関西学院大学リポジトリ
172
0
0
全文
(2) Contents List of Symbols and Abbreviations ••••••••••••••••••••••••••''•••• I-II. General Introduction •••••••••••••••••••••••••••••••••••••e•••••••1. Chapter 1: FTIR Study on Hydrogen Bonding Interactions in Biodegradable Polymer Blends of Poly(3-hydroxybutyrate) and Poly(4-vinylphenol). Ab stract• ••••••••••••••••••••••••••••••••••••••••••••••••• 31. Introduction••••••••••••••••••••••••••••••••••••••••••••••32 Experimental Section••••••••e••••••••••••••••••••••••••••••35 Results and Discussion •••••••••••••••••••••••••e•••e•••e••• 37. Conclusions ••••••••••••••••••••••••e•••••e••••••••••••••• 44. References and Notes•••••••••••e••••••••••••••••••••••••••45. Chapter 2: Thermally Induced Exchanges of Hydrogen-Bonding. Interactions and Their Effects on Phase Structures of Poly(3-hydroxybutyrate) and Poly(4-vinylphenol) Blends. Abstract•••••••••••••••••••••••••••••••••••••••••••••••••58 Introduction••••••••••••••-•••••••••••••••••••••••••••••••59 Experimental Section •••••••''''•''''''''''''•••••••••••••••61 Results and Discussion ••••••'''••''''''''''•••••••••••••••• 62 Quantitative Analyses of Temperature Dependence of C=O Stretching. i.
(3) Vibration Bands in Relation to Phase Structure of the Blends•••••••70. Conclusions •••••••••••••••••••••••••••••••••••••••••••••• 75 Reference ••••••••••••••t••••••••-••••••••••••••••••e••••• 76. Appendix ••••••••••••••••••••••••••••••••••••••••••••••• 95. Chapter 3: Multistep Crystallization Process Involving Sequential Formations of Density Fluctuations, "Intermediate Structures", and. Lamellar Crystallites: Poly(3-hydroxybutyrate) as Investigated by. Time-Resolved Synchrotron SAXS and WAXD Ab stract •••••••••••••e••••••••••••••••••••••••••••••••••• 99. Introduction•••••••••••••••••••••••••••••••••••••••••••••100 Experimental Methods •••••••••••••••••••••••••••••••••e••• 103. Results•••••••••••••••••••••••••••••••••••••••••••••••••107 Analysis and Discussion•••••••••••••••••••••••••••••••••••110 Conclusions •••••••••••••••••••••••••••••••••e••••••••••• 125 References •••••••••••••••••••••••••••••••••••••••••••••• 127. Appendix ••••••••••••••••••••••••••••••••••••••••••••••• 145. Acknowledgements •••••••••••••••••••••••••••••••••••••••••••• 163. List of Publications ••••••••'''''••''''''''••'••''•••••••••••••• 165. ii.
(4) List of Symbols and Abbreviations. 2D-COS. two-dimensional correlation spectroscopy. DSC. differential scanning calorimeter. EFA. evolving factor analysis. freeC=O. free C=O groups ofPB. fk. (k =free, inter, intra) average fractions of C==O groups in a PB chain which.free C=O, intra C=O, and inter C=O. FTIR. Fourier-transform infrared. HB(s). hydrogen bonding(s). intra PHB. intramolecular HBs within PHB. intra PVPh. intramolecular HBs within PVPh. intra C=O. intramolecularly hydrogen-bonded (HBed) C=O groups within PHB. intra CH. intramolecularly hydrogen-bonded (HBed) CH groups within PHB. inter. intermolecular HBs between PHB and PVPh. inter C=O. intermolecularly HBed C=O groups in PHB with PVPh. inter OH. intermolecularly HBed OH groups in PVPh with PHB. Icrys. WAXD profiles ofwell-developed lamellar crystallites ofPHB. Iinter. WAXD profiles of intermediate structures having the mesomorphic. orders ofPB. I.
(5) m. infrared. MCR-ALS. multivariate curve resolution-alternating least squares. PHB. poly(3-hydroxybutyrate). PVPh. poly(4-vinylpX-henol). SAXS. small-angle X-ray scattering. self-OH. HBs between OH groups ofPVPh. SVD. singular value decomposition. Tcc. onset temperature of cold-crystallization. Tg. glass transition temperature. Tm. temperature ofthe melting point by DSC. Tms. temperature above which melting starts to occur. Tmc. temperature above which melting completes. Tani-meso. transformation from amorphous phase to mesomorphic phase. Tmeso-ery. transformation from mesomorphic phase to the lamellar crystals. WAXD. wide-angle X-ray diffraction. Mcrys. weight fraction of well-developed lamellar crystallites of PHB in. non-amorphous phase. PVinber. weight fraction of intermediate structures having the mesomorphic. orders ofPHB in non-amorphous phase. II.
(6) WPVPh. weight percentage ofPVPh in PHBMVPh blends. Xc,app. apparent crystallinity composed of intermediate struetures and lamellar crystallites. Xcrys. true crystallinity ofwell-developed PHB lamellar crystallites. Xinter. weight fraction ofmesomorphic layers. III.
(7) General Introduction. 1. Scope of This Thesis. This thesis is mainly concerned with three aspects. One is the investigation on hydrogen. bonding interactions, crystallization behavior, and miscibility of biodegradable poly(3-hydroxybutyrate) (PHB) blended with poly(4-vinylphenol) (PVPh) as a function of composition by using Fourier transform infrared (FTIR) spectroscopy, wide-angle X-ray diffraction (WAXD), and differential scanning calorimetry (DSC). Another one is the research. on thermally induced variations of hydrogen bondings and phase structure of PHBrpVPh blends as a function of composition and temperature during the crystallization and melting processes by using FTIR and WAXD. The final one is the study of multistep crystallization process involving the sequential formations of density fluctuations, intermediate structures,. and lamellar crystallites during the isothermal crystallization process of PHB by using the. time-resolved synchrotron small-angle X-ray scattering (SAXS) and WAXD in combination with two-dimensional (2D) correlation and multivariate curve resolution-alternating least. squares (MCR-ALS) analyses. The study on hydrogen bonding interaction, crystallization behavior, and phase structure of. polymers is one of the most important research topics in polymer science and has been focused on for a long time. For the practical application of polymers, the appropriate modification for their propenies is very necessary in order to suit the condition of application. environment. For polymer modification, copolymerization and blending are the most popular methods to improve the propenies ofpolymers. In this thesis, blending method was employed for the investigation ofthe hydrogen bonding interactions, thermal behavior, phase structures,. and miscibility ofPHBMVPh blends. It is well-known that the enthalpic contribution to the. free energy of mixing usually controls the miscibility of polymer blends because polymer. blends have small translational (combinatorial) entropy of mixing. Hydrogen bonding interactions, ifthere are, are considered to play a key role in the enthalpic contribution due to. 1.
(8) its diversity and strength. And many hydroxyl group containing polymers are reported as a. good counter polymer to mix with the polyesters because these blends have been found to form totally or partially miscible systems driven by the intermolecular hydrogen bonding interactions. Moreover, the crystal structure and morphological structure are responsible for. many properties ofthe final propenies ofpolymers, so as that the knowledge ofcrystallization behavior is very crucial for designing the polymeric materials with the required properties.. Therefore, a deeply fundamental understanding of the structural development of polymers during the crystallization process is very significative for improving the physical and mechanical properties so as to widen their potential applications.. In the first two chapter of this thesis, we systemically investigated the hydrogen bonding. interactions, thermal behavior and phase structures of PHBMVPh blends as function of composition and temperature. The intermolecular hydrogen bonding interactions between the. C=O groups of PHB and the OH groups of PVPh has been proved by FTIR spectroscopy.. Meanwhile, the miscibility and of the crystal structures of PHB/PVPh blends were investigated by differential scanning calorimetry (DSC), and WAXD, respectively. For the. PHBMVPh blends, the C==O groups of PHB exist at three different states: (i) the intermolecular hydrogen-bonded C=O groups of PHB with the OH groups of PVPh (inter C =O) at amorphous state, (ii) the intramolecular hydrogen-bonded C=O groups ofPHB with one ofthe C-H groups in CH3 ofPHB (intra C=O) at crystalline state, (iii) the free C==O groups ofPHB at amorphous state. Through the curve-fitting analysis, the IR spectra in the. C=O stretching vibration region were decomposed into componential spectra attributed to inter C=O groups, intra C=O groups, and free C=O groups, respectively, so as to determine the respective fractions of.fi.t., fmtra, and ffree as a function of composition and temperature.. Based on the quantitative analysis of the C=O groups of PHB, the transition of phase structures in PHBMVPh blends has been investigated during the crystallization and melting. processes. Meanwhile, the temperature dependence of lattice parameters a and b were calculated by WAXD patterns to study the structures ofPHB crystal in the PHB/PVPh blends.. 2.
(9) In the final chapter of this thesis, the isothermal crystallization behavior of PHB has been. investigated by using time-resolved SAXS and WAXD. Through the application of 2D correlation and MCR-ALS techniques for the analyses of SAXS and WAXD profiles, it is found that density fluctuations are first built up in the molten bulk PHB, the mesomorphic layers composed of the intermediate structures are transformed from the density-rich region,. and then the mesomorphic layers are eventually transformed into the well-ordered lamellar crystallites. Combined with the characteristic parameters calculated by the one-dimensional. correlation functions, structural development of PHB during the isothermal crystallization process was detailedly illuminated in the chapter 3 ofthis thesis.. The originality and novelty ofthis thesis can be described as follows:. 1. Through the analyses of the composition-dependent IR spectra and WAXD patterns of PHBMVPh blends, we proposed that the crystal structure formed in the PHBMVPh blends is almost same as that ofneat PHB, and the competing ofhydrogen bonding interactions of. C=O groups in PHB with the CH3 groups of PHB and with the OH groups of PVPh is a crucial physical factor underlying the basic physics of the blends with respect to ordering. via crystallization. Moreover, the exchange between intra- and inter-molecular hydrogen bonding occurs through the creation ofthe free C==O groups and hence it crucially depends. on the mobility ofthe surrounding media where free C=O groups exist.. 2. Through the analyses of temperature-dependent IR spectra and WAXD patterns, we elucidated that the crystallizable PB chains and noncrystallizable Pyph chains are found. to be incorporated in polymer networks formed by physical crosslinking points composed. of intermolecular hydrogen bondings between the C=O groups of PHB and the OH groups. of PVPh and intramolecular hydrogen bondings within PVPh. The miscibility of the. PHBMVPh blends is enhanced but the crystallization rate of PHB in the blends is suppressed with the increasing weight fraction ofPVPh. We also found the fact that both the crystallization and the melting ofthe PHBMVPh blends occur in a two-step process.. 3. By the successfu1 application of2D-COS and MCR-ALS techniques for the analyses ofthe. 3.
(10) time-resolved WAXD and SAXS profiles, a multistep crystallization process was elucidated, which involves the sequential formations of density fluctuations, intermediate structures, and lamellar crystallites. Meanwhile, the structural entity of the intermediate. structures having the mesomorphic orders between the melts and well-developed lamellar crystallites was identified by the elemental WAXD profiles, whose lattice spacings are larger than those ofwell-developed crystallites.. 2. Introduction ofBiodegradable PHB. Polymer is a kind of widely used materials with large molecular composed of repeating structural units, and these subunits are typically connected by covalent chemical bands. Although the term ofpolymer is sometimes taken to refer to plastics, it actually encompasses a large class of natural and synthetic materials with a wide variety of properties. Polymeric. materials derived from petrochemicals are always important and indispensable to modern economic development and human life. However, they have met serious challenges, not only. due to their production shortage but also the tremendously environmental problems, especially their disposal processes. Therefore, biodegradable polymers show the strong. potential contributions for environmental protection. Biodegradable polymers are the polymers that can be broken down and lost their initial integrity, which are used in many fields such as medical suture and orthopedic fixation devices to avoid a second operation to remove them, drug carrier to gradually release, common application ofplastic, and so on.. Polyhydroxyalkanoates (PHAs) are linear polyesters produced in nature by bacterial fermentation of sugar or lipids to store carbon and energy, and more than 150 different monomers can be combined within this family to give the materials with extremely different. propenies.i-i6 PHAs are biodegradable under aerobic and anaerobic conditions with thermoplasticity and mechanical properties similar to the synthetic polymers. Additionally,. they are hydrophobic, which makes them superior to their biodegradable competitors like starch and proteins in moisture resistance. The illustration of the biodegradation and. 4.
(11) biologically synthesis ofenvironment-friendly polymers, PHAs, are briefly schemed in Figure 1 part (b).. poly(3-hydroxybutyrate) eB), as a kind of PHAs, is a well known biodegradable polymer belonging to the polyester class that was first isolated and characterized in 1925 by. Maurice Lemoignei, which is produced by various micro-organisms in nature apparently in response to conditions of physiological stress.i'2'i7-22 The chemical structure of pHB is presented in Figure 1 part (a). PHB is optically active aliphatic polyester with a relative low. glass transition temperature around O OC and a high melting temperature around 175 OC. Moreover, PHB is the most abundant polyester found in bacteria and has similar thermal and. mechanical properties to isotactic polypropylene.i9'23 As a result of its stereochemical R-configuration, PHB can achieve a relatively high degree of crystallinity usually between 55-800/o,24 whose crystal is orthorhombic structure with lattice parameters a =: 5.76 A, b = 13.20 A, and c = 5.96 A (fiber repeat distance), containing two-left handed helical molecular chains in antiparallel orientation.25'26 Recently, on the basis ofthe molecular weight studies on. depolymerase-treated single crystals of PHB, Marchessault et al.27'28 suggested that PHB single crystals create a multiple erosion surface in the folding plane direction, i.e., parallel to. the long axis (a-axis ofunit cell). They suggested that the antiparallel chain packing ofPHB, which provides a relatively stable tubular ribbon in the first stage ofchain folding, encourages. subsequent self-assembly of precursor elements. PHB creates the initial self-folding chains from random coil at the first stage of the crystallization process, and then the precursor is. formed and self-assembled. The kinetics for the growth rate of PHB spherulites have been investigated at various crystallization temperatures, and the growth rate shows a maximum. value at around 90 OC. The overall rate of crystallization of pure PHB is relative low. compared to that of common synthetic polymers, showing a maximum in the temperature range of 50-6o OC.24'29 It was illuminated that the low overall crystallization rate of PHB centers on the nucleation process rather than the subsequent crystal growth process. Indeed, it. has been shown that PB has a low level ofheterogeneous nuclei.30. 5.
(12) In PHB crystals, it has been found C-H•••C=O hydrogen bondings between the C=O groups in one helix and the C-H groups of CH3 in the other helix exist in PHB lamellae along the a-axis,3i'36 and the lamellar structure and the helix structure of PHB crystals are briefly. sketched in Figure 2 part (a) and (b), respectively. Evidences for proposing the C-H"'C=O. hydrogen bondings in PHB crystals come from the following several facts: (1) a C-H. stretching band appears at above 3000 cm-i, an unusually high frequency, and the corresponding C=O stretching band shows a low frequency shift in IR spectra; (2) the distance between the O atom ofC =O groups and the H atom of one ofthe C-H bands of CH3 groups is shorter than that of the Van der Waals separation (2.72 A); (3) quantum chemical. calculation of dimer model compounds of PHB revealed that the molecular structure with a C-H"'C==O hydrogen bonding is the most stable structure.3i'34. For the practical application ofPHB as environment-friendly polymeric materials, there is still a large technical barrier, which is attributed to that PHB is rigid and brittle due to its. excessively high crystallinity. Meanwhile, the melting temperature of PHB is close to its. thermal decomposition temperature, causing the thermally unstable behavior during the conventional melt-processing. Therefore, it is necessary to improve the properties ofPHB for. practical application, and there are some approaches through the polymer modification. methods of copolymerization and blending. For copolymerization, PHB has been copolymerized with many different monomers to compensate for these drawbacks, like poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (P(HB-co-HV)),37'50 poly(3-hydroxybutyrate -co-3-hydroxyhexanoate) e(B-co-HHx)),7'39'5i-57 and poly(3-hydroxybutyrate-co-4-hydroxy butyrate) e(3HB-co4-HB))58-62. Besides, these PHB-based copolymers show a wide range of the physical and mechanical properties depending on the copolymerized units. Meanwhile, many polymer blends with PHB have been studied, such as poly(L-lactic acid) (pLLA),63-66 poly(vinyl alcohol) (pvA),67-70 poly(vinyl acetate) (PVAc),7i poly(methyl acrylate) (pMA),72. poly(ethylene oxide) (pEO),73'76 poly(butylenes succinate) (PBS)77 and pvph78'82. Among them, the PHBMVPh blends are of special interest due to the strong intermolecular hydrogen. 6.
(13) bondings between them. While, there is still continuing interest in the investigations on the. phase structures and thermal behavior ofPHBMVPh blends.. PVPh is a kind of amorphous polymer with wide applications. Due to the OH groups and the phenyl groups ofPVPh molecular chains, there are two different interactions within PVPh:. self-associations between the OH groups and the z-associations between the OH groups and the phenyl groups. Meanwhile, it is quite reasonable that the C=O groups of PHB can also. form the intermolecular hydrogen bondings with the OH groups ofPVPh. In this thesis, we deeply investigated the phase structures and the thermal behavior ofPHBMVPh blends from the viewpoint ofthe exchanges of intra- and inter-molecular hydrogen bondings as a function of composition and temperature.. 3. Crystallization and Melting Behaviors of PHB. Polymers crystallized in the bulk, however, are never totally crystalline as a consequence of. their long-chain nature and subsequent entanglements, and the melting temperature is always higher than the glass transition temperature, so as that the polymers may be either hard and. rigid or flexible. The development of crystallinity in polymers depends on the regularity of their structures. Thus isotactic and syndiotactic polymers are usually crystallizable, whereas. atactic polymers, with a few exceptions, are noncrystallizable. It is well known that crystallization plays an important role in the physical properties and the biodegradability for. the biodegradable polymers. Meanwhile, the crystal structure and the morphological structure of semicrystalline polymers are also influenced greatly by their thermal history. Much more attention should be directed to the study on crystallization behavior since it affects not only the crystalline structure and morphological structure of semicrystalline polymers but also the final physical properties and biodegradability for the biodegradable polymers.. The melting behavior ofpolymers may be observed by any ofseveral experiments, which is. another important physical property for polymers. Meanwhile, the melting behavior can reflect the structural information of polymers and their blends. Nowadays, DSC is a kind of. 7.
(14) popular methods for the analysis of polymer melting behavior, since it can give the heat of fusion as well as the melting point. One of another important ways of observing the melting. point is to measure the WAXD pattern. The sharp WAXD pattern characteristic ofcrystalline. polymers shows only amorphous halos at the melting temperature. Besides, WAXD has been often used not only to determine the crystalline structure of polymers but also to explore thermally induced changes in the unit cell dimensions as a result of thermal expansion. In. addition, FTIR spectroscopy is also an important method, which can clearly show the variations of typical crystal bands and amorphous bands as a function of temperature, and hence the melting behavior can be investigated thermally. The crystallization and melting behaviors ofPHAs and other polymers have been extensively studied, which are very helpfu1 for us to better understand the physical and mechanical properties to widen their practical applications in many fields. Especially for the isothermal crystallization process, there are. many reports about various polymeric materials by different techniques, such as Dsc,83-86. FTIR spectroscopy,36'87"89 polarized optical microscopy (pOM),84'90'93 and time-resolved. Synchrotron SAXS and wAxD.94-iOi The intense IR bands attributed to the C=O and CH groups assigned to crystalline and amorphous states are exhibited as the characteristic bands of various PHAs, and hence IR spectroscopy has been successfu11y applied to characterize the crystallization and melting. behaviors of PHAs. Besides, IR spectra also can show the variations of intermolecular interactions in polymer blends, if there are. For the PHBMVPh blends, Iriondo et al. has. reported the existence of intermolecular hydrogen bondings between the C=O groups ofPHB and the OH groups of PVPh by the IR spectra in the C==O stretching vibration region.78'79 Meanwhile, Xing et al.80 has reported that PHBMVPh blends are miscible at all compositions. through the single Tg evaluated by DSC thermograms and that the negative value of the segmental interaction parameter determined from the equilibrium melting points depression. supports the miscibility and strong hydrogen bonding interactions between PHB and PVPh.. However, the investigation on the phase structures and the variations of the hydrogen. 8.
(15) bondings within PHB and between PHB and PVPh is still lacking. In this thesis, we aim to. advanced the investigation ofPHBMVPh blends as a function ofcomposition and temperature through the quantitative analysis of C=O groups of PHB component in the blends, so as to. deeply understand the thermally induced transitions of phase structures and exchanges. between inter- and intra-molecular hydrogen bondings in PHBMVPh blends during the crystallization and melting processes.. Based on the broad and detailed evidences from a large variety of experiments on several polymer systems carried out by other research groups and themselveS, Strobli02 proposed a novel concept for understanding the crystallization process of polymers, which indicate that the formation and growth of the lamellar crystallites is a multi-step process passing over the. intermediate states. Through the investigation by infrared (IR) and two-dimension (2D) correlational IR spectroscopy, Zhang et al.i03 presented that the existence of the 1731 cm-i. band in M spectra during the isothermal crystallization process of PHB is suggested by the second derivative and 2D correlational IR results, but the direct evidence relating the 1731 cm'i band to the intermediate structure or intermediate phase is still lacking. Therefore, it is. necessary to further investigate the structural development of PHB during the isothermal crystallization process by using other techniques, especially for the study on the transitions of. amorphous melts, intermediate struetures, and lamellar crystallites with crystallization time, and X-ray technique is a kind ofideal methods for the study on these topics.. In this thesis, by using the time-resolved SAXS and WAXD methods, we further investigated the sequential formations of density fluctuations, intermediate structures, and. lamellar crystallites of PHB during the isothermal crystallization process. We directly evidenced the existence of intermediate structures having the mesomorphic orders with larger. lattice spacings before the formation of lamellar crystallites of PHB by the WAXD profiles.. The structural development of PHB during the isothermal crystallization process was systemically and detailedly discussed.. 9.
(16) 4. Time-Resolved SAXS and VVAXD Techniques. Polymers, which are indispensably important materials in our daily lives as well as in modern industry, exist in a variety ofphysical states that include semicrystalline, amorphous. and glassy states. And the morphological structure is a key point to affect the physical properties ofthe polymers, such as crystallinity, lamellar structure, mesophase structure, and. so on, whose scales are larger than that of atoms and molecules, but much smaller than the macroscopic scale ofthe materials. Hence, the required observational scale for morphological. structure is from about 10-i to 104 nm. X-ray scattering is the most ideal technique to investigate the structure of polymers in this scale, which has a combination of SAXS and. WAXD measurements. Usually, the growth of density fluctuations and the characteristic morphological parameters can be determined by SAXS patterns, and WAXD patterns can tell us the growth of the crystallinity and dimensional parameters of the crystal unit. All of the. information observed from SAXS and WAXD methods is significant to study the polymer morphological structure. Therefore, the simultaneous SAXS and WAXD measurements are widely used for the research ofpolymeric phase structure.. For physical chemistry studies, time-resolved experiment technique is a kind of powerfu1 methods to investigate the dynamic processes in materials or chemical compounds. Usually, the time-resolved technique is carried out with spectroscopy, such as FTIR, near-infrared (NIR), and so on. Nowadays, depending on the strong light sources of synchrotron radiation technique, the intensity of X-ray beam can be much increased so as that the time-resolved. SAXS and WAXD experiments also can be successfu11y carried out with a short data recording time to focus on transient structure of polymers. And hence, time-resolved SAXS. and WAXD techniques are widely applied to investigate the structural development of polymers during the crystallization process, and the intermediate structures were reported during the isothermal and non-isothermal crystallization process. 94'iOi However, the study on. the intermediate structures and the mesomorphic layers during the crystallization process is. 10.
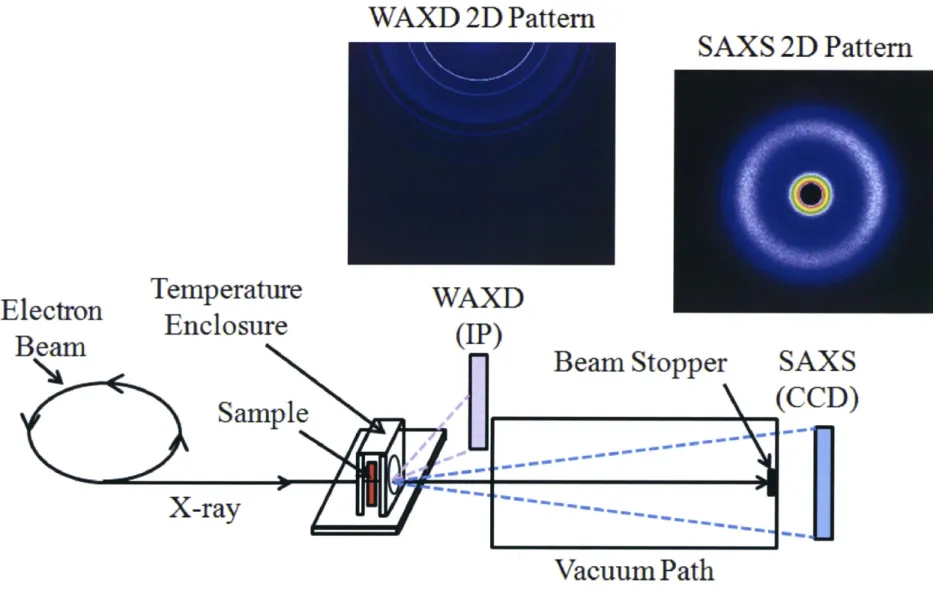
(17) still not deep enough. Besides, the correlative variations between SAXS and WAXI) profiles have not been investigated in detail.. In this thesis, time-resolved synchrotron SAXS and WAXD were employed for the study of struetural development of PHB during the isothermal crystallization process, especially for. the transformation of amorphous melts, intermediate structures, and lamellar crystallites,. through the BL03XU beamline which was constructed at SPring-8 Japan synchrotron radiation research institute. Meanwhile, the correlative analysis between SAXS and WAXD profiles were canied out, and the sequence of density fluctuations, formation of intermediate structures and lamellar crystallites was analyzed.. The beamline used in this study is equipped with the two-dimensional image intensifiers of. a CCD and an imaging plate (IP) system for SAXS and WAXD measurements, respectively, as briefly sketched by the structure ofthis beamline in Figure 3. The two-dimensional SAXS. and WAXD patterns can be recorded by these two detectors, and the SAXS and WAXD profiles can be observed by circularly averaging their two-dimensional patterns as a function. of scattering vector g. Depending on the advanced equipments ofthis beamline and the strong. intensity ofincident beam at SPring-8, the two-dimensional SAXS and WAXD patterns can be measured with a short exposure time such as microsecond, so as that we can carry out the. time-resolved investigation on the structural development of PHB during the isothermal. crystallization process. Meanwhile, the BL03XU beamline can synchronously record the. two-dimensional SAXS and WAXD patterns.. 5. 2D Correlation Analysis. The basic concept of perturbation-based 2D correlation spectroscopy applicable to FTIR. was proposed first by Noda in 1986.i04'i05 In a generalized 2D correlation spectroscopy. experiment, a series of perturbation-induced dynamic spectra are collected first in a systematic manner, e.g. in a sequential order during a temperature-, concentration-, or time-dependent process. Such a set of dynamic spectra is then transformed into a set of 2D. 11.
(18) correlation spectra by a cross-correlation analysis,i04'i05 The general scheme for obtaining the. 2D correlation spectra is briefiy described as shown in Figure 4, in which the experimental. data matrix is spread along the second dimension, and the synchronous and asynchronous maps are observed with the cross-peaks. The positive synchronous cross-peak, O(vi, v2), means that the intensity at vi and v2 synchronously increase or decrease. The negative Åë(vi, v2). suggests the different intensity changes at vi and v2. When Åë(vi, v2) is positive, the asynchronous cross-peak, 'Y(vi, v2), becomes positive if the intensity change at vi occurs predominantly before that at v2 in the sequential order oftime, or it becomes negative ifthe change at vi occurs predominantly after that at v2. However, this sign rule is reversed if Åë(vi,. v2) < O. The rule described above is the Noda rule for the sequential order analysis of the bands obtained in the perturbation-induced dynamic spectra.. A very intriguing possibility found in 2D correlation spectroscopy is the idea of heterospectral 2D correlation analysis, where two completely different types of spectra. obtained for a system using multiple spectroscopic probes under a similar external perturbation are compared.i06'i08 The heterospectral 2D correlation may be divided into two. types. One type is concerned with the comparison between closely related spectroscopies, such as IRINIR and RamanlNIR spectroscopy.i07'i09'iiO In this case, the correlation between. bands in two kinds of spectroscopy can be investigated. And hence it becomes possible to. make band assignments and resolution enhancement by heterospectral 2D correlation. The other type of heterospectral 2D correlation is the hetero-correlation between completely different types of spectroscopy or physical techniques such as IR and X-ray scattering.i06 This. type of heterospectral 2D correlation is usefu1 to investigate the structural and physical properties ofmaterials under a particular external perturbation.. There are many advantages ofthe 2D correlation spectroscopy, such as (1) simplification of. complex spectra consisting ofmany overlapped peaks, and enhancement of spectral resolution. by spreading peaks over the second dimension; (2) establishment of unambiguous assignments through correlation of bands; (3) probing the specific sequential order of spectral. 12.
(19) intensity variations taking place during the measurement; (4) truly universal applicability of. the technique, which is not limited to any type ofspectroscopy, or even any form ofanalytical technique, e.g., chromatography, microscopy, and so on.. The above-mentioned features in 2D correlation spectroscopy are ideally suited for the. research of polymer spectroscopy during melting and crystallization processes. And 2D correlation M spectroscopy has be widely and successfu11y used to investigate the transition frOm crystalline to amorphous or from amorphous to crystalline of some pHAs.52,88,89,i03,iii. These results suggested that the transition from amorphous phase to crystalline phase undergoes an intermediate phase corresponding to the IR bands around 1731 cm"i. However, assignment of the IR band around 1731 cm'i to the intermediate phase is still week without further powerfUl evidence. It is still quite necessary to further investigate the phase transitions. of PHAs during the isothermal crystallization process, especially for the transition of intermediate structure, and X-ray scattering technique is a very suitable tool to support this. information. Therefore, we canied out the time-resolved SAXS and WAXD experiments to study the phase transitions during the isothermal crystallization process ofPHB in this thesis.. Meanwhile, we employed the 2D correlation analysis for the time-resolved WAXD and SAXD profiles, which support the existence of intermediate mesomorphic structure and the sequential transformation of density fluctuations and the formation of intermediate structures as discussed in Chapter 3.. 6. MCR-ALS Analysis. Multivariate Curve Resolution (MCR) has become a popular and powerfu1 chemometrics method and is widely used for the resolution of multiple component responses in the unknown. unresolved mixtures. The basic assumption of this method is that the measured data with. multiple components can be decomposed into the sum of individual contributions coming from different sources of components. Among MCR methods, the one based on alternating least squares (MCR-ALS), which was first introduced by Karjlainenii2 and later expanded by. 13.
(20) Taulerii3-ii6, has been widely and popularly applied in recent studies for the analyses of COMplex multicomponent system.ii7'i24. The primary purpose of the MCR-ALS analysis is to transform the theoretical solution obtained by the experimental data matrix D into matrices C and ST, which has a real physical significance, according to the following equationii6•i25'i27:. D=CST+E (1) in which matrix C (n Å~ c) has column vectors corresponding to the concentration ofthe c-th. pure component that are presented in matrix D, the row vectors of matrix ST (c Å~ m) correspond to the profiles of the c-th pure component, and E is the residual matrix. The general scheme for obtaining the relative concentrations (matrix C) and individual component. (matrix S') by MCR-ALS analysis is briefly shown in Figure 5, and the operating procedure. ofMCR-ALS technique is schemed in Figure 6. Within this algorithm, the number of species for the MCR-ALS analysis has to be first determined, which can be carried out by evolving factor analysis (EFA) thechnique.i28 The number of significant EFA factors is an indication of. the sources of variability in the data and can be correlated with the number of species that exist in experimental data matrix D. The matrix rank is evaluated by using the singular value. decomposition algorithm in both directions (forwards and backwards) in the EFA analysis.i29'i30 And all of these algorithms can be carried out within the calculation. environment ofMATLAB software. By using MCR-ALS technique to analyze the time-resolved WAXD profiles as discussed in Chapter 3, we successfu11y separated the pure WAXD profiles of the intermediate structure. having the mesomorphic orders and the lamellar crystallites of PHB during the isothermal crystallization process, indicating that the characteristic spacings of the intermediate structure. are larger than those lamellar crystallites. Meanwhile, time-dependent relative weight fractions of the intermediate structure and the lamellar crystallites were also calculated by MCR-ALS analyses, indicating that the intermediate structures increase prior to the lamellar crystallites, which also can be consisted with the 2D correlation results ofWAXD profiles.. 14.
(21) 7. 0utRine of Each Chapter. The outline ofthe present studies will be described as follows. This thesis consists ofthree chapters.. Chapter 1 describes the exchanges of hydrogen bonding interactions in PHBMVPh blends as a function ofcomposition by using FTIR spectroscopy, crystal structure and crystallinity of. them by using WAXD, and their thermal behavior by using DSC. The intermolecular. hydrogen bondings between the C=O groups of PHB and the OH groups of PVPh were explored not only in C=O stretching vibration region but also in OH stretching vibration region. It is found that the competing hydrogen bonding interactions of C=O groups in PHB. with the CH3 groups of PHB and with the OH groups of PVPh is a crucial physical factor underlying the basic physics of the blends with respect to ordering via crystallization and. miscibility. The exchange between inter- and intra-molecular hydrogen bondings in PHBMVPh blends occurs through the first creation ofthe free C=O groups at amorphous state (via dissociations of inter- and intra-molecular hydrogen bonding interactions), while the. created free C=O groups are trapped in amorphous region of PHB surrounded by the crystalline part and hence cannot be associated with the OH groups ofPVPh. Therefore, the. transformation between inter- and intra-molecular hydrogen bondings crucially depends on the mobility ofthe surrounding media where free C=O groups exist.. Chapter 2 reports the thermally induced exchanges of various hydrogen bonding interactions and their effects on the phase structures ofPHBMVPh blends as investigated by. FTIR spectroscopy and WAXD as a function of composition and temperature. The crystallizable PHB molecular chains and noncrystallizable PVPh chains are found to be. incorporated in polymer networks formed by physical cross-linking points composed of. intermolecular hydrogen bondings between the C=O groups of PHB and the OH groups of PVPh and intramolecular hydrogen bondings within PVPh chains. Temperature-dependent variations of the physical cross-links are intimately related to the thermally induced. 15.
(22) exchanges of the hydrogen bondings. The miscibility of PHBMVPh blends is enhanced but the crystallization rate of PB in the blends is suppressed by these physical cross-links.. Moreover, both the crystallization and the melting of PHBMVPh blends occur in a two-step. process from the viewpoint of the exchange between intra- and inter-molecular hydrogen bondings in the blends.. Chapter 3 is concerned by the time-resolved SAXS and WAXD measurements of PHB during the isothermal crystallization process, in combination of2D correlation and MRR-ALS analyses. The existence of intermediate structure having the mesomorphic orders and larger characteristic spacings is detailedly confirmed by the elemental WAXD profiles. Moreover,. the 2D correlation analyses were successfu11y carried out between SAXS and WAXD profiles and of SAXS profiles themselves, which indicates that the density fluctuations occur to. increase prior to the scattering peak in SAXS profiles and the diffraction peak in WAXD profiles. We found that, during the isothermal crystallization process of PHB, the density. fluctuations are first transformed from the amorphous melts, and then the intermediate structures increase from the density-rich region and subsequently transformed into the well-developed lamellar crystallites with crystallization time. Combined with the results of. characteristic parameters, the structural development of PHB during the isothermal crystallization including the mesomorphic layers composed of intermediate structures was comprehensively and systematic investigated in this chapter.. 16.
(23) Reference. 1. Lemoigne, M.Ann. Inst. Past. 1925, 34, 144. 2. Doi, Y. Microbial Polyester; VCH Publishers: New York, 1990. 3. Anderson, A. J.;Dawes, E. A. Microbiol. Rev. 1990, 54, 450.. 4. Steinbuchel, A. Polyhydroxylalkanoic acids, In: Byrom D, editor. Biomaterials: novel. materialsfrom biologtcal sources, New York, 1991. 5. Lara, LM.; Gjalt WH. Microbiol. Mol. Biol. Rev. 1991, 63, 21. 6. Barham, P. J.;Barker, P.;Organ, S. FEZVISMicrobiol. Rev. 1992, 103, 289.. 7. Doi, Y.; Kitamura, S.; Abe, H. Macromolecules 1995, 28, 4822. 8. Iwata, T; Doi, Y. Macromol. Chem. Phys. 1999, 200, 2429. 9. Lara, M. L.; Gjalt, H. W Microbiol. Mol. Biol. Rev. 1999, 63, 21. 10. Sudesh, K.; Abe, H.; Doi, Y, Prog. Polym. Sci. 2000, 25, 1503. 11. El-Hadi, A.; Schnabel, R.; Straube, E.; Muller, G.; Henning, S. Polym. Test. 2002, 21,. 665. 12. 0jumu, V. T.; Yu, J.; Solomon, O. B. Aifrican lournal ofBiotechnology 2004, 3, 18.. 13. Ishida, K.; Asakawa, N.; Inoue, Y. Macromol. Symp. 2005, 224, 47.. 14. Vert, M. Biomacromolecules 2005, 6, 538. 15. Chen G. Q.; Wu Q. Biomaterials 2005, 26, 6565. 16. Verlinden, R. A. J.; Hill, D. J.; Kenward, M. A.; Williams, C. D.; Radecka, I. J. Appl.. Microbiol. 2007 IO2 1437.. )). 17. Lemoigne, M. Bull. Soc. Chem. Biol. 1926, 8, 770. 18. Kobayashi, G.; Shiotani, T.; Shima, Y.; Doi, Y. In Biodegradoble Plastics andPolymer,. Elsevier: Arnsterdam, 1994, P410 19. Satkowski, M. M.; Melik, L. A.; Autran, J. P.; Green, P. R.; Noda I.; Schechtman, L. A.. In Biopolymers, Wiley-VCH: Weinheim, 2001, P231. 20. Iwata, T.; Aoyagi, Y.; Fujita, M.; Tamane, H,; Doi, Y.; Suzuki, Y.; Takeuchi, A.; Uesugi,. 17.
(24) K. Macromol. Rapid Commun. 2004, 25, 11OO. 21.. Potter, M.; Steinbuchel, A. Biomacromolecules 2005, 6, 552.. 22.. Bastioli, C. Handbook ofBiodegradoble Polymers: Rapra Technology Limited: UK, 2005.. 23.. Web site: www.nodax.com. 24.. Babel, W.; Sternbuchel, A. Advances in Biochemical Engtneering, Biotechnology: Biopolyestes, Spring-Verlag Berlin Heidelberg, New York, 2001.. 25,. Comibert, J.; Marchessault, R. H. J. Mol. Biol. 1972, 71, 735.. 26.. Yokouchi, M.; Chatani, Y.; Tadokoro, H.; Teranishi, K.; Tani, H. Polymer 1973, 14, 267.. 27.. Marchessault, R. H.; Yu, G. In Biopolymers, Polyesters ll; Doi, Y., Steinbuchel, A.,. Eds.; Wiley-VCH: Wienhiem, 2002, P157. 28.. Marchessault, R. H.; Kawada, J. Macromolecules 2004, 37, 7418.. 29.. Sudech, K.; Abe, H.; Doi, Y. Prog. Polym. Sci. 2000, 25, 1503.. 30.. Koning, G. Can. J. Microbiol. 1995, 41,303.. 31.. Sato, H.; Nakamura, M.; Padermshoke, A.; Yamaguchi, H.; Terauchi, H.; Ekgasit, S.; Noda, I.; Ozaki, Y. Macromolecules 2004, 37, 3763.. 32.. Sato, H.; Murakami, R.; Padermshoke, A.; Hirose, F.; Senda, K.; Noda, I.; Ozaki, Y.. Macromolecules 2004 37 7203.. )). 33.. Sato, H; Dybal, J.; Murakami, R.; Noda, I.; Ozaki, Y. 1. Mol. Struct. 2005, 744-747, 35.. 34.. Sato, H.; Mori, K.; Murakami, R.; Ando, Y.; Takahashi, I.; Zhang, J.; Terauchi, H.; Hirose, F.; Senda, K.; Tashiro, K.; Noda, I.; Ozaki, Y. Macromolecules 2006, 39, 1525.. 35.. Zhang, J.; Sato, H.; Tsuji, H.; Noda, I.; Ozaki, Y. J. Mol. Struct. 2005, 249, 735.. 36.. Hu, Y.; Zhang, J.; Sato, H.; Futami, Y.; Noda, I.; Ozaki, Y. Macromolecules 2006, 39,. 3841. 37.. Bloembergen, S.; Holden, D. A.; Hamer, G. K.; Bluhm, T. L.; Marchessault, R. H.. 18.
(25) Macromolecules1986 I9 2865.. )). 38.. Bluhm, T. L.; Hamer, G. K.; Marchessault, R. H. Macromolecules 1986, I9, 2871.. 39.. Kunioka, M.; Tamaki, A.; Doi, Y. Macromolecules 1989, 22, 694.. 40.. Kamiya, N.; Sakurai, M.; Inoue, Y.; Chujo, R. Macromolecules 1991, 24, 3888.. 41.. Scandola, M.; Ceccoru11i, G.; Pizzoli, M.; Gazzano, M. Macromolecules 1992, 25, 1405.. 42.. Yoshie, N.; Sakurai, M.; Inoue, Y.; Chujo, R. Macromolecules 1992, 25, 2046.. 43.. Mitomo, H.; Morishita, N.; Doi, Y Macromolecules 1993, 26, 5809.. 44.. Moroshi, M.; Morishita, N.; Doi, Y. Polymer 1995, 36, 2573.. 45.. Mansour, A. A.; Saad, G. R.; Hamed, A. H. Polymer 1999, 40, 5377.. 46.. Yamada, S.; Wang, Y.; Asakawa, N.; Yoshie, N.; Inoue, Y. Macromolecules 2001, 34,. 4659. 47.. Yoshie, N.; Saito, M.; Inoue, Y. Polymer 2004, 45, 1903.. 48.. Keen, I.; Broot& P.; Rintoul, L.; Fredericks, P; Trau, M.; (irondahl, L. Biomacromolecules2006 7 427.. J). 49.. Lao, H. K.; Renard, E.; Linossier, I.; Langlois, V.; Vallee-Rehel, K. Biomacromolecules2007 8 416.. )). 50. Jiang, L.; Chen, F.; Qian, J.; Huang, J.; Wolcott, M; Liu, L.; Zhang, J. Ind. Eng. Chem.. Res. 2010 49 572.. )). 51.. Abe, H.; Doi, Y.; Aoki, H.; Akehata, T.; Macromolecules 1998, 31, 1791.. 52.. Wu, Q.; Tain, G.; Sun, S.; Noda, I.; Chen, G. Q. 1. Appl. Polym. Sci. 2001, 82, 934.. 53.. Tain, G; Wu, Q.; Sun, S.; Noda, I.; Chen, G. Q. Appl. Spectrosc. 2001, 55, 888.. 54.. Xu, J.; Guo, B.; Yang, R.; Wu, Q.; Chen, G; Zhang, Z. Polymer 2002, 43, 6893.. 55.. Asrar, J.; Valentin, H. E.; Berger, P. A.; Tran, M.; Padgette, S. R.; Garbow, J. R.. Biomacromolecules2002 3 1006.. 7). 56.. Yoshie, N.; Asaka, A.; Yazawa, K.; Kuroda, Y.; Inoue, Y. Polymer 2003, 44, 7405.. 57.. Alata, H.; Aoyama, T.; Inoue, Y. Macromolecules 2007, 40, 4546.. 19.
(26) 58.. Nakamura, S.; Doi, Y.; Scandola, M. Macromolecules 1992, 25, 4237.. 59.. Nakamura, S.; Yoshie, N.; Sakurai, M. Inoue, Y. Polymer 1994, 35, 193.. 60.. Abe, H.; Doi, Y.; Aoki, H.; Akehata, T. Macromolecules 1998, 3I, 1791.. 61.. Ishida, K.; Wang, Y.; Inoue, Y. Biomacromolecules 2001, 2, 1285.. 62. Zhu, Z.; Dakwa, P; Tapadia, P.; Whitehouse, R. S.; Wang, S. Q. Macromolecules 2003,. 36 4891. ' 63.. Blumm, E.; Owen, A. J. Polymer 1995, 36, 4077.. 64.. Koyama, N.; Doi, Y. Polymer 1997, 38, 1589.. 65.. Yoon, J. S.; Lee, W. S.; Kim, K. S.; Chin, I. J.; Kim, M. N. Kim, C. Eun Polym. J.. 2000 36 435.. )). 66.. Hu, Y.; Sato, H.; Zhang, J.; Noda, I. Ozaki, Y. Polymer 2008, 49, 4204.. 67.. Azuma, Y.; Yoshie, N.; Sakurai, M.; Inoue, Y.; Chujo, R. Polymer 1992, 33, 4763.. 68.. Ikejima, T.; Yoshie, N.; Inoue, Y. Macromol. Chem. Phys. 1996, I97, 869.. 69.. Ikejima, T.; Yoshie, N.; Inoue, Y. Polym. Degrad. Stab. 1999, 66, 263.. 70.. Huang, H.; Hu, Y.; Zhang, J.; Sato, H; Zhang, H.; Noda, I.; Ozaki, Y. X Phys. Chem. B. 2005 I09 19175.. )). 71.. An, Y.; LI, L.; Dong, L.; Mo, Z.; Feng, Z. J. Polym. Sci.: Part B. 1999, 37, 443.. 72,. An, Y.; Dong, L.; Li, G.; Mo, Z.; Feng, Z. J. Polym. Sci.: Part B. 2000, 38, 1860.. 73.. He, Y.;Asakawa, N.; Inoue, Y. Polym. Int. 2000, 49, 609.. 74.. Park, S. H.; Lim, S. T.; Shin, T. K.; Choi, H. J.; Jhon, M S. Polymer 2001, 42, 5737.. 75.. Chee, M. J. K.; Ismail, J.; Kummerlowe, C.; Kammer, H. W Polymer 2002, 43, 1235.. 76.. You, J. W; Chui, H. J.; Don, T. M. Polymer 2003, 44, 4355.. 77.. Qiu, S. B.; Ikehara, T.; Nishi, T. Polymer 2003, 44, 2503.. 78.. Iriondo, P; Iruin, J. J.; Fernandez-Berridi, M. J. Polymer 1995, 36, 3235.. 79.. Iriondo, P.; Iruin, J. J.; Fernandez-Benidi, M. J. Macromolecules 1996, 29, 5605.. 80.. Xing, P.; Dong, L.; An, Y.; Feng, Z.; Avella, M.; Martuscelli, E. Macromolecules 1997,. 30 2726. ). 20.
(27) 81.. Zhang, L.; Goh, S. H.; Lee, S. Y. 1. Appl. Polym. Sci. 1999, 74, 383.. 82.. Alata, H.; Zhu, B.; Inoue, Y. J. Appl. Polym. Sci. 2007, I06, 2025.. 83.. Vilanova, P. C.; Rjbas, S. M.; Guzman, G M. Poljy7ner 1985, 26, 423.. 84.. Peng, S.; An, Y.; Chen, C.; Fei, B.; Zhuang, Y.; Dong, L. Eur Polym. J. 2003, 39, 1475.. 85.. Qiu, Z. B.; Yang, W T.; Ikehara, T.; Nishi, T. Polymer 2005, 46, 11814.. 86.. Qiu, Z. B.; Yang, W T. Polymer 2006, 47, 6429.. 87.. Zhang, J.; Tsuji, H.; Noda, I.; Ozaki, Y. Macromolecules 2004, 37, 6433.. 88.. Padermshoke, A.; Katsumoto, Y.; Sato, H.; Ekgasit, S.; Noda, I.; Ozaki, Y. Polymer. 2004 45 6547.. )). 89.. Padermshoke, A.; Katsumoto, Y.; Sato, H.; Ekgasit, S.; Noda, I.; Ozaki, Y. Spectrochimica Acta PartA 2005, 6I, 541.. 90.. Jiang, Y.; Zhou, J. J.; Li, L.; Xu, J.; Guo, B. H.; Zhang, Z. M.; Wu, Q.; Chen, G. Q.;. Weng, L. T.; Cheung, Z. L.; Chan, C. M. Langmuir 2003, I9, 7414. 91.. Pan, P; Liang, Z.; Zhu, B.; Dong, T. Inoue, Y Macromolecules 2008, 4I, 8011.. 92. Rahman, N.; Kawai, T.; Matsuba, G.; Nishida, K.; Kanaya, T.; Watanabe, H.; Okamoto,. H.; Kato, M.; Usuki, A.; Matsuda, M.; Nakajima, K.; Honma, N. Macromolecules. 2009 42 4739.. )). 93.. Vasanthan, N.; Ozkaya, S.; Yaman, M.; 1. Phys. Chem. B 2010, 114, 13069.. 94.. Baldrian, J.; Horky, M.; Sikora, A.; Steinhart, M.; Vlcek, P.; Amenitsch, H.; Bernstorff,. S. Polymer 1999, 40, 439. 95.. Hsiao, B. S.; Wang, Z.; Yeh, F.; Gao, Y.; Sheth, K. C. Polymer 1999, 40, 3515.. 96.. Sasaki, S.; Tashiro, K.; Kobayashi, M.; Izumi, Y.; Kobayashi, K. Polymer 1999, 40, 7125.. 97.. Lisowski, M S.; Liu, Q.; Cho, J.; Runt, J.; Yeh, F.; Hsiao, B. S. Macromolecules 2000,. 33 4842. ' 98.. Xu, J. T.; Fairclough, J. P.; Mai, S. M.; Ryan, A. J.; Chaibundit, C. Macromolecules. 21.
(28) 2002 35 6937.. )). 99.. 100.. Hama, H; Tashiro, K. Polymer 2003, 44, 6973. Heo, K.; Yoon, J.; Jin, K. S.; Jin, S.; Sato, H; Ozaki, Y.; Satkowski, M. M.; Noda I.;. Ree, M. J. Phys. Chem. B2008, I12, 4571. 101.. Reddy, K. R.; Tashiro, K.; Sakurai, T.; Yamaguchi, N.; Sasaki, S.; Masunaga, H.; Takata, M. Macromolecules 2009, 42, 4191.. 102.. Strobl, G. R. Eur Phys. J. E2000, 3, 165.. 103.. Zhang, J; Sato, H.; Nodeq I.; Ozaki, Y. Macromolecules 2005, 38, 4274.. 104.. Noda, I. Bull. Am. Phys. Soc. 1986, 520, 31.. 105.. Noda, I. J. Am. Chem. Soc. 1989, III, 8166.. 106.. Noda I. Chemtract-Macromol. Chem. 1990 1 89.. 107.. Barton II, F. B.; Himmelsbach, D. S.; Duckworth, J. H.; Smith, M. J. Appl. Spectrosc.. ,. 1992 46 420.. )). 108.. Noda, I.; Ozaki, Y. 2D Nodu book Two-Dimensional Correlation Spectroscopy; John Wiley & Sons: Chichester, UK. 2004.. 109.. Ren, Y.; Matsushita, A.; Matsukawa, K.; Inoue, H.; Minami, I.; Noda, I.; Ozaki, Y. vab. Spectrosc. 2000, 23, 207.. 110.. Matsushita, A.; Ren, Y.; Matsskawa, K.; Inoue, H.; Minami, I.; Noda, I.; Ozaki, Y. vab. Spectrosc. 2000, 24, 171.. 111.. Padermshoke, A.; Sato, H.; Katsumoto, Y.; Ekgasit, S.; Noda, I.; Ozaki, Y. vab. Spectrosc. 2004, 36, 241.. 112.. Karjlainen, E. J. Chemom. Intell. Lab. Syst. 1989, 7, 31.. 113.. Tauler, R.; Kowalski, B.; Fleming, S. Anal. Chem 1993, 65, 2040.. 114.. Tauler, R. Chemom. Intell. Lab. Syst. 1995, 30, 133.. 115.. Tauler, R.; Smidile, A. K.; Kowalski, B. J. Chemom. 1995, 9, 31.. 116.. Juan D'Tauler R.Anal. Chim.Acta2003 500 195.. 117.. Larrechi, S. M.; Rjus, X. F. Appl. Spectrosc. 2004, 58, 47.. ) .) ). 7). 22.
(29) 118.. Marquez-Alvarez, C.; Rodriguez-Ramos, I.; Guerrero-Ruiz, A.; Haller, G. L.; Fernandez-Garcia, M. J. Am. Chem. Soc. 1997, 119, 2905.. 119.. Ganido, M.; Lazaro, I.; Larrechi, S. M.; Rius, X. F. Anal. Chim. Acta 2004, 515, 65.. 120.. Spegazzini, N.; Ruisanchez, I.; Larrechi, S. M.; Cadiz, V.; Canadell, J. Analyst 2008,. I33 1028. ' 121.. Perera, P.; Wyche, M.; Loethen, Y.; Ben-Amotz, D. 1. Am. Chem. Soc. 2008, 130, 4576.. 122.. Spegazzini, N.; Ruisanchez, I.; Larrechi, S. M. Anal. Chim. Acta 2009, 642, 155.. 123.. Blobel, J.; Bernado, P.; Svergun, I. D.; Tauler, R.; Pons, M. 1. Am. Chem. Soc. 2009,. 13I 4378. ' 124.. Iglesias-Juez, A.; Kubacka, A.; Fernandez-Garcia, M.; Michiel, M. D.; Newton, M. A.. J. Am. Chem. Soc. 2011 133 4484.. )p. 125.. Jiang, J. H.; Liang, Y. Z.; Ozaki, Y. Chemom. Intell. Lab. Syst. 2004, 71, 1.. 126.. Jaumot, J.; Gargallo, R.; Juan, A.; Tauler, R. Chemom. Intell. Lab. Syst. 2005, 76, 101.. 127.. Juan A.' Tauler R. Crit. Rev. Ana.lChem. 2006 36 163.. 128.. Maeder, M.Anal. Chem. 1987, 59, 527.. 129.. Golub, G. H.; Loan, F. V. Matrix Computations, Johns Hopkins University Press:. )). 7). '. Baltimore MD 1983.. )). 130.. 7he Mathwork, Natick, MA MATLAB version 6.5. 2002.. 23.
(30) o. H3C. c. o. fi2. n. PB (a). Biodegradation. Polymer. S13iii. Processing. Polymeric Product. Biologically Environment synthesis by Friendly Polymer Microorganism. Conversionthit of ..s-. Plant Materials Plants Photosynthesis (b) Figure 1: The general chemical structure ofPHB (a) and scheme for the biodegradation and. biologically synthesis ofenvironment friendly polymer (b).. 24.
(31) (a). a. /. Nlt. 't"tJt:vl. -:'''i/iil ''te. /. twxN..... tt. ti-l-1t-t. ""'<x..LQ. 'Jt -le. lt. tI' t't'. ,x .e'. ./.. l.... F". .....••i. tlii-. ri. i-tl-ttl-s-. tl!. i"ltt ttlvt--l-t. --p---r-. jVtt. --e tV;-"a-4t. tl--. t. a. e :C atom. "tw :O atom. (b). o :H atom. x. t . . .e. H/sVc,.,, r.x. x. -v. H'. -. e. ... •••••••••b. es. t. x. i9. >.'. '. H. ,. H Figure 2: Schematic illustration ofthe lamellar crystal structure (a) and helix structure (b) of. PHB crystals (Reproduced from reference 32).. 25.
(32) WAXD2DPattern. E1ectron. Beam. Temperature Enc1osure. SAXS 2D Pattern. WAXD (P). SAXS. Bearn Stopper. ""•••)tbL. (CCD). Samp1e ., "f pt :pt. X-ray. -k a- --. -" "- -t pt de "P -" -" ". -. "" ei. n- pt -p tpt e-" "-1 --t ---. --J. -ij k -lt in -- dta -k -- .-,. - -k "- - -" - -e -- ij- -b tk ts. Vacuum Path Figure 3: Schematic illustration ofthe time-resolved synchrotron SAXS/WAXD measurement system.. 26.
(33) 20 40 60 80. li.liiI'll,,,,,fk,i,2.ll',)lilllllii'i"''i'//1'lii'1111il'ii'g''.i/ill,,]ii.11'L-,'si'\•'`,i,,... 2D correlation. 20 i•g. 80 60 40 20. 80 60 40 20. 3 2 1 o -1. 40 an • 's. BO. Original Spectra. ICD -2. 100. eo ,. ee '. co 4e '20 va. synchronous Figure 4: General scheme for the two-dimensional correlation spectroscopy.. 27. 1co. 6[l et). oo. asynchronous.
(34) Pure Component lnformation. Mixed lnformation. wavelengths. =. time. D. : : : 1 : 1 : : 1 Cl :. l l l. i i i l/ Cn. Sl --e------el---------------. ee-------------------. Sn. ST. 1. c. $ g. `Åí.Iii. g. {. .. 1.;% in -. "N•. i.]i,i•itt:/E,illi•iiiii-i,k•'ii. i.'z.lxN/5-g. ,.?. --. u'v .. , Retenbon times. Retembon times Wavelengths Figure 5: General scheme for the MCR-ALS analysis (Reproduced from reference 116).. 28. Wavelengths. +E.
(35) D. Detection of pure variable. EFA NO component. Initial estimates. Local rank information. Non-negativity. ALS. Closed system. C. ST. Figure 6: The operating procedure ofMCR-ALS technique.. 29. +. E.
(36) Chapter 1. FTIR Study on Hydrogen Bonding Interactions in Biodegradable. Polymer Blends of Poly(3-hydroxybutyrate) and. Poly(4-vinylphenol). 30.
(37) ABSTRACT: The hydrogen bonding interactions and the crystallinity of biodegradable polymer poly(3-hydroxybutyrate) (PHB) blended with poly(4-vinylphenol) (PVPh) were studied on the as-cast films as a function of the blend composition wpvph by using Fourier-transform infrared spectroscopy, differential scanning calorimetry, and wide-angle X-ray diffraction.. The intermolecular hydrogen bondings between the C=O groups ofPHB and the OH groups ofPVPh (designated as inter) were explored not only in C=O stretching vibration region but. also in OH stretching vibration region. The bands in OH stretching vibration region were assigned. The spectra in the C=O stretching vibration region was decomposed into elemental spectra attributed to inter, the free C=O group, and the intramolecularly hydrogen bonded. C=O group between the C=O group and one ofthe C-H group in CH3 ofPHB (designated as .free and intra, respectively) to determine the respective fractions,,iinter,ffre,, andfmtra. We found. thatfks (k == intra, inter and.free) exhibit a double-step change with wpvph. (i) Step 1 with O S. wpvph S O.5:ffree is unchanged with wpvph and almost identical toffree of neat PHB, whilefintr. andfmt.r decreases and increases, respectively. This reveals that intra and inter exchange each. other when wpvph is changed via the following two elemental transformations: (a) the transformation from intra to.free, which destabilizes the lamellar crystals, decreasesfntr. and. increasesffr,.; (b) that from.free to inter, which decreasesffree and increasesfmt,r. The two. counterbalancing processes cause ffree unchanged with wpvph. (ii) Step 2 with O.5 < wpvph where X-ray crystallinity tends to vanish:ffr,e starts to decrease andfmter starts to increase even. more largely with wpvph than the case of (i), implying that the exchange from.free to inter is. suppressed by the presence of the crystallinity in step 1, while that from .free to inter is enhanced in the absence ofthe crystallinity in step 2.. 31.
(38) Introduction. Polyhydroxyalkanoates (PHAs) are well-known as naturally made biodegradable high molecular weight aliphatic polyesters.i'` Among PHAs, poly(3-hydroxybutyrate) (PHB) is the. most abundant polyester found in bacteria and also most extensively studied.i'3 However, there is still a large technical barrier for practical applications ofPHB as environment-friendly polymeric materials, because PHB is rigid and brittle due to its excessively high crystallinity,. and it is also thermally unstable during the conventional melting processing due to the high melting temperature (Tm).. The crystal structure ofPHB is orthorhombic with lattice parameters a = 5.76A, b = 13.20A,. and c = 5.96A (fiber repeat distance).5'6 PHB crystals have the intramolecular hydrogen bondings (C=O•'•H-C) (designated hereafter as intra) between the C==O groups in one helix and the CH3 groups in the other helix along the a axis.7-iO. Moreover, intra can stabilize the chain folding in the lamellar structure ofPHB.9. There are some approaches to improve the property of PHB-based polymeric materials, such as copolymerizationii'i6 and blendingi7-2i. Many hydroxyl group containing polymers are reported as a good counter polymer to be mixed with polyesters, because those blends have been found to form totally or partially miscible systems driven by the intermolecular hydrogen bonding interactions.i3'22'24 We would like to further advance the study along this. line by one step deeper and point out that the exchange ofthe hydrogen bondings between the intermolecular one (between the C =O group in the polyester and the OH group in the counter polymer) (designated hereafter as inter) and intra, and vice versa, would give crucial effects on both crystallization and miscibility. In a more rigorous sense, this exchange between intra. and inter involves two elemental transformation processes: that between intra and .free and that between.free and inter as well. Here `tfree" designates the free C=O group. The inter is. expected to influence the glass transition temperature (Tg), increment of heat capacity, crystallinity, and crystal lattice parameters ofthe blends as well.. 32.
(39) In this chapter, poly(4-vinlphenol) (PVPh) was selected as the hydroxyl-group-containing. polymer and blended with PHB. Figure 1-1 shows the chemical structures of PHB (A) and. PVPh (B), respectively Due to the existence of C=O and OH groups in the constituent polymers (A) and (B), respectively, the blends are possible to form inter. Let us briefly review. the works reported on PHBMVPh blends below. Xing et al.25 reported that this particular blend system is miscible at all compositions. through the single Tg evaluated by DSC thermograms and that the negative values of the segmental interaction parameter determined from the equilibrium melting point depression. support the miscibility and strong hydrogen bonding interactions between PHB and PVPh. Iriondo et al.24'26 reported the existence of inter in Pyph blends with tactic and atactic PB. through the FTIR spectra in the C=O stretching vibration region. For the tactic PHB blends. which are crystallizable, the specimens were heated above melting temperature and the spectra were recorded in different cooling temperatures in order to avoid the influence ofthe. crystallinity on the crystalline carbonyl band. Using the association model of Painter and Coleman27, they reported the difference in the equilibrium constants ofthe association for the. two blend systems with the tactic and atactic PHBs, respectively, based upon the evaluated values offmter andffr,. only under the conditions offmtra = O.. In this chapter, we aim to advance the investigation of this blend by one step further. through systematically investigating the following parameters for the same samples: the. thermal behavior with DSC, crystal structure and crystallinity with wide-angle X-ray diffraction (WAXD) and DSC, and intra, .free, and inter with FTIR spectroscopy. The investigation was focused on the following aspects: (1) further reinforcement of existence of. inter through the assignment of the OH stretching vibration bands in addition to the C==O stretching vibration bands; (2) decomposition of the vibrational spectra in the C=O stretching. region into fundamental vibration spectra reflecting inter, intra, and .free in order to systematically study the transformations among them when wpvph is varied; (3) determination ofX-ray crystallinity X. and crystal lattice parameters. In item (2) described above, we should. 33.
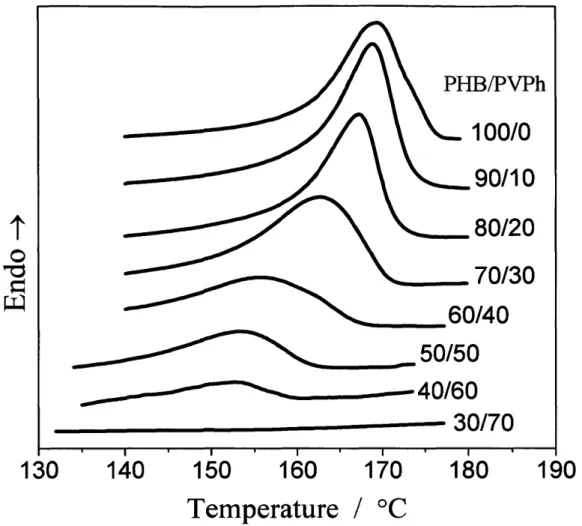
(40) note that the decomposition process was generalized beyond the level ofIriondo et al.24'26 and Gonzalez et al.28 to include intra associated with crystallinity ofPHB phase.. 34.
(41) Experimental Section. Materials and SampRe Preparation Procedures. The bacterially synthesized PHB with number-averaged molecular weight M. = 6.5Å~105 was purchased from Aldrich Chemical Corp., Inc. PVPh, which is amorphous with glass transition temperature (Tg) equal to 118.2 OC. and with weight-averaged molecular weight Mw = 8.0Å~103, was purchased from Aldrich Chemical Corp., Inc. PHB and PVPh were separately dissolved in chloroform and 2-butanone to prepare homogenous solutions having concentrations 1 and 4 wtO/o, respectively. The two solutions were then mixed into a homogeneous solution and casted on CaF2 substrates at 80 OC for 10 minutes. Subsequently, the cast films were kept in a vacuum oven at 60 OC for 24 hours in order to evaporate the solvent completely, and then cooled to room temperature. The. samples thus prepared were designated hereafter as "as-prepared" samples and held in the vacuum oven at room temperature until measurements.. FTIR Spectroscopy. The transmission FTIR spectra were measured at 30 OC for the. as-prepared samples of the PHB/PVPh blends using a Nicholet NEXUS 870 Fourier transform IR spectrometer (Waltham, Massachusetts) equipped with a liquid-nitrogen-cooled. mercury-cadmium-telluride detector. A total of 256 scans were accumulated for signal-averaging of each IR spectral measurement to ensure a high signal-to-noise ratio with a. 2 cm'i resolution. The CN4400, OMEGA thermoelectric device (Boulder, Colorado) was used as a temperature controller with an accuracy Å}O. 1 OC.. Differential Scanning Calerimetry (DSC). DSC measurements of the PHBIPVPh blends were performed with a Perkin-Elmer Pyris6 DSC system (Waltham, Massachusetts, USA) at heating and cooling rate of 1O OCImin under a nitrogen purge. Separately from the as-prepared. samples described above, the sample films for the DSC measurement were prepared by casting the solution on aluminum pans at 80 OC for 30 minutes and then sealed in them. The samples were heated from 30 to 190 OC (first heating process) and then maintained at 190 OC. for 2 min before cooling to -30 OC. Subsequently, the samples were reheated to 463 K. 35.
(42) (second heating process). The value ofthe peak point in the first heating process was taken as the Tm.. VVide-Angle X-ray Diffraction ('WAXD). The WAXD patterns were measured for the as-prepared samples of PHBMVPh blends at 30 OC in the scattering-angle range of 2e =. 2400 by using RJGAKU RINT2000 X-ray diffractometer (Tokyo, Japan) with CuKct radiation (wavelength, 1.5418A) and with an X-ray generator ofpower 50kV and 40mA. The WAXD patterns ofthe blends were recorded as a function ofwpvph at the scanning rate of2e = O.50 per minute at room temperature.. 36.
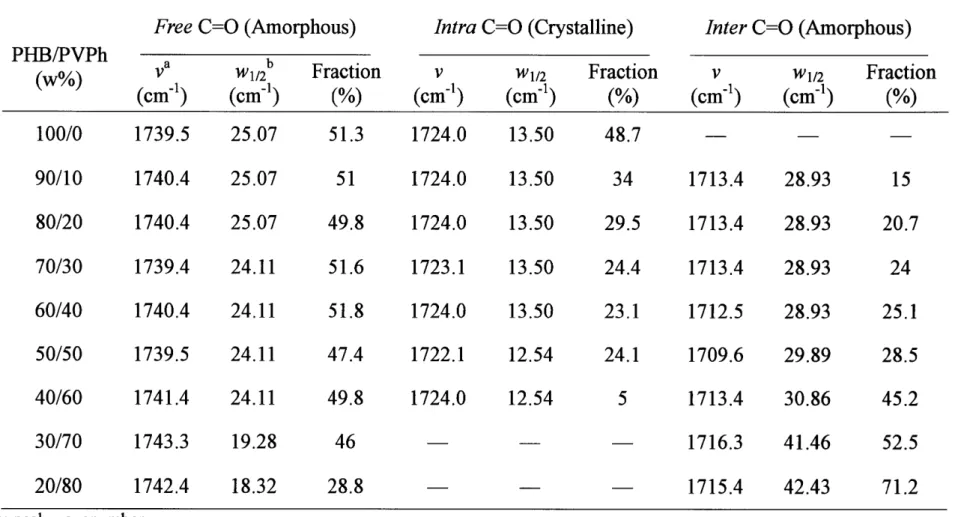
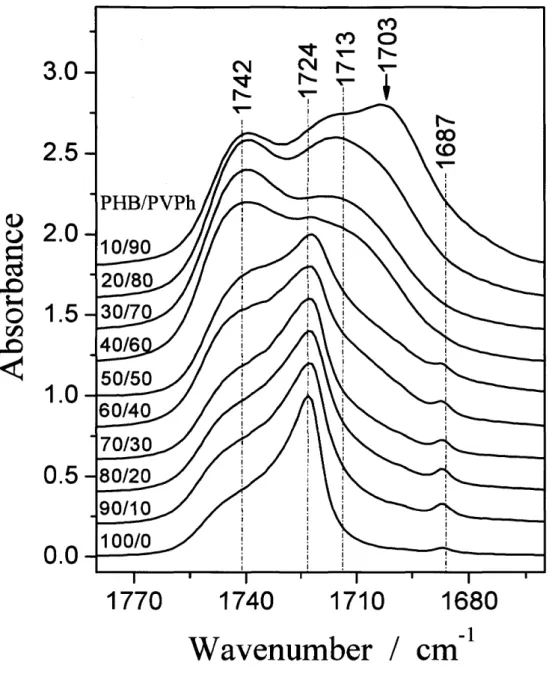
(43) Results and Discussion. Thermal Anatysis. Figure 1-2 shows the thermograms ofthe PHB/PVPh specimens during the first heating process. The temperature at the peak was taken as melting point (Tm). It is. clear that, compared with pure PHB, the Tm and melting enthalpy ofthe blends decrease with wpvph, indicating that the blend system is miscible in the molten state and the crystallinity is. depressed with wpvph, consistent with the results reported by Xing et al.25 Moreover, the DSC curves ofthe blends with wpvph ) 70 wtO/o do not exhibit the melting peak, indicating that the. crystallization hardly occurs in these blends prepared as the above description. The disappearance ofcrystallinity will also be discussed in the following FTIR and WAXD parts.. Composition-Dependent FTIR Spectra in the C==O Stretching Region. Figure 1-3 shows the FTIR spectra obtained at room temperature for the as-prepared specimens of pure PHB and the PHBMVPh blends with increasing wpvph in the C =O stretching vibration region. Two bands centered at 1742 cm-i and 1724 cm-i are due to.free and intra, respectively, each. of which corresponds to the amorphous and crystalline states of PHB for the reasons as clarified later. For the spectrum with wpvph = 10 wtO/o, the peak around 1700 cm-i (shown by the arrow) is due to a residual amount of the solvent (2-butanone) in the as-prepared films, which is difficult to be evaporated completely under the heat treatment employed in this work.. The composition-dependent FTIR spectra ofthe as-prepared specimens show the following trends with wpvph. The sharp and dominant intra band around 1724 cm-i gradually broadens. and eventually disappears when wpvph ) 70 wtO/o, where crystallinity as observed by the melting exotherm also disappears as shown in the DSC results (Figure 1-2). Besides, the absorption intensity of.free around 1742 cm-i increases with wpvph. Thus we can reasonably assess intra and.free to crystalline and amorphous bands, respectively. The second derivatiye ofthe spectra in Figure 1-3 was calculated and shown in Figure 1-4. For the blend with wpvph. = 70 wtO/o, the crystalline C=O band (or intra) totally disappeared. Moreover, this phenomenon can be verified also by the disappearance ofa minor band centered at 1687 cm-i. 37.
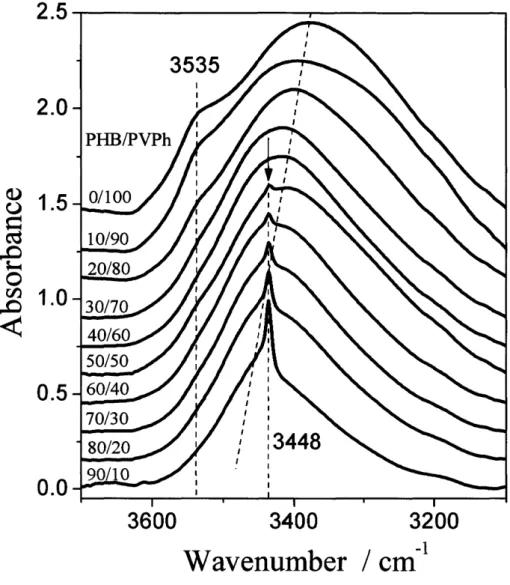
(44) when wpvph ) 70 wtO/o as shown in Figure 1-3 and Figure 1-4. This band has been reported to be undoubtedly a crystalline band, although its spectral origin is not yet assigned.i7 All of. these suggest that PHB is no longer able to crystallize in the as-prepared PHBMVPh blends,. when wpvph ) 70 wtO/o. This point will be further confirmed by the WAXD study to be discussed later.. Accompanied by the increased breadth of the band at 1724 cm"i with wpvph, a new band appears at the lower-wavenumber side around 1713 cm'i as shown in Figures 1-3 and 1-4.. However, this peak does exist neither in the pure PHB spectrum nor in the pure PVPh spectrum. Therefore, this is a new band inherent in the PHBneVPh blend system and is expected to reflect inter. In general, the hydrogen bonding in the region from 1700 to 1720. cm-i is quite well-known in many hydrogen-bonded polymer blends containing C==O groups and oH groups.22'23 Actually, Iriondo et al.24'26 also have reported existence of inter in the. C=O stretching vibration region at approximately 1709cm'i for the PHB/PVPh blend specimens cooled down from the melts to 110 OC. They reported the contribution of inter increases with wpvph. Moreover, we found that the increase of inter with wpvph occurs in parallel to the decrease of intra and increase offree. These results assure the formation of inter.. Intra can stabilize the chain folding in the lamella strueture and hence brings about the high. crystallinity ofPHB.9 On the other hand, the OH group ofPVPh can hold the C=O group of PHB from the amorphous phase through inter, so as to depress the crystallinity ofPHB. With. the increase of wpvph, intra appears to be exchanged into inter, which suppresses the crystallizability of PHB. Therefore, intra and inter can influence each other through the exchange between them, which will be discussed in the following part.. Composition-Dependent FTIR Spectra in the OH Stretching Region. Figure 1-5 shows the FTIR spectra in the OH stretching vibration region of pure PVPh and the PHBIPVPh blends with decreasing wpvph measured at room temperature. As shown in the spectrum of pure PVPh, it exhibits two obvious stretching vibration bands, and we assign them due to the. 38.
(45) z-associated OH band around 3535 cm-i and the self-associated OH band around 3380 cm-i. The z--association is the interactions between the hydroxyl and phenyl groups ofPVPh, and the self-association is the interactions among the OH groups ofPVPh.. A narrow and sharp band centered around 3448 cm-i observed in the FTIR spectra (shown by the line and the arrow), gradually decreases with wpvph and disappears when wpvph ) 60 wtO/o. The wavenumber ofthis band (3448 cm-i) is the double than that ofthe crystalline C=O band (intra at 1724 cm'i). Therefore, the band at 3448 cm-i should originate from the first. overtone ofthe crystalline C=O stretching vibration at 1724 cm-i ofPHB. However, as shown in the C=O stretching vibration region (Figure 1-3), the intra C=O band disappears for the blends having wpvph ) 70 wtO/o. At wpvph = 60 wtO/o, the disappearance of the band around 3448cm-i is due to the low intensity ofthis band and the strong OH stretching vibration. This. broad OH stretching vibration tends to cover the first overtone of the crystalline C=O band when the crystallinity is low. However, we can also detect the decrease ofcrystallinity ofPHB, through the intensity reduction ofthis first overtone band.. The second derivatives of the spectra in Figure 1-5 (wpvph ) 60 wtO/o), which were not affected by the first overtone ofthe crystalline C=O band, were shown in Figure 1-6. For pure. PVPh spectrum, there is a peak around 3600 cm-i which is not observed in original spectrum.. The peak around 3600 cm'i is reasonable due to the free OH groups ofPVPh, which is just shown as a quite small shoulder in Figure 1-5. We can clearly find another new band centered. around 3460 cm-i for the PHBIPVPh blends with wpvph = 60 wtO/o. Therefore, we propose that. this new band is the inter OH band and combined with the self-associated OH band in the. blend which is shifted from the neat PVPh band around 3380 cm'i toward a higher wavenumber with the PHB content. This shift is in turn caused by inter. However, a quantitative assessment of the OH band in this region is quite difficult, because of an extensive and intricate overlap of some bands.. Curve-Fitting Analysis for the Fraction of ffree, fmtra, and .flmter. The analysis of. composition-dependent FTIR spectra in the C=O and OH stretching vibration regions. 39.
図
+7





関連したドキュメント
[r]
Introduction to Japanese Literature ② Introduction to Japanese Culture ② Changing Images of Women② Contemporary Korean Studies B ② The Chinese in Modern Japan ②
3 学位の授与に関する事項 4 教育及び研究に関する事項 5 学部学科課程に関する事項 6 学生の入学及び卒業に関する事項 7
[r]
[r]
[r]
[r]
[r]
