平成 28 年度 博士論文
ex vivo 処置を介さない新規ゲノム
編集マウス作製法「GONAD」の開発
Development of a novel genome editing method “GONAD”
that does not require ex vivo handling of embryos
指導教授
東京農業大学大学院 生物産業学研究科
生物産業学専攻
動物資源管理学研究室
目次 序章 ··· 1 第 1 章 エレクトロポレーション法による卵管内 2 細胞期胚への RNA 導入法の確立 ·· 6 第 1 節 緒論 ··· 6 第 2 節 材料および方法 ··· 8 1. マウス ··· 8 2. マウスの過排卵処置 ··· 8 3. RNA 合成 ··· 9 4. 卵管注入用 RNA 溶液の調製 ··· 12 5. 卵管への RNA 溶液の注入 ··· 12 6. in vitro / in situ エレクトロポレーション··· 15 7. eGFP 蛍光観察 ··· 18 8. 変異解析 ··· 18 第 3 節 結果 ··· 20 1. in vitro エレクトロポレーションによる卵管内 2 細胞期胚への RNA 導入 ··· 20 2. in situ エレクトロポレーションによる卵管内 2 細胞期胚への RNA 導入··· 23 3. in vitro エレクトロポレーションによる卵管内 2 細胞期胚への CRISPR 系 RNA
の導入 ··· 27
4. in situ エレクトロポレーションによる卵管内 2 細胞期胚への CRISPR 系 RNA の導入 ··· 34 第 4 節 考察 ··· 38 第 2 章 GONAD 法による KO マウスの作製 ··· 40 第 1 節 緒論 ··· 40 第 2 節 材料および方法 ··· 42 1. マウス ··· 42 2. マウスの過排卵処置 ··· 42 3. RNA 合成 ··· 42 4. 卵管注入用 RNA 溶液の調製 ··· 42 5. 卵管への RNA 溶液の注入 ··· 42 6. in situ エレクトロポレーション ··· 43 7. eGFP の蛍光観察 ··· 43 8. 変異解析 ··· 43 第 3 節 結果 ··· 45 1. in situ エレクトロポレーションによる eGFP KO 妊娠中期胎仔の作出 ··· 46 2. in situ エレクトロポレーションによる eGFP KO 妊娠後期胎仔の作出 ··· 50
第 4 節 考察 ··· 54 総括 ··· 56 参考文献 ··· 58 謝辞 ··· 66 要約 ··· 67 Summary ··· 69
1 序章
トランスジェニック(Transgenic, Tg)マウスの作製は、1980 年に Gordon らによって初め て報告された[Gordon et al., 1980]。Tg マウスはマウス受精卵の前核へプラスミド DNA を顕 微注入し、注入卵を偽妊娠雌マウスへ移植することにより作出される。導入遺伝子(トラ ンスジーン)はゲノム DNA に挿入され、受精卵の分裂とともに体細胞すべてに分布し、さ らにトランスジーンはメンデル則に従い、子孫へ遺伝する。その後、ラット由来の成長ホ ルモン遺伝子(Growth hormone 1, Gh1)をマウスへ導入した Tg マウス(スーパーマウス) が樹立され、このトランスジーンから転写・翻訳された成長ホルモンが高濃度で血中から 検出された。スーパーマウスは通常の 2 倍以上の生育速度を示す大型のマウスとなり、導 入された遺伝子産物が in vivo で機能することが示された[Palmiter et al., 1982]。これらの報 告以降、ウサギ、ヒツジ、ブタおよびウシなどにおいても Tg 動物が作製されるようになっ た[Hammer et al., 1985 ]。現在、これらの Tg 技術は、遺伝子の機能解析などの基礎生物学的・ 医学的研究のみならず、家畜育種への応用や、さらには人類に有用な物質生産のために利 用され、それを用いた研究が盛んに進められている。 これまで、Tg マウスの作製の基幹となる遺伝子導入法には、いくつかの手法が考案・実 用化されており、透明帯を除去した初期胚にレトロウイルスベクターを感染させる遺伝子 導入法[Jaenisch 1976; Stewart et al., 1987; Tsukui et al., 1995; Tsukui et al., 1996; Lois et al.,
2 2002]、トランスジーンが導入された ES 細胞を胚盤胞期胚へ移植し、作出されたキメラマ ウスをもとに交配によって Tg マウス系統を樹立する方法[Capecchi, 2005]などがあるが、前 述した顕微注入法(マイクロインジェクション)が最も簡便で効率の良い、確実な手段と して多くの研究で用いられている[Sato et al., 2016]。顕微注入法による Tg マウス作製では、 受精卵前核に顕微注入された DNA が染色体上にランダムに組み込まれることを利用して遺 伝子を導入する。この方法では、導入した DNA がゲノム上のランダムな位置に挿入される ものの、トランスジーン内に cDNA 発現を駆動するための遺伝子発現制御領域(プロモー ター)、遺伝子発現増強因子(エンハンサー)、および mRNA 合成停止シグナルである(ポ リ A 付加シグナル)などを配置することで、目的遺伝子(cDNA あるいはゲノム遺伝子) の発現を誘導することが可能である[Potts et al., 2000; Giralod et al., 2003]。しかしながら、導 入される遺伝子のコピー数を制御できないこと、導入された遺伝子が染色体上の位置に影
響を受けること(位置効果)、トランスジーンのランダム挿入により内在性遺伝子の機能が
破壊されること、多コピートランスジーンのタンデム挿入でトランスジーンの遺伝子サイ レンシングが誘導されることなどの問題も存在する[Gordon et al., 1980; Brinster et al., 1981; Jasin et al., 1996; Garrick et al., 1998]。一方で ES 細胞を介する場合、相同組換え(Homologous recombination)に基づく遺伝子ターゲティング法によって、部位特異的に遺伝子を破壊 (Knock-out, KO)または導入(Knock-in, KI)することが可能である[Hopper et al., 1987]。し かし、遺伝子ターゲティング法は相同組換えの頻度が低いうえに、ターゲティングベクタ
3
ーの構築、ターゲティングされた ES clone のスクリーニング、ES 細胞 clone の未分化状態 での維持、キメラマウスの作出および繁殖などに多大な時間、コスト、労力を要し、必ず しも目的の遺伝子を導入・破壊した遺伝子改変マウスが得られるとは限らないことも報告 されている[Jasin et al., 1996; Bronson et al., 1996; Sorrell et al., 2005]。
近年、ゲノム編集(Genome editing)技術と呼ばれる遺伝子改変技術が注目されている。 その一つである Clustered Regularly Interspaced Short Palindromic Repeats / Crispr associated protein 9(CRISPR/Cas9)は、従来の ES 細胞ベースの KO や KI マウスの作製法よりも、簡 便かつ短期間で KO/KI マウスを作出することを可能とした[Fujii et al., 2013; Harms et al., 2014]。CRISPR は細菌のもつ獲得免疫機構であり、1 度目の感染の際に外来性 DNA を断片 化して自らのゲノムに組み込み、2 度目の感染時に組み込まれた外来性 DNA 断片から転写 された 2 種の小分子 RNA を外来性 DNA に認識させ、その配列を Cas9 ヌクレアーゼによっ て切断することで外来性 DNA を不活化する[Barrangou et al., 2007]。この仕組みを利用した CRISPR/Cas9 によるゲノム編集は、Cas9 をコードする mRNA または DNA と標的遺伝子と 相補的な塩基配列を有する single guide (sg) RNA(2 種の小分子 RNA のキメラ)を受精卵へ 顕微注入することで、標的ゲノム DNA を配列特異的に Cas9 ヌクレアーゼが切断し、この 二本鎖 DNA 切断(double strand break, DSB)、続く DNA 修復機構によって生じる欠失/挿入 変異 (insertion/deletion mutation, indel mutation)を標的遺伝子に発生させる技術である[Wang
4
注入することで、遺伝子 KI を生じさせることも可能となっている[Yang et al., 2013; Hsu et al., 2014]。さらに、CRISPR 系の核酸を受精卵へと顕微注入する場合、従来のように受精卵前 核ではなく細胞質への導入が効果的であることが示され、顕微注入の技術にも簡便化が図 られている[Horii et al., 2014]。しかしながら、CRISPR/Cas9 による遺伝子 KO/KI を実施する には、1) 過排卵処理して雄マウスと交配させた雌マウスからの受精卵の採卵、2) 核酸 溶液の受精卵への顕微注入、3) 顕微注入受精卵の偽妊娠マウスへの移植、などの従来の Tg マウス作出法と同様の ex vivo 処理が不可欠である[Harms et al., 2014]。
ex vivo 処置をバイパスするための方法として、Sato らは in vivo エレクトロポレーション
を用いたアプローチである gene transfer to the oviductal epithelium(GTOVE)法を報告した [Sato, 2005; Sato et al., 2012]。この方法は、レポーター遺伝子(EGFP または β-galactosidase) 発現カセットを含むプラスミド DNA 溶液を妊娠マウス卵管内へ注入し、直後に卵管全体に エレクトロポレーションを実施することで卵管内の胚(2 細胞期胚)に遺伝子導入を果たす ものである。しかし、導入されたトランスジーンの発現は一過的で、さらにゲノム DNA へ の挿入も確認されなかった。そこで筆者は、GTOVE 法が卵管内の受精卵に DNA を導入可 能であるという点に着目し、CRISPR/Cas9 をこの実験系に適用させることを考えた。GTOVE 法は、導入核酸が胚ゲノムへ挿入こそされないが、導入 DNA の発現カセットから転写され た RNA が機能性タンパク質として翻訳される。したがって、GTOVE 法に CRISPR 系 RNA を用いれば、それから機能性タンパクが発現され、着床前胚(2 細胞期胚)のゲノム編集が
5
可能となることが期待される。言い換えれば、GTOVE 法に CRISPR 系 RNA を用いれば、 一切の ex vivo 処置を必要とせずに遺伝子改変マウスを作出できることになる。
本研究では、筆者はこの GTOVE 法に CRISPR 系 RNA を適用させた系を新たなゲノム編 集マウス作製法と捉え、ex vivo 処置を介さない新規ゲノム編集マウス作製法として 「Genome-editing via Oviductal Nucleic Acids Delivery(GONAD)法」と命名した。本論文で は、第 1 章では、卵管を介した in vitro / in situ エレクトロポレーションにより卵管内 2 細胞 期胚への RNA 導入が可能であること、CRISPR 系 RNA の導入によるゲノム編集も可能であ ることを述べる。第 2 章では、GONAD 後の胚発生能およびゲノム編集マウス作出の可能性 を検討した。
6
第1章 エレクトロポレーションによる卵管内 2 細胞期胚への RNA 導入法の確立
第 1 節 緒論
ゲノム編集技術には、第 1 世代の Zinc Finger Nucleases(ZFNs)、第 2 世代の Transcription Activator-Like Effector Nuclease(TALEN)、そして第 3 世代の技術として CRISPR/Cas9 があ る。特に CRISPR/Cas9 系では、マウス受精卵に CRISPR 関連 DNA/RNA または sgRNA と Cas9 タンパク質のリボ核タンパク質(Ribonucleoprotein, RNP)複合体を導入するだけで遺 伝子改変マウスを作出可能であり[Wang et al., 2013; Chen et al., 2016]、さらに、ヒト細胞 [Jinek et al., 2012]、酵母[DiCarlo et al., 2013]、植物[Li et al., 2013]、ゼブラフィッシュ[Hwang
et al., 2013]、ショウジョウバエ[Yu et al., 2013]、線虫[Friedland et al., 2013]など多くの生物種
にも適用可能であることから、ゲノム編集におけるもっとも一般的な方法となりつつある [Hsu et al., 2014]。また、近年、顕微注入法ではなく in vitro エレクトロポレーションを介し てラット着床前胚でのゲノム編集を可能とした方法が報告された [Kaneko et al., 2014; Kaneko and Mashimo, 2015]。さらに、この後、同様の報告がマウスでもなされた[Hashimoto et
al., 2015; Qin et al., 2015]。これらの研究は、CRISPR 系 RNA 溶液が含まれたチャンバー型電
極内で受精卵にエレクトロポレーションを実施したもので、顕微注入ステップのみの代替 法である。したがって、胚操作や胚移植などは従来通り実施する必要があり、ex vivo 処置 を必要としないゲノム編集マウス作製法の開発という観点では、まだ不足感がある。筆者
7
は、GTOVE 法を用い卵管内受精卵への RNA 導入が可能であれば、ex vivo 処置が不要とな り、より簡便なゲノム編集マウスの作製が可能となると考えた。
今回、GONAD 法の開発に向け、エレクトロポレーションによる卵管内着床前胚への RNA 導入が可能かを検討した。この場合、1 細胞期胚を取り囲む卵丘細胞が核酸の導入を困難に することから、2 細胞期胚を対象とした[Sato et al., 2012]。具体的には、Enhanced GFP(eGFP)
mRNA の卵管内注入、続く in vitro/in situ エレクトロポレーション、Cas9 mRNA および sgRNA
8 第 2 節 材料および方法
1. マウス
実験に用いたマウス ICR(10 – 18 週齢)および C57BL/6N(8 – 12 週齢)は、実験動物生
産・販売業者(日本クレア)から購入し、東海大学医学部動物施設 Center of Genetic Engineering
for Human Diseases(CGEHD)で飼育維持された。eGFP Tg マウス系統は、大塚らによって 樹立・維持されたもので、Rosa26 遺伝子座位に eGFP 発現カセットが 1 コピー導入されて いる(Ohtsuka et al., 2010)。動物実験は東海大学動物実験委員会規定のガイドラインに従っ て実施された(認可番号: #143037)。
2. マウスの過排卵処置
妊馬血清性性腺刺激ホルモン(PMSG; Nippon Zenyaku Kogyo Co., Ltd., Fukushima, Japan) およびヒト絨毛性ゴナドトロピン(hCG; Nippon Zenyaku Kogyo)は、生理食塩水を用いて それぞれ 7.5 IU/0.2 ml となるように調製後、-80 °C で保存した。PMSG は、16:00 に 0.2 ml を 雌マウスに腹腔内投与し、48 時間後、hCG 0.2 ml 腹腔内投与した。hCG 投与後、雄マウス と交配させ、翌日、膣栓が観察された日を受胎 0.5 日(Day 0.5)とした。膣栓確認マウスは、 その翌日(Day 1.5)に供試した。
9 3. RNA 合成
Cas9 mRNA および eGFP mRNA の合成には、それぞれ pBGK(Addgene plasmid number: #65796)[Harms et al., 2014]および pcDNA3.1EGFP-poly(A83)[Yamagata et al., 2005]を用い た。両プラスミドは、Xba I(TaKaRa Biomedicals Co., Shiga, Japan)により直鎖化後、フェノ ー ル /クロロホルム/イソアミルアルコールおよびエタノール沈殿によって精製し、 RNase-free water(Ambion Co., Austin, TX, USA)に溶解した。RNA 合成には mMESSAGE mMACHINE T7 Ultra Kit(Ambion)を用い、精製後の直鎖化プラスミド DNA 1 g を鋳型に 付属プロトコルに従い、in vitro Transcription にて mRNA を合成した。合成後の mRNA は MEGAclear Kit(Ambion)によって精製した。
eGFP を標的配列とする sgRNA は先行研究による sgGFP.NT2 を基に設計した[Gilbert et al., 2013]。また、hypoxanthine guanine phosphoribosyl transferase(Hprt)および eukaryotic translation
elongation factor 2(eEF2)遺伝子における標的配列は CRISPR design(http://crispr.mit.edu/)
[Ran et al., 2013; Haems et al., 2014]または CHOPCHOP(https://chopchop.rc.fas.harvard.edu/) [Montague et al., 2014]を用いてデザインし、高い特異性を示す sgRNA を選択した。sgRNA 合成用の鋳型は、pUC57-sgRNA vector(Addgene plasmid number: #51132)[Shen et al., 2014] を用いて KOD-Plus-Neo(TOYOBO Co., Ltd, Tokyo, Japan)による PCR 反応により増幅した。 プライマーは Table 1 に記載したセットを用い、PCR 産物は電気泳動による分離後、切り出 したゲル片をフェノール抽出およびエタノール沈殿によって精製した。sgRNA 合成は、鋳
10
型 DNA(400-500 ng) 用い MEGAshortscriptTM T7 Kit(Ambion)にて合成し、MEGAclear Kit
11 4. 卵管注入用 RNA 溶液の調製
12
卵管内へ注入する eGFP mRNA、CRISPR 系 RNA は、Table 2, 3, 4, 5 に記載された最終濃 度となるよう RNase -free water(Nacalai Tesque, Inc., Kyoto, Japan)を用いて調製した。これ らの溶液は卵管注入を容易にモニターできるトリパンブルー(Nacalai tesque)を最終濃度 0.05%となるよう混合した。混合後の RNA 溶液は 4 l ずつ 1.5 ml チューブに分注して、使 用直前まで-80 °C で保存した。 5. 卵管への RNA 溶液の注入 導入麻酔はプロピレングリコールで 20%に希釈したイソフルラン入りの麻酔瓶に短時間 妊娠マウス(Day 1.5;13:00 頃;2 細胞期胚に相当)を置き、麻酔が効くまで放置した。導 入麻酔後、維持麻酔用器具をマウスの鼻(顔全体)にセットし、37 °C の Micro-warm-plate (KM-1; Kitazato Co., Tokyo, Japan)上へ移して保温した(Fig. 1A)。次に、Fig. 1B および 1C に示した手順に従って体外に卵管を露出させた。RNA 溶液の注入は、萎縮した卵管膨大部
から卵管采にかけての領域に対して、ガラスピペット(Fig. 2A)を用い実体顕微鏡下(SZ11;
Olympus Co., Tokyo, Japan)で注入した(Fig. 1D)。また、ガラスピペット(Glass: # GDC-1;
Narishige Co., Tokyo, Japan)は、Puller(P-97/IVF; Sutter Instrument Co., Novato, CA, USA) を用いて Heat: 790; Pull: 35; Velocity: 50; Pressure: 500; Time: 100 の条件で作製し、先端約 1-5 mm を眼科用ハサミで実体顕微鏡下切断した。
13
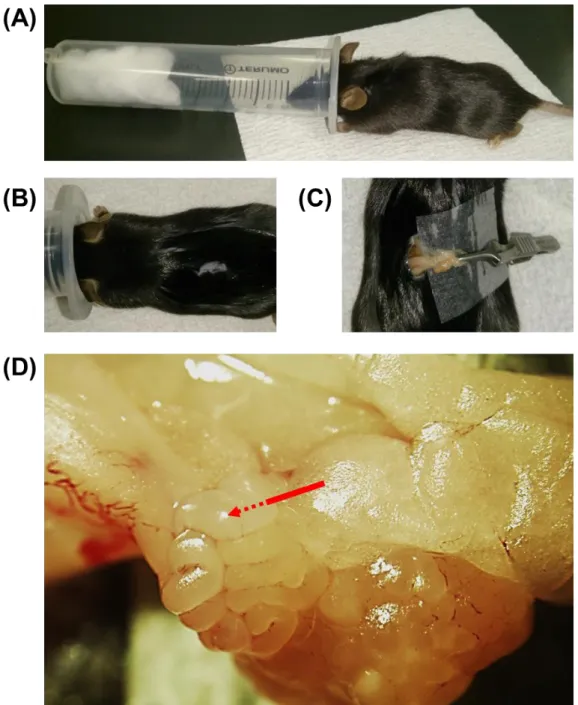
Fig. 1. Surgical exposure of oviducts for GONAD procedure.
(A) Disposable 35-ml syringe (right top) and the anesthetized mouse from the jar transferred to the syringe photographed just prior to surgery. (B) Using the scissors, make a dorsal incision (5 to 10-mm long) at the central portion of the back. (C) Grasped adipose tissue the adipose tissue surrounding the ovary with the forceps and pull the tissue outside of the incision. (D) Higher magnification of the exposed tissues showing the point of instillation in a red arrow, and schematic of the instillation needle is shown as a dotted line from the point of instillation.
14 Fig. 2. Intraoviductal instillation of nucleic acids.
(A) Higher magnification of the part of the installation needle filled with nucleic acids solution with trypan blue dye. (B) Oviduct showing the correctly instilled nucleic acids solution.
15 6. in vitro / in situ エレクトロポレーション
エレクトロポレーターは Square-wave pulse generator(BTX Electroporation Cuvettes Plus; BTX Genetronics Inc., San Diego, CA, USA)を用いた。in vitro エレクトロポレーションのス
テップでは、RNA 溶液を卵管に注入後 (Fig. 2B)、卵管全体を切除し、80 l の Hepes-buffered saline(HBS; Applichem Co, Darmstadt, Germany)が入った 1-mm Gap のキュベット(BTX Electroporation Cuvettes Plus; BTX Genetronics)(Fig. 3A)に移し、エレクトロポレーション を実施した。エレクロポレーション条件は、電圧 50 V、Pulse On 5 msec、Pulse Off 1 sec を 8 回リピートした。エレクトロポレーション直後に、M2 培地(Sigma-Aldrich Co., St Louis, MO,
USA)を用いた卵管潅流によって着床前胚を回収し、CO2 インキュベーターで平衡化した
KSOM 培地(ARK Resource, Kumamoto, Japan)中で 2 日間培養した。
in situ エレクトロポレーションは、RNA 注入直後(Fig. 2B)に卵管全体を 1x Phosphate
buffered saline(PBS)で浸した 100 x 100 mm サイズのキムワイプ片(Kim wipe; Jujo-Kimberly Co. Ltd., Tokyo, Japan)で被覆し、次いで、オーダーメイドのロッド型の電極(#CT-234; NEPA
GENE Co. Ltd., Ichikawa, Chiba, Japan)で卵巣/キムワイプを挟み(Fig. 3B)、直ちに、エレ
クトロポレーションを実施した。パルス設定は、前述した in vitro エレクトロポレーショ ンと同じ条件を用いた。エレクトロポレーション後、Suture wound clips(MikRon 9-mm autoclip; Becton Dickinson and Co, Franklin Lakes, NJ, USA)で皮膚切開部を閉じ、麻酔覚醒 後、安静な環境下で飼育した。その後、Day 2.5 でマウスを安楽死させ、卵管切除後、M2
16
培地による潅流によって胚を回収。平衡化した KSOM 培地へ胚を移し、CO2 インキュベ
17 Fig. 3. Electrodes used for electroporation.
(A) Schematic of 1-mm gap cuvette. (B) Tissues held between the electrodes showing the oviduct instilled with nucleic acids solution and covered with 1 to 2 layers of pre-cut KimWipe towels soaked in 1xPBS.
18 7. eGFP 蛍光観察
エレクトロポレーション後の切除卵管および培養した着床前胚における蛍光観察は、蛍 光顕微鏡(BIOREVO BZ-9000; KEYENCE Co., Osaka, Japan)を用いて行った。また、eGFP 陽性卵管は+と表記した(Table. 3)。なお、過排卵処理と交配のみを行った個体を未処置コ ントロールとし、eGFP 蛍光の有無は蛍光強度をこの未処置コントロールと比較することで 判断した。
8. 変異解析
着床前胚からのゲノム DNA は、All-In-One Mouse Tail Lysis Buffer(ABP-PP-MT01500; KURABO Co., Osaka, Japan)を用い、60 °C で 3 時間以上インキュベートした後、95 °C で
10 min 加熱することで抽出した。1st PCR は、全量 10 l の反応系として、1X PCR buffer、
0.25 U/ l TaKaRa LA-Taq(TaKaRa Biomedicals)および 1 l 鋳型 DNA 溶液を含むように調 製した。また、プライマーは Table 1 に記載したセットを用い、PCR 反応は、96 °C 1 min で 初期変性、96 °C 30 sec および 66 °C 1.5 min の 2 ステップを 24 サイクル行い、68 °C 7 min で最終伸長させた。つづく Nested PCR は、全量 10 l の反応系として、1X PCR buffer、0.25 U/ l TaKaRa r-Taq(TaKaRa Biomedicals)および 1 l 1st PCR 産物を含むように調製した (eEF2 を標的とした Nested PCR では、鋳型 DNA 溶液を D.W.で 100 倍希釈して使用)。 ま た、プライマーは Table 1 に記載したセットを用い、PCR 反応は、95 °C 5 min で初期変性、
19
95 °C 45 sec、58 °C 30 sec および 72 °C 1 min の 3 ステップを 35 サイクル行い、72 °C 5 min で最終伸長させる条件で実施した。
Surveyor assay は、SURVEYOR Mutation Detection Kit(Transgenomic, Omaha, NE, USA)を 用いて、5 l の Nested PCR 産物を付属のプロトコルに従って解析した。T7 endonuclease 1 (T7E1)assay は、5 μl Nested PCR 産物に 1 l 10X NEB Buffer 2(New England Biolabs Japan Inc., Tokyo, Japan)、0.2 l T7E1(2 unit; NEB)および 3.8 l の D.W.を加えて混合し、37 °C で 30 min 間反応させた。 反応後は、2%アガロースゲルによる電気泳動を行い、SYBR Gold (Life Technologies, Inc., Grand Island, NY, USA)で染色後、泳動図を記録した。
20 第 3 節 結果
1. in vitro エレクトロポレーションによる卵管内 2 細胞期胚への eGFP mRNA 導入 まず、卵管内に注入された eGFP mRNA が透明帯を有する 2 細胞期胚内に導入され、eGFP 蛍光タンパクが発現されるかを検討した。Fig. 1A-D に示した手順によって、1 個体の雌マ ウスに eGFP mRNA を含む溶液を卵管内に注入後(Fig. 2B)、直ちに、卵管全体を切除し、 これをキュベットに投入し、in vitro エレクトロポレーションを実施した(Fig. 3A)。エレク トロポレーション後、卵管潅流によって胚を回収し、2 日間培養後、eGFP 蛍光を観察した。 エレクトロポレーション後回収した胚の総数は 27 個であり、そのうち 16 個の胚が正常に 胚盤胞期胚まで発生し、その 25%(4/16)において eGFP 蛍光が観察された(Fig. 4; Table 2)。 また、8-16 細胞期胚での eGFP 蛍光は、モザイク的発現であった(Fig. 4)。
以上から、in vitro エレクトロポレーションにおいても卵管内着床前胚に RNA を導入でき、 導入された RNA は機能性タンパク質として発現されることが判った。
22
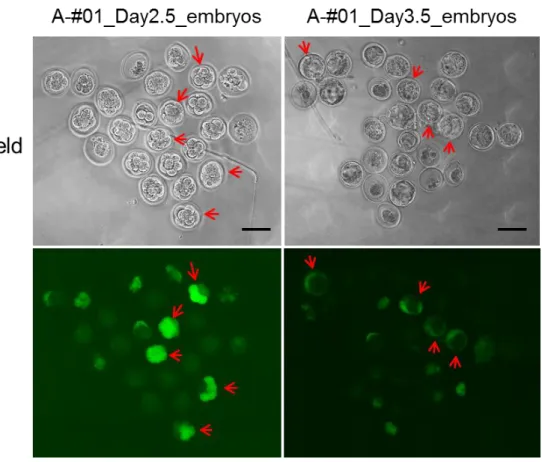
Fig. 4. Enhanced GFP expression in day 2.5 to day 3.5 stage embryos recovered from the dissected oviducts that were in vitro electroporated with eGFP mRNA.
The embryos were derived from A-#01 mouse (C57BL/6N strain) shown in Table 2. The embryos exhibiting fluorescence throughout are shown with red arrows. Scale bar = 100 μm.
23
2. in situ エレクトロポレーションによる卵管内 2 細胞期胚への RNA 導入
次に、卵管を切除せずに in situ の条件下で卵管内 2 細胞期胚に対して RNA を導入できる かを検討した。前項と同様、卵管内に eGFP mRNA を含む RNA 溶液を注入後、ロッド型電 極を用いた in situ エレクトロポレーションを実施した(Fig. 3B)。実験は 6 個体のマウスを 用い(計 12 卵管)、エレクトロポレーションの翌日(Day 2.5)に卵管から胚(4~8 細胞期胚 相当)を回収した(Table 3)。まず、卵管での eGFP 蛍光を観察した結果、蛍光強度、蛍光 発現部位は卵管毎に多少異なっていたが、すべての卵管に明確な蛍光が観察された(Fig. 5; Table 3)。また、回収された胚 52 個(総数)の内 23 個が正常な形態を示し、残る 29 個は異 常な形態であった。23 個の正常胚の内 6 個(26.1%)は eGFP 蛍光を示したが、その蛍光強 度は個々の胚で差異があった(Fig. 6; Table 3)。Fig. 6A には蛍光強度の異なる胚が示される。 赤矢印は強い蛍光を示す胚、青矢印は中程度の蛍光を示す胚を示している。また、Day 3.5 (胚盤胞期相当)での観察では、内部細胞塊(ICM)に均一な蛍光が見られた(Fig. 6B)。 一方、コントロール群では、eGFP 蛍光は全く観察されなかった(Fig. 6C)。以上から、in situ エレクトロポレーションによって外部から投じた mRNA を卵管内の 2 細胞期胚に導入する ことが可能であることが示された。
25
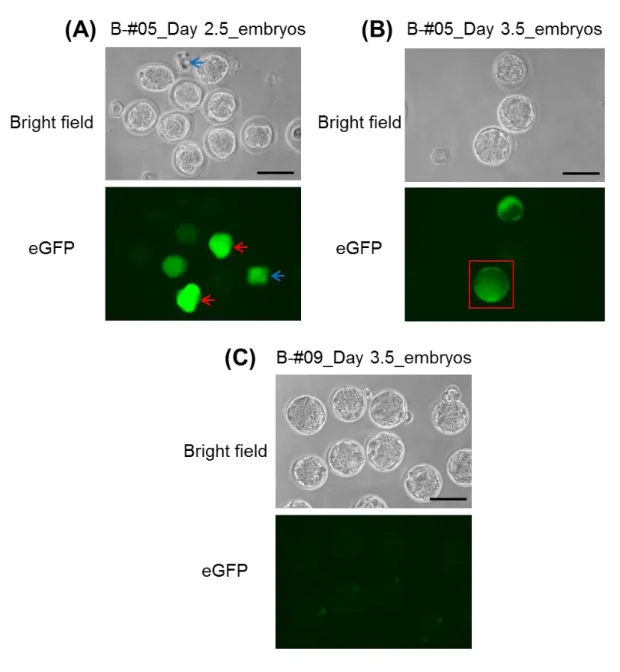
Fig. 5. Enhanced GFP expression in the oviducts following the GONAD procedure.
eGFP fluorescence in the oviducts dissected and analyzed one day after the procedure (from mice B-#05, see Supplementary Table 3). Scale bars = 1 mm (left side panel), 100 μ m (right side panel).
26
Fig. 6. Enhanced GFP expression in the embryos following the GONAD procedure.
(A) eGFP fluorescence in 8- to 16-cell stage embryos recovered from dissected oviducts shown in Fig. 5. The microscopic field shows the representative zygotes with bright (red arrows) and moderate (blue arrows) eGFP fluorescence. (B) eGFP fluorescence in blastocysts derived from embryos in (A) after one day-culturing, showing a normal looking embryo with uniform fluorescence (red box). (C) eGFP fluorescence in morula to blastocyst stage control embryos recovered from a super-ovulated female not subjected to GONAD procedure (B-#09). Scale bars = 100 m.
27
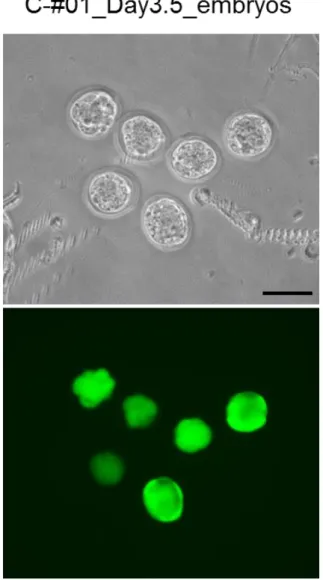
3. in vitro エレクトロポレーションによる卵管内 2 細胞期胚への CRISPR 系 RNA の導入 次に、CRISPR 系 RNA 導入による着床前胚におけるゲノム編集の可能性について検討を 行った。CRISPR 系 RNA、eGFP mRNA を含む RNA 溶液を前項と同様の方法で卵管内に注 入し、その後、卵管を切り出し、キュベット内での in vitro エレクトロポレーションを実施 した(Fig. 3A)。Hprt および eEF2 遺伝子を内在性遺伝子の標的としたほか、eGFP Tg マウ スが有する eGFP 遺伝子も標的とした(Table 4)。Hprt 遺伝子および eEF2 遺伝子について
は、sgRNA をそれぞれ 2 種設計し、eGFP については 1 種を設計した。交配に用いた雄の eGFP
Tg マウスは、広範囲の胎性組織で eGFP 遺伝子が発現する[Ohtsuka et al., 2010]。エレクトロ ポレーション後、卵管から回収した胚は胚盤胞期胚まで培養し、eGFP 蛍光、胚の形態を観 察した。その後、ゲノム DNA を胚盤胞期胚から抽出し、T7E1 または Surveyor assay によっ て indel 変異をスクリーニングし、シークエンスによって標的遺伝子領域の塩基配列を決定 した。その結果、変異解析から indel 変異は各標的遺伝子について最低 1 種確認された(Fig.
8, 10, 11; Table 4)。また、eGFP 遺伝子については eGFP 蛍光が検出された胚のみに indel 変
異が検出された(Fig. 8)。さらに、eGFP 遺伝子を標的とした胚では、eGFP 蛍光の減弱が観 察され(Fig. 11A)、シークエンス解析からゲノム編集の成功を示唆する indel 変異が検出さ れた(Fig. 11B)。
以上から、CRISPR 系 RNA を注入された卵管の in vitro エレクトロポレーションにより卵 管内 2 細胞期胚でのゲノム編集が可能であることが示された。
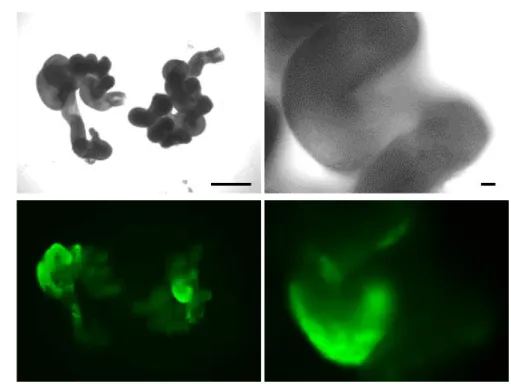
29 Fig. 7. Enhanced GFP fluorescence in the blastocysts.
One day after culturing of embryos recovered from an in vitro electroporated mouse with eGFP mRNA (used as an indicator of electroporation), Cas9 mRNA and Hprt_Cr1 sgRNA (C-#01 mouse: Table 4). Scale bar = 100 m.
30
Fig. 8. CRISPR/Cas9-mediated disruption of Hprt locus achieved through in vitro electroporation of oviducts.
(A) Agarose gel electrophoresis of a few Surveyor-treated PCR products derived from eGFP fluorescence-positive embryos (C-#01_No.36, 42, 43 and 44). The red arrows indicate the cleavage products generated in Surveyor assay and the wild-type sized band is indicated by a black arrow M: 100-bp DNA ladder markers. N.C: a Negative Control embryo derived from a non-instilled and non-electroporated mouse. (B) Direct sequencing of the PCR products from wild-type mouse or a mutated blastocyst (C-#01_No.42, shown in (b). The black arrow below the electropherogram shows overlapping peaks indicative of indel mutations.
31
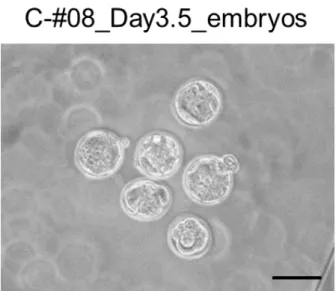
Fig. 9. Bright field micrograph of embryos collected and cultured for two-days after in vitro electroporation of CRISPR/Cas9 RNAs against the eEF2 gene.
eGFP mRNA, as an indicator of electroporation, was not included in this experiment. The embryos from two different mice (C57BL/6N strain) are shown: the upper panel embryos were recovered from the C-#08 mouse (Table 4) and the bottom panel embryos were recovered from the C-#09 mouse. Scale bars = 100 m.
32
Fig. 10. CRISPR/Cas9-mediated disruption of eEF2 locus achieved through in vitro electroporation of oviducts.
(A) Agarose gel electrophoresis of T7E1-treated PCR products derived from the embryos shown in Fig. 9. The red arrows indicate the cleavage products generated in T7E1 assay and the wild-type sized band is indicated by a black arrow. D.W.: distilled water used as negative control. M: 100-bp DNA Ladder. (B) Direct sequencing of the PCR products of the target region amplified from samples C-#09_No.14 and _No.16. The black arrow below the electropherogram shows overlapping peaks indicative of indel mutations.
33
Fig. 11. CRISPR/Cas9-mediated disruption of eGFP transgene achieved through in vitro electroporation of oviducts.
(A) eGFP fluorescence in the blastocysts, two-days after culturing of embryos recovered from an in
vitro electroporated mouse (C57BL/6N strain mated with hemizygote eGFP Tg males) with Cas9
mRNA, and eGFP sgRNA. Typical embryo from two different mice are shown: the upper panel embryo showing normal eGFP fluorescence recovered from the C-#12 mouse (Table 4) and the bottom panel embryo showing greatly reduced fluorescence was recovered from the C-#13 mouse. Scale bars: 100 m. (B) Direct sequencing of the PCR product amplified from the target region from bottom panel’s embryo in (A). The black arrow below the electropherogram shows overlapping peaks indicative of indel mutations.
34
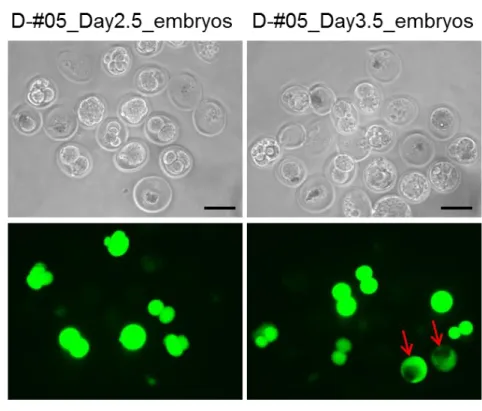
4. in situ エレクトロポレーションによる卵管内 2 細胞期胚への CRISPR 系 RNA の導入 次に、in situ エレクトロポレーションによる卵管内 2 細胞期胚のゲノム編集が可能である かを検討した。この場合、indel 変異誘導効率を高めるため、RNA 濃度を約 2 倍に調製した (Table 5)。in situ エレクトロポレーションを実施した翌日(Day 2.5)に胚を回収し、胚盤 胞期まで培養した。胚からはゲノム DNA を抽出し、T7E1 による変異のスクリーニングと シークエンスによる塩基配列の決定を行った。
計 6 個体のマウスに Cas9 mRNA、Hprt_Cr1_sgRNA、eGFP mRNA を含む RNA 溶液を導入 した結果、5 個体から回収された胚に eGFP 蛍光が検出された(Table 5)。また、正常に発生 した 49 個の胚(桑実胚または胚盤胞期胚)の内 6 個(12%)に eGFP 蛍光が観察された(Fig.
12; Table 5)。さらに、T7E1 assay、塩基配列決定から蛍光を示した 6 個の胚の内 5 個(83%)
に標的遺伝子における indel 変異が検出された(Fig. 13)。これまでの実験結果と同様に、eGFP 蛍光を示す胚のみに indel 変異が検出された(Table 5)。
36
Fig. 12. Enhanced GFP expression in the embryos following the GONAD procedure.
eGFP fluorescence at the 8-cell to 16-cell embryos (left side panels) and blastocyst embryos (right side panels) that were collected from GONAD procedure performed using eGFP mRNA (used as an indicator of electroporation), Cas9 mRNA and Hprt_Cr1 sgRNA (Table 5). Scale bars = 100 m.
37
Fig. 13. CRISPR/Cas9 mediated disruption of Hprt locus using the GONAD system.
(A) Agarose gel electrophoresis of T7E1-treated PCR products derived from 7 selected blastocysts. The red arrows indicate the cleavage products generated in T7E1 assay and the wild-type sized band is indicated by a black arrow. M: 100-bp DNA ladder marker. (B) Direct sequencing of PCR products amplified from the target region from wild-type, D-#05_No.5, _No.6, and _No.7 samples. The black arrow below the electropherogram shows overlapping peaks indicative of indel mutations.
38 第 4 節 考察 in vitro エレクトロポレーション法を用いたマウス着床前胚への遺伝子導入は、Grabarek et al.(2002)によって初めて報告された。一般的に、透明帯を除去しない場合、遺伝子導入 効率は低下する。しかし、遺伝子導入された透明帯除去胚を更に発生させるため偽妊娠仮 親卵管に移植しても、このような胚、特に胚盤胞期前までの分割期胚は、卵管内壁に吸着 されることから妊娠率が低下する[Modliński, 1970; Bronson and McLaren, 1970]。そこで
Grabarek らは(2002)、酸性タイロード液(Acidic Tyrode’s solution)を用いて透明帯を脆弱
化した上でエレクトロポレーションを行うことにより核酸導入効率の改善を図った。しか し近年、透明帯に特別な処理を行わずにラット受精卵を in vitro エレクトロポレーションに 付した結果、生存率 91%、ZFNs によるゲノム編集の成功率 73%という成果が報告され、こ の方法は Technique for Animal Knockout system by Electroporation(TAKE:テイク法)と命名さ れた[Kaneko et al., 2014; Kaneko and Mashimo, 2015]。テイク法では NEPA 21(Nepa Gene)と
いう新型のエレクトロポレーターを用い、この機器では、3 ステップ(Poring pulse, 1st transfer
pulse, 2nd transfer pulse)の特別なパルス条件が設定されていた。この一連の条件は、エレク
トロポレーション時の加熱による検体へのダメージ低減を企図したものである。しかしな がら、この電気的条件は、NEPA21 エレクトロポレーターの性能に依存しているため、すべ てのエレクトロポレーターでこの条件を再現することは困難である。一方、Hashimoto and Takemoto(2015)は、BEX 社の CUY21 EDIT II エレクトロポレーターを用い ICR、B6D2F1 由来マウス胚での in vitro エレクトロポレーション時の電圧・パルス回数と CRISPR/Cas9 に
39 よるゲノム編集効率との関係性を検討した。その結果、良好な胚生存率と高い変異導入効 率は、電圧 30 V、7 回のパルス回数により達成されると結論付けている。本研究は、Sato(2012) の GTOVE 法に準じた条件(電圧 50 V、8 回のパルス回数)で実施された。因みに、Hashimoto and Takemoto(2015)に依れば、電圧 50 V での条件は胚の生存率が約 50%、パルス回数は 7 回を超えると生存率が低下するという。実際に本研究では、エレクトロポレーション後回 収された胚の中で明らかに異常な形態を示す胚が数多く観察された(Table 2, 3, 4)。したが って、GONAD 法におけるエレクトロポレーションのプロセスは、胚に物理的な損傷を引き 起こし、この損傷が最終的に遺伝子改変マウス作製効率低下を招くと推測された。しかし、 一方で、エレクトロポレーション後に回収した時点で正常と判断された胚の多くは、胚盤 胞期胚まで正常に発生した(Table 4)。これはエレクトロポレーションによる一時的なダメ ージを乗り越えれば、胚は正常に発生し得ることを示唆する。本研究では、エレクトロポ レーション条件の最適化を行っていないので、より最適化された条件を用いれば、胚の生 存率向上、CRISPR/Cas9 によるゲノム編集効率向上も可能であると考えられる。
40 第2章 GONAD 法による KO マウスの作製
第 1 節 緒論
遺伝子 KO マウスの作出を行う場合、従来の ES 細胞を介した遺伝子ターゲティング法に 代わり、近年、受精卵の細胞質/前核への CRISPR/Cas9 系核酸の顕微注入によるゲノム編集 法が広く用いられるようになってきた[Wang et al., 2013; Li et al., 2013; Harms et al., 2014]。し かし、このような斬新なテクノロジーを用いる場合でも、採卵、核酸の顕微注入、注入受 精卵の偽妊娠雌マウスへの移植という胚の ex vivo 操作が求められる。上述のように、これ らの ex vivo 操作は、マイクロマニュピレーターなどの高価な設備を必要とし、さらに顕微 注入法、胚操作、胚移植と言った高度で熟練した技術も求められる[Harms et al., 2014]。こ のような複雑な操作は、簡便にしかも効率的に遺伝子改変マウスを作製する場合のボトル ネックとなる。従って、上述のような高価な設備、技術に依存しないより簡便なゲノム編 集マウス作製法の開発が望ましい。 今回、第 1 章に記載した実験結果から、卵管内への核酸注入、続く卵管全体へのエレク トロポレーションは卵管内にある着床前胚への核酸導入に有効であることが判明したが、 回収胚の多くは異常発育を示した(Table 3, 5)。このようなエレクトロポレーションに起因 するであろう胚損傷は、正常な妊娠率を阻害する一つの要因となりえる [Grabarek et al., 2002; Hashimoto et al., 2015; Qin et al., 2015]。さらに、本研究に用いた C57BL/6 由来マウス
41
受精卵は、一般的に他の系統よりも外部からのストレスに弱いとされる[Auerbach et al., 2003; Fielder et al., 2010]。これらの点を踏まえ、エレクトロポレーション処理後の胚がどの 程度の発生能を示すのかについての検討が必要であると考えた。そこで、本研究の第 2 章 では、Cas9 mRNA、eGFP_sgRNA を用いた in situ エレクトロポレーション後の発生段階中 期(Day 13.5)、妊娠末期(Day 19.5)の胎仔に焦点を当てて、胎仔への発生能、ゲノム編集 効率を検討した。
42 第 2 節 材料および方法 1. マウス 実験に用いたすべてのマウスは、第 1 章第 2 節に記載した内容に従い、購入あるいは飼 育した。また、すべての動物実験は東海大学動物実験委員会規定のガイドライン(認可番 号: #143037)に従って実施された。 2. マウスの過排卵処置 過排卵処置の条件および方法は、第 1 章第 2 節記載の内容に準拠した。 3. RNA 合成
Cas9 mRNA、eGFP に対する sgRNA は、第 1 章第 2 節記載の内容に従い、作製した。
4. 卵管注入用 RNA 溶液の調製
卵管内へ注入する CRISPR 系 RNA は、第 1 章第 2 節記載の内容に従い、調製した。
5. 卵管への RNA 溶液の注入
RNA 溶液の注入ならびにガラスピペットの作製は、第 1 章第 2 節に記載した手順に従っ て実施した。
43 6. in situ エレクトロポレーション in situ エレクトロポレーションは、第 1 章第 2 節に記載した方法と同様の手順と条件で実 施し、Day 13.5、19.5 に安楽死させたマウスを開腹し、胎仔を回収した。また、Day19.5 胎 仔はナンバーの前に(#)と記載することで、Day 13.5 のものと区別した。 7. eGFP 蛍光観察
マウス胎仔における eGFP 蛍光観察は、蛍光実体顕微鏡(M165FC; Leica Microsystems GmbH, Wetzlar, Germany)を用いて観察した。
8. 変異解析
胎仔由来ゲノム DNA 抽出は、胎仔の一部を切り出し、これを All-In-One Mouse Tail Lysis Buffer(KURABO)を用い、60 °C で 3 時間以上インキュベートした後、95 °C で 10 分間加 熱することにより抽出した。PCR は、全量 10 l の反応系として、1X PCR buffer、0.25 U/ l TaKaRa r-Taq(TaKaRa Biomedicals)および 1 l の鋳型 DNA を含むように調製した。また、 プライマーは Table. 1 に記載したセットを用い、PCR 反応は、95 °C 5 min で初期変性、95 °C 45 sec、58 °C 30 sec および 72 °C 1 min の 3 ステップを 35 サイクル行い、72 °C 5 min で最 終伸長させる条件で実施した。T7E1 assay およびダイレクトシークエンスは、第 1 章第 2 節と同様の手順で実施した。また、Fig. 15 の胎仔 2(eGFP_No.2)および胎仔 3(eGFP_No.2)
44
における indel 変異を正確に決定するため、TOPO® TA Cloning Kit(Life Technologies)を用 いて PCR 産物を TA cloning vector にクローニングし、18 サンプル(胎仔 2)および 10 サン プル(胎仔 3)について標的配列を決定した(Fig. 19)。
45 第 3 節 結果
1. in situ エレクトロポレーションによる eGFP KO 妊娠中期胎仔の作出
エレクトロポレーションに起因する胚損傷が着床・胎仔発生へ及ぼす影響を検討するた め、Cas9 mRNA + eGFP sgRNA を含む RNA 溶液を予め eGFP Tg 雄マウスと交配させた妊娠 ICR 雌マウス卵管に注入後、直ちに in situ エレクトロポレーションを行い、その後、Day 13.5 まで妊娠を継続させて胎仔を回収した。3 個体の処置雌マウスの内 1 個体(1/3)で妊娠が 確認され、外貌的に正常な計 14 体の胎仔を回収することができた(左子宮角から 8 匹、右 子宮角から 6 匹)。この結果はエレクトロポレーション後、胚の着床、胚発生が正常に進行 することを示唆する。また、左子宮角から回収された 8 匹の胎仔はすべて強く均一な eGFP 蛍光を示した。これは、エレクトロポレーションの不発または eGFP 遺伝子改変の失敗を示 唆する。他方、右子宮角から回収された 6 匹の胎仔では、胎仔 1 と胎仔 4 が eGFP 蛍光の完 全な欠失、胎仔 2 と胎仔 3 が蛍光の減弱を示し、胎仔 5 と胎仔 6 は均一で明確な蛍光を示 した(Fig. 14A)。T7E1 assay では、胎仔 1 と胎仔 4 を除くすべての胎仔で eGFP 蛍光の減弱 と一致する結果が得られた(Fig. 14B)。加えて、胎仔 2 と胎仔 3 のゲル電気泳動図からは、 108、122-bp と推測される位置に切断された DNA 断片が検出され、さらに塩基配列決定に よって得られた波形は複数の混合したピークが検出され、indel 変異がモザイクに導入され ていた(Fig. 14B, 15, 16)。胎仔 5 と胎仔 6 には蛍光の変化が認められず、T7E1 assay の結果 も陰性で(Fig. 14B)、標的配列上の変異も検出されなかった(Fig. 15)。一方、完全な蛍光
46
欠失を示した胎仔 1、胎仔 4 では、T7E1 assay による変異は検出されなかったものの、塩基 配列決定から各々4-bp、3-bp の欠失が検出された(Fig. 15)。
47
Fig. 14. CRISPR/Cas9 - mediated genome editing using the GONAD system (day 13.5 fetuses) (A) The fetuses were isolated from one mouse at day 13.5 and analyzed for eGFP fluorescence using a fluorescence stereomicroscope. The fetuses 1 and 4 that lost eGFP expression completely (white arrows), the fetuses 2 and 3 exhibited greatly reduced eGFP expression and fetuses 5 and 6 had no loss of eGFP expression. (B) Agarose gel electrophoresis of T7E1-treated PCR products derived from fetuses shown in (a). The red arrows indicate the cleavage products generated in T7E1 assay and the wild-type sized band is indicated by a black arrow. D.W.: distilled water used as a negative control. M: 100-bp DNA ladder marker.
48
Fig. 15. Direct sequencing of PCR products amplified from the eGFP target region of all six fetuses (day 13.5).
The black arrows below the electropherogram (in samples 2 and 3) show overlapping peaks indicative of indel mutations. Note that there are three kinds of sequences: clear deletions (samples 1 and 4), mixed indels (samples 2 and 3) and no mutations (samples 5 and 6).
49
Fig. 16. Mutated eGFP alleles possessed in founder fetuses.
The changes in the nucleotide sequence are shown in red and the type of changes (insertions: + or deletions: Δ) are indicated on the right-side of the sequences. All fetuses had only one type of mutation except the fetus 7 of Figure 17 which was a mosaic with two mutant alleles (shown in a box).
50 2. in situ エレクトロポレーションによる eGFP KO 妊娠後期胎仔の作出 最後に、GONAD 処置胚が妊娠期間全体を通し生存・発生できるかを検証するため、妊娠 満期に近い Day 19.5 に帝王切開で胎仔を取り出し、解析を行った。なお、Day 19.5 胎仔は 前項の Day13.5 胎仔と区別するため、サンプル番号の前に#という記号を置いた。計 4 匹の 雌マウスに対して前項と同様に GONAD を行ったところ、1 匹(1/4)が妊娠を維持してお り、この雌マウスから外貌的に正常な Day 19.5 胎仔を回収した。得られた胎仔数は合計 11 体(左子宮角から 2 体、右子宮角から 9 体)であり、左子宮角から回収された胎仔はエレ クトロポレーションまたは遺伝子改変の失敗を示唆する eGFP 蛍光の安定的な発現が認め られた。一方、右子宮角から得られた 9 体の胎仔の内 2 体(#7 および#9)は、蛍光が完全 に失われており、胎仔#3 は蛍光が減弱していた(Fig. 17)。さらに T7E1 assay による解析か ら、胎仔#3 と胎仔#7 に変異が検出された(Fig. 18A)。しかし、胎仔#9 は当初 PCR 増幅産 物が認められなかったため、標的配列からさらに離れた領域を増幅しうるプライマーセッ トを用いたところ、標的領域の PCR 増幅に成功し、胎仔#9 は約 200-bp 程度の欠失を有して いることが判った(Fig. 18B)。塩基配列決定から、胎仔#3 と胎仔#7 は indel 変異をモザイ クに含んでいた(Fig. 19)。この点を更に解析すべく、PCR 断片をサブクローニングし、そ の clone を用いた塩基配列決定から、胎仔#3 には 3 塩基欠失および野生型配列のアレルが検 出され、胎仔#7 には野生型配列を欠いた 4 塩基欠失および 1 塩基挿入のアレルが検出され た(Fig. 16)。
51
Fig. 17. CRISPR/Cas9-mediated genome editing using the GONAD system (day 19.5 fetuses). The fetuses were isolated from one mouse at day 19.5 and analyzed for eGFP fluorescence using a fluorescence stereomicroscope. The fetuses 7 and 9 that lost eGFP expression completely (white arrows), the fetus 3 exhibited reduced eGFP expression and other fetuses had no obvious loss of eGFP expression.
52 Fig. 18. Analysis of CRISPR/Cas9-induced mutations.
(A) Agarose gel electrophoresis of T7E1-treated PCR products derived from fetuses shown in Fig. 17. The red arrows indicate the cleavage products generated in T7E1 assay and the wild-type sized band is indicated by a black arrow. D.W.: distilled water used as a negative control. M: 100-bp DNA ladder marker. (B) Agarose gel electrophoresis of PCR product amplified with primer set (M026: GGTGGTGCAGATGAACTTCAG and #125: CGGGATCCATTGCCTTTTATGGTAATCG) using genomic DNAs isolated from fetus 9 and eGFP transgenic mouse as template.
53
Fig. 19. Direct sequencing of PCR products amplified from the eGFP target region of fetuses. The black arrows below the electropherogram show overlapping peaks indicative of indel mutations.
54 第 4 節 考察
GONAD 法を実施後、妊娠中期(Day 13.5)、妊娠末期(Day 19.5)の胎仔を調査した結果、
双方のステージにおいて外貌的に正常な胎仔を得ることができ、うちいくつかの個体では、
eGFP 遺伝子の KO が認められた(Fig. 14, 17)。自然分娩の直前のステージで標的遺伝子の
KO 個体が得られたことから、GONAD 法によって実際にゲノム編集マウスが作製できるだ ろうと考えられた。一方、Day 13.5、19.5 のいずれの胚でも複数の変異アレルがモザイクの 形で検出された。本研究において、CRISPR により生じた変異のスクリーニングにもちいた T7E1 assay は、二本鎖 DNA のミスマッチを認識して切断することから、前述の複数の変異 アレルが体細胞に分布するモザイク変異を検出するには有効な解析法である。しかし、Fig. 14 の胎仔 1 と胎仔 4 は、eGFP 蛍光の完全な欠失を示し、さらに塩基配列決定から変異の存 在が明らかとなったにも関わらず、T7E1 assay による解析結果は陰性であり、変異を検出で きなかった。標的配列決定によりこの 2 つのサンプルが、それぞれ 4 塩基または 3 塩基欠 失を有する変異体であることが示されたが、このように PCR 産物に 1 種類の変異アレルし か含まれない場合、ミスマッチする配列が存在しないため、T7E1 assay では indel 変異を検 出できない。この問題を解決する手段として、あらかじめサンプルの PCR 産物に野生型の PCR 産物を混合することが、有効な方法であると考えられる。この方法であれば、1 種類の 変異アレルしか含まない場合であっても T7E1 assay で検出が可能となる。したがって、T7E1 assay は GONAD 法のように複数の変異アレルが発生し易い場合に加え、1 種の変異アレル
55
のみでも変異を検出可能となることが期待されることから、GONAD 法で作出された複数の 変異アレルのスクリーニングには強力なツールになると考えられる。
56 総括
本研究では、従来の顕微注入法による遺伝子改変マウス作製に必須な胚の ex vivo 操作と いうステップを経ずにゲノム編集マウスを作製できる「GONAD 法」と呼ぶ新規手法の開発 を進めた。第 1 章では、in vitro/in situ エレクトロポレーションによって卵管内の 2 細胞期胚 に mRNA が導入可能であり、その mRNA から機能性タンパク質が翻訳されることを明らか にした。また、CRISPR 系 RNA を用いることで、Cas9 ヌクレアーゼによる配列特異的な標 的遺伝子のゲノム編集が可能であることを示した。一方でエレクトロポレーションによる 胚へのダメージが示唆され、胚の着床・発生能についても検証が必要であると考えられた。 そこで第 2 章では、in situ エレクトロポレーションにより CRISPR 系 RNA が導入された胚 が正常な着床と妊娠維持を行うことができるかどうかを検討するため、2 つの胎仔期におけ る胎仔の表現型を解析した。その結果、妊娠中期(Day 13.5)ならびに妊娠末期(Day 19.5) の双方のステージにおいて、標的遺伝子を KO された外貌的に正常な胎仔を得ることに成功 し、「GONAD 法」が実用化される可能性を見出した。 GONAD 法は、1)mRNA 導入により目的のタンパク質を卵管内に存在する着床前胚で発 現させることが出来る、2)CRISPR/Cas9 関連 RNA の導入によりゲノム編集マウスを従来 法に較べより簡便に作製できる、という 2 つの特徴を持つ。したがって、導入 RNA として Cre mRNA または FLP mRNA を用いれば、in vivo で標的遺伝子の組換え(floxed アレルまた は FRTed アレルを除去するなど)も可能となることから、conditional KO などの他領域への
57
応用性も期待される。また、GONAD 法は胚の ex vivo 処置を必要としないので、従来の顕 微注入法で使われる供試マウスの数を大幅に減らすことができる。例えば、従来法では繁 殖用雄マウス、採卵のための複数の雌マウス、胚移植のための偽妊娠雌マウスおよび精管 結紮雄マウスが必要となる[Harms et al., 2014]。一方、GONAD 法は繁殖用雄および雌マウス の 1 組のペアのみで実施が可能であるため、偽妊娠雌マウス、精管結紮雄マウスは不要と
なる。したがって、動物福祉で重要視される3R’s(Replacement, Refinement and Reduction)
にも十分叶った手法と言える。さらに GONAD 法はラット、ブタ、ヒツジ、ヤギ、ウシ等 の他の生物種にも適用できるポテンシャルを有する。卵管を外科的に処置することができ れば、CRISPR / Cas9 システムの適用によるゲノム編集動物作製の可能性が大いに期待でき る。過去 30 年にわたり、Tg 技術はマウスを中心に進化しており、世界中の研究機関の多く の施設で各種モデルマウスが作製されている。しかし、旧来法では胚の ex vivo 操作を伴う。 これはマウス以外の動物種を扱う場合でも例外ではない。しかし、一般的に、マウス以外 の動物種における胚の ex vivo 操作は様々な困難を伴う。故に、採卵、胚操作、顕微注入お よび偽妊娠動物への胚移植などのステップをバイパスできる GONAD 法は、他種動物での 遺伝子改変に光明を与える手技となり得るかもしれない。
58 参考文献
1. Auerbach, A.B., Norinsky, R., Ho, W., Losos, K., Guo, Q., Chatterjee, S. and Joyner, A.L. 2003. Strain-dependent differences in the efficiency of transgenic mouse production. Transgenic Res. 12: 59-69.
2. Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D.A. and Horvath, P. 2007. CRISPR provides acquired resistanceagainst viruses in prokaryotes.
Science. 315: 1709-1712.
3. Brinster, R.L., Chen, H.Y., Trumbauer, M., Senear, A.W., Warren, R. and Palmiter, R.D. 1981. Somatic expression of herpes hymidine kinase in mice following injection of a fusion gene into eggs. Cell. 27: 223-231.
4. Bronson, R.A. and McLaren, A. 1970. Transfer to the mouse oviduct of eggs with and without the zona pellucida. J. Reprod. Fertil. 22: 129-137.
5. Bronson, S.K., Plaehn, E.G., Kluckman, K.D., Hagaman, J.R., Maeda, N. and Smithies, O. 1996. Single-copy transgenic mice with chosen-site integration. Proc. Natl. Acad. Sci. U S A. 93: 9067-9072.
6. Capecchi, M.R. 2005. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat. Rev. Genet. 6: 507-512.
59
7. Carlson, D.F., Fahrenkrug, S.C. and Hackett, P.B. 2012. Targeting DNA with fingers and TALENs. Mol. Ther. Nucleic. Acids. 1: e3.
8. Chen, S., Lee, B., Lee, A.Y., Modzelewski, A.J. and He, L. 2016. Highly Efficient Mouse Genome Editing by CRISPR Ribonucleoprotein Electroporation of Zygotes. J. Biol. Chem. 291 :14457-14467.
9. DiCarlo, J.E., Norville, J.E., Mali, P., Rios, X., Aach, J. and Church, G.M. 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41: 4336-4343.
10. Fielder, T.J., Barrios, L. and Montoliu, L. 2010. A survey to establish performance standards for the production of transgenic mice. Transgenic Res. 19: 675-681.
11. Friedland, A.E., Tzur, Y.B., Esvelt, K.M., Colaiácovo, M.P., Church, G.M. and Calarco, J.A. 2013. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods. 10: 741-743.
12. Fujii, W., Kawasaki, K., Sugiura, K. and Naito, K. 2013. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41: e187. 13. Garrick, D., Fiering, S., Martin, D.I. and Whitelaw, E. 1998. Repeat-induced gene silencing in
mammals. Nat. Genet. 18: 56-59.
14. Gilbert, L.A., Larson, M.H., Morsut, L., Liu, Z., Brar, G.A., Torres, S.E., Stern-Ginossar, N., Brandman, O., Whitehead, E.H., Doudna, J.A., Lim, W.A., Weissman, J.S. and Qi, L.S. 2013.
60
CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 154: 442-451.
15. Giraldo, P., Rival-Gervier, S., Houdebine, L.M. and Montoliu, L. 2003. The potential benefits of insulators on heterologous constructs in transgenic animals. Transgenic Res. 12: 751-755. 16. Gordon, J.W., Scangos, G.A., Plotkin, D.J., Barbosa, J.A. and Ruddle, F.H. 1980. Genetic
transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. U S
A. 22: 693-702.
17. Grabarek, J.B., Plusa, B., Glover, D.M. and Zernicka-Goetz, M. 2002. Efficient delivery of dsRNA into zona-enclosed mouse oocytes and preimplantation embryos by electroporation.
Genesis. 32: 269-276.
18. Hammer, R.E., Pursel, V.G., Rexroad, C.E. Jr., Wall, R.J., Bolt, D.J., Ebert, K.M., Palmiter, R.D. and Brinster, R.L. 1985. Production of transgenic rabbits, sheep and pigs by microinjection.
Nature. 315: 680-683.
19. Harms, D.W., Quadros, R.M., Seruggia, D., Ohtsuka, M., Takahashi, G., Montoliu, L. and Gurumurthy, C.B. 2014. Mouse genome editing using the CRISPR/Cas system. Curr. Protoc.
Hum. Genet. 83: Unit 15. 7. 1-27.
20. Hashimoto, M. and Takemoto, T. 2015. Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci. Rep. 5: 11315.
61
21. Hooper, M., Hardy, K., Handyside, A., Hunter, S. and Monk, M. 1987. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 326: 292-295.
22. Horvath, P. and Barrangou, R. 2010. CRISPR/Cas, the immune system of bacteria and archaea.
Science. 327: 167-170.
23. Horii, T., Arai, Y., Yamazaki, M., Morita, S., Kimura, M., Itoh, M., Abe, Y. and Hatada, I. 2014. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci. Rep. 4: 4513.
24. Hsu, P.D., Lander, E.S. and Zhang, F., 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 57: 1262-1278.
25. Hwang, W.Y., Fu, Y., Reyon, D., Maeder, M.L., Tsai, S.Q., Sander, J.D., Peterson, R.T., Yeh, J.R. and Joung, J.K. 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat.
Biotechnol. 31: 227-229.
26. Jaenisch, R. 1976. Germ line integration and Mendelian transmission of the exogenous Moloney leukemia virus. Proc. Natl. Acad. Sci. U S A. 73: 1260-1264.
27. Jasin, M., Moynahan, M.E. and Richardson, C. 1996. Targeted transgenesis. Proc. Natl. Acad.
Sci. U S A. 93: 8804-8808.
62
programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337: 816-821.
29. Kaneko, T. and Mashimo, T. 2015. Simple Genome Editing of Rodent Intact Embryos by Electroporation. PLoS One. 10: e0142755.
30. Kaneko, T., Sakuma, T., Yamamoto, T. and Mashimo, T. 2014. Simple knockout by electroporation of engineered endonucleases into intact rat embryos. Sci. Rep. 4: 6382.
31. Li, D., Qiu, Z., Shao, Y., Chen, Y., Guan, Y., Liu, M., Li, Y., Gao, N., Wang, L., Lu, X., Zhao, Y. and Liu, M. 2013. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system.
Nat. Biotechnol. 31: 681-683.
32. Li, J.F., Norville, J.E., Aach, J., McCormack, M., Zhang, D., Bush, J., Church, G.M. and Sheen, J. 2013. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 31: 688-691.
33. Lois, C., Hong, E.J., Pease, S., Brown, E.J. and Baltimore, D. 2002. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295:868-872. 34. Modliński, J.A. 1970. The role of the zona pellucida in the development of mouse eggs in vivo.
J Embryol. Exp. Morphol. 23: 539-547.
35. Montague, T.G., Cruz, J.M., Gagnon, J.A., Church, G.M. and Valen, E. 2014. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic. Acids. Res. 42: 401-407.
63
36. Ohtsuka, M., Ogiwara, S., Miura, H., Mizutani, A., Warita, T., Sato, M., Imai, K., Hozumi, K., Sato, T., Tanaka, M., Kimura, M. and Inoko, H. 2010. Pronuclear injection-based mouse targeted transgenesis for reproducible and highly efficient transgene expression. Nucleic. Acids. Res. 38: e198.
37. Palmiter, R.D., Brinster, R.L., Hammer, R.E., Trumbauer, M.E., Rosenfeld, M.G., Birnberg, N.C. and Evans, R.M. 1982. Dramatic growth of mice that develop from eggs microinjected with met
allothionein-growth hormone fusion genes. Nature. 300: 611-615.
38. Potts, W., Tucker, D., Wood, H. and Martin, C. 2000. Chicken beta-globin 5’HS4 insulators function to reduce variability in transgenic founder mice. Biochem. Biophys. Res. Commun. 273: 1015-1018.
39. Qin, W., Dion, S.L., Kutny, P.M., Zhang, Y., Cheng, A.W., Jillette, N.L., Malhotra, A., Geurts, A.M., Chen, Y-G. and Wang, H. 2015. Efficient CRISPR/Cas9-mediated genome editing in mice by zygote electroporation of nuclease. Genetics. 200: 423–430.
40. Ran, F.A., Hsu, P.D., Wright, J., Agarwala, V., Scott, D.A. and Zhang, F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8: 2281-2308.
41. Sato, M. 2005. Intraoviductal introduction of plasmid DNA and subsequent electroporation for efficient in vivo gene transfer to murine oviductal epithelium. Mol Reprod Dev. 71: 321-330. 42. Sato, M., Ohtsuka, M., Watanabe, S. and Gurumurthy, C.B. 2016. Nucleic acids delivery
64
methods for genome editing in zygotes and embryos: the old, the new, and the old-new. Biol.
Direct. 11:16.
43. Shen, B., Zhang, W., Zhang, J., Zhou, J., Wang, J., Chen, L., Wang, L., Hodgkins, A., Iyer, V., Huang, X. and Skarnes, W.C. 2014. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 11: 399-402.
44. Sorrell, D.A. and Kolb, A.F. 2005. Targeted modification of mammalian genomes. Biotechnol.
Adv. 23: 431-469.
45. Stewart, C.L., Schuetze, S., Vanek, M. and Wagner, E.F. 1987. Expression of retroviral vectors in transgenic mice obtained by embryo infection. EMBO. J. 6: 383-388.
46. Tsukui. T., Kanegae, Y., Saito, I. and Toyoda, Y. 1996. Transgenesis by adenovirus-mediated gene transfer into mouse zona-free eggs. Nat. Biotechnol. 14: 982–985.
47. Tsukui, T., Miyake, S., Azuma, S., Ichise, H., Saito, I. and Toyoda, Y. 1995. Gene transfer and expression in mouse preimplantation embryos by recombinant adenovirus vector. Mol. Reprod.
Dev. 42: 291–297.
48. Wang, H., Yang, H., Shivalila, C.S., Dawlaty, M.M., Cheng, A.W., Zhang, F. and Jaenisch, R. 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153:910-918.
65
generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154 :1370-1379.
50. Yu, Z., Ren, M., Wang, Z., Zhang, B., Rong, Y.S., Jiao, R. and Gao, G. 2013. Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics. 195: 289-291.
66 謝辞 本論文の作成にあたり、終始ご助言を賜りました本研究の指導教授である東京農業大学 大学院生物産業学研究科・教授・亀山祐一博士(生物産業学)ならびに東京農業大学名誉 教授・横濵道成農学博士に厚く御礼を申し上げる。また、実験遂行と論文執筆について多 大なるご指導を頂きました東京農業大学生物産業学部・准教授・和田健太博士(生物産業 学)に深く感謝の意を表する。また、本研究における GONAD 法の考案と開発を主導され、 実験系の立ち上げ、実験指導、学術論文の執筆について多くのご指導を頂きました、東海 大学医学部基礎医学系・准教授・大塚正人博士(理学)ならびに鹿児島大学医用ミニブタ 先端医療開発研究センター・遺伝子発現制御分野・教授・佐藤正宏博士(医学)に厚く御 礼を申し上げる。また、実験遂行にあたり多大なるご協力を頂きました、東海大学医学部
基礎医学系・研究員・三浦浩美博士(医学)、米国 University of Nebraska Medical Center・