第 84 回日本感染症学会総会座長推薦論文
ノロウイルスのゲノム解析と流行発生のしくみ
1)国立感染症研究所病原体ゲノム解析研究センター第二室,2)同 ウイルス第二部,
3)国立医薬品食品衛生研究所,4)堺市衛生研究所
本村 和嗣
1)横山 勝
1)岡 智一郎
2)片山 和彦
2)野田 衛
3)田中 智之
4)佐藤 裕徳
1)Norovirus Surveillance Group of Japan
(平成 24 年 5 月 1 日受付)
(平成 24 年 6 月 1 日受理)
Key words : norovirus, antigenic drift, genome recombination
要 旨
我々は,ヒトノロウイルス感染症の流行予測とワクチン開発の基盤情報を得る目的で,2006 年 5 月〜2010 年 3 月の間に全国 19 の道府県,20 カ所の拠点衛生研究所で収集した感染者糞便中の Group II の中の遺伝 子型 4 型(以下 GII.4)全ゲノム配列を調べた(約 7.5kb,n=277).糞便試料から核酸を抽出し,RT-PCR により重複する 2 種のゲノム断片(5.3,2.5kb)を増幅し,ダイレクトシークエンシング法で一感染者から 1 つのゲノム全長の配列情報を得た.ゲノム配列の進化系統,近縁関係は最尤法により解析した.流行株の アミノ酸の特徴を同定し,分子モデリング法を用いて,カプシド蛋白質で立体配置を視覚化した.解析期間 内,2006b 亜株が圧倒的に優勢な GII.4 単系統群として存続した.一方,他にも GII.4 単系統群が 8 種類発 生したが,劣勢群として局地的流行に留まった.2006b 亜株は,2006〜2010 秋冬期にかけて,全長にわた り,8 カ所アミノ酸置換が生じていた.カプシド蛋白質に生じた変異は,立体構造上,ループに位置してい た.この変異により,抗原性が変化すると推察された.高い変異率,感染力,増殖能が組み合わさり,ヒト 社会では,日々膨大な数の変異ウイルスが発生していると推察される.抗原性が大きく変化したウイルスが 出現すれば,ヒト社会の中で感染が広がり易いことが推定された.流行の変動に,ヒト集団のカプシド突端 部への免疫が関与している可能性がある.
〔感染症誌 86:563〜568,2012〕
序 文
ヒトノロウイルスは,わが国において秋冬季を中心 に流行する感染性胃腸炎の主要な原因ウイルスのひと つである1)2).経口摂取後,小腸(空腸,回腸)上皮細 胞に感染し,40〜48 時間潜伏時間を経て発症する3). ヒトには嘔吐,下痢などの急性胃腸炎症状を起こすが,
致死的ではなく,その多くは数日の経過で自然に回復 する.糞便中には,7〜10 日排出されることが報告さ れている3).また,ヒトノロウイルスは集団施設に持 ち込まれると集団発生を引き起こし,食品を汚染する と食中毒の原因となる.その結果,学校,レストラン 等の食品取扱施設,医療施設,高齢者施設,米国では
クルージング客船内での発生が報告されており3),広 汎な被害をもたらす.現代社会は,航空機,車,電車 などの輸送網や経済網が高度に発達している.ヒトノ ロウイルスに汚染された物資やヒトが移動すること で,国内はもとより大陸を超えた感染の拡大が示唆さ れる.よって,社会環境の変化が新興再興感染症を生 み出す 1 例と言える.
現在,ノロウイルスは,カプシド遺伝子配列の類似 性をもとに,Group I〜V の 5 つの遺伝子グループに 分類されている4).Group I,Group II,Group IV が ヒトに感染するが,Group I,Group II が主流となっ ている5)6).このなかで,Group II 4 型(GII.4)株は,
感染性胃腸炎の世界的な流行をおこすウイルスとして 注目されている3).1990 年代後半以降,少なくとも 4 回(1995!96,2002!03,2004!05,2006!07),世 界 的 原 著
別刷請求先:(〒208―0011)東京都武蔵村山市学園 4―7―1 国立感染症研究所病原体ゲノム解析研究セン
ター第二室 本村 和嗣
大流行をきたしたことが知られている7).これらの流 行では,例外無く GII.4 新亜株が出現していた.最近 で は,2006!07 秋 冬 期 に GII.4 新 亜 株(2006a 亜 株,
2006b 亜株)が流行し,日本を含めた世界の感染性胃 腸炎の散発事例と集団発生事例が激増した7).我が国 でも,感染症発生動向調査や病原微生物検出情報によ ると感染症例報告数は前年度の 4〜5 倍に達し,10 月〜2 月の間の集団発生事例報告数も過去最高となっ た2)8).2007!08 秋冬期〜2009!10 秋冬期の感染症例報 告数は,例年通りになっている2).
我々は,科学的知見に基づくヒトノロウイルスの感 染症対策に役立てるために,全国の衛生研究所と国立 感染症研究所(ウイルス第二部と病原体ゲノム解析研 究 セ ン タ ー)が 協 力 し て「Norovirus Surveillance Group of Japan」を立ち上げ,2006 年より国内で流 行したヒトノロウイルスの全ゲノム情報の収集と解析 を実施している9)10).解析情報には,流行株の性質を 規定する全ゲノムの配列情報に加えて,流行株の遺伝 的特徴,蛋白質の構造と機能の特徴,自然界での経時 変化の特徴などが含まれる.これらの情報の継続的取 得は,ヒトノロウイルスの流行発生のしくみや生存戦 略を解き明かすのに寄与するのみならず,流行株の検 出,サーベイランス,リスク評価,創薬やワクチン開 発等に幅広く役立つ重要な科学基盤を提供すると期待 される.
今回,我々は 2006!07 秋冬期〜2009!10 秋冬期に日 本で流行した株の遺伝学的分類と流行の動態,対象期 間中,優勢株であった 2006b 亜株全長配列の推移と カプシド蛋白質の構造解析に焦点を当てて報告する.
対象と方法 1.解析対象
2006 年 5 月 15 日から 2010 年 3 月 10 日の間に,19 の道府県で発生し,20 カ所の拠点衛生研究所にてヒ トノロウイルス感染症と確定した 330 症例を対象とし た.
2.ゲノム全長のシークエンシング
糞 便 に PBS(−)を 加 え 10% 懸 濁 液 を 作 成 し,
11,000×g,20 分間遠心の後,その上清を RNA 抽出 用検体とした.この遠心上清より,QIAamp Viral RNA Mini Kit(QIAGEN)を用いて,ヒトノロウイルス RNA を抽出した後,G2SKF プライマーと Oligo dT30SXN プライマーを用いて cDNA を合成した5).cDNA を template にして,4 種の GII.4 特異的プライマーを用 いて相互に重複するヒトノロウイルスゲノム cDNA 断 片 2 種(約 5.3kb,2.5kb)を PCR 増 幅 し た.ABI 3730 xl(Applied Biosystems)を用い,direct sequenc- ing 法により,塩基配列を決定した.277 の糞便試料 について,GII.4 ゲノム全長(約 7.5kbps)の塩基配列
を得た.
3.系統樹作成およびコンセンサス配列作成 配列情報をもとに,MEGA(Molecular Evolutionary Gene Analysis)ver. 4.0 プログラムを用いて,最尤法 による系統樹解析を行った11).新亜株の定義は,系統 樹解析結果より,ブートストラップ(信頼)値が 90 以上で単系統群を形成し,過去の流行株と比較して特 徴的なアミノ酸を有するものとした.
また,配列情報をもとに,GeneFisher2(http:!!bi biserv.techfak.uni-bielefeld.de!genefisher2!welcome.h tml)プログラムを用いて,各流行シーズンの 2006b 亜株コンセンサス配列を作成した12).
4.全長ゲノム構造の解析
カプシドタンパク質二量体のモデルは,1995〜1996 年に流行した GII.4 株の P domain の X 線結晶構造
(PDB:2OBS)を鋳型として,Molecular Operating Environment(MOE)の解析ツールを用いてホモロ ジーモデリング法により構築した13).カプシド蛋白質 に特徴的なアミノ酸変異の立体的位置を知るために構 造上表示した.
結 果 1.新型ヒトノロウイルス亜株と動態
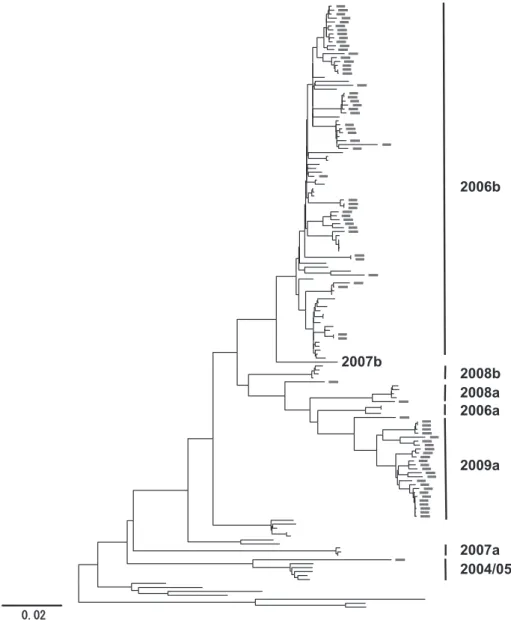
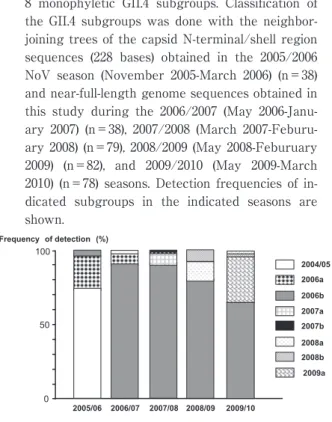
近年,日本で流行しているヒトノロウイルスの多く は,ゲノム断片を用いた予備的解析により,世界的に 流行している GII.4 と呼ばれる株であることが判明し ている.そこで我々は,2006 年から 2010 年春の間に 日本で流行したヒトノロウイルス GII.4 株のゲノム全 長の塩基配列(N=277)を取得し,最尤法による系 統樹解析を実施することで,GII.4 のさらなる分類と 流行の主流となる GII.4 亜株の同定を試みた.その結 果,調査期間の間に,(1)少なくとも 8 種類の GII.4 亜株が日本で流行したこと(Fig. 1),(2)その中で も特定の株(2006b 亜株)が,4 シーズンにわたって 日本国内で大流行したこと(222!277:80.7%)(Fig.
2),などがわかった.我が国でヒトノロウイルスが大 流行した 2006!07 秋冬期に,2006b 亜株がそれまでの 流行株(2004!05 亜株)と入れ替わって全国規模で大 流行し,その後も全国各地の流行の原因となったこと がわかった.また,2006b 亜株は,2005!06 秋冬期に も,限局的ではあるが日本国内に存在していたことも わ か っ た.2006b 亜 株 は,そ の 後 2007!08〜2009!10 秋冬期にも引き続き優勢な変異株として国内に流行し た.しかし,2006!07 秋冬期に比べると流行の規模は 徐々に縮小していた.2007!08 秋冬期に新たな GII.4 新 亜 株(2007a と 2007b),2008!09 秋 冬 期 に 新 た な GII.4 新 亜 株(2008a と 2008b),2009!2010 秋 冬 期 に は,新しく GII.4 2009a 新亜株が出現していた.2009a 新亜株は,2009 年米国で報告された New Orleans̲
Fig. 1 Phylogenetic classification of the GII.4 variant subgroups in Japan during 2006-2010. The maximum likelihood tree was constructed with the near-full-length genome sequences ( 〜 7.5 kb) obtained from 20 sites in Japan between May 2006 and March 2010 in this study (n=277), GII.4 reference sequences from past NoV epidemics in the world. These specimens collected in 2009/2010 were marked grey. The 8 monophyletic GII.4 variant subgroups were identified in Japan.
2009̲US と単系統群を形成しており,2009!10 年秋冬 期に 33.7%(26!77)を占めていた(Fig. 1,2).年々,
2006b 亜株の流行が減少する一方で,新型亜株の発生 数が増加し,日本各地で検出されるようになっていた ことがわかった.
2.過去 4 シーズンにわたる 2006b 亜株全長配列の 推移
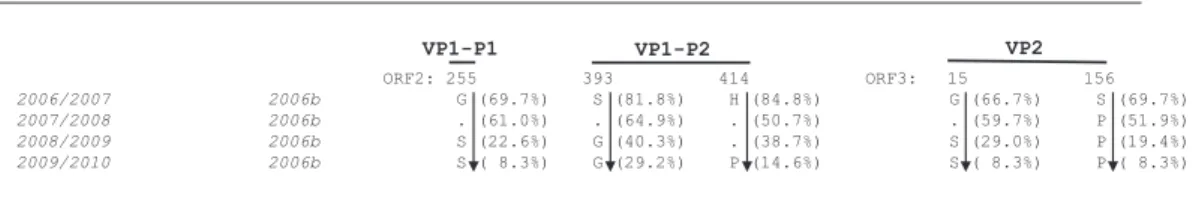
2006〜2009 年の間,優勢な流行株である 2006b 亜 株において全長配列の推移を調べるために,各シーズ ン毎に流行した 2006b 亜株のコンセンサス配列を作 成し比較した.更に,アミノ酸置換が生じている座位 をゲノム全長にわたって抽出したところ,8 カ所アミ
ノ酸置換が生じていた.3A 蛋白質に 1 カ所,Vpg 蛋 白質に 1 カ所,3D polymerase 蛋白質に 1 カ所,カプ シド蛋白質の P1 ドメインに 1 カ所,P2 ドメインに 2 カ 所,VP2 蛋 白 質 に 2 カ 所 存 在 し て い た(Fig. 3).
以上の結果から,2006b 亜株は,4 シーズンにわたっ て,全長にわたり変異を蓄積し,免疫感受性,複製―
増殖能のバランスを保ちながら,優勢な流行株として 存続している可能性が高いと考えられた.
3.2006b 亜株のカプシド蛋白質に生じる変異の立 体配置
ノロウイルスのカプシド蛋白質は,ウイルス粒子の 最外殻に存在し,ウイルスの感染力と抗原性を規定す
Fig. 2 Temporal dynamics of the 8 GII.4 variant subgroups in Japan. A. Detection frequency of the 8 monophyletic GII.4 subgroups. Classification of the GII.4 subgroups was done with the neighbor- joining trees of the capsid N-terminal/shell region sequences (228 bases) obtained in the 2005/2006 NoV season (November 2005-March 2006) (n=38) and near-full-length genome sequences obtained in this study during the 2006/2007 (May 2006-Janu- ary 2007) (n=38), 2007/2008 (March 2007-Feburu- ary 2008) (n=79), 2008/2009 (May 2008-Feburuary 2009) (n=82), and 2009/2010 (May 2009-March 2010) (n=78) seasons. Detection frequencies of in- dicated subgroups in the indicated seasons are shown.
る.ウイルスの性質変化を推察するために重要な蛋白 質の一つである.そこで,流行株のカプシド蛋白質の 分子モデルを構築し,上述したカプシド蛋白質に生じ た特徴的な変異を立体構造上に可視化した.興味深い ことに,特徴的な変異はカプシド蛋白質のループに位 置することがわかった(Fig. 4).これらの変異は,ウ イルス粒子を形成するタンパク質の外郭に位置するこ とから,免疫抗体の認識部位の可能性が示唆された.
2006b 亜株は,ヒトの免疫淘汰圧から逃避するために,
抗原性の変化させヒト集団内で存続している可能性が 示唆される.
考 察
我々の解析により,2006!07 年に日本を含め世界で 大流行した 2006b 亜株は,2007!8 年秋冬期以降も,主 要な国内流行株であった(Fig. 1).4 年間全体の 80.7%
(222!277)を占めていた(Fig. 2).一方で,2006b の 占有率は年々減少している(Fig. 2).減少している 理由には,2006b 亜株は,過去 4 シーズンにわたり,
ヒト集団内で大流行しているため,ヒト集団免疫の強 化に因る可能性が考えられる.今後,ヒト集団の免疫 淘汰圧から逃避できた,遺伝子型や新亜株の流行,そ れに伴い遺伝子亜型の置換がおこるかもしれない.
我々は,全て新亜株に生じた特徴的なアミノ酸置換 がカプシド蛋白質の最外殻に位置する P2 ドメインに
集中していることを報告した10).P2 ドメインは,粒 子の表面に露出しており,抗体の攻撃を容易に受ける と考えられる.実際,マウスノロウイルスでは,カプ シド蛋白質 P2 ドメインの 1 カ所の変異は,中和抗体 からの逃避能を高める変異であった14).今回,我々が 抽出した過去 4 シーズンにわたる 2006b 亜株カプシ ド蛋白質のアミノ酸変異蓄積座位の一つ,S393G は抗 原エピトープ領域に存在している(Fig. 3,4)15).ヒ トノロウイルスとマウスノロウイルスのカプシド蛋白 質の構造は類似している.したがって,この領域に生 じる変異の種類によっては抗原性が大きく変化し,ウ イルスの中和抗体感受性が低下する可能性がある.ヒ トノロウイルスの感染受容体は未だ同定されていない が,P2 ドメインは感染受容体候補因子への結合を司 る領域と推測されており,この領域に生じる変異の種 類によっては,感染受容体候補因子への親和性や特異 性,細胞指向性が変化する可能性もある13).
我々は,自然界ではキメラウイルスが頻繁に発生し ていることを報告した10).ゲノム組換え点は,全て,
複製蛋白質と構造蛋白質の境界領域に存在することが 明らかとなった10).これにより,複製蛋白質とカプシ ドの遺伝情報が効率的に交換され,新たな性質(免疫 逃避能と増殖能)を獲得した変異ウイルスが発生し,
その中で免疫逃避能力と高い感染・増殖能力をバラン スよく獲得したウイルスが,ヒト集団内で広がると推 察している.
ヒトノロウイルスが新たな流行発生に寄与する他の 要因として,集団免疫が考えられる.ヒトノロウイル スはリバースジェネティクス法が確立されていないた め,ヒト集団内の中和抗体保有率の解析は行われてい ない.また,感染動物モデルも開発されていないため,
感染防御に局所免疫がどのように関わるのか不明であ る.今後,我々は,カプシド蛋白質から成るウイルス 様粒子(VLP;Virus Like Particle)の構築に取り組 み,ヒト集団における抗カプシド抗体の分布や反応特 異性,それらの経時推移について解析する予定である.
これにより,集団免疫がヒトノロウイルスの流行発生 にどのように関与するのかを明らかに出来ると期待し ている.
謝辞:本研究内容は,第 84 回日本感染症学会総会
(平成 22 年 4 月 5,6 日,国立京都国際会館,京都)ワー クショップ 27 「ウイルス感染症」で発表いたしま した.糞便試料の収集に,御協力いただきました No- rovirus Surveillance Group of Japan の先生方に,厚 く御礼申し上げます.
Norovirus Surveillance Group of Japan:吉澄志磨
(北海道立衛生研究所),三上稔之(青森県環境保健セ ンター),斉藤博之(秋田県健康環境センター),高橋
Fig. 3 Covariation of the 2006/2007 〜 2009/2010 season of GII.4 2006b consensus sequences. The deduced amino acids of NoV GII.4 ORF1s, ORF2s, and ORF3s of the 2006b consensus sequences were aligned with those detected in 2006/2007. Amino acids are each indicated by a one letter code. The positions in the ORFs of the Lordsdale strain are used for the amino acid numbering.
Numbering in parentheses indicate the ratio of 2006b consensus amino acids signature detected in the 2006/07 season. Dots indicate amino acids identical to the 2006b consensus of the 2006/07 season.
Fig. 4 3D locations of unique amino acid substitutions in the capsid P do- main. Structural models of the capsid P domain dimmers were construct- ed by the homology modeling using the X-ray crystal structures of the P domain dimmer. The top and side view of the models were presented as figures. The model was constructed by homology modeling using the X- ray crystal structure of the P domain dimmer of the 1995/96 epidemic GII.4 strain (13). Black letters and arrows indicate locations and types of the unique amino acids in GII.4 2006b. Dot circles: the fucose ring binding sites formed by the P domain dimmer.
朱実,蛇口哲夫,高橋知子(岩手県環境保健研究セン ター),植木 洋(宮城県保健環境センター),田村 務(新潟県保健環境科学研究所),名古屋真弓,滝澤 剛則(富山県衛生研究所),篠崎邦子(千葉県衛生研 究所),吉田徹也(長野県環境保全研究所),小林慎一
(愛知県衛生研究所),東方美保(福井県衛生環境研究 センター),内野清子(堺市衛生研究所),入谷展弘(大 阪市立環境科学研究所),阿部勝彦,伊藤文明(広島
市衛生研究所),福田伸治(広島県立総合技術研究所 保健環境センター),飯塚節子(島根県保健環境科学 研究所),山下育孝,近藤玲子(愛媛県立衛生環境研 究所),増本久人,船津丸貞幸(佐賀県衛生薬業セン ター),松岡由美子(熊本市環境総合研究所),岩切 章(宮崎県衛生環境研究所)
本研究は,平成 19 年度〜平成 21 年度厚生労働科学 研究費補助金・食品の安心・安全確保推進研究事業お
よび平成 20 年度〜平成 22 年度厚生労働科学研究費補 助金・政策創薬総合研究事業の補助を受けた.
利益相反自己申告:申告すべきものなし 文 献
1)Ozawa K, Oka T, Takeda N, Hansman GS:No- rovirus infections in symptomatic and asympto- matic food handlers in Japan. J Clin Microbiol 2007;45(12):3996―4005.
2)国立感染症研究所感染症情報センター:病原微
生物検出情報(IASR).ノロウイルス感染症 h ttp:!!idsc.nih.go.jp!disease!norovirus!index.htm l.
3)Estes MK, Prasad BV, Atmar RL:Noroviruses everywhere : has something changed? Curr Opin Infect Dis 2006;19(5):467―74.
4)Zheng DP, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS:Norovirus classification and proposed strain nomenclature. Virology 2006 15;346(2):312―23.
5)Katayama K, Shirato-Horikoshi H, Kojima S, Kageyama T, Oka T, Hoshino F,et al.:Phyloge- netic analysis of the complete genome of 18 Norwalk-like viruses. Virology 2002 1;299
(2):225―39.
6)Hansman GS, Natori K, Shirato-Horikoshi H, Ogawa S, Oka T, Katayama K,et al.:Genetic and antigenic diversity among noroviruses. J Gen Virol 2006;87 (Pt 4):909―19.
7)Siebenga JJ, Vennema H, Renckens B, de Bruin E, van der Veer B, Siezen RJ,et al.:Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J Virol 2007;81(18):
9932―41.
8)厚生労働省:病因物質別食中毒発生状況.食中
毒統計資料(2)過去の食中毒発生状況 http:!!
www.mhlw.go.jp!topics!syokuchu!04.html..
9)Motomura K, Oka T, Yokoyama M, Nakamura H, Mori H, Ode H,et al.:Identification of mono- morphic and divergent haplotypes in the 2006- 2007 norovirus GII!4 epidemic population by genomewide tracing of evolutionary history. J Virol 2008;82(22):11247―62.
10)Motomura K, Yokoyama M, Ode H, Nakamura H, Mori H, Kanda T,et al.:Divergent Evolution of Norovirus GII!4 by Genome Recombination over 2006-2009 in Japan. j Virol 2010 ; revised.
11)Tamura K, Dudley J, Nei M, Kumar S:MEGA 4 : Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 2007;24(8):1596―9.
12)Giegerich R, Meyer F, Schleiermacher C:
GeneFisher--software support for the detection of postulated genes. Proc Int Conf Intell Syst Mol Biol 1996;4:68―77.
13)Prasad BV, Hardy ME, Dokland T, Bella J, Ross- mann MG, Estes MK:X-ray crystallographic structure of the Norwalk virus capsid. Science 1999 8;286(5438):287―90.
14)Lochridge VP, Hardy ME:A single-amino-acid substitution in the P2 domain of VP1 of murine norovirus is sufficient for escape from antibody neutralization. J Virol 2007;81(22):12316―
22.
15)Debbink K, Donaldson EF, Lindesmith LC, Baric RS:Genetic mapping of a highly variable no- rovirus GII.4 blockade epitope : potential role in escape from human herd immunity. J Virol 2012;86(2):1214―26.
A Mechanism of Norovirus Pandemic Based on Comprehensive Genome Analysis Kazushi MOTOMURA1), Masaru YOKOYAMA1), Tomoichiro OKA2), Kazuhiko KATAYAMA2), Mamoru NODA3), Tomoyuki TANAKA4), Hironori SATO1)& Norovirus Surveillance Group of Japan
1)Pathogen Genomics Center and2)Department of Virology II, National Institute of Infectious Diseases,
3)National Institute of Health Sciences,4)Sakai city Institute of Public Health
Norovirus GII.4 is a major etiological agent of acute viral gastroenteritis worldwide. We examined GII.4 evolution using 277 near-full-length GII.4 genome sequences from human stool specimens collected at 20 sites in Japan between May 2006 and March 2010. We found outbreaks of 8 monophyletic GII.4 subtypes, among which a single subtype, termed 2006b, had continually predominated (222!277 : 80.7%). Four of the 8 GII.4 subtypes were chimera viruses of recently prevalent GII.4 subtypes. Notably, single putative recombi- nation breakpoints with the highest statistical significance were constantly located around the border of open reading frame 1 (ORF) 1 and ORF 2 (P<0.0001), suggesting outgrowth of specific recombinant viruses in the outbreaks. The GII.4 subtypes had many unique amino acids at the time of their outbreaks, especially in the N-term, 3A-like, and capsid proteins. Unique amino acids in the capsid were preferentially positioned on the outer surface loops of the protruding P2 domain. These data and computer-assisted structural study of NoV capsid protein are compatible with a model of antigenic drift with tuning of the structure-functions of multiple proteins for the survival strategy of GII.4 2006b variant.