約 4.8 kb を増 幅して供与核酸とし、コンピテンスセルとした r22 株に形質転換して、エリス ロマイシン耐性を持つ株を複数得た。これらのうち 8 株を選んでゲノム DNA を 抽出してサザン解析を行い、2 回交叉の結果、正しく破壊されている株 1 株を選 んで SKAPRE1 とした。 バシロペプチダーゼ F 遺伝子bpr の遺伝子破壊も、基本的には aprE の遺伝子 破壊と同様にbpr をエリスロマイシン耐性遺伝子 ermC で完全に置換することで 行った。bpr の上流 2 kb については表 1-3 に示した bpr-F1 及び bpr-F2 を、下 流 2 kb については bpr-B1 及び bpr-B2 を用いてそれぞれ増幅した。上流 2 kb
は SphI 及び XbaI で、下流 2 kb は BamHI 及び EcoRI でそれぞれ末端を消化し、
pUC のマルチクローニングサイトに順にクローニングした。ermC 遺伝子は aprE
破壊で用いた両端を XbaI/BamHI で消化したものを、bpr 上流及び下流をクロー
ーを鋳型にプライマーbpr-F1 及び bpr-B2 を用いて増幅し、bpr の上流 2 kb、 ermC 遺伝子 0.8 kb 及びbpr の下流 2 kb を含む約 4.8 kb を得た。この断片を供与核 酸として、コンピテントセルとした r22 株に形質転換し、エリスロマイシン耐 性を持つ株を複数得た。これらのうち 9 株を選んでゲノム DNA を抽出してサザ ン解析を行い、2 回交叉の結果、正しく破壊されている株 1 株を選んで AIBPR1 とした。 epr、nprE、vpr ついても aprE 及び bpr と同様に、各遺伝子のコード領域を薬 剤耐性遺伝子で置換することにより破壊を行った。epr の破壊にはカナマイシン 耐性遺伝子(kan)を、nprE の破壊にはクロラムフェニコール耐性遺伝子(cat)を、 vpr の破壊にはスペクチノマイシン耐性遺伝子(aadⅢ)をそれぞれ用いた。kan は、pDG148 を鋳型に PCR を行って取得したが、pDG148 上のkan のプロモーター からの転写は弱く、菌体内でマルチコピーにしないとカナマイシン耐性を示さ ない。そこでゲノム上のシングルコピーでも耐性を示すようなプロモーターに
変更するため、Marburg168 株のリボソーマル RNA オペロンrrnO プロモーターに
変更した。すなわち、kan コード領域と rrnO プロモーター領域をそれぞれ増幅
し、overlap extension PCR(Horton et al. 1989)と呼ばれる手法で連結した。
まず、pDG148 を鋳型にして kanB1 及び kanB2 を用いて kan コード領域と、
Marburg168 株のゲノム DNA を鋳型にして kanF1 及び kanF2 を用いてrrnO のプロ
モーター領域を増幅した。 ただし、kanF2 プライマーには、rrnO プロモーター と相同な配列が付加してあるため、kan 遺伝子の 5’側に rrnO プロモーターの 3’側の配列が付加された形で増幅されている。これらの 2 つの PCR 産物を混合 して鋳型とし、kanF1/kanB2 プライマーを用いて再度 PCR を行い、rrnO プロモ ーターの下流に kan 遺伝子が連結された PrrnO-kan を取得した。epr 遺伝子の上 流 2 kb、PrrnO-kan、epr 遺伝子の下流 2 kb の順に PCR 産物を連結して、epr 遺
伝子破壊用プラスミドを作製し、epr 破壊株を取得した。cat 遺伝子は pC194 を、 aadII 遺伝子は pIC333 をそれぞれ鋳型にして増幅した。 mpr 遺伝子の破壊については、mpr 遺伝子のコード領域の内部に存在する BsrGI 部位にrrnO プロモーターに連結した cat 遺伝子を挿入することで行った。まず、 mpr 遺伝子のコード領域に存在する BsrGI 部位から上下各 2 kb を増幅するよう に設計した 1 対のプライマーmpr-F/mpr-B を用いてmpr 遺伝子を含む 4 kb 増幅 し、pT7Blue に TA クローニングした。次に、epr 遺伝子の破壊時に作製した PrrnO-kan と同様の方法を用いて、PrrnO-cat を作製した。PrrnO-cat の両端を BsrGI で消化して、あらかじめ mpr 領域をクローニングしたベクターの BsrGI 部 位に挿入して、mpr 遺伝子破壊用プラスミドを得た。これを鋳型として、 mpr-F/mpr-B を用いて PCR を行い、mpr の内部に PrrnO-cat が挿入された 5.7 kb を増幅して供与核酸とし、r22 株に対して形質転換を行い、50 mg/ml のクロラ ムフェニコールに耐性を示す株を得た。 プロテアーゼを多重に破壊した株の取得は、bpr 破壊株 AIBPR1 を親株にして、 上に述べた各単独破壊株の作製方法と同様に vpr,nprE,wprA,epr の順に破壊し SKPROD5 株を得た。SKPROD5 株ではクロラムフェニコールに対し、5 mg/ml で耐 性を示すが、50 mg/ml では耐性を示さない。最後に、SKPROD5 株を親株にして mpr を破壊し、50 mg/ml のクロラムフェニコールに耐性を示す 6 重破壊株 SKPROD6 株を得た。 納豆発酵 まず、納豆発酵に使用する胞子液は以下のように調製した。NB 寒天培地上に 単一コロニーとなるように画線し、37 ℃で終夜培養した。得られた単一コロニ ーを一白金耳かき取り、NB 液体培地(5 ml)に植菌して、終夜培養した。得られ
た培養物 100 ml を胞子形成培地(10 ml)に添加し、更に 24 時間培養して胞子液 とした。液体培養はいずれも 37 ℃にて 150 rpm で振とう培養した。胞子液中の 胞子数は 75℃で 15 分処理した後に得られる耐熱性コロニーの数を計測すること で算出した。 得られた胞子液を適当な濃度になるように脱イオン水で希釈した後、大豆 100 g 当たり 106 /ml となるように添加して、50 g ずつポリスチレン製パックに入れ、 自動納豆製造装置 SY-NO/20(鈴与産業)にて 39 ℃、60 %加湿下で 18 時間発酵 させた。 菌体外プロテアーゼ活性の測定 菌体外プロテアーゼは発酵直後の納豆を用いて、柏原らの方法に従って測定 した(柏原ら 2001)。発酵終了直後の納豆を 5 g 取り、20 mL の 20 mM リン酸緩 衝液(pH 7.0)に 1 分間ボルテックスにて懸濁した後、4 ℃、15000 rpm にて 10 分間遠心した。遠心後の上清を回収し、菌体外プロテアーゼ粗酵素液とした。 粗酵素液 40 mL と、基質である 1 % Hammerstein カゼインを含む 20 mM リン酸 緩衝液 160mL を混合し、37℃で 30 分反応させた後、200 mL の 5 % TCA を添加 して室温にて 30 分放置し、反応を終了させた。反応液を 4 ℃、15000 rpm で 10 分間遠心し、その上清を回収し、A280を測定した。ブランクとしては、反応開始 と同時に TCA を添加したものを用いた。活性は、1 分間に与える、ブランクで得 られた A280と反応後に得られた A280の差DA280を 1 unit として定義し、納豆 1 g 当たりに換算して示した。菌体外プロテアーゼ活性は、各株ごとに 3 つのパッ クから別々に回収した納豆を用いて測定し、その平均と標準偏差を算出した。 g-PGA の定量
納豆からのg-PGA の抽出と定量は、菅野と高松の方法に従って行った(菅野&高 松 1995)。発酵直後の納豆 10 g を 2.5 %TCA 20mL と懸濁した後、50℃ で 10 分間抽出した。TCA でインキュベートされた納豆をナイロン製のメッシュにてろ 過し、ろ液を 4℃、12000 rpm にて 15 分遠心を行った。得られた上清 20 mL の pH を NaOH にて pH7.0 に調整し、水で 25 mL にメスアップした。このうち、5 mL を 20 mL のエタノールと混合して、10 分間氷上で放置し、次に 4℃、12000rpm で 10 分遠心を行った。得られた沈殿を 20 mL の 20mM リン酸緩衝液(pH7.0)に溶 解し、g-PGA 粗精製液とした。これを適当に希釈した後、セチルメチルアンモニ ウムと反応させ、A420の吸収を測定した。予め、濃度のわかっているg-PGA 標品 で検量線を作成しておき、検量線からサンプルのg-PGA 濃度を算出した。g-PGA 濃度も菌体外プロテアーゼ活性と同様に、各株ごとに 3 つのパックから別々に 回収した納豆を用いて測定し、その平均と標準偏差を算出した。 アミノ酸分析 発酵直後の納豆 2 g を乳鉢でよくすりつぶした後、0.2 N の塩酸で懸濁して 50 mL にメスアップした。これを、終夜 4 ℃にてインキュベートしてアミノ酸 を抽出した後、ろ紙を用いてろ過したろ液 25 mL を 3 %のスルホサリチル酸と 混合し、もう一度ろ紙でろ過した。ろ液の pH を LiOH で 2.2 に調整した後、そ の一部を JCL-500/V AminoTacTM アミノ酸分析計にて測定した。得られたアミノ 酸の濃度を納豆 1 g 当たりに換算して示した。アミノ酸濃度も菌体外プロテア ーゼ活性と同様に、各株ごとに 3 つのパックから別々に回収した納豆を用いて 測定し、その平均と標準偏差を算出した。 大豆粉培地上でのg-PGA 生産性評価
予め固めておいた大豆粉寒天培地上に、胞子が 1.0x107となるように塗抹植菌
し、37℃にて 18 時間培養した。得られた培養物をもんじゃ焼き用のコテを用い て回収した。以降の抽出及び定量は、納豆からの場合と同様にして行った。大 豆粉培地上でのg-PGA 濃度は、3 つのプレートから別々に回収した培養物を用い て測定し、その平均と標準偏差を算出した。
1-3 結果 (1)納豆菌における菌体外プロテアーゼ遺伝子セット 納豆菌と分類学上同一の菌とされる枯草菌B.subtilis 168 株には、アルカ リセリンプロテアーゼ遺伝子 aprE、バシロペプチダーゼ遺伝子 bpr、マイナー な菌体外プロテアーゼをコードするepr 及び vpr、金属プロテアーゼ遺伝子 mpr、 中性プロテアーゼnprB 及び nprE、細胞壁に結合する wprA の 8 種が存在してい る。納豆菌にも同様の菌体外プロテアーゼの遺伝子セットが保存されているか どうかを確認するために、r22 株のゲノム DNA を鋳型に、各プロテアーゼ遺伝子 のコード領域を PCR にて増幅した。その結果、aprE、bpr、epr、vpr、mpr、nprE、 wprA の 7 種については 168 株と同様の長さに増幅が確認されたが、nprB につい ては増幅が確認されなかった(図 1-1A)。r22 株の親株である O-2 株ではゲノム 解読が完了しており、O-2 株においてnprB を含む約 5 kb の領域が欠損している ことが明らかとなっていた(データは示さない)。そこで、r22 株でも同様の欠損 が起きていると推定し、欠損している領域に隣接した 2 つの ORF であるyitL 及
びyitS の内部にアニールするプライマーyitL-N 及び yitS-C を設計して PCR を
行ったところ、約 500bp の増幅が確認された。これを pT7Blue に TA クローニン グしてシーケンスしたところ、図 1-1B に示すように r22 株においても nprB を 含む約 5 kb の領域を欠損していることが確認できた。以上から、r22 株には 168 株ゲノムに存在が確認されている 8 種の菌体外プロテアーゼのうち、nprB を除 く 7 種が存在していることが確認された。 (2)各遺伝子破壊株の菌体外プロテアーゼ活性とg-PGA 生産性 各プロテアーゼ遺伝子の納豆発酵における重要性を検討するために、7 種の各
プロテアーゼ遺伝子を破壊した破壊株を作製した。これらはいずれも破壊株を 取得することができ、胞子形成培地における胞子数は親株と同等であった(デー タは示さない)。これら破壊株の胞子液を用いて納豆を作製し、発酵終了直後の 納豆からプロテアーゼ粗酵素液を調製して、カゼインを基質とした菌体外プロ テアーゼ活性を測定した。その結果、bpr、epr、mpr、nprE、vpr、wprA の各破 壊はいずれもプロテアーゼ活性に大きな影響を及ぼさなかったのに対し、主要 アルカリプロテアーゼをコードする aprE の破壊株 SKAPR1 においてのみプロテ アーゼ活性の顕著な減少が確認された(図 1-2A)。次に、g-PGA 生産量を測定した ところ、プロテアーゼ活性と同様に、aprE を除く各破壊株ではg-PGA の生産量に ほとんど影響が見られなかったのに対し、SKAPR1 ではg-PGA の有意な減少が確認 された(図 1-2B)。これらの結果から、AprE は納豆発酵における主要なプロテア ーゼ活性の本体であり、g-PGA の生産にも重要な役割を果たしていることが示唆 された。更に AprE の重要性を確認するために、aprE 以外の 6 種の菌体外プロテ アーゼ遺伝子を全て破壊し、菌体外プロテアーゼとしてaprE のみを持つ 6 重破 壊株 SKPROD6 を作製して、プロテアーゼ活性(図 1-2A)とg-PGA 生産量(図 1-2B) を測定した。その結果、いずれも親株 r22 の約 80 %程度にしか減少せず、これ ら 7 種の菌体外プロテアーゼのプロテアーゼ活性やg-PGA 生産性に対する寄与度 は、AprE 単独に比べて非常に小さいことが確認された。以上の結果から、納豆 発酵において AprE は主要なプロテアーゼ活性を担い、g-PGA を最大量生産する のに必須の役割を果たしていることが示された。加えて、AprE 以外のプロテア ーゼ群はおそらく納豆菌の産生するプロテアーゼ活性やg-PGA 生産にほとんど 関与していないことも示唆された。 (3)プロテアーゼ遺伝子 6 重破壊株及びaprE 破壊株の遊離アミノ酸濃度
納豆発酵が進むにつれて、大豆のアミノ酸がプロテアーゼによって分解され、 納豆中のアミノ酸濃度が増加することが知られている。前項で述べたように、
aprE の破壊によりg-PGA 生産量の減少が確認されたが、それがg-PGA 生産の基質
となるアミノ酸の供給が減少したことに起因しているのであれば、納豆中の遊 離アミノ酸濃度も減少していることが予測される。また、この予測が正しけれ ば、aprE 以外の 6 種の菌体外プロテアーゼ遺伝子を全て破壊した 6 重破壊株 SKDPRO6 では、破壊による遊離アミノ酸濃度に対する影響が少ないと考えられる。 SKPROD6 株で作製した納豆の発酵直後の遊離アミノ酸濃度を測定したところ、蛋 白質を構成する標準アミノ酸 20 種のいずれもが、親株とほぼ同様の濃度である ことが確認された(図 1-3A)。一方、aprE 破壊株を用いて親株と同時間発酵させ た納豆の遊離アミノ酸濃度は、20 種いずれも極端に低いことが判明した(図 1-3B)。以上の結果から、AprE は納豆発酵時にg-PGA 生産に必要なアミノ酸等の 材料を供給する役割を担うことが示唆された。
(4) L-Glu の添加によるaprE 破壊株のg-PGA 生産性に対する効果
AprE が納豆発酵においてg-PGA 生産に必要な材料を供給しているのであれば、
その材料を添加することにより、aprE 破壊株で観察されるg-PGA 生産性不全は回
復することが期待される。まず通常のg-PGA 生産培地に aprE 破壊株 SKAPR1 を植
菌したところ、g-PGA 生産が回復することが確認された(データは示さない)。次 に、納豆試作時にL-Glu を添加して納豆を製造したが、期待に反しg-PGA 生産は 回復しなかった。蒸煮した大豆に添加できるL-Glu の量には限界があり、回復さ せることができなかったと考えられた。更に添加するL-Glu の量を増加させるた めに、L-Glu 溶液を用いて乾燥大豆を浸漬することを試みたが、今度は大豆が脱 水してしまい、納豆を製造することができなかった(いずれもデータは示さな
い)。そこで、試作に使用する大豆を粉末化した大豆粉に寒天を加えて固め、大 豆を模倣した培地を開発することにした。大豆粉はオートクレイブ処理を行う と凝固分離してしまうため、まず培地が作製可能な大豆粉濃度を検討したとこ ろ、10 %が上限であった。そこで、大豆粉 10 %を寒天で固めた培地を作製し、 その表面に納豆菌(r22 株及び SKAPR1 株)を塗抹して培養したところ、r22 株で は粘り気のある培養物が得られたのに対し、SKAPR1 株ではほとんど粘り気のな い培養物しか得られなかった。それぞれの培養物を掻きとってg-PGA の生産量を 比較したところ、r22 株の培養物にはg-PGA が確認されたのに対し、SKAPR1 株の それにはg-PGA はほとんど含まれておらず(図 1-4)、大豆粉培地上でも納豆で確 認されたg-PGA 生産性を反映する結果が得られた。図では、ここで r22 株が生産 したg-PGA の量を 100 %として相対%で示した。次に、大豆粉培地にg-PGA 生産の 基質であるL-Glu を添加した場合のg-PGA 生産量を測定した。最少培地に通常ア ミノ酸を加える際の濃度である 0.2 %と、g-PGA を生産させる培地にL-Glu を加 える濃度である 2 %となるように Glu を添加した大豆粉培地を作製し、g-PGA 生 産量を測定した。その結果、0.2 %添加では SKAPR 株のg-PGA 生産性は回復しな かったのに対し、2 %添加ではほとんど r22 株と同レベルまでg-PGA 生産性が劇 的に回復することが確認された。以上から、AprE は納豆発酵においてg-PGA 生産 に必要なL-Glu などの材料を供給していることが示唆された。 (5)菌体外ペプチダーゼに関する解析 前項で述べたように、AprE はg-PGA 生産に材料を供給していると考えられるが、 その一方で AprE は基質である蛋白質やペプチドをエンド型に切断をすることが 報告されている。また、AprE は基質の疎水性の高い残基やヒスチジン残基を認 識して、その C 末側で切断する。従って、蛋白質やペプチドに AprE を作用させ
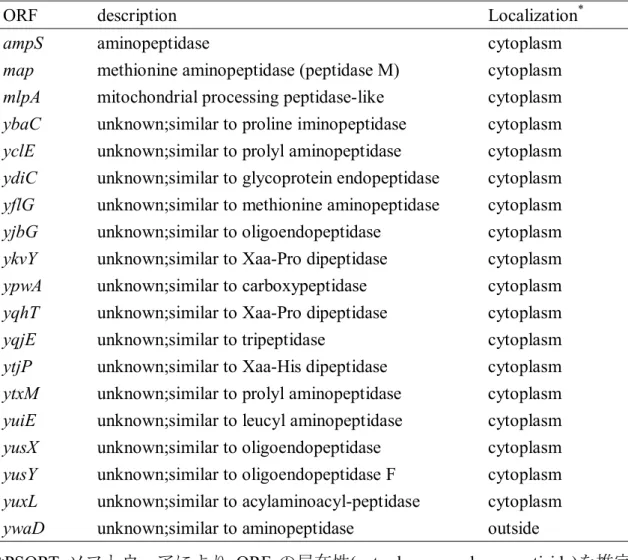
ても Glu はほとんど遊離せず、オリゴペプチドが遊離するものと推察される。 SKAPR1 がオリゴペプチドを供給できないことで、ペプチドからアミノ酸への更 なる分解が滞り、g-PGA 生産不全が引き起こされている可能性も否定できない。 そこで、ペプチドを更に切断してアミノ酸に分解するペプチダーゼがg-PGA 生産 に関与しているのかどうかについて検討を行った。まず、ペプチダーゼ遺伝子 の 検 索 を 行 っ た 。 168 株 の ゲ ノ ム デ ー タ ベ ー ス で あ る Subtilist(http://genolist.pateur.fr./Subtilist/)を用いてペプチダーゼを コードすると予測される ORF を選抜した結果、19 の ORF が見出された(表 1-4)。 こ れ ら の ORF の 推 定 さ れ る 局 在 性 に つ い て PSORT プ ロ グ ラ ム (http://psort.hgc.jp/form.html)を用いて解析したところ、ywaD のみが細胞外 に分泌される可能性が示唆された(表 1-4)。実際、YwaD はプロテオーム解析に より細胞外に分泌されることが報告されており、同じ解析においてその他のペ プチダーゼは菌体外に検出されていなかった(Mäder et al. 2002)。そこで、菌 体外に分泌されるペプチダーゼは YwaD のみであると判断し、ywaD 破壊株 AIYWAD1 を用いて製造した納豆のg-PGA 生産量を測定したが、親株と同等であっ た(図 1-2)。以上から、YwaD はg-PGA 生産における基質供給には関係していない ことが示唆された。
1-4 考察 (1)菌体外プロテアーゼ遺伝子セットとプロテアーゼ活性 図 1-1 に示したように、納豆菌と枯草菌 168 株は同じB.subtilis とされてい るが、納豆菌ではnprB が欠損しており、菌体外プロテアーゼ遺伝子の数として は 1 つ少ない。近年、慶応大学のグループにより、納豆菌 BEST195 株のゲノム 解読が完了しており(Nishito et al.2010)、168 株の nprB 遺伝子座に対応する 領域を解析したところ、r22 株及びその親株である O-2 株と同様に nprB を欠損 していることがわかった。従って、納豆菌においてnprB を欠損していることは 一定程度保存されているものと推察される。なお、BEST195 株において、nprB 以外のプロテアーゼ遺伝子は保存されており、r22 株と同様の遺伝子セットを持 つことが確認された。枯草菌における NprB は菌体外に分泌されると報告(Tran et al. 1991)されてはいるものの、プロテオーム解析では他の菌体外プロテア ーゼが検出されているにも関わらず、NprB は検出されておらず(Mäder et al. 2002)、分泌量が非常に少ないことが予想される。 また、nprB を欠くため、納豆菌 r22 株では枯草菌 168 株に比べてプロテアー ゼ遺伝子の数としては少ないことになるが、大豆表面上でのプロテアーゼ活性 は圧倒的に r22 株の方が高かった(図 1-2A)。168 株では、プロテアーゼ等菌体 外酵素の分泌能が上昇するような点変異がいくつか知られているが、その1つ である degQ プロモーターへの変異 degQ36 が納豆菌にも存在することが見出さ
れている(Ohsawa et al. 2009)。r22 株の degQ36 変異に相当する部位を確認し
たところ、やはり degQ36 変異と相同な変異が見出された。以上から、r22 株で
はプロテアーゼ遺伝子が 168 株に比べて 1 つ少ないにもかかわらずプロテアー ゼ活性が高いのは、degQ36 変異によるものと考えられる。degQ36 変異では、プ
ロモーター領域への変異によりdegQ の転写量が増加しており(Yang et al.1986)、 菌 体 外 酵 素 遺 伝 子 群 の 転 写 を 制 御 す る 二 成 分 制 御 系 DegS-DegU の
hyperactivation を引き起こすことが報告されている(Yang et al. 1986)。また、
同じ hyperactivation を引き起こすdegU32 変異では AprE、Bpr、Mpr、NprE、Vpr
の分泌量が増加することも報告されている(Mäder et al. 2002)。納豆菌はその 利用、馴化の過程において、納豆製造可能な、糸引きの強い菌を選抜した結果 として、プロテアーゼ活性の高い変異株が選ばれてきたと推察される。 (2)g-PGA 生産に材料を供給するプロテアーゼ AprE 図 1-2 に示したように、aprE 破壊株でカゼインを基質としたプロテアーゼ活 性が特異的に著しく減少し、それに伴ってg-PGA の生産量も激減する。また、図 1-3B に示したように同破壊株では遊離アミノ酸濃度も著しく減少しており、大 豆蛋白質を基質とする場合にもプロテアーゼ活性が減少していることが示唆さ
れた。更に、aprE 破壊株の納豆発酵におけるg-PGA 生産不全はL-Glu の添加によ
って回復した(図 1-4)ことから、AprE は納豆発酵において主要なプロテアーゼ として大豆蛋白質を分解して、g-PGA 生産に必要な材料を供給していることが明 らかとなった。AprE は、エンド型プロテアーゼであり、また、基質の疎水性ア ミノ酸やヒスチジン残基を認識して切断する(Kashihara et al.2001;Ichishima et al.1986)ことから、AprE によって生じるのはオリゴペプチドかあるいは疎水 性アミノ酸、ヒスチジンであると考えられ、これがg-PGA 合成の基質となってい ると予測される。しかし、納豆菌が効率よくg-PGA を生産するのはL-Glu 等のグ ルタミン酸族アミノ酸を添加した場合であり、g-PGA 合成酵素である PgsBCA 蛋
白質複合体はL-Glu またはD-Glu を基質としてg-PGA を合成することが、in vitro
プチドや疎水性アミノ酸はL-Glu に変換されて、g-PGA 生産に用いられていると
考えられる。図 1-2 に示したように、この過程で生じたペプチドを更に分解し
うるペプチダーゼ YwaD はg-PGA 生産に影響を及ぼさなかったことから、おそら
く AprE によって生じたオリゴペプチドは、例えば AppABCDF や OppABCDF 等のオ リゴペプチド輸送体(Koide&Hoch 1994)によって細胞内に移入され、表 1-4 に示 したような細胞内のペプチダーゼによってアミノ酸に分解されると考えられる。 蛋白質を構成する 20 種のアミノ酸のうち、グリシン、システイン、セリン、メ チオニン及びセリンを除く実に 15 種のアミノ酸は Glu に変換されうるアミノ酸 であり、アミノ酸レベルまで分解されたペプチドは、その多くがL-Glu に変換さ れている可能性が示唆される。細胞内でいずれのペプチダーゼが細胞外から移 入されたペプチドの分解に関与しているのか、また、実際にL-Glu に変換されて いるのかについては今後の解析が必要であると考えている。 通常、液体培地や固体培地においてg-PGA を生産させる場合には、L-Glu 等を 十分量添加するため、g-PGA 生産において材料を供給する役割を何が担うのかと いう点は全く着目されてこなかった。しかし、B.subtilis は元来土壌や植物表 面に存在する細菌であり、遊離の Glu に富む環境で生活しているわけではない。 今回、納豆発酵というg-PGA 生産にプロテアーゼの活性発揮が必須な現象を解析 することで、積極的にg-PGA を生産するための材料を調達する仕組みを備えてい ることが明らかになったことは大変興味深い。 (3)CSF 成熟化因子としての AprE のg-PGA 生産への関与の可能性 AprE は菌体の密度を感知する CSF を成熟化させる機能を持つため、CSF の生 産を介して Quorum sensing system を制御する因子としても機能している (Lanigan-Gerdes et al.2007)。また、g-PGA は Quorum sensing system によっ
てその合成が制御されている(Tran et al.2000)。従って、AprE が CSF の成熟化 を通してg-PGA の生産を制御している可能性も示唆される。もし、前項で述べた g-PGA 生産に対する材料の供給不足ではなく、CSF が成熟化できないことによっ てg-PGA 生産が低下しているのであれば、aprE 破壊株においても親株と同等の菌 体密度は維持されているはずである。SKAPR1 株で製造した納豆における納豆菌 の数を測定したところ、1x109 /g 納豆であったのに対し、親株のそれは 8x109 /g 納豆であった。また、CSF の未成熟化がg-PGA 生産不全の原因であれば、L-Glu の添加によりg-PGA の生産性不全は回復しないと考えられるが、図に示したよう にL-Glu の添加によりg-PGA の生産性は回復することが確認されている。これら のことから、aprE 破壊株によって引き起こされるg-PGA 生産性不全は、CSF の未 成熟化ではなく、材料の供給不足が主要な原因と考えられる。 (4)AprE 以外のプロテアーゼの納豆発酵における役割 AprE 以外の 6 種のプロテアーゼ遺伝子を全て破壊した株を作製して、プロテ アーゼ活性やg-PGA 生産量について検討したが、aprE のみを破壊した株に比べて 影響は小さいことが確認された。6 重破壊株では、菌体外プロテアーゼとして AprE のみが機能していると考えられるが、プロテアーゼ活性も 80 %程度は残存 しており、他のプロテアーゼが AprE の成熟化に必須であったり、また AprE の 安定性を制御している可能性は低いと考えられる。菌体外プロテアーゼの栄養 源 獲 得 以 外 の 機 能 に つ い て は い く つ か 報 告 が あ る が 、 納 豆 菌 で は な い undomesticated strain において、swarming motility を制御しているという報 告がある(Connelly et al. 2004)。そこでは、納豆菌に存在する 7 種の菌体外 プロテアーゼオルソログを全て破壊した株において swarming motility が著し く低下すること、また swarming motility にはプロテアーゼ活性が必須である
ことも明らかとなっている。納豆菌における 6 重破壊株 SKPROD6 の swarming motility を観察したところ、r22 株に比べて swarming motilty が低下している ことが判明した(図 1-5B)。興味深いことに、aprE 破壊株に比べても、6 重破壊 株では swarming motility が低下していることが確認された。aprE 破壊株と 6 重破壊株とを比較すると、aprE 破壊株においてプロテアーゼ活性が圧倒的に低 下しているにも関わらず、swarming motility への影響は 6 重破壊株のほうが強 いことが確認された。このことから、swarming motilty の制御は単にプロテア ーゼ活性の強弱が重要ではなく、それに関与するプロテアーゼ種が存在してい ることを示唆しているものと考えられる。なお、r22 破壊株と 6 種破壊株の LB 液体培地における生育の差は観察されなかった(図 1-5A)。6 種の菌体外プロテ アーゼのうち、いずれが swarming motiity に強く関与しているかについては、 今後の解析が必要であると考えているが、aprE 以外のプロテアーゼも納豆発酵 において大豆表面での運動性を制御する等の役割を担っている可能性が示唆さ れた。
第2章 納豆菌の 2 種のグルタミン酸ラセマーゼに関する生理学的解析 2-1 背景と目的 第 1 章で述べたように、納豆発酵においては、菌体外プロテアーゼのうち、 主にアルカリプロテアーゼ AprE がg-PGA の生産の基質となるペプチドまたはア ミノ酸を供給している。AprE によって生じたペプチドは、おそらく細胞膜に存 在するペプチド輸送体によって菌体内に取り込まれた後、菌体内のプロテアー ゼやペプチダーゼによってアミノ酸に分解され、蛋白質合成に使用されるほか、 細胞内のアミノ酸代謝経路によって L-Glu などの別のアミノ酸に変換されると 考えられる。生成したL-Glu は、本章で述べるようにグルタミン酸ラセマーゼに よって異性化されてD-Glu となるか、あるいは第 3 章で述べるようにグルタミン 酸脱水素酵素によって 2-OG となってクエン酸サイクルの中間体となるか、ある いは蛋白質合成に用いられると考えられる。 納豆には、天然に存在するL-体のアミノ酸に加えてD-Glu が豊富に含まれてお り、そのD-Glu は納豆の糸の主成分であるg-PGA と、納豆菌細胞壁ペプチドグリ カンのペンタペプチドに局在することがわかっている。これまで、細菌細胞壁 のペンタペプチドに含まれる D-Glu は、アラニンラセマーゼによって生じた D-Ala のアミノ基が D-アミノ酸アミノトランスフェラーゼによって 2-OG に転移
されて D-Glu が生じると考えられてきた(Thorne et al. 1955; 藤井 1963)。実
際、大腸菌や黄色ブドウ球菌、B. stearothermophilus などの細菌の抽出液にグ
ルタミン酸ラセマーゼ活性が検出されないこと、また Marburg 168 株では、
D-アミノ酸アミノトランスフェラーゼが検出される(Soper et al. 1977)ことから
菌 IFO3336 株の細胞抽出液には、L-Glu を直接D-Glu にラセミ化するグルタミン
酸ラセマーゼ活性が検出されることが見出され、活性本体を精製することによ りグルタミン酸ラセマーゼ Glr が単離された(Ashiuchi et al. 1998)。Glr の生
化学的な解析により、L-Glu を基質とした場合に高い Km 値と Vmax 値を持つこと
から、細胞内に多量の Glu プールが存在する際に効率よくL-Glu をD-Glu にラセ
ミ化する活性を持つことが明らかとなった。一方で、168 株のゲノム解析の結果
から 168 株ゲノムには glr 遺伝子オルソログ(racE)に加えて、もう1つグルタ
ミン酸ラセマーゼをコードすると考えられる遺伝子yrpC が存在していることが
判明した(Kunst et al. 1997)。Ashiuchi らは yrpC ホモログが納豆菌にも存在
していると考え、納豆菌ゲノムからyrpC をクローニングして大腸菌にて過剰発 現系を構築し、その酵素活性を測定した(Ashiuchi et al. 1999)。その過程で、 yrpC を過剰発現した場合には大腸菌の生育阻害が起きるのに対し、glr を高発 現した場合には生育阻害が起きないことを見出した。g-PGA を生産しない大腸菌 では、D-Glu の供給先は細胞壁のペンタペプチドのみであることから、不要な D-Glu が YrpC によって大腸菌に供給されたために生育阻害が起きたと考えられ た。大腸菌のグルタミン酸ラセマーゼ遺伝子murI を過剰発現した場合も同様の 生育阻害が報告されており、その表現型と一致していた(Ashiuchi et al.1999)。 一方、Glr はg-PGA 生産時にのみD-Glu を供給するような生化学的特性、すなわ ちL-Glu に対する高い Km 値と Vmax 値を持つため、生育阻害が起きないと考えら れた。加えて生化学的な解析により、YrpC は L-Glu を基質とした場合に低い Km
値と Vmax 値を持ち、少量の L-Glu を少しずつD-Glu に異性化する活性を持つこ
とが判明した。細胞壁のペンタペプチド合成では細胞の分裂に同調しながら少
量の L-Glu を少しずつ D-Glu に変換する必要があることを考え合わせ、Glr は
を、それぞれ供給していると主張されてきた(芦内 2002)。 一方で、168 株のポストゲノム研究の進展により、ゲノム中の遺伝子の必須性 がカタログ化されてきた。その結果、納豆菌のg-PGA 生産に必要なD-Glu を供給 するとされていた glr のホモログ遺伝子である racE が LB 培地での生育におい て必須遺伝子であることが報告された(Kobayashi et al. 2003)。168 株におい てracE 遺伝子が必須遺伝子であるということは、少なくとも細胞壁のペンタペ プチド合成に必要なD-Glu を供給していると考えられる。なぜなら、細胞壁のペ ンタペプチド合成は生育に必須であり、g-PGA 生産は生育に必須ではないからで ある。racE 遺伝子と glr 遺伝子のアミノ酸レベルでの相同性は 99.5 %であり、 これら 2 つの遺伝子産物は極めて似た機能を持つことが予想される。以上のこ とから、芦内らが主張するように、納豆菌における 2 つのグルタミン酸ラセマ ーゼの片方がg-PGA 生産に、もう一方が細胞壁のペンタペプチドに、それぞれ D-Glu を供給しているのか、すなわち機能分化しているのか疑問が持たれた。そ こで、本章では納豆菌の 2 種のグルタミン酸ラセマーゼ遺伝子について、その 生理学的な役割、すなわち明確な機能分化があるのかどうか、またg-PGA 生産に 関与するグルタミン酸ラセマーゼはいずれなのかについて明らかにすることを 目的に分子生物学的な解析を行った。
2-2 材料と方法 使用菌株と培地 本章で使用した菌株を表 2-1 に示した。 納豆菌の各遺伝子破壊株は、第 1 章と同様に B. subtilis r22 株を親株とし て作製した。遺伝子破壊用プラスミドの構築などの遺伝子操作には大腸菌 JM109 株を用いた。 納豆菌の通常の培養には Nutrient broth(NB)培地(1 % ポリペプトン(日本製 薬)、0.5 % NaCl (シグマアルドリッチ)、0.5 % 肉エキス (OXOID))を用いた。 g-PGA 生産培地には、SG 培地(5 % スクロース(関東化学)、1.5 % グルタミン酸 ナトリウム(シグマアルドリッチ)、0.27 % KH2PO4(関東化学)、0.42 % Na2HPO4・
12H2O(和光純薬工業)、0.05 % NaCl(和光純薬工業)、0.05 % MgSO4・7H2O(シグマ
アルドリッチ)、0.1mg/ml ビオチン(シグマアルドリッチ))を用いた。納豆菌の
形質転換には、Spizizen の最少培地(0.25 % グルコース(関東化学)、5 mM MgSO4・7H2O(シグマアルドリッチ)、0.6 % KH2PO4(関東化学)、1.4 % K2HPO4(関東
化学)、0.1 % クエン酸ナトリウム(シグマアルドリッチ)、0.1 mg/mL ビオチン))
を用いた。必要に応じて、クロラムフェニコール(5 mg/ml)、エリスロマイシン
(0.5 mg/ml)、カナマイシン硫酸塩(7.5 mg/ml)を添加した。
大腸菌 JM109 株の培養には、LB 培地を用い(1 % Bacto Tryptone(Becton, Dickinson and Company)、0.5 % Bacto yeast extract(Becton, Dickinson and
Company)、1 % NaCl(シグマアルドリッチ))、必要に応じて 50 mg/mL アンピシ
プラスミド
本章で使用したプラスミドを表 2-2 に示した。
エリスロマイシン耐性遺伝子 ermC はプラスミド pE194(Horinouchi&Weisblum
1982)からBamHI 及び XbaI 処理により切り出して用いた。また、IPTG で誘導で
き る プ ロ モ ー タ ー Pspac を 持 つ 大 腸 菌 - 枯 草 菌 シ ャ ト ル ベ ク タ ー に は pDG148(Stragier et al. 1988)を用いた。更に、b-ガラクトシダーゼアッセイ 用の株の構築には、pDL2(Fukuchi et al. 2000)を用いた。 破壊用プラスミドの作製には、pUC19 及び、PCR 産物を TA クローニングでき るプラスミドベクターpT7Blue(TaKaRa)を用いた。 遺伝子破壊株の作製 遺伝子破壊は、破壊したい遺伝子の上流及び下流を PCR で増幅し、エリスロ マイシン耐性遺伝子ermC の両端に挟んだコンストラクションにより目的遺伝子 を ermC で置換する方法で行った(図 2-1)。遺伝子破壊用プラスミドの構築に使 用したプライマーは表 2-2 に示した。遺伝子破壊用プラスミドの作製に先立ち、
薬剤耐性遺伝子であるermC を pAE194 から BamHI 及び XbaI で処理して切り出し、
pUC19 のBamHI/XbaI サイトにクローニングして pEMR を作製した。
グルタミン酸ラセマーゼ遺伝子 glr の遺伝子破壊用プラスミドの作製は、以
下の方法で行った。表 2-2 に示したプライマーglr-F1 及び glr-F2 を用いてglr
の開始メチオニンから上流 3 kb を PCR にて増幅し、pT7Blue の TA クローニング サイトにクローニングして、pDGLR-UP を得た。次に、glr の終止コドンから下
流 3 kb をプライマーglr-B1 及び glr-B2 を用いて増幅し、末端をBamHI/KpnI で
消化して、上述の pEMR の BamHI/KpnI サイトにクローニングし、pEMRplusDOWN
片を切り出し、 予めSmaI で消化した pDGLR-UP にクローニングすることにより、 glr の遺伝子破壊用プラスミド pDGLR を得た。
グルタミン酸ラセマーゼ遺伝子yrpC の遺伝子破壊用プラスミドの作製は、以
下の方法で行った。表 2-2 に示したプライマーyrpC-F1 及び yrpC-F2 を用いて yrpC の開始メチオニンから上流 3 kb を PCR にて増幅してその両端を SphI で消
化し、pEMR の SphI サイトにクローニングして pEMRplusUP を得た。次に、yrpC
の終止コドンから下流 3 kb をプライマーyrpC-B1 及び yrpC-B2 を用いて増幅し
てその両端をBamHI で消化し、pEMRplusUP の BamHI サイトにクローニングして、
yrpC の遺伝子破壊用プラスミド pDYRPC を得た。
これらの破壊用プラスミドを ScaI により直線化して、r22 株コンピテントセ
ルと混合することにより形質転換を行った。r22 株のコンピテントセルは、 Marburg168 株のコンピテントセル作製方法(Ashikaga et al. 2000)と同様に行 った。目的遺伝子が正しく置換されて破壊が行われていることをサザン解析に より確認した。 また、glr オルソログである racE は Marburg168 株において必須遺伝子である ことが報告されており(Kobayashi et al. 2003)、単純に破壊できない可能性が あるため、glr をクローニングしたプラスミド pDG-GLR を予め r22 株に導入した SKGL1301 株を宿主としても形質転換を行った。なお、pDG-GLR 及び SKGL1310 の 作製は以下の方法により行った。glr のコード領域をプライマーglr-N 及び glr-C を用いて増幅し、末端をHindIII/SphI で消化し、pDG148 の Pspac の直下に挿入 し、pDG-GLR を得た。ただし、glr の SD 配列は非典型的な配列であり、また開 始メチオニンも TTG であるため、プラスミド上で効率的な翻訳を行わせる目的 で典型的な SD 配列(AGGAGG)を付加し、また開始メチオニンを TTG から ATG に変 更した。得られた pDG-GLR を r22 株にプロトプラスト法(Chang&Cohen 1979)を
用いて導入し、SKGL1301 株を得た。 プラスミドキュアリング 前項で述べた pDG-GLR の納豆菌からのキュアリングは以下の方法で行った。 pDG-GLR を持つ SKGL1301 株及び SKDG0501 株(後述)を 2 ml の 2xYT(Sambrook et al. 1989)培地にて 37℃で終夜培養し、得られた培養物を新たな 2xYT 培地に 1/1000 容量となるように植菌し、37 ℃で再度終夜培養した。これを繰り返し行 い、各終夜培養物を 107 倍希釈してカナマイシン硫酸塩を含む LB 寒天培地にて 塗抹培養し、カナマイシン耐性を示すコロニーの数をカウントして、プラスミ ドの保持率を算出した。 g-PGA の定性定量分析 定性定量分析に用いたg-PGA は以下の方法で調製した。各納豆菌株を坂口フラ スコ中、100ml SG 液体培地にて 37℃で 72 時間培養し、得られた培養物を 12000 rpm で 30 分遠心することにより上清を回収した。上清に 4 倍量のエタノールを 添加し、更に 15000 rpm で 10 分遠心することで沈殿を得た。これを 50 mM リン 酸緩衝液(pH 7.0)に溶解し、g-PGA 粗画分とした。 得られたg-PGA 粗画分を用いて前章で述べた菅野と高松の方法により定量を 行った。D/L 比分析では、g-PGA 粗画分を 50 mM リン酸緩衝液を外液として透析 を行い、遊離のグルタミン酸を除去した。遊離のグルタミン酸を除去したg-PGA 粗画分を塩酸により 100 ℃で終夜処理を行い、モノマーのグルタミン酸にまで 加水分解を行った。得られた分解物をキラルカラムである CHIRALPAK MA(+)(ダ イセル)に供し、D/L 比を算出した。
ノザン解析
ノザン解析に使用する全 RNA は SG 培地にて培養した菌体から回収した。RNA の回収方法は Ogura らの方法(Ogura et al. 2001)に従った。
glr 及び yrpC のプローブについては以下の方法により32P-ラベル化した。glr
のコード領域を、プライマーglr-N2 と glr-C2 を用いて増幅し、得られた産物と
32P-dATP を用いて BcaBSET ラベリングキット(TaKaRa)によりラベル化した。YrpC
についても、そのコード領域をプライマーyrpC-N と yrpC-C を用いて増幅し、glr と同様にラベル化した。 得られた全 RNA 10 mg を変性条件下でアガロース電気泳動を行い、Hybond N 上にブロッティングした後、32P-ラベル化された各プローブでハイブリダイズさ せることにより、転写を検出した。 b-ガラクトシダーゼアッセイ b-ガラクトシダーゼアッセイに用いる菌株は以下の方法により作製した。 ノザン解析の結果、glr は 1 つ前の ORF、ysmB と共転写されていることが判明 したため、glr のプロモーターは ysmB の上流に存在することが示唆された。そ こで、glr のプロモーター領域として、ysmB とその上流に位置する gerE の遺伝 子間領域 245 bp を増幅するように設計したプライマーglr-F3 及び glr-F4 を用 いて増幅し、末端をEcoRI/BamHI で処理して pDL2 の EcoRI/BamHI サイトに挿入 し、pDL-GLR を得た。一方、yrpC はモノシストロニックな転写のみが検出され たため、yrpC 上流にプロモーターが存在していることが示唆された。yrpC のプ ロモーター領域として、上流の yrpB との遺伝子間領域 296 bp を増幅するよう に 設 計 し た プ ラ イ マ ー yrpC-F3 及 び yrpC-F4 を 用 い て 増 幅 し 、 末 端 を