Table of Contents 1 非臨床試験計画概略 ... 7 2 薬理試験... 10 2.1 効力を裏付ける試験 ... 10 2.1.1 活性及びジェノタイプ網羅性 ... 10 2.1.2 特異性及び選択性 ... 11 2.1.3 各種細胞における細胞毒性 ... 11 2.1.4 作用機序... 11 2.1.4.1 NS5B ポリメラーゼに対する作用 ... 11 2.1.4.2 耐性 ... 11 2.1.4.3 交差耐性... 12 2.1.5 In vitro 併用試験 ... 12 2.2 安全性薬理... 12 2.2.1 受容体・イオンチャネル結合及び酵素アッセイ ... 13 2.2.2 心血管系(In vitro / In vivo) ... 13 2.2.3 中枢神経系... 14 2.2.4 呼吸系 ... 14 3 薬物動態試験 ... 15 3.1 吸収、バイオアベイラビリティ及び薬物動態 ... 15 3.2 分布 ... 16 3.3 代謝 ... 17 3.4 排泄 ... 19 3.5 蛋白結合... 19 3.6 薬物動態学的薬物相互作用 ... 19 3.7 トキシコキネティクス ... 21 4 毒性試験... 26 4.1 単回投与毒性 ... 27 4.2 反復投与毒性 ... 27 4.2.1 ラット ... 27 4.2.2 イヌ ... 28 4.3 併用投与毒性 ... 29 4.4 遺伝毒性... 29
4.6 生殖発生毒性 ... 30 4.7 その他の毒性 ... 31 4.7.1 光毒性 ... 31 4.7.2 抗原性及び免疫毒性 ... 31 4.7.3 毒性発現の機序に関する試験 ... 32 4.7.4 依存性 ... 32 4.7.5 代謝物の安全性評価 ... 32 4.7.6 不純物の安全性評価 ... 32 5 総括及び結論 ... 33 6 参考文献... 37
List of in-text Tables
Table 1-1: BCV の毒性試験 ... 9 Table 3.3-1: 動物及びヒトに[14C]BCV を単回経口投与したときの in vivo 代謝 ... 18 Table 3.7-1: 主要な毒性試験におけるBCV の AUC 値及びヒト AUC に対する動物
AUC との比 ... 22 Table 3.7-2: 主要な毒性試験におけるBMS-794712 の AUC 値及びヒト AUC に対す
る動物AUC との比 ... 24 Table 4-1: BCVの無毒性量及び主な毒性発現用量における曝露量とヒト曝露量と
用語及び略号一覧
略号 英語 日本語
ADME absorption, distribution, metabolism and
excretion 吸収、分布、代謝及び排泄
A alanine アラニン
ALT alanine aminotransferase アラニンアミノトランスフェラーゼ APD50 action potential duration at 50%
depolarization 50%再分極時までの活動電位持続時間 APD90 action potential duration 90 at 90%
depolarization 90%再分極時までの活動電位持続時間
ASV asunaprevir アスナプレビル
AUC area under the plasma concentration-time
curve 血漿中濃度曲線下面積
BCRP breast cancer resistance protein 乳癌耐性蛋白 BCV NS5B polymerase inhibitor, beclabuvir
(BMS-791325) NS5B ポリメラーゼ阻害薬、ベクラブビル(BMS-791325) BDC bile duct-cannulated 胆管カニューレを挿入した
BID twice daily 1 日 2 回
BMS Bristol-Myers Squibb ブリストル・マイヤーズ スクイブ社 BMS-650032 NS3 protease inhibitor, asunaprevir, ASV NS3 プロテアーゼ阻害薬、アスナプレビ
ル、ASV BMS-790052 NS5A replication inhibitor, daclatasvir,
DCV NS5A複製阻害薬、ダクラタスビル、DCV
BMS-791325 NS5B polymerase inhibitor NS5B ポリメラーゼ阻害薬 BSEP bile salt export pump 胆汁酸塩輸送ポンプ
BVDV bovine viral diarrhea virus ウシウイルス性下痢ウイルス CAC Carcinogenicity Assessment Committee がん原性評価委員会
CC50 50% cytotoxic concentration 50%細胞毒性濃度 Cmax maximum plasma concentration 最高血漿中濃度 CYP cytochrome P450 チトクロームP450
D aspartic acid アスパラギン酸
DAA direct acting antivirals 直接作用型抗ウイルス薬
DCV daclatasvir ダクラタスビル
DNA deoxyribonucleic acid デオキシリボ核酸 EC50 50% effect concentration 50%有効濃度 EMA European Medicines Agency 欧州医薬品庁
用語及び略号一覧
略号 英語 日本語
FDA Food and Drug Administration 米国食品医薬品局 GALT gut-associated lymphoid tissue 消化管関連リンパ系組織
GGT gamma-glutamyl transferase γ-グルタミルトランスフェラーゼ GLP Good Laboratory Practice 医薬品の安全性に関する非臨床試験の実
施の基準
GTP guanosine triphosphate グアノシン三リン酸 HCV hepatitis C virus C 型肝炎ウイルス
hERG human ether-a-go-go-related gene ヒトether-a-go-go 関連遺伝子 HIV human immunodeficiency virus ヒト免疫不全ウイルス HLM human liver microsome ヒト肝ミクロソーム IC50 concentration that causes 50% inhibition 50%阻害濃度
ICH International Conference on Harmonization 日米 EU 医薬品規制調和国際会議 IKr rapidly activating, delayed rectifier cardiac
potassium current 急速活性化遅延整流カリウム電流 Ki inhibitory constant 阻害定数
LC-MS/MS liquid chromatography with tandem mass
spectrometry 液体クロマトグラフィー・タンデム質量分析
L Leucine ロイシン
MRP multiple drug-resistance protein 多剤耐性蛋白
ND not determined 実施せず
NS3 nonstructural protein 3 of HCV HCV 非構造蛋白 3 NS5A nonstructural protein 5A of HCV HCV 非構造蛋白 5A NS5B nonstructural protein 5B of HCV HCV 非構造蛋白 5B NTCP sodium-taurocholate cotransporting
polypeptide タウロコール酸ナトリウム共輸送ポリペプチド OAT organic anion transporter 有機アニオントランスポーター
OATP organic anion transporting polypeptide 有機アニオン輸送ポリペプチド OCT organic cation transporter 有機カチオントランスポーター PAMPA parallel artificial membrane permeability
assay 人工膜透過測定
PDE4 phosphodiesterase 4 ホスホジエステラーゼ4
P proline プロリン
用語及び略号一覧
略号 英語 日本語
PK pharmacokinetics 薬物動態
RNA ribonucleic acid リボ核酸
S serine セリン
SCN5A sodium channel ナトリウムチャネル
T threonine スレオニン
T-HALF elimination half-life 消失半減期
Tmax time to maximum plasma concentration 最高血漿中濃度到達時間
UGT uridine diphosphate glucuronosyl transferase ウリジン二リン酸グルクロン酸転移酵素
V valine バリン
1 非臨床試験計画概略 ベクラブビル塩酸塩[BMS-791325、以下、ベクラブビル(BCV)]は C 型肝炎ウイルス(HCV)非 構造蛋白5B(NS5B)ポリメラーゼを阻害し、NS5B によるウイルスリボ核酸(RNA)の複製を阻害 する非核酸系thumb site 1 阻害薬である。BCV は哺乳類のポリメラーゼに対して活性を示さず、ヒト における本薬の標的分子は知られていない。 BCV は他の直接作用型抗ウイルス薬(DAA)と相加又は相乗的な相互作用を示す。今回、C 型慢性 肝炎患者又はC 型代償性肝硬変患者を対象に、既承認のダクラタスビル塩酸塩(DCV、BMS-790052) 及びアスナプレビル(ASV、BMS-650032)との固定用量配合剤による併用療法として製造販売承認 申請を行う。 BCV の臨床推奨用量は 75 mg の 1 日 2 回(BID)経口投与(150 mg/日)である。臨床推奨用量にお ける定常状態でのBCV のヒト曝露量(以下、ヒト曝露量)は、最高血漿中濃度(Cmax)で 1.49 µg/mL、 血漿中濃度曲線下面積(AUC)で 17.4 µg•h/mL であった。動物での BCV 曝露量のヒト曝露量に対す る比(動物の曝露量 ÷ ヒトの曝露量)は、これらのヒト曝露量を基に算出した。マウス、ラット、イ ヌ、サル及びヒトにおける蛋白結合率は同程度であることから(97%超)、in vivo の曝露量比較では、 BCV 遊離体としての補正は行わなかった。なお、Cmax 及び AUC に性差がなかったことから、曝露 量は雌雄合算平均値で示した。 BCV の in vitro 評価として、HCV NS5B との相互作用における作用強度及び選択性について酵素を用 いたポリメラーゼアッセイを実施し、BCV の効力及び細胞毒性に関しては HCV レプリコンアッセイ を実施した。BCV の NS5B ポリメラーゼ活性に対する 50%阻害濃度(IC50)は、HCV レプリコンアッ セイにおける50%有効濃度(EC50)と良好な相関を示した。 BCV は HCV に対して選択的であり、細胞培養による HCV レプリコンアッセイにおいて、ジェノタ イプ1a 及び 1b のレプリコンに対する EC50値は1.6~9.5 nM であった。BCV は近縁のウシウイルス性 下痢ウイルス[(BVDV)のレプリコン及びウイルス]を含む一連の RNA 及び DNA ウイルスに対し て活性を示さなかった。BCV 代謝物では、主代謝物の BMS-794712(N-脱メチル化代謝物)が BCV と同程度の活性を有し、その他の代謝物は、比較的弱い(BCV の 2~15 倍の EC50値)活性を有する (BMT-110547、BMT-171207 及び BMT-142478)、若しくは非活性[BMS-948158(N-及び O-脱メチ ル化体のO-グルクロン酸抱合体)]であった。BMS-794712 の血漿中濃度はヒト血漿中 BCV の約 25% を占め、BMT-110547、BMT-171207、BMT-142478 及び BMS-948158 の血漿中濃度は微量であった。 これらのことから、BMS-794712 も BCV の薬理活性に寄与しているものと考えられた。 In vitro 及び in vivo の安全性薬理試験により、心血管系、中枢神経系及び呼吸系に及ぼす影響を評価 した。すべての安全性薬理試験は適切な試験計画に基づき、日米EU 医薬品規制調和国際会議(ICH) ガイドラインに準拠して実施した。 高感度かつ特異的な液体クロマトグラフィー・タンデム質量分析(LC-MS/MS)法を用いて、BCV とその代謝物の血漿中濃度を測定した。定量的全身オートラジオグラフィーを用いて、Long-Evans ラット(有色)及びSprague-Dawley(SD)ラット(アルビノ)における BCV の組織内分布を検討し
た。SD ラットにおける BCV の乳汁移行及び胎盤通過を検討した。In vivo における代謝物投与試験は 行わなかった。 各種動物及びヒトを用いて、BCV の吸収、分布、代謝及び排泄(ADME)並びに薬物動態(PK)を 検討した。いくつかのPK 試験から得られた生体試料中の BMS-794712[薬理学的活性(抗 HCV 活性) を有するN-脱メチル化体]及び BMS-948158(N-及び O-脱メチル化体の O-グルクロン酸抱合体)を 測定し、チトクローム P450(CYP)酵素の阻害及び誘導作用並びにトランスポーターの阻害作用を 検討した。すべてのADME 及び PK 試験は ICH ガイドラインに基づき実施した。 人工膜透過測定(PAMPA)法を用いて、膜透過性、Caco-2 細胞を用いた膜透過性、血球移行、ミク ロソーム蛋白との共有結合、各種動物及びヒト由来の血清又は血漿蛋白との可逆的結合、ラット及び ヒト肝細胞内取込み機序を検討した。BCV の主代謝物を生成する酵素を調べるため、ヒト肝ミクロ ソーム及びヒトcDNA 発現 CYP 酵素及びウリジン二リン酸グルクロン酸転移酵素(UGT)を用いた in vitro 試験を実施した。BCV の CYP 酵素阻害作用及び誘導作用をそれぞれヒト肝ミクロソーム及び ヒト肝細胞を用いて検討した。また、ヒト肝ミクロソームを用いてBCV の UGT1A1 阻害作用を検討 した。BCV、BMS-794712 及び BMS-948158 が、P 糖蛋白(P-gp)、乳癌耐性蛋白(BCRP)、有機ア ニオン輸送ポリペプチド(OATP)1B1、OATP1B3、有機アニオントランスポーター(OAT)1、OAT3、 有機カチオントランスポーター(OCT)2、多剤耐性蛋白(MRP)2、胆汁酸塩輸送ポンプ(BSEP) 及びタウロコール酸ナトリウム共輸送ポリペプチド(NTCP)等のトランスポーターの基質又は阻害 剤であるかどうかをin vitro 試験系で評価した。 BCV の毒性を評価するための非臨床毒性試験として、単回投与毒性試験(マウス、ラット、イヌ)、 反復投与毒性試験(ラット最長6 ヵ月間、イヌ最長 9 ヵ月間)、DCV 及び ASV との併用投与毒性試 験(ラット及びイヌ1 ヵ月間)、遺伝毒性試験(in vitro 及び in vivo)、がん原性試験[トランスジェ ニック(Tg-rasH2)マウス、ラット]、生殖発生毒性試験(ラット、ウサギ)、光毒性試験(in vitro 及びin vivo)を実施した。重要な試験はいずれも GLP 適合下で ICH ガイドラインに準拠して実施し た。用量設定や毒性発現機序解明のための探索的試験では、試験の一部を非 GLP 下で実施した。が ん原性試験は、BCV を含む HCV 療法の臨床投与期間は 12 週間であり、「がん原性試験ガイドライ ン」で評価が必要とされる6 ヵ月間より短期間であるため必要とされないが、本剤の開発計画時に臨 床投与期間が48 週間と予測されていたため実施した。それぞれのがん原性試験の投与量は、Tg-rasH2 マウスと同腹の非トランスジェニックマウスを用いた 28 日間投与用量設定試験及びラット 6 ヵ月間 反復投与毒性試験の成績に基づいて設定し、米国FDA のがん原性評価委員会(CAC)の承認を得て いる。 肝臓が治療の標的器官であることから、主要な反復投与毒性試験では BCV 及び代謝物 BMS-794712 の肝臓中濃度を測定した。また、サルにおいて125 mg/kg/day の 1 週間投与で胆嚢上皮の分裂像の増 加がみられたため、発現機序検討のための3 ヵ月間投与試験を実施したが、本所見は再現されなかっ た。BCVは波長290~700 nmの光を吸収することから、マウス線維芽細胞及び有色ラット(Long-Evans) を用いて光毒性を評価した。
重要な試験の投与量は用量設定試験又は探索的試験の結果に基づいて設定した。併用投与毒性試験の 投与量は明らかな毒性が発現する用量よりも臨床におけるヒト曝露量に関連性のあるAUC が得られ る用量を設定した。併用投与毒性試験の投与期間はICH M3(R2)ガイダンス及び FDA の HCV ガイダ ンス案1 に基づき、個々の被検薬の長期投与毒性試験の方がヒト曝露量に関連した AUC が得られる 用量で実施した併用投与毒性試験よりも安全性の評価に有用と考えられることから、最長1 ヵ月間と した。 特記する場合を除き、毒性試験の投与経路は臨床での投与経路と同様の経口投与とし、BCV を媒体 の0.1 M Tris 緩衝液(pH 8.5)に溶解して投与した。 毒性試験に用いる動物種には、げっ歯類はラット、非げっ歯類はイヌを選択した。ラットは毒性試験 の標準的動物種で背景値が豊富であることから選択した。イヌはBCVの経口バイオアベイラビリティ (85%)がサル(16%)より高く、高い全身曝露量が得られることから選択した。併用投与毒性試験 に用いる動物種の選択及び試験期間は、各薬物の単剤での毒性及び動物とヒトとの代謝プロファイル の類似性に基づいて設定した。 実施したBCV の毒性試験を Table 1-1 に示す。 Table 1-1: BCV の毒性試験 試験の種類及び期間 投与経路 試験系 単回投与毒性 経口 マウス、ラット、イヌ 反復投与毒性 2 週間投与毒性試験 経口 ラット 1 ヵ月間投与毒性試験 経口 ラット、イヌ 6 ヵ月間投与毒性試験 経口 ラット 9 ヵ月間投与毒性試験 経口 イヌ 併用投与毒性 BCV + ASV 1 ヵ月間投与毒性試験 経口 ラット BCV + DCV 1 ヵ月間投与毒性試験 経口 イヌ BCV + ASV + DCV 1 ヵ月間投与毒性試験 経口 イヌ 遺伝毒性
復帰突然変異試験 In vitro S. typhimurium, E. coli
染色体異常試験 In vitro CHO 細胞
小核試験 経口 ラット
がん原性
26 週間投与がん原性試験 経口 Tg-rasH2 マウス
Table 1-1: BCV の毒性試験 試験の種類及び期間 投与経路 試験系 生殖発生毒性 受胎能及び着床までの初期胚発生に関する試験 経口 ラット 胚・胎児発生に関する試験 経口 ラット、ウサギ 出生前及び出生後の発生並びに母体の機能に関する試験 経口 ラット その他の毒性 毒性発現の機序に関する試験 経口 サル 光毒性試験 In vitro Balb/c 3T3 マウス線維芽細胞 単回投与光毒性試験 経口 ラット 以上のBCV の非臨床薬理試験、薬物動態試験及び毒性試験の成績は、C 型慢性肝炎患者における BCV のDCV 及び ASV との併用療法の有効性及び安全性を裏付けるものと考えられる。 2 薬理試験 2.1 効力を裏付ける試験 2.1.1 活性及びジェノタイプ網羅性 BCVはヌクレオシド基質とRNAテンプレートに対して非競合的にNS5Bポリメラーゼ活性を阻害し、 HCV RNA の合成開始を阻止する。共結晶構造解析及び耐性マッピングにより、BCV が NS5B ポリメ ラーゼのthumb 領域(thumb site 1)に結合するアロステリックな阻害薬であることが確認された。 BCV の活性を評価する in vitro 試験として、HCV NS5B との相互作用における強度及び選択性につい ては酵素を用いたポリメラーゼアッセイを実施し、BCV の効力及び細胞毒性に関しては HCV レプリ コンアッセイを実施した。BCV の NS5B ポリメラーゼ活性阻害の IC50値は、HCV レプリコンアッセ イにおけるEC50値と良好な相関を示した。 BCV による NS5B ポリメラーゼ阻害の IC50値は、ジェノタイプ1a、1b、3a 及び 5a では 1.8~4.8 nM、 ジェノタイプ4a 及び 6a では 7.6~61.6 nM、ジェノタイプ 2a 及び 2b では 164~228 nM であった。ま た、HCV レプリコンアッセイにおいて、ジェノタイプ 1a 及び 1b の実験室株並びに C 型慢性肝炎患 者由来のNS5B配列を基に作製したハイブリッドレプリコンに対するEC50値は1.6~9.5 nMであった。 ジェノタイプ3a、4a 及び 5a の患者由来の NS5B 配列を基に構成したハイブリッドレプリコンに対す るBCV の EC50値は0.8~18 nM の範囲であった。ジェノタイプ 6a に対する EC50値は8.6~79.5 nM の 範囲であり、低い感受性を示す値(79.5 nM)は、V494A の変異が存在する場合に認められた。血中 主代謝物のBMS-794712 及びヒト胆汁及び糞中代謝物の BMT-110547、BMT-171207 及び BMT-142478 もジェノタイプ1b のレプリコンに対して阻害活性を示した(EC50値 4~50 nM)。BCV の抗ウイル ス活性は 40%ヒト血清の存在下で軽度に抑制され、ジェノタイプ 1b レプリコンアッセイでは EC50
値が4.4 ± 1.5 倍に増加した。しかし、in vitro での血清の存在による抗ウイルス活性低下の臨床にお ける抗HCV 活性との関連性については不明である。 2.1.2 特異性及び選択性 BCV は HCV NS5B ポリメラーゼに特異性を示す。本薬は哺乳類のポリメラーゼ α、β 及び γ 並びに近 縁のペスチウイルスであるBVDV ポリメラーゼに対して活性を示さなかった。BCV はヒト免疫不全 ウイルス(HIV)の逆転写酵素及び HIV インテグラーゼに対して活性を示さなかった。BCV のデオ キシリボ核酸(DNA)及びRNAに対する非特異的な結合は認められなかった。主代謝物のBMS-794712 は同解析においてBCV と同程度の特異性を示した。 BCV の抗ウイルス選択性について、BVDV を含む一連の RNA 及び DNA ウイルスに対する活性の測 定により評価した。その結果、BCV は対照のジェノタイプ 1b の HCV を除き、いずれのウイルスに 対しても活性を示さなかった(IC50 =4 µM 超)。本結果から、BCV が HCV に対する選択的な阻害薬 であることが示された。 2.1.3 各種細胞における細胞毒性 BCVのジェノタイプ1bレプリコンを導入したHuh-7細胞における細胞毒性は、50%細胞毒性濃度(CC50) が20 µM であり、in vitro 細胞毒性に対する治療係数は 3300 超(EC50 = 0.006 µM)であった。BCV の CC50値を種々の組織由来細胞株でも算出した。各細胞をBCV と共に最長 5 日間培養した結果、本薬 のCC50値は試験に用いたすべての細胞で14 µM以上であった。BCV代謝物(BMS-794712、BMT-110547、 BMT-171207 及び BMT-142478)の CC50値もBCV の値と同程度であった。 2.1.4 作用機序 2.1.4.1 NS5B ポリメラーゼに対する作用 BCV の作用機序試験より、本薬が HCV RNA の合成開始を時間依存的に阻害することが示された。 RNA テンプレート又はグアノシン三リン酸(GTP)に対する見かけの Ki(阻害定数)値のプロット により、本薬のテンプレート及びGTP に対する阻害がそれぞれ Ki 値 1.1 ± 0.1 nM 及び 2.6 ± 0.4 nM の 非競合的様式であることが確認された。結合試験より、BCV と HCV NS5B ポリメラーゼの相互作用 は可逆的で二段階[Kd* = Kd/(1+k3/k4)]の緩徐な結合であることが示された。 2.1.4.2 耐性 耐性試験において、ジェノタイプ1a 及び 1b の HCV レプリコン細胞を、野生型レプリコンでの EC50 値の5~30 倍高濃度の BCV と共に培養すると、本薬に対する感受性が低下した細胞が得られた。耐 性レプリコンの遺伝子解析の結果、NS5B の 495 番のアミノ酸残基における置換が明らかとなった2。 野生型NS5B ポリメラーゼの 495 番のプロリン(P)は、ジェノタイプ 1a の耐性細胞ではセリン(S)、 アラニン(A)、ロイシン(L)又はスレオニン(T)に、また、ジェノタイプ 1b では S、A 又は L に置換していた。これらの個々の置換を野生型の遺伝子背景を有するレプリコン細胞に導入すると、 BCV に対する EC50値が15~64 倍の耐性が生じた。概してレプリコンの複製効率は耐性の程度と逆相 関し、耐性が高い変異はgenetic barrier が高いことが示唆された。
BCV 単剤を単回投与した第 1 相臨床試験(AI443002 試験)のすべての BCV 投与群において、ウイル スRNA の急速(数時間以内)かつ持続的な減少がみられたが、BCV の 100、300 及び 600 mg 投与群 では、in vitro レプリコンアッセイでみられた耐性変異の増幅はみられなかった。900 mg 投与群では、 ジェノタイプ1a の被験者 1 例に投与後 24 時間の 1 時点で P495L/S の耐性変異が認められたが、その 後はウイルス量の顕著な低下(3.4 log10)がみられた。これらの変異が投与後24 時間の 1 時点のみで 検出されたことから、投与前から微量に存在していた可能性が示唆された。P495L/S の耐性変異は投 与後24 時間以降には検出されなかったことから、これらの変異の複製能は低く、野生型ウイルスに 置き換わった可能性が示唆された。 以上の結果から、ジェノタイプ1a 及び 1b において選択された最も頻度が高く耐性の程度が高い変異 は495 番のアミノ酸であった。In vitro 試験における耐性に関連した NS5B アミノ酸部位の自然発生多 型をEuropean Hepatitis C Virus Database のアミノ酸配列を用いて HCV ジェノタイプ 1 について検討し た結果、P495 はジェノタイプ 1 のすべての配列に保存されていた。本解析結果から、BCV の結合部 位はジェノタイプ1a 及び 1b を通して保存されていることが示された。
2.1.4.3 交差耐性
各種HCV 標的阻害薬及び各種 HCV NS5B 阻害薬に特異的な耐性変異は各薬剤によって異なる。ジェ ノタイプ1a 又は 1b レプリコンを用いた一過性複製試験では、NS5B の P495 にいずれの置換が存在し てもASV 及び DCV に対する感受性は変化しなかった2。同様に、P495 のいずれの置換も palm site 2 に結合する非核酸系NS5B 阻害薬(BMS-929075)に対する感受性は変化しなかった。
NS5B ポリメラーゼの thumb site 1 に結合することから予測されるように、BCV は NS5B 核酸系阻害 薬(ソホスブビルなど)、thumb site 2 並びに palm site 1 及び 2 に結合する非核酸系阻害薬に特異的な 耐性変異、更にDCV 及び ASV に特異的な耐性変異に対しても活性を維持している。臨床使用に相当 する濃度のBCVをDCV及び ASVと併用することにより、DCV及びASVにそれぞれ耐性を示す NS5A (L31M-Y93H)及び NS3[アスパラギン酸(D)168V]のアミノ酸置換を有するジェノタイプ 1b の 変異レプリコンが排除された。BCV が他の NS5A 耐性変異体(Q30E/K/D、Y93N/H、M28A-Q30R、 M28T-Q30H、Q30R-L31M、Q30H-Y93H 及び Q30R-H58D)も野生型と同様に阻害することを示す更 なるin vitro 試験の結果は、DCV の申請資料(DCV CTD モジュール 2.6.2、表 2.3.2-3)に記載した。 これらの結果から、BCV を ASV 及び DCV と併用することが耐性に対しても有用であることが示さ れた。 2.1.5 In vitro 併用試験 HCV レプリコンシステムを用いた併用試験において、BCV は DCV、ASV、NS5B 核酸系阻害薬、NS5B 非核酸系阻害薬等の異なるHCV 蛋白を標的とする低分子化合物との併用で相加又は相乗作用を示し た。いずれの併用でも細胞毒性の明らかな増強はみられなかった2。 2.2 安全性薬理 ICH S7A ガイドラインで推奨される心血管系、中枢神経系及び呼吸系の評価を、主要な BCV の反復 投与毒性試験の一部として実施した。また、一連のin vitro 及び in vivo 安全性薬理試験を実施した。
In vitro 試験では BCV 及び BMS-794712 の受容体及びイオンチャネルのリガンド結合並びに酵素活性 の相対的阻害に及ぼす影響について評価した。心血管系については、hERG/IKr 電流及び心筋イオン チャネル、ウサギプルキンエ線維の活動電位、摘出ウサギ心臓に及ぼす影響をin vitro 試験で評価し、 麻酔下ウサギ及びイヌを用いたテレメトリー試験によりBCV 単回投与の心血管系パラメータに及ぼ す影響をin vivo で評価した。BCV の曝露量がサルに比べてイヌの方が高かったことから、安全性薬 理試験にはイヌを用いた。In vitro の安全性薬理評価は試験系に血液中の蛋白を含まないことから、 これらの試験における曝露量比は、[in vitro 濃度 ÷ 臨床推奨用量におけるヒトの BCV 遊離体(非結 合型)のCmax(ヒト遊離体 Cmax、0.018 µg/mL)]として算出した。 2.2.1 受容体・イオンチャネル結合及び酵素アッセイ 多様な受容体、酵素及びイオンチャネルのリガンドとの結合に対するBCV の阻害作用を in vitro で評 価した。37 種類の薬理学的標的(受容体、酵素、イオンチャネル)のアッセイパネルを用いて BCV 及びBMS-794712 を濃度 6.6 μg/mL で評価した結果、BCV 及び BMS-794712 の明らかな作用(50%以 上の阻害)は、ヒトのホスホジエステラーゼ 4(PDE4)酵素の阻害(それぞれ 57%及び 71%阻害) のみであった。U937 細胞(ヒトリンパ腫由来細胞)から単離したヒト PDE4 酵素を用いたフォロー アップ試験の結果、BCV の IC50値は1.3 μg/mL であり、本薬の PDE4 阻害活性は弱いことが示唆され た。TNF-α の産生を測定した細胞の機能アッセイ 3では、BCV は弱い PDE4 酵素阻害薬(6.6 μg/mL で 31%阻害、IC50値 6.6 μg/mL 超)であった。本質的に蛋白を含まないこれらの試験における BCV の最低IC50値(1.3 μg/mL)はヒト遊離体 Cmax の 72 倍以上であることから、ヒトにおける標的外作 用の可能性は低いと考えられた。ヒトでは一過性の軽度な消化管症状が認められたのみであった。 BCV の PDE4 酵素阻害作用が高い曝露量比で認められた弱い作用であること、その他の PDE4 に関連 する所見(抗炎症作用や炎症誘発作用、白血球異常調節など)3,4 がみられなかったことから、ヒトで の消化管症状は本薬のPDE4 阻害作用によるものではないと考えられた。BMS-794712 については、 単離酵素や細胞の機能アッセイによる評価を実施しなかった。 2.2.2 心血管系(In vitro / In vivo) BCV の心筋カリウムチャネル(hERG/IKr)、ナトリウムチャネル(SCN5A)及びウサギプルキンエ 線維活動電位に及ぼす影響をin vitro で検討した。BCV は心筋 hERG/IKr カリウムチャネル(IC50値 = 8.2 μg/mL)及び SCN5A ナトリウムチャネル(1 Hz 及び 4 Hz でそれぞれ 62.9%及び 66.3%阻害)を中 等度に阻害した。ウサギプルキンエ線維アッセイでは、BCV は 6.6 及び 19.8 μg/mL の濃度で APD50 を用量に依存して軽微に増加させ(それぞれ10.1%及び 14.4%)、最大立ち上がり速度を軽微に減少 させた(それぞれ6.4%及び 7.8%)。APD90及び他の活動電位パラメータに影響はみられなかった。 摘出ウサギ心臓を用いた電気生理学的試験では、BCV の濃度 0.7 µg/mL 以上で濃度に依存した心拍数 の軽度な減少(4%~18%)及び洞結節回復時間の軽微な延長(7%~22%)が認められた。In vitro で 心血管系パラメータに影響のみられた濃度の0.7~19.8 µg/mLは、ヒト遊離体Cmaxの 39倍以上であっ た。 麻酔下のウサギを用いたin vivo 心臓電気生理学的試験において、BCV の最大濃度 15 mg/kg まで本薬 に関連した心電図パラメータへの影響はみられなかったが、3 mg/kg(血漿中濃度:13.5 μg/mL)以上
で一過性の動脈血圧の軽度~中等度の上昇(投与前値との比較で9%~19%)及び心拍数の減少(−20% ~−7%)が認められた。血圧及び心拍数への影響に関する無作用量は 1 mg/kg(血漿中濃度:7.4 μg/mL) であった。無作用量におけるCmax のヒト Cmax との比は、心電図への影響に関して 42 倍、血圧及 び心拍数への影響に関して5 倍であった。 イヌにおける単回経口投与心血管系テレメトリー試験では、BCV の投与量 53 mg/kg(投与 4 時間後 の雌雄平均血漿中濃度63.9 μg/mL)で血圧(全身動脈血圧、左心室収縮期圧及び拡張末期圧、左心室 圧)、心拍数、心電図パラメータ、身体活動及び深部体温に変化はみられなかった。無作用量での Cmax はヒト Cmax の 43 倍であった。 イヌの反復経口投与毒性試験でもBCV の心血管系に及ぼす影響を検討した。その結果、BCV を最長 9 ヵ月間、最高用量 25 mg/kg/day(ヒト曝露量と比較して Cmax で 43 倍、AUC で 65 倍)を投与した イヌにおいて本薬投与に関連した心拍数及び心電図の変化は認められなかった。 更に、BCV を DCV 及び ASV と併用投与したイヌの反復併用投与毒性試験において、ヒト AUC の最 大8.7 倍で本薬投与に関連した心血管系への影響は認められなかった。 以上より、BCV は hERG 及びナトリウムチャネルの中等度の阻害とプルキンエ線維アッセイにおけ る軽微な影響を示したのみであった。In vivo 試験において、ヒト曝露量より高曝露量でも心血管系に 懸念される影響は認められなかったことから、BCV がヒトの心血管系に影響を及ぼす可能性は低い と考えられた。 2.2.3 中枢神経系 BCV の中枢神経系に及ぼす影響は毒性試験の一部として評価し、独立した安全性薬理試験は実施し なかった。有色Long-Evans ラット及び白色 Sprague Dawley ラットに[14C] BCV を投与した試験におい て、本薬の脳組織及び脊髄内濃度は低濃度又は定量下限未満であった。マウスの単回経口投与毒性試 験において、非致死量の 125 mg/kg では中枢神経系に関連した変化はみられなかったが、375 mg/kg 以上の用量では死亡例がみられ、一般症状として活動性低下、振戦、間代性痙攣、円背位あるいは横 臥位が認められた。イヌの単回経口投与トキシコキネティクス及び忍容性試験、ラットの2 週間経口 投与毒性試験、ラットの最長6 ヵ月間及びイヌの最長 9 ヵ月間の主要な反復経口投与毒性試験では、 ヒトAUC の最大 79 倍の曝露量でも神経学的な臨床症状や神経組織の組織学的所見に BCV 投与に関 連した変化は認められなかった。 また、BCV と DCV 及び ASV をラット及びイヌに併用投与した試験においても、ヒト AUC の最大 11 倍の曝露量で BCV 投与に関連した中枢神経系の影響は認められなかった。 以上より、BCV がヒトの中枢神経系に影響を及ぼす可能性は低いと考えられた。 2.2.4 呼吸系 BCVの呼吸系に及ぼす影響は毒性試験の一部として評価し、独立した安全性薬理試験は実施しなかっ た。マウスの単回経口投与毒性試験において、非致死量の125 mg/kg では呼吸系に関連した変化はみ られなかったが、375 mg/kg 以上の用量では、死亡例に努力性呼吸が認められた。ラットの最長 6 ヵ
月間及びイヌの最長9 ヵ月間の反復経口投与毒性試験では、ヒト AUC の最大 79 倍の曝露量でも BCV 投与に関連した呼吸器系への影響は認められなかった。 また、BCV と DCV 又は ASV をラット又はイヌに併用投与した試験においても、ヒト AUC の最大 11 倍の曝露量で BCV 投与に関連した呼吸系への影響は認められなかった。 以上より、本薬がヒトの呼吸系に対して影響を及ぼす可能性は低いと考えられた。 3 薬物動態試験 BCV の非臨床 PK は、一連の in vitro 試験及びマウス、ラット、ウサギ、イヌ及びサルを用いた in vivo 試験で評価した。更に、BMS-794712 及び BMS-948158 の PK、CYP 酵素阻害及び誘導作用、並びに トランスポーター阻害作用についても評価した。これらの動物由来の各種生体試料中の BCV、 BMS-794712 及び BMS-948158 を LC-MS/MS 法を用いて分析した。分析法は、高感度で、精度良く、 正確であることを確認した。 動物において、BCV は経口で生物学的に利用可能であり、体内に広範に分布した。BCV の in vivo 代 謝物プロファイルはすべての種で質的に類似し、ヒトに特有の代謝物は検出されなかった。BMS-794712 は放射能検出によりヒト血漿中に検出された唯一の代謝物であり、この代謝物は主に CYP3A4 を介 して生成された。その他に痕跡量の血漿中代謝物が質量分析法により検出された。BCV は主に代謝 により消失し、代謝物は動物及びヒトの糞便中及び胆汁中に排泄された。また、胆汁中排泄も BCV の消失経路の一つであった。腎クリアランスは、BCV の主要な消失経路ではなかった。In vitro にお いて、BCV は複数の CYP 酵素及びトランスポーターの基質、阻害剤及び誘導剤であったことから、 薬物動態学的薬物相互作用の発現が予測されたが、臨床試験の結果、BCV、ASV 及び DCV の間で臨 床的意義のある薬物相互作用は認められなかった。 3.1 吸収、バイオアベイラビリティ及び薬物動態 PAMPA 法及び Caco-2 細胞モデルにおける BCV の膜透過性は高かったことから、ヒトでの高い吸収 性及び広範な組織内分布が示唆された。ヒトにおける絶対的バイオアベイラビリティは 66%であっ た。溶液として経口投与したときのBCV の吸収は、ラット及びイヌよりもマウス及びサルの方が速 やかで、最高血漿中濃度到達時間(Tmax)は、マウス、ラット、イヌ及びサルでそれぞれ 1.0、6.0 ~6.7、5.3 及び 1.3 時間であった。P-gp 及び BCRP は BCV の経口吸収及び組織内分布を抑制し、この 抑制作用はBCV の高い膜透過性により軽減されるものの、BCV はヒトの P-gp 及び BCRP の基質で あることが示された。 マウス、ラット及びイヌにBCV を経口投与したときの絶対的バイオアベイラビリティは、それぞれ 100%超、42%~47%及び 86%であったが、サルでは 15%と低い値であった。イヌに DCV(30 mg)、 ASV(200 mg)及びBCV(75 mg)を単剤として又はこれらの3種類の薬物の配合錠(以下、DCV/ASV/BCV 配合錠と記載する)として経口投与したとき、BCV の曝露量及び血漿中濃度-時間プロファイルは両 投与群間で類似していた(p > 0.05)。 単回静脈内投与後のBCV は、ラット及びイヌよりもマウス及びサルで速やかに消失した[消失半減 T-HALF):ラット及びイヌでそれぞれ 8.4 及び 13.8 時間、マウス及びサルでそれぞれ 3.2 及び
1.4~2.1 時間]。ラット及びイヌの T-HALF 値は、ヒトの T-HALF 値(7.7~13.1 時間)と類似してい た。マウス、ラット、イヌ及びサルの総血漿クリアランスは、それぞれ4.7、2.4、0.34及び4.7~8.5 mL/min/kg で、報告されている各種動物の肝血流量 5 のそれぞれ 5.22%、4.35%、1.10%及び 10.8%~19.5%に相 当することから、BCV の肝除去率は低いことが示された。同様に、ヒトにおける全身クリアランス の幾何平均値(93.2 mL/min)は、報告されている肝血流量5の約6%に相当したことから、BCV の肝 除去率は低いことが示された。 3.2 分布 マウス、ラット、イヌ及びサルにBCV を静脈内投与したときの定常状態分布容積(Vss)は、それぞ れ1.3、1.5、0.4 及び 0.1~0.6 L/kg であった。なお、ヒトの Vss 値は 35.7 L であった。 マウス及びラットのVss値は、報告されている総血液量及び全身水分量5よりも大きかったことから、 これらの動物種ではBCV が血管外に分布することが示唆された。サルに BCV を 1 及び 2 mg/kg の用 量で静脈内投与したときのVss 値はそれぞれ 0.1 及び 0.6 L/kg であったことから、投与量と Vss 値の 関係に矛盾が生じたものの、その原因は不明である。しかしながら、サル及びイヌにおけるVss 値は 各動物種の総血液量の値5よりも大きいことから、これらの動物種においてBCV は血管外に分布す ることが示唆された。このことは、サル及びイヌにおけるBCV の肝臓中濃度/血漿中濃度比が高値 (それぞれ24 及び 2)であることと一致する。 BCV[濃度 1 μM(0.660 μg/mL)]の血液中濃度/血漿中濃度比は、ヒトで 0.67~0.73、ラット、イ ヌ及びサルではそれぞれ0.61~0.78、1.48~1.70 及び 0.99~1.25 であった。BCV の血液中濃度/血漿 中濃度比が2 未満と低い値であったことから、BCV の血球移行は顕著ではないことが示唆された。 Long-Evans ラット(有色)及び SD ラット(アルビノ)に[14C]BCV を 10 mg/kg の用量で単回経口投 与したところ、[14C]BCV 由来の放射能は速やかに吸収され、体内に広範に分布し、投与後 168 時間 までに完全に消失した。放射能の組織内分布はLong-Evans ラットと SD ラットで類似しており、放射 能は副腎、肝臓(薬効の標的器官)、胃、小腸及び盲腸で高レベルであった。Long-Evans ラットの 中枢神経系組織中及び眼レンズ中の放射能は、定量下限未満(< 0.072 μg-equivalent/g)又は定量下限 付近(0.077~0.088 μg-equivalent/g)であった。Long-Evans ラットの有色皮膚中及び眼ブドウ膜中の 放射能濃度(≤ 2.04 μg-equivalent/g)は、SD ラットの眼ブドウ膜中の放射能濃度よりも低く、投与後 48 時間では定量下限未満であったことから、BCV とメラニン含有組織との特異的結合はないことが 示唆された。雄性 SD ラットに[14C]BCV を 20 mg/kg の用量で 1 日 1 回 14 日間反復投与したとき、 [14C]BCV 由来の放射能の蓄積傾向は認められなかった。 SD ラットを用いた更に詳しい組織内分布試験において、[14C]BCV 由来の放射能の組織内分布は雌雄 間で類似していることが示された。放射能の組織内濃度/血漿中濃度比は、大部分の組織及び採取時 点で1 未満であったが、肝臓では投与後 24 時間まで 9.49~17.5 の範囲であった。[14C]BCV 由来の放 射能は血液脳関門を通過せず、投与後48 時間までに雌雄ラットの消化管及び包皮腺を除く大部分の 組織から完全に消失した。
上述の[14C]BCV を用いた組織内分布試験の結果は、非標識 BCV を用いた組織内分布試験の結果と一 致した。非標識BCV を用いた組織内分布試験において、マウスに BCV を経口投与後 6~24 時間の肝 臓中濃度/血清中濃度比は 3.1~5.1、ラット、イヌ及びサルに BCV を投与後の肝臓中曝露量/血漿 中曝露量比(AUC 比)はそれぞれ 15、2 及び 24 であったことから、BCV はマウス、ラット、イヌ及 びサルの肝臓へ選択的に移行し、マウスにBCV を経口投与後 6~8 時間の脳中濃度/血清中濃度比は 0.01、ラットに BCV を経口投与後の脳中曝露量/血漿中曝露量比(AUC 比)は 0.05 であったことか ら、マウス及びラット脳へのBCV の移行は限定的であることが示された。また、野生型マウス及び P-gp ノックアウトマウスを用いた試験において、BCV を経口投与後の脳中濃度/血清中濃度比がそ れぞれ0.01 及び 0.17 であったことから、P-gp は BCV の脳への移行を抑制することが示唆された。マ ウスにBCV を経口投与後 6~24 時間における BMS-794712 の肝臓中濃度/血清中濃度比は 8.4~12.6 であり、また、ラット、イヌ及びサルにBCV を経口投与後の BMS-794712 の肝臓中曝露量/血漿中 曝露量比(AUC 比)はそれぞれ 26.1、3.9 及び 114 であり、BCV よりも高い値を示した。 ヒト及びラット肝細胞を用いたBCV 肝取込み試験結果から、主として受動輸送が BCV の肝取込みに 関与することが示唆された。この試験結果は、BCV がヒト OATP1B1 及び OATP1B3(ヒト肝取込み トランスポーター)の基質ではないことを示したin vitro 試験結果と一致した。 妊娠 SD ラットに[14C]BCV を経口投与したところ、[14C]BCV 由来の放射能は胎盤を通過し、胎児組 織中に検出された。また、授乳中のラットに[14C]BCV を経口投与したところ、[14C]BCV 由来の放射 能が乳汁中に検出された[放射能の乳汁中曝露量/母動物血漿中曝露量比:0.277(Cmax 比)、0.258 (AUC 比)]。これらの試験結果から、BCV の投与を受けている女性の胎児及び乳児は BCV とそ の代謝物に曝露される可能性が示唆された。 3.3 代謝 ヒト肝ミクロソームを用いた反応表現型解析試験より、BCV は主として CYP3A4 により代謝され、 CYP3A5 がわずかながら BCV の代謝に寄与することが示された。
BCV の in vitro 及び in vivo 代謝により、多様な酸化代謝物が生成した。In vitro 及び in vivo 代謝物プ ロファイルはすべての動物種で質的に類似し、ヒトに特有の代謝物は検出されなかった。BCV の主 要な代謝反応として、BMS-794712 を生成する N-脱メチル化(スルホンアミド窒素原子に結合したメ チル基の脱離)、BMT-110547 を生成する N-脱メチル化(二環性ピペラジン窒素原子に結合したメチ ル基の脱離)、BMS-974194 を生成する O-脱メチル化及び M4、M5、M26 及び M27 を生成するモノ ヒドロキシ化が挙げられ、N-脱メチル化がヒトにおける主要な代謝反応であった。更に、BMS-794712 のN-脱メチル化により BMT-171207 及び BMT-142478(いずれも BCV の N,N-脱メチル化体)が生成 され、BMT-110547 の N-脱メチル化によっても BMT-142478 が生成された。反応表現型解析試験の結 果より、BMS-794712は主としてCYP3A4及びCYP3A5により代謝され、BMT-171207及びBMT-142478 を生成することが示唆された。 In vitro及びin vivoのいずれにおいてもグルタチオン付加物は検出されなかった。[14C]BCVとラット、 イヌ、サル及びヒトの肝ミクロソーム蛋白をインキュベートした結果、ラット、イヌ及びヒトでは肝
められ、サルでは中等度(312 pmol/mg protein/30 min)にみられた。これらの結果は反応性代謝物の 生成を示唆したが、臨床推奨用量のAUC より高い曝露量で評価したラット及びイヌの長期安全性試 験で肝毒性が認められなかったことから、臨床的意義はないと考えられた。 動物(マウス、ラット及びイヌ)及びヒトの血漿中に検出されたBCV 由来の主要化合物は、BCV 及 びBMS-794712 であり(Table 3.3-1)、その他の代謝物もわずかに検出された(血漿中総放射能の 0.8% 以下)。健康被験者に BCV を反復投与した結果、BMS-794712 の明らかな蓄積は認められず、その 他の代謝物は微量であった。ヒトでは、BCV の AUC 値に対する BMS-794712 の AUC 値の比が単回 投与(0.23~0.27)及び反復投与(0.15~0.30)で同程度であった。なお、血中唯一の主代謝物である BMS-794712 の安全性については、忍容性がある用量で実施した BCV の非臨床安全性試験で、 BMS-794712 に十分量曝露された条件下[ラット:ヒト AUC の 0.4 倍(20 mg/kg/day、6 ヵ月間)、 イヌ:ヒトAUC の 56 倍(25 mg/kg/day、9 ヵ月間)]で評価した(3.7 項参照)。 Table 3.3-1: 動物及びヒトに[14C]BCV を単回経口投与したときの in vivo 代謝 動物種 投与量 血漿中の割合(%) 回収された代謝物の 投与量に対する割合 (%) BCV BMS-794712 マウス 30 mg/kg 93.4 - 96.1 1.3 - 3.2 49.4 ラット 20 mg/kg 96.2 - 97.8 ≤ 1.3 52.5 ウサギ 60 mg/kg 73.3 - 89.0 MS ND イヌ 5 mg/kg 87.1 - 91.0 4.6 - 7.0 52 胆管カニューレ挿入サル 125 mg/kg 66.4 - 80.3 19.7 - 31.8 41.2 ヒトa 800 mg 67.9 - 86.1 10.3 - 24.6 76.1 MS:LC-MS/MS 法でのみ検出、ND:測定せず a 胆汁を採取したヒトでは、BCV、BMS-794712、BMT-110547、BMT-171207 及び BMT-142478(糞便中 及び胆汁中の主化合物)はそれぞれ胆汁中に投与量の6.8%、1.3%、1.3%、0.9%及び 2.4%、糞便中に 5.4%、 6.3%、3.2%、4.7%及び 13.5%を占めた。胆汁を採取しなかったヒトでは、BCV、BMS-794712、BMT-110547、 BMT-171207 及び BMT-142478 は糞便中にそれぞれ投与量の 6.9%、11.7%、9.3%、10.9%、22%を占めた。 BCV をヒトに反復投与した結果、微量代謝物である BMS-948158(BCV の N,O-脱メチル化体の O-グ ルクロン酸抱合体)の曝露量の増加がみられたが、総薬物曝露量の10%未満であった[総 AUC の 1.1% ~2.9%(単回投与後)又は 8.5%(反復投与後)]。BMS-948158 はラット及びイヌの血漿中で検出さ れなかったが、125 mg/kg/day で 1 ヵ月間投与したサルの血漿中で検出された(AU C:5 µg•h/mL、健 康被験者にBCV を 150 mg の用量で 1 日 2 回、14 日間投与したときの AUC の 4.5~4.9 倍)。ヒト及 び非臨床試験で用いた動物種における BCV 及び血中代謝物の曝露量の詳細な比較検討は、4 項に記 載した。 ヒトの糞便中では、BCV が薬物由来の主化合物であり(投与量の 6.9%)、代謝物として BMT-142478、 BMS-794712、BMT-171207 及び BMT-110547(それぞれ投与量の 22.0%、11.7%、10.9%及び 9.3%)
が検出された。糞便中の主要代謝物は、胆管カニューレを挿入した(BDC)サルでは BMS-794712、 BMT-110547 及び BMT-142478、マウス、ラット及びイヌでは BMS-794712、BMT-110547、M4、 BMS-974194 及び BMT-142478 であった。ヒトに[14C]BCV を経口投与し、胆汁を採取(投与後 3~4、 4~6 及び 6~8 時間に採取)した結果、胆汁中で薬物由来の主化合物の BCV(投与量の 6.8%)と共 に、様々な代謝物(投与量の 2.4%以下)が検出され、糞便中に検出された代謝物はすべて胆汁中に も検出された。ヒトの胆汁中に微量の BMS-948158 が検出されたが、糞便中には検出されなかった。 なお、ヒトで生成された代謝物は、非臨床試験で用いた動物種の最低1 種以上でも生成された。 HCV レプリコンアッセイでは、BMS-794712 は BCV と同等の抗 HCV 活性を有した。BMT-171207、 BMT-110547 及び BMT-142478 の活性は BCV の 1/2~1/15 程度であり、BMS-948158 はほとんど又は まったく活性を示さなかった。また、NS5B ポリメラーゼを用いた試験の結果より、BMS-794712、 BMT-171207、BMT-110547 及び BMT-142478 は BCV と同様の活性を有することが示唆された。微量 の BMT-171207、BMT-110547 及び BMT-142478 がヒト血漿中に検出された。これらの結果より、 BMS-794712 は薬理作用全般に寄与する可能性があることが示唆された。他の代謝物の寄与について は、肝臓での曝露量を測定していないため不明である。 3.4 排泄 動物及びヒトでは、代謝クリアランスが経口投与後のBCV の主要な消失経路であり、次いで代謝物 の糞便中及び胆汁中排泄であった。代謝クリアランスはマウス、ラット及びイヌ(投与量の 49%~ 53%)よりヒト(投与量の 76%)で大きかった。マスバランス試験では、[14C]BCV 由来の放射能の 大部分は糞便中に排泄され(動物:投与量の92%~95%、ヒト:投与量の 89%)、動物及びヒトにお いてそれぞれ投与量の20.3%~31.1%及び 6.9%が未変化体として回収された。糞便中の未変化体の一 部は、経口投与後体内に吸収されなかったBCV である可能性が考えられた。また、動物及びヒトで は、投与量の0.05%~0.25%が尿中で回収されたことから、腎クリアランスは BCV の主要な消失経路 でないことが示唆された。[14C]BCV を経口投与後胆汁中に排泄された放射能は、BDC ラット、BDC サル及びヒト(投与後3~8 時間に胆汁を採取した)でそれぞれ投与量の 67.2%、41.9%及び 18.5%を 占めた。なお、BDC ラット、BDC サル及びヒトの胆汁中の BCV は、それぞれ投与量の 35.3%、0.7% 及び6.8%であり、これらの動物種では胆汁中排泄も BCV 消失経路の一つであることが示唆された。 3.5 蛋白結合 BCV(10 µM)の in vitro 血清蛋白結合率は、マウス、ラット、イヌ、サル及びヒトでそれぞれ 98.8%、 98.7%、97.8%、97.9%及び 98.8%であり、いずれの動物種でも高値を示した。BMS-794712 の血清蛋 白結合率は 97.2%~98.9%であった。すべての動物種で蛋白結合率が同程度の値を示したことから、 曝露量を比較検討する際にBCV の遊離型分率で補正しなかった。なお、BCV 及び BMS-794712 の in vitroヒト血漿蛋白結合率は0.1~10 µMの範囲で濃度依存性がなく1 µMではそれぞれ99.4%及び99.0% であった。 3.6 薬物動態学的薬物相互作用 ヒトでは、代謝クリアランスがBCV の主要な消失経路である(投与量の 76%を代謝物として排泄)。
謝に寄与することが示唆された。そのため、CYP3A4 阻害剤又は誘導剤と BCV を併用投与した場合、 薬物動態学的薬物相互作用が生じる可能性が考えられた。
BCV はヒト P-gp 及び BCRP の基質であるが、OATP1B1 及び OATP1B3 の基質ではなかった。BCV はPAMPA 及び Caco-2 モデルで膜透過性が高いことから、P-gp 及び BCRP の阻害剤又は誘導剤と BCV の併用投与が薬物相互作用をもたらす可能性は低いと考えられた。
In vitro で、BCV、BMS-794712 及び BMS-948158 は CYP 酵素、UGT1A1 及びトランスポーターの阻 害剤であることが示された。BCV 及び BMS-794712 は軽度~中等度の阻害作用を有し、可逆的及び 弱い時間依存性のCYP3A4 阻害剤であることが示された(IC50:9.6~33.4 µM)。更に、BCV は CYP2C8 の軽度~中等度の可逆的阻害剤であることが示された(IC50:23.7 µM)。評価した他の CYP 酵素に ついては、IC50値は40 µM超であった。ヒト肝細胞では、BCV及びBMS-794712はCYP1A2及びCYP2B6 のmRNA レベルをほとんど又はまったく増加させなかったが、用量依存的に CYP3A4 の mRNA レベ ルを増加させた。FDA の薬物相互作用に関するガイダンス 6 に従い、ベーシックモデルを用いてこ れらのデータを評価した結果、BCV 及び BMS-794712 は CYP1A2 及び CYP2B6 の有意義な誘導剤で はなかったが、CYP3A4 を誘導すると予測されたことから、BCV を反復投与した場合、CYP3A4 の誘 導及び阻害が同時に起こる可能性が考えられた。しかしながら、臨床試験で健康被験者にBCV(150 又は300 mg の用量で 1 日 2 回 14 日間)と CYP3A の基質であるミダゾラムを併用投与したところ、 ミダゾラムの曝露量が減少した。このことから、BCV の投与は相対的には CYP3A を誘導すると考え られた。また、BCV による CYP2C8 阻害の R1値(R1:1.5、ベーシックモデル、IC50値及びC 型慢性 肝炎患者にBCV を 75 mg の用量で 1 日 2 回反復投与したときの血漿中 Cmax 値 2.25 µM に基づく) は推奨カットオフ値の1.1 より高いため、BCV との併用投与により CYP2C8 基質の曝露量が増加する 可能性が示唆された。しかしながら、BCV(75 mg、1 日 2 回)の追加投与の有無に関わらず、 DCV/ASV/BCV 配合錠(DCV:30 mg、ASV:200 mg、BCV:75 mg)の投与が CYP2C8 基質である モンテルカストの曝露量に与える影響は軽微であり、BCV は臨床で使用されている他の CYP2C8 基 質のPK に影響を及ぼす可能性は低いと考えられた。 BCV はヒト肝ミクロソームの UGT1A1 を阻害した(IC50:12.6 µM)。しかしながら臨床試験におい て、75 又は 150 mg の用量で 1 日 2 回、12 週間以上投与された C 型慢性肝炎患者又は最高 300 mg の 用量まで1 日 2 回反復投与された健康被験者では、総ビリルビン又は間接型ビリルビン(UGT1A1 の 基質)の顕著な増加はみられなかったため、in vitro の UGT1A1 阻害作用は臨床的に意義が低いと考 えられた。 BCV、BMS-794712 及び BMS-948158 は、複数のトランスポーターを阻害した。BCV は Caco-2 細胞 のジゴキシン輸送、P-gp、BCRP、OATP1B1、OATP1B3、OAT1、NTCP 及び BSEP を阻害したが(IC50: 1.6~42.9 µM)、MRP2、OAT3及びOCT2は阻害しなかった。BMS-794712はBCRP、OATP1B1、OATP1B3、 OAT1、NTCP 及び BSEP を阻害したが(IC50:1.4~79.6 µM)、P-gp、MRP2、OAT3 及び OCT2 は阻 害しなかった。BMS-948158 は、OATP1B1(IC50:10.2 µM)、OATP1B3(IC50:30.0 µM)、MRP2 (IC50:17.1 µM)及び BSEP(IC50:> 50 µM、50 µM で 35%阻害)を阻害したが、NTCP を阻害しな かった。
FDA6及びEMA7 ガイダンスに従い、in vitro のトランスポーター阻害データから in vivo でのプロファ イルを予測した結果、BCV は P-gp、BCRP、OATP1B1、OATP1B3 及び NTCP の基質と薬物相互作用 を起こす可能性が考えられた。また、BCV と OAT1、OAT3 及び OCT2 基質との相互作用は、臨床推 奨用量で起こる可能性が低いと考えられた。DCV/ASV/BCV 配合錠及び BCV(75 mg BID)とロスバ スタチン(OATP1B1、OATP1B3 及び BCRP 基質 8)を併用投与した結果、ロスバスタチンの Cmax 及びAUC 値がロスバスタチンの単独投与時よりもそれぞれ 9 及び 3 倍増加した。別の臨床試験で、 DCV/ASV/BCV 配合錠及びプラバスタチン(OATP 基質)を BCV(75 mg BID)と併用して又は併用 せず投与したところ、プラバスタチンのAUC(INF)値がそれぞれ 81%及び 68%増加した。同一試験で、 ジゴキシン(P-gp 基質)の AUC(INF)値は、それぞれ 23%及び 17%増加した。これら臨床試験の結果 は、BCV の OATP1B1、OATP1B3、BCRP 及び P-gp 阻害作用の予測と一致した。
BSEP 及び MRP2阻害作用の in vitro 試験結果を in vivo の予測に外挿するための方法は、FDA 及び EMA から提案されていないため、文献9 で報告されている手法を用いて BSEP 及び MRP2 阻害に起因した 薬物相互作用の可能性について検討した。その結果、BCV 及び BMS-794712 による BSEP 又は MRP2 阻害の可能性は低いことが示唆された。しかしながら、BMS-948158 による MRP2 阻害は否定できず、 BCV を 75 mg の用量で 1 日 2 回投与した C 型慢性肝炎患者で薬物相互作用が生じる可能性が考えら れた。
BCV は、DCV 及び ASV を併用して投与される。DCV 及び ASV は、主に CYP3A4 又は CYP3A を介 した代謝により消失する。また、DCV 及び ASV は P-gp 基質であり、CYP3A の誘導剤かつ阻害剤で あり、P-gp 及び OATP の阻害剤である。BCV が主に CYP3A を介して代謝されること、P-gp の基質 かつ阻害剤であること、CYP3A4 の誘導剤かつ阻害剤であることから、BCV、DCV 及び ASV の 3 剤 間に薬物相互作用が生じる可能性が考えられた。しかしながら、BCV、DCV 及び ASV 間の薬物相互 作用を薬物相互作用試験で検討しなかったものの、第2 相試験(AI443014 試験)の PK データとこれ までに得られたPK データ(他の BCV、DCV 及び ASV 投与試験の PK データ)との比較検討から、3 種類の薬物間に臨床的意義のある薬物相互作用が起こる可能性は低いと考えられた。 3.7 トキシコキネティクス 非臨床毒性試験に用いたいずれの動物種においても、BCV 経口投与後の BCV 曝露量はおおむね用量 依存的に増加した。ICH M3(R2) ガイダンス10 に基づき、雄及び雌における曝露量(AUC 又は Cmax) の差が2 倍未満であれば顕著な差はないものと考えられ、主要な毒性試験の動物種(ラット及びイヌ) では、BCV の曝露量に顕著な性差がなかったことから、BCV の AUC 及び Cmax は雌雄を合わせた平 均値として表記した。主要な毒性試験におけるBCV の投与量、AUC 値並びにヒトの臨床推奨用量で のAUC 値(17.4 µg•h/mL)に対する動物の AUC 値の比を Table 3.7-1 に示す。ラット、イヌ及びサル における最高投与量でのAUC 値は、ヒトの臨床推奨用量での AUC 値の約 5~80 倍であった。妊娠 ラット及びウサギのAUC 値は、ヒトの臨床推奨用量での AUC 値のそれぞれ 85 及び 29 倍であった。 BCV は動物及びヒト血漿中の主要化合物であった。ヒトに特有の代謝物は検出されず、BMS-794712 がヒト血漿中の主要代謝物であった。ヒトの臨床推奨用量では、BMS-794712 の Cmax 及び AUC はそ れぞれ0.314 µg/mL 及び 4.16 µg•h/mL であった。BMS-794712 の安全性については、ヒトの臨床推奨
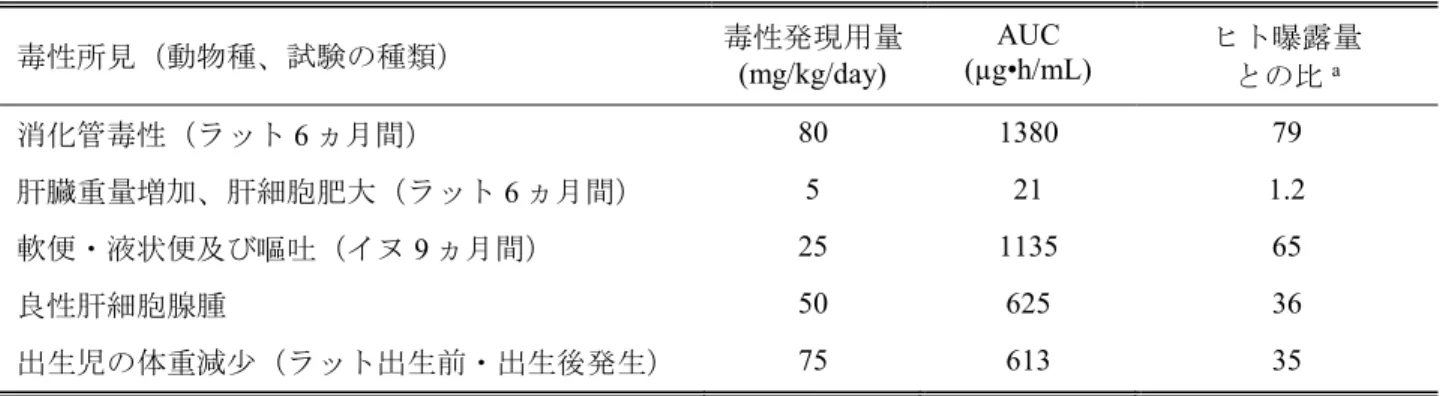
用量におけるAUC と同程度又はそれ以上の曝露条件下でラット、ウサギ、イヌ及びサルの非臨床毒 性試験を実施しているため、適切に評価できていると考えられた。主要な毒性試験におけるBMS-794712 のAUC 値及びヒトの臨床推奨用量での BMS-794712 の AUC 値に対する動物の AUC 値の比を Table 3.7-2 に示す。 肝臓が薬効の標的器官であるため、いくつかの反復投与試験で血漿中及び肝臓中の BCV 及び BMS-794712 濃度を測定した。各種動物における肝臓中薬物濃度の個体差は大きかったものの、BCV 及び BMS-794712 は高濃度に検出された(血漿中濃度に対する肝臓中濃度、BCV:1~17 倍、 BMS-794712:2~71 倍)。ラットにおいて肝臓重量の増加と相関して認められた、適応性変化であ る肝細胞の過形成を除き、最長6 ヵ月間投与したラット及び 9 ヵ月間投与したイヌのいずれの試験に おいても、肝臓の組織学的変化は認められなかった。それぞれの試験の代謝物濃度の概略については、 毒性の概要表に記載した。なお、サルの探索毒性試験においても、血漿中、肝臓中、胆嚢中及び胆汁 中のBCV 及び BMS-794712 濃度を測定した。 臨床試験(AI443003 試験)において、BCV を 900 mg の用量で 1 日 1 回、2 週間反復投与後、肝内胆 汁鬱滞並びにBMS-794712 及び BMS-948158(グルクロン酸抱合体)の血漿中濃度の高値が認められ た被験者がいたため、BCV 及びその代謝物の毒性プロファイルを明らかにする目的で反復投与トキ シコキネティクス試験を実施した。ラット及びイヌの2 週間反復投与試験並びにサルの 1 ヵ月間反復 投与試験では、上述の被験物質に加え、血漿中、尿中、胆汁中、糞便中及び肝臓中の BMS-974194(O-脱メチル化体)及びBMS-942348(O,N-脱メチル化体)などの新たな代謝物について評価した。BCV 及びBMS-794712 が主化合物として血漿中、尿中、胆汁中、糞便中及び肝臓中に検出され、他の被験 物質はBCV より低い濃度(0.7 倍以下)で検出された。なお、BMS-948158 については、ラット及び サルにおいて検出されたいずれの生体試料中でも BCV の 0.3 倍以下の濃度であり、イヌでは少数例 の尿中から低濃度で検出されたのみであった。
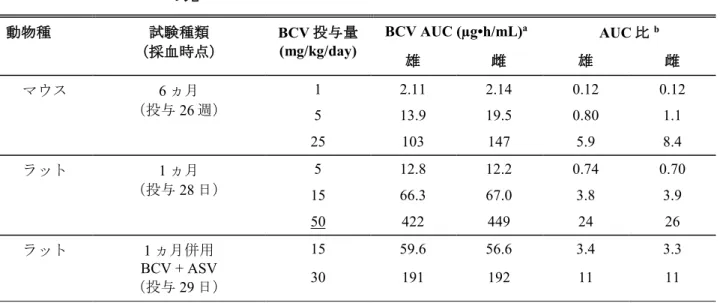
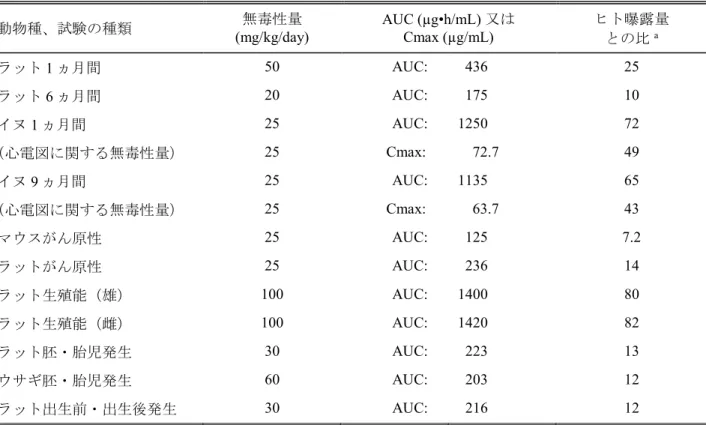
Table 3.7-1: 主要な毒性試験におけるBCV の AUC 値及びヒト AUC に対する動物 AUC と の比 動物種 試験種類 (採血時点) BCV 投与量 (mg/kg/day) BCV AUC (µg•h/mL) a AUC 比b 雄 雌 雄 雌 マウス 6 ヵ月 (投与26 週) 1 2.11 2.14 0.12 0.12 5 13.9 19.5 0.80 1.1 25 103 147 5.9 8.4 ラット 1 ヵ月 (投与28 日) 5 12.8 12.2 0.74 0.70 15 66.3 67.0 3.8 3.9 50 422 449 24 26 ラット 1 ヵ月併用 BCV + ASV (投与29 日) 15 59.6 56.6 3.4 3.3 30 191 192 11 11
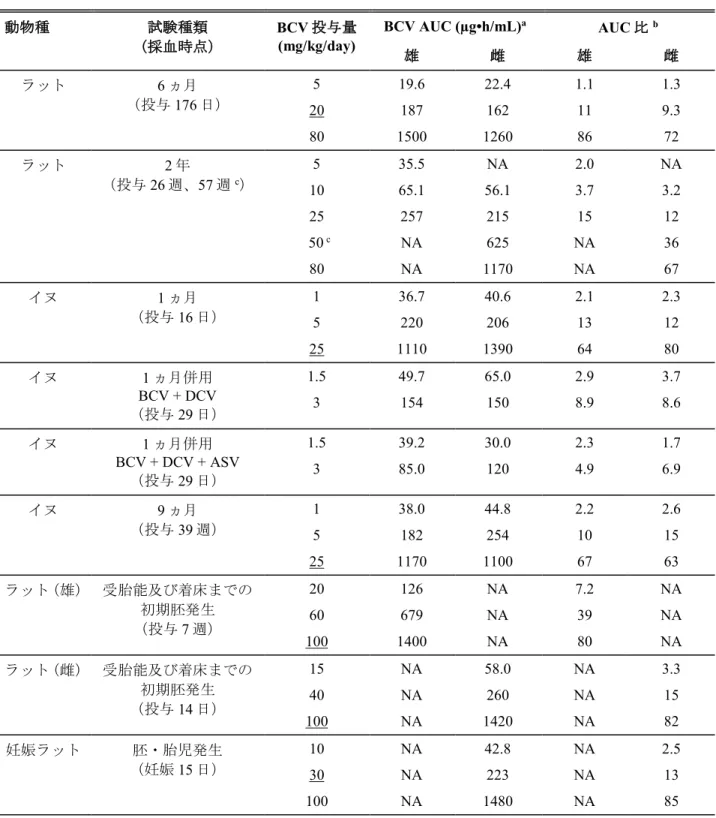
Table 3.7-1: 主要な毒性試験におけるBCV の AUC 値及びヒト AUC に対する動物 AUC と の比 動物種 試験種類 (採血時点) BCV 投与量 (mg/kg/day) BCV AUC (µg•h/mL) a AUC 比b 雄 雌 雄 雌 ラット 6 ヵ月 (投与176 日) 5 19.6 22.4 1.1 1.3 20 187 162 11 9.3 80 1500 1260 86 72 ラット 2 年 (投与26 週、57 週c) 5 35.5 NA 2.0 NA 10 65.1 56.1 3.7 3.2 25 257 215 15 12 50 c NA 625 NA 36 80 NA 1170 NA 67 イヌ 1 ヵ月 (投与16 日) 1 36.7 40.6 2.1 2.3 5 220 206 13 12 25 1110 1390 64 80 イヌ 1 ヵ月併用 BCV + DCV (投与29 日) 1.5 49.7 65.0 2.9 3.7 3 154 150 8.9 8.6 イヌ 1 ヵ月併用 BCV + DCV + ASV (投与29 日) 1.5 39.2 30.0 2.3 1.7 3 85.0 120 4.9 6.9 イヌ 9 ヵ月 (投与39 週) 1 38.0 44.8 2.2 2.6 5 182 254 10 15 25 1170 1100 67 63 ラット(雄) 受胎能及び着床までの 初期胚発生 (投与7 週) 20 126 NA 7.2 NA 60 679 NA 39 NA 100 1400 NA 80 NA ラット(雌) 受胎能及び着床までの 初期胚発生 (投与14 日) 15 NA 58.0 NA 3.3 40 NA 260 NA 15 100 NA 1420 NA 82 妊娠ラット 胚・胎児発生 (妊娠15 日) 10 NA 42.8 NA 2.5 30 NA 223 NA 13 100 NA 1480 NA 85
Table 3.7-1: 主要な毒性試験におけるBCV の AUC 値及びヒト AUC に対する動物 AUC と の比 動物種 試験種類 (採血時点) BCV 投与量 (mg/kg/day) BCV AUC (µg•h/mL) a AUC 比b 雄 雌 雄 雌 授乳ラット 出生前及び出生後の 発生 (授乳4 日) 10 NA 33.5 NA 1.9 30 NA 216 NA 12 75 NA 613 NA 35 妊娠ウサギ 胚・胎児発生 (妊娠19 日) 30 NA 105 NA 6.0 60 NA 203 NA 12 100 NA 510 NA 29 NA: 該当なし 下線を施した投与量は無毒性量を示す。 a 投与後 0 時間から最終測定可能時間(概ね 8~24 時間)までの血漿中濃度曲線下面積
b 臨床推奨用量における BCV の AUC(17.4 µg•h/mL)に基づき算出(動物 AUC ÷ ヒト AUC) c 投与 57 週は投与量 50 mg/kg/day の測定値(投与 52 週まで 80 mg/kg/day を投与した後に減量)
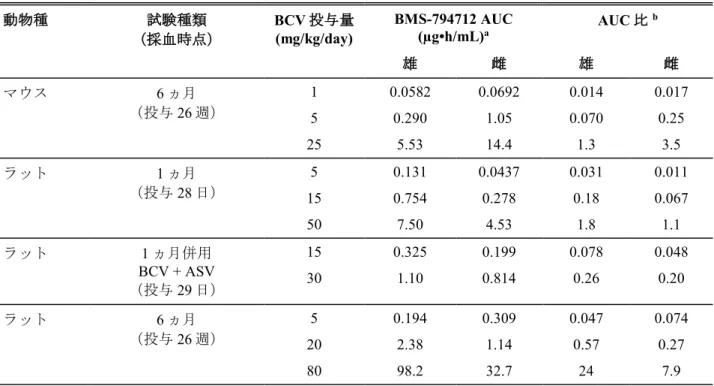
Table 3.7-2: 主要な毒性試験におけるBMS-794712 の AUC 値及びヒト AUC に対する動物 AUC との比
動物種 試験種類
(採血時点)
BCV 投与量
(mg/kg/day) BMS-794712 AUC (µg•h/mL)a AUC 比
b 雄 雌 雄 雌 マウス 6 ヵ月 (投与26 週) 1 0.0582 0.0692 0.014 0.017 5 0.290 1.05 0.070 0.25 25 5.53 14.4 1.3 3.5 ラット 1 ヵ月 (投与28 日) 5 0.131 0.0437 0.031 0.011 15 0.754 0.278 0.18 0.067 50 7.50 4.53 1.8 1.1 ラット 1 ヵ月併用 BCV + ASV (投与29 日) 15 0.325 0.199 0.078 0.048 30 1.10 0.814 0.26 0.20 ラット 6 ヵ月 (投与26 週) 5 0.194 0.309 0.047 0.074 20 2.38 1.14 0.57 0.27 80 98.2 32.7 24 7.9
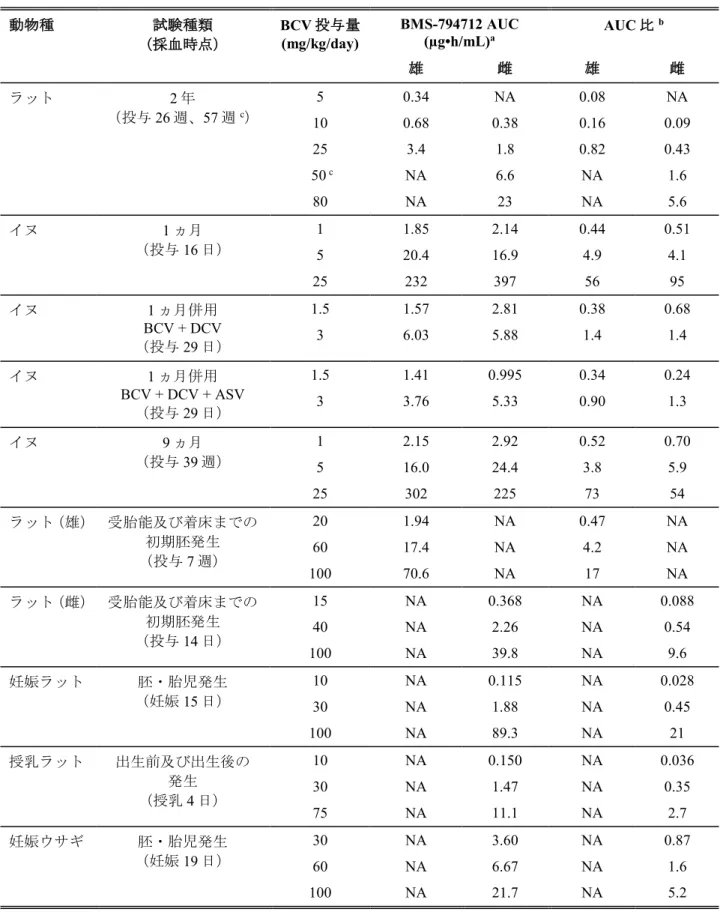
Table 3.7-2: 主要な毒性試験におけるBMS-794712 の AUC 値及びヒト AUC に対する動物 AUC との比
動物種 試験種類
(採血時点)
BCV 投与量
(mg/kg/day) BMS-794712 AUC (µg•h/mL)a AUC 比
b 雄 雌 雄 雌 ラット 2 年 (投与26 週、57 週c) 5 0.34 NA 0.08 NA 10 0.68 0.38 0.16 0.09 25 3.4 1.8 0.82 0.43 50 c NA 6.6 NA 1.6 80 NA 23 NA 5.6 イヌ 1 ヵ月 (投与16 日) 1 1.85 2.14 0.44 0.51 5 20.4 16.9 4.9 4.1 25 232 397 56 95 イヌ 1 ヵ月併用 BCV + DCV (投与29 日) 1.5 1.57 2.81 0.38 0.68 3 6.03 5.88 1.4 1.4 イヌ 1 ヵ月併用 BCV + DCV + ASV (投与29 日) 1.5 1.41 0.995 0.34 0.24 3 3.76 5.33 0.90 1.3 イヌ 9 ヵ月 (投与39 週) 1 2.15 2.92 0.52 0.70 5 16.0 24.4 3.8 5.9 25 302 225 73 54 ラット(雄) 受胎能及び着床までの 初期胚発生 (投与7 週) 20 1.94 NA 0.47 NA 60 17.4 NA 4.2 NA 100 70.6 NA 17 NA ラット(雌) 受胎能及び着床までの 初期胚発生 (投与14 日) 15 NA 0.368 NA 0.088 40 NA 2.26 NA 0.54 100 NA 39.8 NA 9.6 妊娠ラット 胚・胎児発生 (妊娠15 日) 10 NA 0.115 NA 0.028 30 NA 1.88 NA 0.45 100 NA 89.3 NA 21 授乳ラット 出生前及び出生後の 発生 (授乳4 日) 10 NA 0.150 NA 0.036 30 NA 1.47 NA 0.35 75 NA 11.1 NA 2.7 妊娠ウサギ 胚・胎児発生 (妊娠19 日) 30 NA 3.60 NA 0.87 60 NA 6.67 NA 1.6 100 NA 21.7 NA 5.2