2.5 臨床に関する概括評価の目次 2.5.1 製品開発の根拠 ... 5 2.5.1.1 疾患の背景 ... 5 2.5.1.1.1 加齢黄斑変性(AMD)の疫学的側面及び臨床的側面 ... 5 2.5.1.1.2 AMD の診断... 6 2.5.1.1.3 眼内血管新生を仲介する VEGF 及び P1GF... 7 2.5.1.2 滲出型 AMD の治療法 ... 8 2.5.1.2.1 抗 VEGF 療法導入以前の治療法 ... 8 2.5.1.2.2 抗 VEGF 療法 ... 9 2.5.1.2.3 滲出型 AMD の治療法の改善の必要性 ... 10 2.5.1.3 VEGF Trap:新規抗血管新生薬 ... 11 2.5.1.3.1 化合物の特性 ... 11 2.5.1.3.2 VEGF Trap-Eye の臨床開発プログラム... 13 2.5.1.3.3 民族的要因の考察 ... 15 2.5.1.4 各国規制当局との相談の経緯 ... 17 2.5.2 生物薬剤学に関する概括評価 ... 19 2.5.3 臨床薬理に関する概括評価 ... 21 2.5.3.1 薬物動態 ... 21 2.5.3.2 薬力学 ... 23 2.5.3.3 曝露量と反応の関連 ... 24 2.5.3.4 免疫原性 ... 24 2.5.3.5 結論 ... 24 2.5.4 有効性の概括評価 ... 26 2.5.4.1 有効性評価のための臨床開発計画 ... 26 2.5.4.1.1 初期の臨床開発 ... 26 2.5.4.1.2 有効性評価における民族的要因の考察 ... 26 2.5.4.1.2.1 内因性要因 ... 26 2.5.4.1.2.2 外因性要因 ... 29 2.5.4.1.2.3 まとめ ... 30 2.5.4.2 第Ⅲ相ピボタル試験(VIEW 1 試験及び VIEW 2 試験)... 30 2.5.4.2.1 方法 ... 30 2.5.4.2.2 結果及び考察 ... 33 2.5.4.2.2.1 有効性の主要評価項目 ... 34 2.5.4.2.2.2 有効性の副次評価項目 ... 35
2.5.4.2.2.3 有効性の追加の評価項目 ... 39 2.5.4.2.2.4 その他の有効性評価 ... 39 2.5.4.2.3 日本人部分集団(VIEW 2 試験)における有効性... 40 2.5.4.3 有効性の結論 ... 42 2.5.5 安全性の概括評価 ... 44 2.5.5.1 安全性評価の計画 ... 44 2.5.5.2 被験者の内訳及び曝露量 ... 45 2.5.5.3 TEAE の全体的な発現状況... 45 2.5.5.4 死亡及び重篤な TEAE ... 46 2.5.5.5 試験中止に至った TEAE ... 46 2.5.5.6 注目すべき有害事象 ... 47 2.5.5.7 臨床検査値の異常及びその他の全般的な安全性パラメータ ... 48 2.5.5.8 副作用 ... 48 2.5.5.9 プール 1 において 2 年間でみられた有害事象 ... 50 2.5.5.9.1 プール 1 の 2 年間における被験者の内訳及び曝露量 ... 51 2.5.5.9.2 有害事象の全体的な発現状況 ... 51 2.5.5.9.3 死亡及び重篤な有害事象 ... 51 2.5.5.9.4 試験中止に至った有害事象 ... 51 2.5.5.9.5 注目すべき有害事象 ... 52 2.5.5.10 プレフィルドシリンジ製剤 ... 52 2.5.5.11 日本人部分集団(VIEW 2 試験)における安全性... 53 2.5.5.11.1 日本人部分集団における 1 年目の安全性 ... 53 2.5.5.11.2 日本人部分集団における 2 年間の安全性 ... 54 2.5.5.12 安全性の結論 ... 54 2.5.6 ベネフィットとリスクに関する結論 ... 56 2.5.6.1 ベネフィット ... 56 2.5.6.2 リスク ... 57 2.5.6.3 バランス ... 58 2.5.6.4 考察 ... 59 2.5.6.5 結論 ... 59 2.5.7 参考文献 ... 61
略語一覧
略語 英語名称 日本語名称
ADA Anti-drug antibodies 抗薬物抗体
AMD Age-related macular degeneration 加齢黄斑変性
BCVA Best corrected visual acuity 最高矯正視力
BLA Biologic License Application 生物学的製剤承認申請書
CHMP Committee for Medicinal Products for Human Use
ヒト用医薬品委員会
CI Confidence interval 信頼区間
CNV Choroidal neovascularization 脈絡膜新生血管
CR/LT Central retinal/Lesion thickness 網膜中心部(病変)の厚さ
CRT Central retinal thickness 中心網膜厚
CSR Clinical study report 治験総括報告書
ELISA Enzyme-linked immunosorbent assay ELISA 法、酵素結合免疫吸着法
EMA European Medicines Agency 欧州医薬品庁
ETDRS Early Treatment Diabetic Retinopathy Study
糖尿病網膜症早期治療試験
EU European Union 欧州連合
FA Fluorescein angiography 蛍光造影
FAS Full analysis set 最大の解析対象集団
FDA Food and Drug Administration 米国食品医薬品局
IV Intravenous 静脈内
IVT Intravitreal 硝子体内
LOCF last observation carried forward 最終評価スコア外挿法 MAA Marketing Authorisation Application 販売承認申請
MPA Medical Products Agency スウェーデン医薬品庁
NEI VFQ-25 National Eye Institute 25-item Visual Function Questionnaire
米国国立眼病研究所の 25 項目からなる視 覚機能についてのアンケート
OCT optical coherence tomography 光干渉断層撮影
PD Pharmacodynamics 薬力学
PDT Photodynamic therapy 光線力学療法
PK Pharmacokinetics 薬物動態
PlGF Placental growth factor 胎盤増殖因子
PMDA Pharmaceuticals and Medical Devices Agency
独立行政法人医薬品医療機器総合機構
PPS Per protocol set 治験実施計画書に適合した対象集団
PRN As needed (pro re nata) 必要に応じ、随時
R ranibizumab ラニビズマブ
RPE Retinal pigment epithelium 網膜色素上皮
SD Standard deviation 標準偏差
SPA Special Protocol Assessment 特別プロトコル査定
TEAE Treatment-emergent adverse event 試験治療下で発現した有害事象
US(A) United States (of America) 米国
VA Visual acuity 視力
VEGF Vascular epithelial growth factor 血管内皮増殖因子
序文 この文書の目的 「中心窩下脈絡膜新生血管を伴う加齢黄斑変性」の治療を予定効能・効果とする VEGF Trap-Eye の開発プログラム中に集積された臨床データを分析し、有効性及び安全性について包括的に 評価した結果を提示し、当該適応症に対して本剤を用いる妥当性と用法・用量を示すことを目的 とする。 用語 VEGF Trap-Eye は、販売名「アイリーア硝子体内注射液 40mg/mL」及び「アイリーア硝子体内 注射用キット 40mg/mL」として販売される予定の製剤である。その有効成分は aflibercept (INN)/アフリベルセプト(遺伝子組換え)(JAN)であり、本文書中では「VEGF Trap」と表記 した。「VEGF Trap-Eye」は、硝子体内投与するために、特別に精製した VEGF Trap を用いて等 張液とした製剤である。
2.5.1 製品開発の根拠 2.5.1.1 疾患の背景 2.5.1.1.1 加齢黄斑変性(AMD)の疫学的側面及び臨床的側面 AMD は最も一般的に見られる網膜黄斑部の変性疾患であり、先進国における法的盲の主要な原 因となっている。AMD は高齢者に特有の疾患であり、65~74 歳の 10%、75~85 歳の 30%に AMD の臨床徴候がみられることが報告されている1)。
AMD には萎縮型及び滲出型の 2 つの型に大別される。萎縮型 AMD は AMD の全症例の 90%を占め るが、進行が緩徐で視力予後も比較的良好なため、視力喪失にまで進行するのは萎縮型 AMD のう ちの 10%である。一方、滲出型 AMD は AMD 患者の 10%を占めるにすぎないが、進行が速く、無 治療では急速に重度の視覚障害を来し、法的盲に至る。
AMD の病態生理学の概要
AMD は視細胞及び網膜色素上皮(Retinal pigment epithelium:RPE)の疾患である。RPE は外 側血液網膜関門を形成し、視細胞に栄養を供給する重要な役割を担っている。また、RPE は視細 胞外節を貪食し、視細胞の再生・維持に寄与することで視覚サイクルの一部を成している。
高齢者の眼では、遺伝的素因、光刺激により RPE への酸化ストレス、あるいは炎症などの原因 により、ブルッフ膜の組成が変化し、RPE の機能が低下する。RPE の機能が低下すると、RPE と ブルッフ膜の間にドルーゼンと呼ばれる沈着物が蓄積する。ドルーゼンは初期の AMD に見られる 臨床所見であり、検眼鏡検査で観察される。ドルーゼンには、photophore A2E やその他の有害 な代謝物を含むリポフスチンが含まれる。このリポフスチンが、RPE の機能に悪影響を与え、有 害な代謝産物が蓄積し、ドルーゼンの形成が更に促進されるという悪循環をもたらす。 萎縮型 AMD 萎縮型 AMD では滲出型 AMD と異なり、網膜下腔に異常な血管形成は起こらず、したがって、異 常な血管からの漏出も生じない。ドルーゼンが黄斑の内部や周辺に集積し、時間の経過と共に増 加し、拡大していく。 ドルーゼンのみからなる萎縮型 AMD は通常は無症状であるが、霧視を訴える場合もある。 ドルーゼンだけでなく、RPE 細胞が変性しアポトーシスが生じた場合は地図状に萎縮が起こり、 これが中心窩に及んでいれば近見視力と遠見視力が大幅に低下することがある。最終的には RPE が萎縮し、その結果、視細胞の機能が失われ視力障害に至る。地図状萎縮のある患者は、読書や 人の顔の判別に困難を感じることがある。また、視野の中心に盲点が認められることもある。盲 点は、発症初期は小さくとも、時間の経過と共にゆっくりと増大する可能性がある。 萎縮型 AMD の治療法は現時点では存在しない。抗酸化剤及びビタミン剤にわずかながら AMD の 進行のリスクを軽減する予防効果があることが、一部の研究者によって示されている2)。
滲出型 AMD
萎縮型 AMD は滲出型 AMD に進行することがある。滲出型 AMD は新生血管型 AMD としても知られ ており、萎縮型 AMD より有病率が低い。滲出型の患者は AMD 患者の約 10%にすぎないが、AMD に よる重度の視力低下を来す患者の 80~90%は滲出型 AMD 患者である。滲出型 AMD では、局所的 な炎症反応に伴ってブルッフ膜が破損し、VEGF が放出され、脈絡膜新生血管(CNV)の形成が誘 発される。CNV は漏出を起こしやすい異常血管であり、脈絡膜からブルッフ膜の破損部を超えて、 RPE 及び網膜の下まで増殖することがある。この機能的に未熟な新生血管からは、脂質、水分、 血液が漏出する。CNV は網膜の浮腫及び漿液性剥離の原因となり、霧視及び視界の歪みをもたら す。滲出型 AMD における視覚機能障害は急激に発症し、数週間以内に進展する。特に、出血があ る場合は更に急速に進行する。網膜下の出血あるいは持続性の浮腫がある場合、中心視力の喪失 は永久的なものとなる。患者には、視野の中心に大きな暗点が見える。通例、患者は読書をした り、テレビを見たり、自動車を運転したり、あるいは人の顔を識別したりすることが困難になる。 CNV は、蛍光造影(FA)で観察される CNV の描出パターンによって、occult 型あるいは classic 型のサブタイプに分類することができる。occult 型 CNV 病変は通常は RPE 下腔に限局さ れており、視力低下の程度も classic 型 CNV より軽度である。一方、classic 型 CNV 病変はしば しば色素上皮を貫き、網膜下腔まで増殖する。 日本では、AMD は、法的盲を含む視力障害の原因の第 4 位を占める 3)。住民を対象とした最近 の疫学調査により、AMD の有病率は約 1.0%であり4),5),6)、増加傾向にある7), 8)ことが明らかと なっている。したがって、現在 AMD は、日本の高齢者の視機能をおびやかす重大な網膜疾患とし て認識されている。また、日本では、AMD の 2 つの病型のうち、一般に滲出型 AMD 患者の方が萎 縮型 AMD 患者より多い傾向にある4 ), 7),8)。
厚生労働省の特定疾患研究グループ(Specified Disease Study Group)の滲出型 AMD の疫学 調査(1987 年と 1993 年)の結果から、滲出型 AMD 患者の年間増加率(約 8.9%)、1993 年の受 療患者数(14,400 例)をもとに算定した結果、2010 年における国内の滲出型 AMD の受療患者数 は、約 60,000 例と推定される。 2.5.1.1.2 AMD の診断 AMD の初期症状は、霧視、変視症、あるいは読字が困難である等の患者の自覚的な視覚異常で あることが多い。滲出型 AMD の臨床症状は通常徐々に進行するが、網膜下の CNV の出血により急 速に視力を喪失することがある。片眼が侵されることもあれば、両眼が同時にあるいは順に侵さ れることもある。 以下に AMD の診断に重要な検査法について簡単に説明する。本開発プログラムの 2 本のピボタ ル試験(VIEW 1 試験及び VIEW 2 試験)では、AMD の診断に FA を用いた。
眼底検査
診断にはまず、網膜の立体視検査を含む眼底検査を行うが、確定診断のためにはさらなる検査 が必要である。
蛍光造影(FA) FA では、水溶性のフルオレセインナトリウムを、通常は前肘部の静脈から血中に注入する。 フルオレセインナトリウムは青色光(波長 465~490nm)に励起され、緑黄色の蛍光(波長 520~ 530nm)を発する。適切なフィルターを用いた眼底写真により、眼底の血管系とその他種々の異 常が明らかとなる。 FA によって CNV 病変型(classic 型あるいは occult 型)、境界(明瞭あるいは不明瞭)、構 成(predominatly classic 型、minimally classic 型、classic CNV を伴わない occult 型)、 CNV の中心窩に対する位置(中心窩の外側、傍中心窩、あるいは中心窩下)が決定できるため、 FA は滲出型 AMD の初期診断にも用いられる。以前はこれらの蛍光造影所見が滲出型 AMD の診断、 治療及び追跡治療に重要であったが、抗 VEGF 療法時代となった現在では、CNV のすべてのサブ タイプが抗 VEGF 療法に反応することが示されているため、こうしたサブタイプ分類は以前ほど 重視されていない。 FA は、初期診断だけでなく治療成績の検討にも用いることができる。治療が奏功した場合、 FA 上の漏出は減少し、最終的には消失する。 光干渉断層撮影(OCT) OCT は非侵襲性の画像解析法であり、この数十年間で、黄斑を侵すさまざまな網膜の疾患の診 断及び治療効果の評価に使用されるようになってきた。OCT では、光の特異的な反射を利用して 網膜横断面の平面図が描出される。滲出型 AMD の病態解析では、OCT は網膜内、網膜下、あるい は RPE 下の滲出液の同定に有用である。一般的に網膜厚が、OCT の主要なパラメータとして用い られる。OCT は、FA の診断を裏付ける補助手段として有益であることから、診断の裏付け、特に 治療効果の評価、患者モニタリングにおける FA の施行回数の減少に役立っている。AMD の診断 には、一般的に OCT と FA の両方が用いられる。 2.5.1.1.3 眼内血管新生を仲介する VEGF 及び P1GF 1990 年代以降、臨床試験及び動物試験の結果から、眼内血管新生における VEGF の重要な役割 を裏付けるエビデンスが集積されてきた。その結果、VEGF は新規治療の格好の標的となった (2.5.1.2.2 参照)。 血管新生とは、さまざまな抑制因子及び促進因子の相互作用を含む複雑なプロセスを経て、新 しい血管が形成されることである。VEGF は内皮細胞の血管形成を促進する。また、VEGF は CNV の透過性を亢進させ、血管拡張物質として作用する9),10)。生物学的活性を有する二量体型 VEGF のそれぞれの末端には、受容体の結合部位があり10)、VEGF 受容体 1 及び VEGF 受容体 2 と結合す る。血管新生を促進する経路が活性化されると、血管内皮細胞が増殖・遊走し、脆弱で透過性の 亢進した異所性の血管が形成される。この異常な新生血管が、滲出型 AMD の病態に深く関わって いる。 霊長類モデル及びマウスモデルなどを用いた in vivo 試験で11),12),13)、VEGF の発現量が眼内血 管新生と関連することが示された。さらに、VEGF を正常霊長類の眼内に投与したところ、虹彩 に血管新生、血管新生緑内障及び網膜微小血管症が惹起された14),15)。脈絡膜新生血管(レーザー 照射による誘発 CNV など)及び網膜新生血管(streptozotocin により誘発された糖尿病網膜症 など)のモデルを含む上記の動物モデルでは、VEGF を薬理学的に阻害することによって眼内血 管新生が抑制された 12)(2.6.2.2)。
ヒトを対象とした臨床試験においても、VEGF の発現と病的な眼内血管新生との関連性が確認 された。硝子体内の VEGF 濃度の測定により、活性型の増殖性糖尿病網膜症の被験者は、異常な 血管新生がみられないその他の網膜障害の被験者と比べて VEGF 濃度が有意に高いことが明らか となった16 )。 胎盤増殖因子(P1GF)は、VEGF 受容体 1 を介して血管新生を促進する増殖因子である。PlGF の作用機序は十分に解明されていないが 17)、最近の研究では、特に病的な状況下での病的血管新 生並びに血管透過性の亢進における PlGF の関与が示唆されている 18), 19)。 P1GF は VEGF 受容体 1 を介して炎症細胞に対する化学誘導物質としても作用し、障害された網 膜への炎症細胞の遊走と、VEGF 及びその他の炎症性メディエーターの放出を亢進させると考え られている。滲出型 AMD では、VEGF の局所産生の増加によって惹起された血管新生と炎症が P1GF によって更に加速される。 VEGF と P1GF に関する詳細については、2.6.1 及び 2.6.2 を参照すること。 2.5.1.2 滲出型 AMD の治療法 滲出型 AMD の治療方針は、近年の抗 VEGF 療法の導入以降大きく変化した。その結果、以前の 治療法は限定された症例のみに適用されるようになった。 2.5.1.2.1 抗 VEGF 療法導入以前の治療法 熱レーザー光凝固 熱レーザー光凝固は、滲出型 AMD の治療に広く用いられた最初の治療法であり、レーザーを用 いて網膜内の CNV を凝固する。このアプローチの重大な欠点は、正常な網膜周囲又は上層組織に まで損傷を与えることである。このため熱レーザー光凝固は、特に黄斑中心部の病変には使用が 推奨されない。新規治療法が導入される前は、レーザー照射を行い経過観察する方法が、あらゆ るサブタイプの進行性滲出型 AMD に対する治療の中心であった20)。新規治療法の導入により、熱 レーザーの使用は新生血管の病変が黄斑中心部に及んでいない症例の治療に限られるようになっ た。 光線力学療法(PDT) ベルテポルフィンを用いた PDT では、熱レーザー光凝固を多少上回る改善が得られる。PDT は、 光感受性色素ベルテポルフィンの静脈内投与と、CNV に対する低出力のレーザー治療を組み合わ せたものである。PDT 用の色素は角膜を通して照射されたレーザーエネルギーによって活性化さ れ、放出されたフリーラジカルが異常な脈絡膜の血管を閉塞させる。PDT は、特定の進行性滲出 型 AMD の CNV の治療に有益であることが示されている21), 22)。PDT は病的な血管組織を選択的に破 壊し、上層にある正常な組織を損傷しないため、黄斑中心部を侵す病変では従来のレーザー光凝 固より優れている。PDT を施行した患者の多くでは、治療後に年 3~4 回の血管造影を行った際 に、治療部位における持続性漏出の有無に応じて、再治療が必要となる。PDT を施行した患者の 多くは視力が安定するが、臨床的に意味のある視力の改善はまれにしかみられない。
外科的治療 AMD に対する外科的治療は、まだ実験的治療の域をでていない。外科的治療としては、経毛様 体扁平部硝子体切除術による CNV の除去などが行われている。治療選択肢として、RPE が正常な 領域へ黄斑を移動させたり、RPE の移植を行う眼科医もいる。しかし、現時点では、外科的治療 は、例外的な症例のみに施行されている。滲出型 AMD に対する外科的治療は、ある程度の成果を 挙げており、また過去には他に有効な治療選択肢がなかったにもかかわらず、侵襲性が高いとい う理由から一部の患者集団に限定的に施行されるのみであった。 2.5.1.2.2 抗 VEGF 療法 抗血管新生療法(抗 VEGF 療法)は、血管新生の阻害因子の作用を促進すること、あるいは血 管新生の促進因子を抑制することによって滲出型 AMD の病態の進展に影響を与える23)。VEGF は内 皮細胞の血管形成を促進するばかりでなく、CNV の透過性を亢進し、血管拡張物質として作用す る9 ),10 )。 現在、抗 VEGF 療法〔特にラニビズマブ(下記参照)〕が滲出型 AMD の標準的治療となってい る。抗 VEGF 療法の利点は、外科的治療に比べて侵襲性の低い治療であることである。抗 VEGF 療 法は、滲出型 AMD の病態の主要因である VEGF のシグナル情報伝達経路を直接遮断することに よって多くの患者で視力を改善し、ほとんどの患者で視力を安定させる。CNV を一時的に閉塞さ せる PDT とは異なり、この抗 VEGF 療法は新たな血管新生を予防し 23)、既に存在する異常新生血 管からの漏出を抑制する。 米国食品医薬品局(FDA)、欧州医薬品庁(EMA)及びその他の国々において滲出型 AMD の適応 症が承認されている抗 VEGF 薬は、ペガプタニブナトリウム(以下、ペガプタニブと略す)とラ ニビズマブの 2 剤のみであるa)。 ペガプタニブ(マクジェン、Pfizer 社)は、2004 年に FDA により、2006 年にヨーロッパにお いて承認された23)。6 週ごとに硝子体内注射される RNA アプタマー、ペガプタニブは、硝子体内 において VEGF-A165アイソフォームと結合する 24 ), 25 ),10)。第Ⅲ相試験である VEGF Inhibition Study in Ocular Neovascularization(VISION)では、ペガプタニブの硝子体内投与によって滲 出型 AMD の視力低下を遅延させた。BCVA 文字数が 15 文字以上増加した患者の割合は、ペガプタ ニブ群では 6%であったのに対し、対照群では 2%であった 26)。 ラニビズマブ(ルセンティス、Genentech-Roche/Novartis 社)は、VEGF-A のすべてのアイソ フォームと結合する硝子体内投与用の抗体フラグメントであり 27)、AMD 治療法の発展における重 要で画期的な薬剤であった。ラニビズマブは卓越した有効性を示し、米国、EU 諸国、スイス、 オーストラリア、日本及びその他の国々において滲出型 AMD 治療薬として承認された 28),29)。 ラニビズマブのピボタル試験 2 試験(MARINA 試験 30)及び ANCHOR 試験31 ),32))では、ラニビズマ ブが 1 カ月ごとに硝子体内投与された。両試験において 12 カ月後に視力を維持していた被験者 (15 文字未満の視力低下と定義)の割合は、ラニビズマブ群では約 95%であったのに対し、対 照群では 62%(MARINA 試験の偽注射群)と 64%(ANCHOR 試験の PDT 群)であった。重要な点は、 それ以前の治療法とは異なり、ラニビズマブは実際に多くの患者で低下した視力を改善させたこ とである。両ピボタル試験では、ラニビズマブを投与した結果、3 分の 1 以上の被験者の視力が
改善した(15 文字以上増加)。糖尿病網膜症早期治療試験(ETDRS)チャートの文字数における 視力の改善は、MARINA 試験では平均 6.5~7.2 文字(偽注射群では -10.4 文字)、ANCHOR 試験 では 8.5~11.3 文字(PDT 群では -9.5 文字)であった。 PDT 群では平均視力が低下したのに対し、ラニビズマブ群では平均視力が改善したという結果 は、AMD の治療において画期的であり、抗 VEGF 療法の将来が有望であることを示していた。 本邦においては、2008 年にペガプタニブが、2009 年にラニビズマブが承認され、この 2 剤が、 抗 VEGF 薬として滲出型 AMD の治療に用いられている。AMD 治療の第一選択薬としては、すべて の VEGF-A アイソフォームを阻害するラニビズマブが、その視力改善効果の高さから、広く用い られている。 2.5.1.2.3 滲出型 AMD の治療法の改善の必要性 ラニビズマブによる治療では、MARINA 試験及び ANCHOR 試験結果から明らかなように、月 1 回 の投与で高い効果が得られる。しかしながら、月 1 回の治療を生涯に渡って継続することは、毎 回の硝子体内投与に伴う重大なリスクに加えて、患者、介護者、眼科医への負担を大きくし、医 療費も増大させる。ラニビズマブの月 1 回投与は、現在最も有効な治療法と認識されているが、 治療に伴う負担を軽減するために投与回数が減らされることが多く、有効性を著しく低下させて いる可能性がある 33)。 ベネフィットとリスクのバランスを改善する必要性から治療の個別化が試みられており、この ことはラニビズマブの添付文書b)の用量に関する記載にも反映されている: 「ラニビズマブ(遺伝子組換え)として 0.5mg(0.05mL)を 1 カ月ごとに連続 3 カ月間 (導入期)硝子体内投与する。その後の維持期においては、症状により投与間隔を適宜調節 するが、1 カ月以上の間隔をあけること。 維持期においては、1 カ月に 1 回視力等を測定し、その結果及び患者の状態を考慮し、本 剤投与の要否を判断すること。また、定期的に有効性を評価し、有効性が認められない場合 には漫然と投与しないこと。」〔本邦の添付文書(2011 年)の記載〕 再投与基準に従い、必要に応じ、随時(PRN)投与に切り替えることによって、ラニビズマブ の投与間隔を延長し、AMD 治療の投与頻度を減らそうとする試みは、以下の 2 つの重要な所見に より、限界があることがわかった: 1) 投与間隔を延長すると、ラニビズマブの有効性は ANCHOR 試験及び MARINA 試験で行われた月 1 回の固定用量投与に比べて低下する。例えば、ラニビズマブを、月 1 回、連続 3 カ月間投 与(導入期間)した後に、12 週ごとの投与を行った PIER 試験では、導入期間の投与後に得 られた平均視力が徐々に低下し、次の 1 年でほぼ投与前の視力になった34)。
b : EMA によって当初承認されたラニビズマブの添付文書(The summary of product characteristic)では、5 文字を超える視力低下が認められた場合にのみ再投与を行う PRN 用法が記載されていた。しかしながら、2011 年 9 月に、EU 諸国の添付文書は改訂され、視力の改善が最大となるまで毎月の投与を継続する、すなわち、患者の 視力が 12 週連続して安定していることが確認されるまで毎月の投与を継続することを推奨するように変更され た。
2) PRN 投与において再投与の必要性を判断するために、毎月のモニタリングが必要である。再 投与基準の一つである 1 カ月間に 5 文字の視力低下という定義は、不可逆性の視力低下を予 防するモニタリングの方法としては十分確立していない。こうした毎月のモニタリングには OCT 検査が含まれることがほとんどであり、FA による評価を要する場合もある35)。視力検査、 FA や OCT などによる高頻度の定期的モニタリングは、患者、医師、そして医療制度に大き な負担となっている。 本邦における滲出型 AMD の治療法の改善の必要性 ラニビズマブの MARINA 試験及び ANCHOR 試験の結果から、初回投与後に視力が最も顕著に改善 し、更に 2 回の追加投与を行った 3 カ月目までに視力の改善はほぼプラトーに達することが示さ れた。この時期を導入期(induction phase)と呼び、この期間に行われる 1 カ月ごとの 3 回投 与が視力の改善に寄与すると考えられている。その後の維持期(maintenance phase)に毎月投 与を継続することでラニビズマブの視力改善効果が示された。この投与方法で 2 年の経過後も治 療開始後 3 カ月目でみられる視力改善効果のピークをほぼ維持できたことが確認されている。 ただ実際には、AMD の治療は長期にわたるため、ラニビズマブの硝子体内投与を受けるために は、毎月来院しなければならないことから、患者、医療関係者及び医療制度の観点からみて大き な負担となる。 したがって、本邦の滲出型 AMD の治療において、有効性としての視力の改善が確実に達成でき、 更に長い投与間隔にもかかわらず、改善した視力を維持できることが医療ニーズとして挙げられ る。再投与の必要性を決定するために毎月のモニタリングが必要な用法ではなく、更に長い投与 間隔で定期的に投与を継続することにより、患者や医療関係者の負担を減らことも大きな医療 ニーズであると認識されている。 2.5.1.3 VEGF Trap:新規抗血管新生薬 2.5.1.3.1 化合物の特性
VEGF Trap は、水溶性の完全ヒト型遺伝子組換え VEGF デコイ受容体である。VEGF Trap は、異 なる 2 種の VEGF 受容体(VEGF 受容体 1 及び VEGF 受容体 2)の細胞外ドメインを、IgG1 の定常 領域(Fc ドメイン)に遺伝子工学的に融合させた組換え融合たん白質である(図 2.5- 1) 36)。
a b c
VEGF VEGF VEGF
receptor-1 receptor-2 Trap
図 2.5- 1 VEGF Trap の化学構造
VEGF 受容体 1(a)及び VEGF 受容体 2(b)は、細胞外の 7 つの Ig ドメインと細胞内のチロ シンキナーゼドメインを有する受容体ファミリーである。
VEGF Trap(c)は、VEGF 受容体 1 の第 2 Ig ドメインと VEGF 受容体 2 の第 3 Ig ドメインを IgG1 Fc に融合したものである。
VEGF Trap は血中及び血管外の VEGF 及び PlGF と高い親和性で結合し、これらを不活性化させ る特異的阻害薬である。
硝子体内投与に適する添加剤を用いて調製した製剤を VEGF Trap-Eye と呼ぶ。VEGF Trap を眼 内に注射することによって、全身への曝露を最小限に抑えながら治療量を標的組織に到達させる ことができる。
現在利用可能なラニビズマブ及びペガプタニブなどの抗 VEGF 薬とは異なり、VEGF Trap は VEGF-A に加え、PlGF 及び VEGF-B にも結合する(2.6.2.2.1.1 参照)。
リガンド結合モデルにおいて示されているように、VEGF-A に対する VEGF Trap の高い結合親 和性によって低濃度でも VEGF の活性を遮断しうることから、投与間隔を延長することが可能と なるかもしれない37)。また、理論的には、VEGF-A に加え、PlGF 及び VEGF -B に対しても VEGF Trap が結合することにより、抗 VEGF 活性の持続時間を延長するだけでなく、視力改善にも影響 を及ぼす可能性がある。
さまざまな眼障害の動物モデルを用いた VEGF Trap の研究により、VEGF Trap が網膜と脈絡膜 の血管新生だけでなく、網膜浮腫の形成を抑制することが示された(2.6.2.2.2)。VEGF Trap は、眼の血管新生疾患における網膜浮腫、虚血及び出血の原因となる新生血管の増殖を防ぐ。
すなわち、VEGF Trap は、高い結合親和性で VEGF と結合する。さらに他の抗 VEGF 薬とは異な り、VEGF Trap は PlGF 及び VEGF-B にも結合する。これらの特性から作用持続時間が延長し、そ
の結果、投与間隔が月 1 回より延長されることが期待され、従来の標準的治療に比べて視力が改 善される可能性があった。 2.5.1.3.2 VEGF Trap-Eye の臨床開発プログラム 本申請に用いた VEGF Trap-Eye の臨床開発プログラムに係る臨床試験の一覧を、表 2.5- 1に 示す。 表 2.5- 1 臨床試験一覧 試験 治験実施 計画書番号 試験デザイン 及び対照の種類 対象 治験薬の投与方法及び投与経路 参考/ 評価 第Ⅰ相 VGFT-OD-0502 Part A オープン ラベル 滲出型 AMD 0.05mg、0.15mg、0.5mg、1.0mg、2.0mg 及び 4.0mg を 単回硝子体内投与 評価 同上 Part B 無作為化 二重盲検 滲出型 AMD 2.0mg を硝子体内投与、 対照としてペガプタニブナトリウム 0.3mg を硝子体 内投与 同上 Part C 無作為化 二重盲検 滲出型 AMD 0.15mg、4.0mg を硝子体内投与 VGFT-OD-0603 無作為化 二重盲検 滲出型 AMD ITV-1/4mg/100μL 及び ITV-2/4mg/100μL、 ITV-2/4mg/50μL(オープンラベル)を硝子体内投与 評価 VGFT-OD-0512 オープン ラベル DME 参考 VGFT-OD-0305 二重盲検プラセ ボ対照用量漸増 滲出型 AMD 0mg/kg(生食)、0.3mg/kg、1mg/kg 又は 3mg/kg を 1 時間かけて静脈内投与 参考 VGFT-OD-0306 オープン ラベル 滲出型 AMD 0.3mg/kg 又は 1.0mg/kg を 1 時間かけて静脈内投与 参考 VGFT-OD-0307 二重盲検プラセ ボ対照用量漸増 DME 参考 PDY6655 無作為化オープ ンラベル、 クロスオーバー 健康男性 被験者 2.0mg/kg を 1 時間かけて単回静脈内投与及び単回皮 下投与 参考 PDY6656 無作為化二重盲 検プラセボ対照 用量漸増 健康男性 被験者 0mg/kg(0.9%NaCl)、1mg/kg、2mg/kg 又は 4mg/kg を 1 時間かけて静脈内投与 参考 第Ⅱ相 VGFT-OD-0508 無作為化 二重盲検 滲出型 AMD 0.5mg、2.0mg、4.0mg のいずれかの用量で 4 週又は 12 週ごとに 12 週間硝子体内投与(固定量投与期 間)した後、16~52 週目まで PRN 投与 評価
表 2.5- 1 臨床試験一覧(続き) 試験 治験実施 計画書番号 試験デザイン 及び対照の種類 対象 治験薬の投与方法及び投与経路 参考/ 評価 第Ⅲ相 VGFT-OD-0605 (VIEW 1) 無作為化二重 盲検実薬対照 滲出型 AMD ・4 週ごとに 0.5mg を硝子体内投与(0.5Q4) ・4 週ごとに 2mg を硝子体内投与(2Q4) ・ 最初の3 回は 4 週ごとに 2mg 投与、その後、8 週 ごとに2mg を硝子体内投与(2Q8)、中間の 4 週 ごとの来院時(治験薬を投与しない時)に偽注射 ・ 比較対照として、4 週ごとにラニビズマブ 0.5mg を硝子体内投与(RQ4) 2 年目は、1 年目と同じ用量で、再投与基準に従って PRN 投与を行う。ただし、投与間隔は 12 週を超え ないものとする。 評価 311523 (VIEW 2) 日本参加 無作為化二重 盲検実薬対照 滲出型 AMD ・4 週ごとに 0.5mg を硝子体内投与(0.5Q4) ・4 週ごとに 2mg を硝子体内投与(2Q4) ・ 最初の3 回は 4 週ごとに 2mg 投与、その後、8 週 ごとに2mg を硝子体内投与(2Q8)、中間の 4 週 ごとの来院時(治験薬を投与しない時)に偽注射 ・ 比較対照として、4 週ごとにラニビズマブ 0.5mg を硝子体内投与(RQ4) 2 年目は、1 年目と同じ用量で、再投与基準に従って PRN 投与を行う。ただし、投与間隔は 12 週を超え ないものとする。 評価 その他 の臨床 試験 VGFT-OD-0702 単盲検無作為化 オープンラベル (第Ⅱ相 延長試験) 滲出型 AMD プレフィルドシリンジ製剤の投与容量50μL(2mg)又 はバイアル製剤の投与容量50μL(2mg)を PRN 投 与 参考 VGFT-OD-0910 オープン ラベル (第Ⅲ相 延長試験) 滲出型 AMD capped-PRN で 2mg を硝子体内投与(少なくとも 12 週間に1 回は投与する) 参考 VGFT-OD-0706 (DAVINCI) 無作為化二重 盲検対照 (第Ⅱ相試験) DME ・52 週目まで 4 週ごとに 0.5mg を硝子体内投与 (0.5Q4) ・52 週目まで 4 週ごとに 2mg を硝子体内投与 (2Q4) ・ 最初の3 回は 4 週ごとに 2mg を硝子体内投与、 その後は52 週目まで 8 週ごとに硝子体内投与 (2Q8) ・ 最初の3 回は 4 週ごとに 2mg 硝子体内投与、そ の後は必要に応じて2mg(2PRN) ・ 対照治療として、レーザー光凝固術 参考 VGFT-OD-0819 (COPERNICS ) 無作為化二重 盲検対照 (第Ⅲ相試験) CRVO 参考 14130 (GALILEO) 無作為化二重 盲検対照 (第Ⅲ相試験) CRVO 参考
AMD:Age-related Macular Degeneration(加齢黄斑変性) DME:Diabetic Macular Edema(糖尿病黄斑浮腫)
CRVO:Central Retinal Vein Occlusion(網膜中心静脈閉塞症による黄斑浮腫) PRN:As needed (pro re nata)(必要に応じて、随時)
これらのうち、本申請効能・効果である滲出型 AMD の評価に用いた主な臨床試験は、以下の とおりである。 第Ⅰ相試験: 502 試験及び 603 試験 第Ⅱ相試験: 508 試験 第Ⅲ相試験: VIEW 1 試験及び VIEW 2 試験 これら第Ⅲ相ピボタル試験のデザインの概要を、図 2.5- 2 に示す。 図 2.5- 2 第Ⅲ相ピボタル試験(VIEW 1 試験、VIEW 2 試験)の概要 これらの第Ⅲ相試験(VIEW 1 試験及び VIEW 2 試験)は、申請時適応症を裏付けるピボタル試 験としてデザインされた。これらの第Ⅲ相試験の 1 年間(52 週)の結果から、各投与群の有効 性及び安全性について臨床的に意味のある情報が得られると考える。また、各国規制当局との合 意に基づき、投与開始後 1 年目で得られたデータをもって主要評価を行い申請した。VEGF Trap-Eye の各投与群と対照薬(ラニビズマブ)の 4 週ごと 0.5mg 投与群を、試験1年目に直接比較し た。主要評価項目の評価後、試験は更に 1 年間継続された。投与 2 年目には、投与間隔の延長と 再投与基準に基づく PRN 投与が、1 年目に達成された視力及び形態学的な変化の維持にどのよう な影響を及ぼすかについての評価が行われた。本邦における申請後に、これらの 2 年目(96/100 週)の結果が得られたため、2 年目終了時までの有効性及び安全性についても評価し、2.5 及び 2.7 に追記した。 2.5.1.3.3 民族的要因の考察
本剤の第Ⅲ相臨床試験(VIEW 2 試験)(2.5.1.3.2 VEGF Trap-Eye の臨床開発プログラム参 照)を、日本人を含む国際共同治験として計画するにあたり、民族的要因について以下の検討を 行った。
内因性民族的要因 加齢黄斑変性(AMD)は欧米をはじめとし、本邦においても成人の中途失明の主な原因のひと つとなっている。民族差を検討するにあたって、各国で実施された疫学的調査結果を用いて、 AMD の有病率及び発症率の比較を行った。日本人の AMD の有病率の検討には、1998 年に福岡県糟 屋郡久山町で実施した眼科検診の結果の報告4)及びその 5 年後の追跡調査の結果7)を用いた。 1998 年の調査における AMD の有病率は 0.9%で、その後の追跡調査では、最近 5 年間の日本人 の AMD の発症率は欧米における発症率に近づいており、今後も高齢化にともない、AMD 患者数 は増加傾向にあると予想された。日本人において、有病率、発症率とも男性が高く、喫煙率の差 が影響している可能性が報告されている38)。 また、AMD は遺伝的背景に環境因子が加わり発症する多因子疾患と考えられており、遺伝的要 因を解明する研究が進められている。白人に多く日本人に発現頻度が少ない遺伝子の報告がある 一方、白人とアジア人に共通してみられる SNP も報告されており、遺伝的要因の影響について は、今後の研究の課題と考えられた39)。 このように、疫学の観点からは、AMD の発症率に日本人と外国人では大きな差はなく、AMD 発 症の危険因子は、すべての民族に共通する加齢と喫煙であった。 また、本剤は、硝子体内投与されることから、局所における薬物動態に日本人と外国人で差は ないと考えられ、全身曝露量も極めて低く、遺伝子多型を有する薬物代謝酵素による消失経路を たどらないたん白製剤であることなどから、薬物動態学的な要因による民族差の影響は考えにく い。 外因性民族的要因 環境要因のひとつである疾病の定義と診断、治療法について比較を行った。本邦において、厚 生労働省のワーキンググループにより策定された「AMD の分類と診断基準」(2008 年)40)は、欧 米 に お け る The international ARM epidemiological study group に よ る 「 International classification and grading system for age-related maculopathy and age-related macular degeneration」(1995 年)を基盤としており、国内外で疾病の診断において問題となるような 差は見出されなかった。 また、治験開始時点において、海外では、既に抗 VEGF 薬が AMD 治療薬として販売されていた が、国内では AMD の適応で承認された抗 VEGF 薬はなかった。しかしながら、国内の学会発表 (2007 年日本眼科学会)では、抗腫瘍薬として承認されているベバシズマブの硝子体内投与に 関して、日本人においても抗 VEGF 薬の AMD に対する効果が報告されており、本邦においても、 抗 VEGF 薬が今後の AMD の治療薬の中心となるものと推察された。 これらの検討結果を踏まえ、 と判断した。この開発計画について、独 立行政法人医薬品医療機器総合機構(PMDA)と対面助言( 相談)を行い、その 助言を踏まえて、VIEW 2 試験を本邦においても開始した(2.5.1.4参照)。 上述の検討内容、及びその後に得られた知見に基づき、民族的要因について考察した結果の詳 細は、2.5.4.1.2 有効性評価における民族的要因の考察に記載した。
2.5.1.4 各国規制当局との相談の経緯
Regeneron Pharmaceutical Inc.(以下 Regeneron 社)は、20 年に第Ⅰ相試験を、20 年に 第Ⅱ相試験をそれぞれ開始した。第Ⅲ相ピボタル試験である VIEW 1 試験及び VIEW 2 試験の 2 試 験は、Bayer Schering Pharma AG(現 Bayer Pharma AG/BHC group、以下 BHC 社)と Regeneron 社の緊密な協力のもとにそれぞれ 20 年及び 20 年に開始された。
第Ⅲ相試験は、各国の規制当局の科学的助言に基づいてデザインされた。表 2.5- 2に、滲出 型 AMD の適応症に関する VEGF Trap-Eye の開発に係わる規制当局との主な会議とその目的及び結 果を示す。 日本における本剤の開発戦略が適切であることを確認するために、20 年 月、PMDA と対面 助言( 相談)を行い、 相談を行った。 合意を得た。本対面 助言では、PMDA は 、20 年 月に を行い、 了承を得た。(対面助言議事録について は、1.13 参照。) 表 2.5- 2に示した各国の規制当局からの科学的助言に基づき、国際共同第Ⅲ相試験のデザイ ン及び臨床データパッケージが決定された。
表 2.5- 2 EU 諸国、米国及び日本における規制当局との主要な会議
Regio
n Date Regulatory milestone Outcome
EU 20 MPA MPA 20 20 CHMP CHMP 20 20 MAA 20 EMA MAA 20 MAA US 20 SPA FDA 20 SPA FDA
20 BLA FDA BLA FDA
Japan
20 PMDA PMDA
20
BLA: Biologic License Application
CHMP: Committee for Medicinal Products for Human Use EMA: European Medicines Agency
MAA: Marketing Authorisation Application
PMDA: Pharmaceuticals and Medical Devices Agency SPA: Special Protocol Assessment
2.5.2 生物薬剤学に関する概括評価
VEGF Trap の標的組織は網膜であり、本剤は硝子体内に直接投与される。したがって、標的器 官におけるバイオアベイラビリティはほぼ 100%と考えられる。VEGF Trap は硝子体内投与され るため、全身バイオアベイラビリティのような通常の生物薬剤学的アプローチは VEGF Trap-Eye の評価には適当ではない。
VEGF Trap-Eye の開発に際しては、3 種類の製造方法(IVT P1、P2 及び P3)により VEGF Trap (原薬)を製造した。2 種類の製剤処方(ITV-1 及び ITV-2)の硝子体内投与用製剤が開発され、 臨床試験に用いられた。初期製剤 ITV-1 は、安定性を改善するために第Ⅱ相試験中に改良され、 現在の処方である ITV-2 が開発された。第Ⅲ相試験を通して、同じ原薬製造方法(IVT P3)及び 同じ製剤処方(ITV 2)により製造された VEGF Trap-Eye が用いられた。第Ⅲ相試験用製剤と市 販予定製剤の処方は同じである。開発プログラムを通して用いられた各種原薬(製造方法)と製 剤(処方)の概要を、表 2.5- 3に示す。
表 2.5- 3 早期及び後期の開発プログラムにおいて使用された原薬製造方法と製剤の概要
Development phase Study Manufacturing process
(drug substance)
Formulation (drug product)
Phase 1 VGFT-OD-0502 IVT P1 ITV-1
IVT P2 ITV-1
VGFT-OD-0603 IVT P2 ITV-1
IVT P3 ITV-2
VGFT-OD-0512 IVT P2 ITV-1
Phase 2 VGFT-OD-0508 IVT P2 ITV-1
VGFT-OD-0702 long-term safety IVT P2 ITV-1
IVT P3 ITV-2
VGFT-OD-0702 PK substudy IVT P3 ITV-2
Phase 3 VIEW 1 IVT P3 ITV-2
VIEW 2 IVT P3 ITV-2
現在用いられている製造方法(IVT P3)の詳細は 3.2.S.2.2 に、他の原薬の製造方法(IVT P1 及び P2)との比較は 3.2.S.2.6 に、主な変更の概要は 2.7.1 に記載した。また、両硝子体内製 剤の処方(ITV-1 及び ITV-2)の特性は 3.2.P.2.2 に、処方の相違点の概要は 2.7.1.1.3 に記載 した。 第Ⅲ相試験で用いた製剤の処方は市販予定製剤と同じであるが、別の原薬製造方法及び製剤処 方 を 用 い て 調 製 さ れ た 製 剤 に つ い て も 、 薬 物 動 態 ( Pharmacokinetics : PK ) 、 薬 力 学 (Pharmacodynamics:PD)並びに免疫原性を評価した。分析法の詳細は、2.7.1.1.5 に記載した。 原薬製造方法と製剤処方の変更により生じうる影響を評価するためには、網膜中心部(病変) の厚さ(central retinal/lesion thickness:CR/LT)のような臨床的意義のある PD 作用が、適 したパラメータであると考えられた。また、安全性に関連して、本剤の全身曝露量と免疫原性の
比較評価も行った。VEGF Trap-Eye の原薬製造方法と製剤処方の違いから生じうる影響を評価す るために、関連するロットを同じ投与量にて VEGF Trap-Eye の硝子体内投与を受けた被験者を対 象に、CR/LT、遊離型 VEGF Trap 及び結合型 VEGF Trap の血漿中濃度を比較した(2.7.1.2 参 照)。さらに、各製剤の免疫原性の評価も実施した。
試験結果の比較から、原薬製造方法あるいは製剤処方の違いは、VEGF Trap-Eye 投与後の PD に大きな影響を及ぼさない(ベースラインからの CR/LT の変化により判定)ものと考えられた。
VEGF Trap-Eye を硝子体内投与した後の遊離型 VEGF Trap 及び結合型 VEGF Trap の体循環血漿 中濃度から、原薬製造方法及び製剤処方の違いは VEGF Trap-Eye の PK には影響を及ぼさないこ とが示唆された(2.7.1.3 参照)。
初期の硝子体内投与の臨床試験では、最初に確立された ELISA 法を用いて抗薬物抗体(ADA: Anti-drug antibody)を測定することにより免疫原性を検討したが、いずれの被験者においても VEGF Trap の免疫原性は検出されなかった(2.7.1.3.3 参照)。第Ⅲ相試験(VIEW 1 試験及び VIEW 2 試験)では、当初の ADA 分析法に比べて 40 倍もの感度を有する ADA ブリッジングイムノ アッセイ法により免疫原性を検討しており、その結果を2.5.3.4で概説する。VIEW 1 試験と VIEW 2 試験の免疫原性のデータの詳細の概要は、2.7.2.3.4 に記述した。 結論 第Ⅲ相試験を通して、同じ原薬製造方法(IVT P3)及び同じ製剤処方(ITV-2)により製造さ れた VEGF Trap-Eye が用いられた。第Ⅲ相試験用製剤と市販予定製剤の処方は同じである。 総じて、複数の臨床試験において観察された結果から、臨床試験中に用いられた VEGF Trap-Eye の原薬製造方法及び製剤処方の変更は、薬剤の PD、PK 及び免疫原性に影響を及ぼさないこ とが示された。なお、VEGF Trap-Eye の開発プログラム中に、すべての原薬製造方法と製剤処方 において製造された製剤を用いて実施された臨床試験から、一貫した結果が得られている。
2.5.3 臨床薬理に関する概括評価
VEGF Trap-Eye の開発は、当初は静脈内(intravenous:IV)投与製剤を用いたプログラムで 開始された。その後、硝子体内における治療濃度を最も高め、その一方で全身曝露量と全身への 影響を最小限とするため、投与経路を硝子体内投与へと変更した。IV 投与によるプログラムの 初期の臨床試験のデータから有益な情報が得られており、これらは全身における PK と PD の関係 の評価に用いると共に、VEGF Trap-Eye の硝子体内投与が局所の臨床的有効性を最大にしながら、 かつ全身的な薬理学的作用を最小化するという機序を理解する基盤となっている。
臨床薬理に関する検討においては、投与物質である遊離型 VEGF Trap と結合型 VEGF Trap(内 因性 VEGF と結合して形成される不活性の VEGF:VEGF Trap 複合体の VEGF Trap 当量)の血漿中 濃度に基づいて VEGF Trap の PK を評価し、IV 投与及び硝子体内投与後の VEGF Trap の全身性の 作用と眼に対する作用に関連する PD パラメータを区別して評価した。VEGF Trap の眼に対する PD 作用については、CR/LT と CNV 病変面積の測定値を用いて評価した。両投与経路について、血 圧(一部の試験では 24 時間血圧測定)を全身性作用のサロゲートマーカーとした。臨床薬理学 的な検討の目的は以下のとおりであった:
硝子体内投与あるいは全身(IV あるいは皮下)投与後の VEGF Trap の PK 特性を明ら かにする 第Ⅱ相試験及び第Ⅲ相試験の用量設定の根拠となる、硝子体内投与の用量と PD 反応 の関連を立証する VEGF Trap の全身への影響について、PK と PD の関係を理解する 全身性の PD 作用が生じることが判明している曝露量と、硝子体内投与後の全身曝露 量との関係を明らかにする 2.5.3.1 薬物動態
複数の臨床試験において、IV 投与後、皮下投与後及び硝子体内投与後の遊離型 VEGF Trap 及 び結合型 VEGF Trap の血漿中濃度を測定した(2.7.2.1.2 及び 2.7.2.3.1 参照)。VEGF Trap-Eye の硝子体内投与後、投与された本薬の一部は眼内において内因性遊離型 VEGF と高い親和性 をもって結合し、安定した VEGF-VEGF Trap 複合体を形成すると考えられる。過剰な遊離型 VEGF Trap は、眼内で新たに合成された VEGF あるいは体循環中の内因性遊離 VEGF との結合に供され る。硝子体内投与後、VEGF Trap は眼から体循環血中にゆっくりと放出され、体循環血中では活 性を有さない、安定した VEGF-VEGF Trap 複合体としてより多く存在する。
多時点で検体採取を行った薬物動態サブスタディにおける遊離型 VEGF Trap の最高血漿中濃度 (maximum concentration:Cmax)は低く、2mg の硝子体内投与後 1~3 日目における Cmaxの平均濃 度は約 0.02μg/mL(0~0.054μg/mL の範囲)であり、投与 2 週後にはほとんどの被験者で検出 されなかった([参考]5.3.3.2-1 702.PK 試験)。4 週ごとの硝子体内投与後、血漿中に VEGF Trap の蓄積は認められなかった(5.3.5.1-3 VIEW 2 試験)。硝子体内投与後の遊離型 VEGF Trap の絶対的バイオアベイラビリティは、約 15~30%と推定された(2.7.2.3.1.1 参照)。
体循環中に局在する高分子たん白質と同様、IV 投与後の遊離型 VEGF Trap の分布容積は約 6L であり([参考]5.3.4.1-1 PDY6655 試験、[参考]5.3.4.1-2 PDY6656 試験)、これは血漿中の
総容積(2~3L)をやや上回っている程度である。VEGF Trap-Eye はたん白質製剤であるため、 代謝試験は実施していないc)。
遊 離 型 VEGF Trap の PK は 、 飽 和 性 の 標 的 介 在 性 の 薬 物 動 態 ( target-mediated drug disposition)のため、非線形性を示す(2.7.2.3.1.2 参照)。遊離型 VEGF Trap は、VEGF と の比較的速かな特異的かつ飽和性の高親和性結合によって、またこれより緩徐な非飽和性のクリ アランス機序によって消失することがデータから示唆される。後者の非飽和性のクリアランス機 序は、他の高分子たん白質と同様、たん白質分解による異化作用であると考えられており、遊離 型 VEGF Trap 及び結合型 VEGF Trap の消失に寄与している。遊離型 VEGF Trap の消失半減期 (terminal half-life: t1/2)は投与量の増量に伴って延長し、0.3mg/kg を IV 投与後では約 1.9 日であるが、2~4mg/kg を IV 投与後の t1/2 は 5~6 日に延長すると推定される([参 考 ]5.3.3.2-9 305/306 試 験 統 合 PK 報 告 書 、 [ 参 考 ]5.3.4.1-1 PDY6655 試 験 及 び [ 参 考]5.3.4.1-2 PDY6656 試験)。2~4mg/kg を IV 投与後の t1/2は、新たに産生される内因性 VEGF (遊離型 VEGF Trap との結合により、活性を有さない安定した複合体を形成)の産生速度を反映 するものと思われる。硝子体内投与後の血漿中遊離型 VEGF Trap 濃度の消失については、投与 14 日目以降の血漿中遊離型 VEGF Trap 濃度は大半の被験者において定量下限未満であったため、 詳細な評価はできなかった〔[参考]5.3.3.2-1 702.PK 試験、5.3.3.2-2 502 試験パート A、 5.3.3.2-4 502 試験パート C 及び 5.3.3.2-5 603 試験〕。 IV 投与された試験では、硝子体内投与(単眼につき最大 4mg)よりはるかに高用量の VEGF Trap が用いられた(最大 4mg/kg)。VEGF Trap を IV 投与した場合、体循環中の内因性 VEGF の 多くが VEGF Trap に結合すると考えられる。体循環中の内因性 VEGF が VEGF Trap とほぼ完全に 結合したときの特徴として、結合型 VEGF Trap 濃度が一定濃度を維持し、用量依存的な増加を認 めず、遊離型 VEGF Trap 濃度が結合型 VEGF Trap 濃度を上回って用量依存的に増加している状態 が挙げられる(2.7.2.3.1.1 参照)。1mg/kg 以上を IV 投与あるいは皮下投与したとき、体循 環中 VEGF の結合は飽和状態又はそれに近い状態となった([参考]5.3.3.2-7 305 試験、[参 考]5.3.4.1-1 PDY6655 試験及び[参考]5.3.4.1-2 PDY6656 試験)。IV 投与後には全身性 PD 作 用(特に血圧変化)が認められた。対照的に、単眼に最大で VEGF Trap-Eye 4mg を硝子体内投与 したときの結合型 VEGF Trap 濃度は、IV 投与後に認められる濃度の約 20 分の 1 に過ぎなかった (2.7.2.3.1.1 参照)。以上より、検討した投与量において、VEGF Trap-Eye 硝子体内投与後 の体循環中には未結合の内因性遊離 VEGF がかなり残存していること、硝子体内投与後の早期に 観察される結合型 VEGF Trap 濃度を上回る遊離型 VEGF Trap 濃度の一時的な上昇は、複合体の形 成速度が遅いことに起因することが示唆される(2.7.2、図 2.7.2- 3 及び図 2.7.2- 5)。遊離 型 VEGF Trap の Cmaxは、動物モデルにおいて体循環中 VEGF の生物学的活性を 50%阻害するため に要した VEGF Trap 濃度の約 50~500 分の 1 である(2.7.2.3.1.4 参照)。被験者に 2mg を硝 子体内投与した後の遊離型 VEGF Trap の平均 Cmaxは、最大 VEGF 結合能の半分に達するのに必要 な VEGF Trap 濃度の 100 分の 1 未満と推測される41), 42)。このため、全身性の PD 作用はないと考 えられる。
腎機能障害を有する患者を対象とした VEGF Trap-Eye の試験は特に実施しなかった。約 40% に腎機能障害があった VIEW 2 試験の部分被験者集団(25%が軽度、15%が中等度、1%が高度) の薬物動態解析では、VEGF Trap-Eye を 4 週ごとあるいは 8 週ごとに硝子体内投与した後の血漿 中の遊離型 VEGF Trap 濃度あるいは結合型 VEGF Trap 濃度に腎機能障害の程度による差は認めら れなかった(5.3.5.1-3 VIEW 2 試験、2.7.2.2.4.5 参照)。
c :日米 EU 医薬品規制調和国際会議(ICH)のトピック S6(バイオテクノロジー応用医薬品の非臨床における安
VEGF Trap の消失機序及び低い全身曝露量を考慮すると、肝機能障害が PK に特異な影響を及 ぼす可能性は低いと考えられるため、肝機能障害を有する患者を対象とした VEGF Trap-Eye の試 験は特に実施しなかった。現在得られたデータからは、腎機能障害あるいは肝機能障害を有する 患者に対して VEGF Trap-Eye の用量調節の必要はないと考えられる(5.3.5.1-2 VIEW 1 試験及 び 5.3.5.1-3 VIEW 2 試験 参照)。
単眼に VEGF Trap-Eye(VEGF Trap として)2mg を投与(以下、同様)した 335 例の部分被験 者集団の探索的解析から、年齢、性別、体格指数、又は被験者集団(ヨーロッパ地域と日本)は、 遊離型 VEGF Trap あるいは結合型 VEGF Trap の血漿中濃度に対して臨床的に問題となる影響を及 ぼさないことが示された(5.3.5.1-3 VIEW 2 試験、2.7.2.2.4.5 参照)。 食物あるいはその他の薬剤と VEGF Trap-Eye との相互作用を検討した標準的な薬物相互作用試 験は実施されていないd)。 2.5.3.2 薬力学 網膜及び網膜下の網膜内嚢胞様浮腫及び網膜下液、AMD 病変の厚さの測定には、OCT を用いた。 CR/LT は、再現性よく抗 VEGF 療法に対する PD 応答を示し、迅速に反応する評価項目であること が知られている16), 43)。このため CR/LT は、被験者によっては有効性の主な評価項目である最高矯 正視力(best corrected visual acuity:BCVA)と相関しない場合もあることが知られているも のの、十分に容認可能な反応マーカーとされている(5.3.5.1-3 VIEW 2 試験) 44), 45)。また、FA を用いて CNV 病変面積を測定し、標的部位の PD 作用を形態学的にも検討した。 第Ⅰ相試験及び第Ⅱ相試験の CR/LT 及び CNV 病変面積の測定値(2.7.2.3.2 及び 2.7.3.2 参 照)は、第Ⅱ相試験の有効性の主要評価項目であった視力(2.7.3.2 参照)と共に第Ⅲ相試験 の用量設定の根拠とした。 VEGF Trap-Eye を硝子体内投与後、用量依存的な CR/LT の減少が認められた。0.15mg 及び 0.5mg 投与時にも効果がみられたが、単眼に 2mg 又は 4mg を投与した際に最大効果に達し、単眼 に 2mg あるいは 4mg を投与したときの改善は同程度であった(5.3.3.2-2 502 試験パート A、 5.3.3.2-4 502 試験パート C、5.3.5.1-1 508 試験、2.7.2.3.3.1 参照)。これらの結果、最 大の CR/LT の減少は単眼に 2mg を投与した際に達成されること、より高用量の VEGF Trap-Eye を 硝子体内投与する必要はないことが示唆された。CNV 病変面積の変化は、明確ではなかった。 第Ⅱ相 508 試験から得られたデータから、VEGF Trap-Eye の 4 週ごとの投与は持続的な PD 作 用を示したが、投与開始時に 12 週ごとに投与した場合には PD 作用は十分に維持されないことが 示された。 以上より、第Ⅰ相及び第Ⅱ相臨床プログラムにおける CR/LT 及び CNV 病変面積の減少に関する PD 結果は、第Ⅲ相試験における用量設定の根拠となるものであった。設定用量の最終決定は、 第Ⅱ相試験の主要パラメータであった視力の測定値に基づくものであるが、主要な有効性のパラ メータ(視力及び視力の維持)の詳細については、2.7.3.2.2.1 に記載した。 d :日米 EU 医薬品規制調和国際会議(ICH)のトピック S6(バイオテクノロジー応用医薬品の非臨床における安 全性評価、1997 年 7 月/ 同 2000 年 2 月 22 日付 医薬審 第 326 号)
2.5.3.3 曝露量と反応の関連 VEGF Trap-Eye の硝子体内投与の効果に関する探索的検討において、用量依存的な CR/LT の減 少が認められており、0.15mg 投与時から効果がみられ、2mg と 4mg 投与時の改善は同程度であっ た(2.7.2.3.3.1 参照)。このことから、CR/LT の最大の減少は 2mg の投与時に達成され、よ り高用量(単眼につき 4mg)を投与してもさらなる改善はみられないことが示唆された。また、 12 週ごとの投与(治療開始時に投与)では、次の投与まで網膜厚の改善を維持するには不十分 であり、至適投与間隔は 12 週未満であることが示唆された。第Ⅱ相試験の視力のデータ (2.7.3.2.2.1 参照)は、0.5mg ではなく、 2mg 投与によってこうしたベネフィットの最大化 が得られることを示している。 VEGF シグナル伝達経路を阻害あるいは干渉する薬剤を全身投与した際に認められる拡張期血 圧と収縮期血圧の上昇は、体循環中の遊離型 VEGF が減少するためと考えられる。血漿中遊離型 VEGF Trap 濃度(全身投与時)と全身性の PD 作用(血圧変化)には用量依存的な相関性が認め られる(2.7.2.3.3.3 参照)。硝子体内投与時には、VEGF Trap による拡張期血圧あるいは収 縮期血圧の上昇は認められなかったが、硝子体内投与後の血漿中遊離型 VEGF Trap と結合する体 循環中の内因性 VEGF はわずかであることから、十分に予測される結果であった。 2.5.3.4 免疫原性
VEGF Trap-Eye の開発プログラムを通し、以下の 2 つの ADA 分析法が開発され、バリデーショ ン試験が実施された。(i)初期の第Ⅰ相試験と第Ⅱ相試験に用いられた最初の ELISA 法による ADA 分析法、(ii)第Ⅲ相試験に用いられた、開発当初の分析法に比べて約 40 倍の感度を有す るブリッジング ADA イムノアッセイ法(2.7.2.1.4 参照)。AMD 被験者 223 例の試料の評価に 用いられた開発当初の ELISA 法では、ADA の陽性反応は検出されなかった。第Ⅲ相試験 2 試験 (5.3.5.1-2 VIEW 1 試験及び 5.3.5.1-3 VIEW 2 試験)に用いられたブリッジング ADA イムノ アッセイでは、弱い陽性反応が認められた(2.7.2.3.4 参照)。投与前の VEGF Trap への免疫 反応率は、投与群全体で 1~3%であった。52 週間にわたる VEGF Trap-Eye 投与後の抗 VEGF Trap 抗体の検出率は、ラニビズマブ群を含む全投与群で同じく 1~3%であった。抗薬物反応が 認められた被験者と認められなかった被験者の間に、有効性あるいは安全性の差は認められな かった。ブリッジング ADA イムノアッセイにおける陽性反応の多くは被験者の既存の免疫反応性 によるものであり、VEGF Trap への免疫反応によるものではないことが示唆された。また、VIEW 2 試験の日本人被験者において、ブリッジング ADA イムノアッセイにおける陽性反応を示した症 例はみられなかった。
また、中和抗体は、被験者 1 例の 1 検体にのみ認められた。VIEW 1 試験及び VIEW 2 試験の免 疫原性に関するデータの詳細な概要については、2.7.2.3.4 に記載した。
2.5.3.5 結論
多時点で検体採取を行った薬物動態サブスタディにおける血漿中遊離型 VEGF Trap の Cmaxは低 く、2mg を硝子体内投与後 1~3 日目における Cmaxの平均濃度は約 0.02μg/mL(0~0.054μg/mL の範囲)であり、投与 2 週後にはほとんどの被験者で検出されなかった。2mg を硝子体内投与後 の体循環中遊離型 VEGF Trap の平均 Cmaxは、最大 VEGF 結合能の半分に達するのに必要な VEGF Trap 濃度の 100 分の 1 未満であると推測される。臨床試験にて検討した投与量範囲において、 VEGF Trap-Eye を硝子体内投与後の血漿中遊離型 VEGF Trap 濃度あるいはその推移は、体循環中 の内因性 VEGF に臨床的意義のある減少をもたらすまでには達していない。
治療に用いられるすべてのたん白質製剤と同様に、VEGF Trap に対しても免疫反応が生じる可 能性がある。第Ⅲ相試験では、投与前の VEGF Trap-Eye に対する免疫反応率は投与群全体で 1~ 3%であった。52 週間にわたる VEGF Trap-Eye 投与後、抗 VEGF Trap 抗体の検出率は、ラニビズ マブ群を含む全投与群で同じく 1~3%であった。
AMD 患者に対して VEGF Trap-Eye を硝子体内投与後、CR/LT の用量依存的な減少が認められた。 単眼に 0.5mg を投与した群で既に効果がみられ、2mg 投与群では最大効果に達した。なお、4mg 投与群では、それ以上の改善は認められなかった。これらのデータから、CR/LT の最大の減少は 単眼に 2mg を投与した際に得られ、より高用量の VEGF Trap-Eye を投与する必要はないことが示 唆された。また、第Ⅱ相試験のデータから、12 週ごとの投与では次の投与まで薬力学的改善が 持続せず、至適投与間隔は 12 週間未満であることが示唆された。
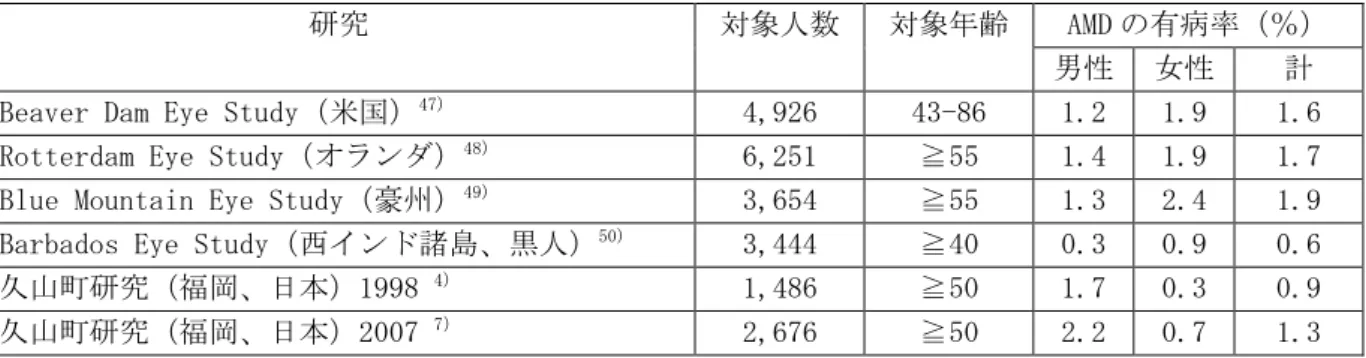
2.5.4 有効性の概括評価 2.5.4.1 有効性評価のための臨床開発計画 2.5.4.1.1 初期の臨床開発 第Ⅰ相試験 502 試験(5.3.3.2-2,-3,-4 502 試験)が滲出型 AMD の治療を目的とした VEGF Trap-Eye の開発の始まりとなった。0.05mg、0.1mg、0.5mg、1mg、2mg 又は 4.0mg を単回硝子体 内投与したこの試験からは、投与された VEGF Trap-Eye の生物学的作用に用量反応性を示す最初 のエビデンスが得られ、0.5mg 未満の VEGF Trap-Eye 単回投与では十分な効果が得られないこと が示された。一方、これより高用量の VEGF Trap-Eye が単回投与された場合には、視力及び脈絡 膜新生血管(CNV)病変の形態学的特性が改善した。このような視力及び形態学的な改善は単回 投与後少なくとも 4 週間は認められた。この試験から、滲出型 AMD 患者に対する VEGF Trap-Eye の硝子体内投与の効果は持続的なものであるという最初のエビデンスが得られた。 第Ⅱ相 508 試験(5.3.5.1-1 508 試験)では、VEGF Trap-Eye 0.5mg 及び 2mg を 4 週ごと又は 12 週ごと、更に 4mg を 12 週ごとに硝子体内投与した。投与後 1 週目に、すべての投与群に視力 の改善が認められ、この改善は 12 週目まで維持された(有効性の主要評価項目)。視力の改善 は 16 週目から 52 週目(PRN 期)まで持続し、この期間の平均追加投与回数はわずか 2 回であっ た。最初に 4 週ごとに、連続して 3 回投与する方が単回投与するよりも、視力改善に関して良好 な経過が示された。また、この試験の 2mg 投与の 2 つの群(4 週ごと又は 12 週ごとの投与)に おいて、8 週目における視力の改善はほぼ同程度であった。この結果から、第Ⅲ相試験での投与 間隔を 8 週ごとと設定しても、期待される VEGF Trap-Eye の効果は維持され、有効性が損なわれ ることはないと予想した。 12 週目の主要評価の時点で、2mg を 12 週ごとに投与する群(2Q12 群)と 0.5mg を 4 週ごとに 投与する群(0.5Q4 群)の全例(100%)が視力を維持していた(ベースライン値から 15 文字未 満の視力低下)。PRN 期に視力を維持した被験者の割合は 2mg 投与群の方が 0.5mg 投与群よりも 高く、0.5mg 投与は 2mg 投与に比べて投与間隔が延長された場合に効果が減弱することが示され た。また、高用量の 4mg 投与群において、2mg 投与群を上回る有効性は認められなかった。 第Ⅱ相 508 試験の結果を考慮して、第Ⅲ相試験における用法用量群(4 週ごと 0.5mg を投与、 4 週ごと 2mg を投与、8 週ごと 2mg を投与)が設定された。第Ⅲ相試験では、8 週ごと 2mg の投 与群では VEGF Trap-Eye 2mg を初めに 4 週ごとに 3 回投与を行った後に、8 週ごとに投与した。 2.5.4.1.2 有効性評価における民族的要因の考察 2.5.4.1.2.1 内因性要因 AMD の有病率及び発症率 AMD は欧米をはじめとした先進国で成人の中途失明の主な原因となっており、本邦においても 近年、中途失明の原因として、糖尿病網膜症、緑内障に次ぐものとなっている。各国で一般住民 を母集団とした population-based study として行われた AMD の疫学研究(Rotterdam Eye




![表 2.5- 6 VIEW 1 試験及び VIEW 2 試験における投与群の概要[国内被験者 ]](https://thumb-ap.123doks.com/thumbv2/123deta/6575781.677215/32.892.129.768.179.332/表256VIEW1試験及びVIEW2試験における投与群の概要国内被験者.webp)