1
魚類細胞性免疫機構における IFNγrel の機能解明
日本大学大学院獣医学研究科獣医学専攻 博士課程
柴﨑 康宏
2013
2
目次
第1章 緒論 4
第 2 章 細胞性免疫応答における T細胞サブセットの動態 (移植片対宿主反
応をモデルとして) 7
2.1. 序論 8
2.2. 材料および方法 10
2.3. 結果 14
2.3.1. GVHRの誘導と組織傷害 14
2.3.2. GVHR誘導時におけるT細胞サブセットの動態 15
2.3.3. GVHD誘導におけるドナー細胞からの CD8陽性T 細胞除去の影響
16
2.4. 考察 17
第3章 魚類特有のインターフェロン、IFNγrelの同定 26
3.1. 序論 27
3.2. 材料および方法 30
3.3. 結果 37
3.3.1. 遺伝子の単離と構造解析 37
3.3.2. IFNγrelの抗ウイルス活性の測定 38
3.3.3. IFNγrelの分類(シグナル伝達経路) 39
3
3.4. 考察 41
第4章 細胞性免疫におけるIFNγrelの機能解明および産生細胞の同定 58
4.1. 序論 59
4.2. 材料および方法 61
4.3. 結果 68
4.3.1. 細胞性免疫反応におけるIFNγrelの発現解析 68
4.3.2. IFNγrelの投与効果 69
4.3.3. IFNγrel産生細胞の同定 69
4.4. 考察 71
第5章 総括 86
謝辞 93
参考文献 94
4
第 1 章
緒論
5
ワクチンの普及により、ブリのα溶血性レンサ球菌症などの被害が減少しつ つある(Takahashi et al., 2011)。しかし、イリドウイルス病などのウイルス性疾病 や、エドワジエラ症やノカルジア症などの細胞内寄生細菌による疾病が増加し ている。(農林水産省平成20年度家畜衛生をめぐる年報)。これらの病原体に対 しては、抗生物質や抗菌薬は効果を示さず予防が重要であるが、有効なワクチ ン開発は遅れている。その原因の一つとして、これらの病原体の排除に重要と 考えられる細胞性免疫に関する知見が魚類では著しく不足し、細胞性免疫機構 を活性化するワクチンの開発が遅れていることが挙げられる。
獲得免疫は細胞性免疫と液性免疫に大別される。細胞性免疫においては、CD4 陽性T細胞(Th1細胞)が産生するサイトカインの働きによりCD8陽性細胞傷 害性T細胞(CTL)が活性化し、抗原特異的に感染細胞や非自己抗原を発現す る標的細胞を傷害する。細胞性免疫機構が重要な役割を担う免疫反応として、
ウイルスや細胞内寄生菌等の細胞内異物に対する感染防御反応、移植片対宿主 反応や移植片拒絶反応などのアロ抗原に対する反応および腫瘍に対する拒絶反 応があげられる。
インターフェロンγ (IFNγ)は、Th1細胞や活性化したCTL、NK細胞より産生 され、抗ウイルス活性を誘導するとともに、Th1細胞への分化促進、Th2細胞の 増殖抑制、免疫プロテアソーム関連タンパク質の合成促進によるMHC分子の発 現上昇、感染局所へのT細胞遊走の促進に関わっている。また、マクロファー ジの活性化による貪食能の増強や貪食した異物の消化を促進するなど多様な役 割を担い、細胞性免疫の活性化に中心的な役割を担うサイトカインである(Abbas et al., 1996; Schoenborn and Wilson, 2007; Stark et al., 1998; Van den Eynde and Morel,
6
2001)。
魚類の細胞性免疫機構に関する研究は、アロ抗原に対する拒絶反応やGVHR といったin vivoの研究(Nakanishi and Ototake, 1999; Shibasaki et al., 2010)、あるい はアロ抗原やウイルス感染細胞に特異的なex vivoあるいはin vitroにおける細胞 性免疫反応(Somamoto et al., 2000, 2006; Stuge et al., 2000; Toda et al., 2009, 2011b) がギンブナ(Carassius auratus langsdorfii)、アメリカナマズ(Ictalurus punctatus) およびニジマス(Oncorhynchus mykiss)を中心に研究されてきた (Fischer et al., 2006; Nakanishi et al., 2011)。
魚類において、高等脊椎動物に相同なIFNγに加えて魚類特有のIFNγrel遺伝 子の存在が明らかとなっており、これまでにIFNγについては抗ウイルス活性や 免疫調節作用がギンブナ、キンギョ、ゼブラフィッシュ等数魚種で明らかとな っている(Aggad et al., 2010; Grayfer et al., 2010; Yabu et al., 2011)。しかし、IFNγrel については、インターフェロンの定義として重要な抗ウイルス活性やその作用 機序は不明である。
そこで、本研究では魚類細胞性免疫研究の優れたモデルとなっているコイ科 魚類のクローンギンブナを用いて、生体内における細胞性免疫の代表的な反応 である移植片対宿主反応(Graft Versus Host Reaction; GVHR)をモデルとして GVHR誘導に伴うT細胞サブセットの動態を解析した。次に、魚類細胞性免疫
におけるIFNγrelの機能を解明するため、IFNγrelの生物活性やその受容体およ
び細胞内シグナル伝達機構の解明を試みるとともに、魚類細胞性免疫反応にお けるT細胞の関与について検討した。
7
第 2 章
細胞性免疫応答における T 細胞サブセットの動態
(移植片対宿主反応をモデルとして)
8
2.1 序論
移植片対宿主反応(Graft Versus Host Reaction: GVHR)は、T細胞が主要な 役割を担う細胞性免疫の代表的な反応であり、レシピエントがドナーを拒絶出 来ない状態で、ドナーの移植組織中に含まれる免疫担当細胞がレシピエントの 主要組織適合遺伝子複合体(MHC)や副組織適合抗原などの非自己抗原を認識し て、レシピエントの組織を攻撃する反応である。この反応は脊椎動物に広く認 められ、両生類や爬虫類においてもGVHRが引き起こされることが知られてい る( Saad and El Ridi, 1984; Nakamura, 1985)。特に、哺乳類や鳥類において、
この反応には細胞傷害性 T リンパ球(CTL)が関与していることが明らかとなっ ている( Glick, 1986;Shlomchik, 2007)。
ヒトにおいては、骨髄細胞移植時にGVHRにより起こる移植片対宿主病
(Graft Versus Host Disease; GVHD)が問題となっており、その発症機構が盛 んに研究されている。急性GVHRは、1)ドナーT細胞のレシピエントのアロ 抗原に対する認識および活性化、2)活性化したTh1細胞が産生するIFNγを はじめとするサイトカインの影響による、エフェクター細胞としてのCTLの増 殖、3)多量のCTLの標的組織への浸潤と、それに伴うPerforin/Granzyme経路、
Fas/Fasリガンド(FasL)経路による細胞傷害や炎症性サイトカインを介した炎
症反応、の3つのステップを経ることが知られている(Krenger and Ferrara, 1996; New et al., 2002; Jaksch and Mattsson, 2005)。
マウスにおけるGVHR誘導モデルとして、ドナーに純系マウスを用い、レシ ピエントにはドナーと同系統のマウスに他の系統の純系マウスを掛け合わせて
9
得たヘテロマウスを用いて移植を行うものが知られている(Lin et al., 1998; Murai et al., 2003)。この移植モデルでは、レシピエントのヘテロマウスは、共通の組織 抗原を有するドナー由来細胞を自己とみなし許容するのに対し、ドナー細胞は 他系統の組織抗原を有するレシピエントの細胞を非自己と認識し攻撃する。
魚類においても、クローンギンブナ(Carassius auratus langsdorfii)やクロ ーンアマゴ(Oncorhyncus maso)において、これまでにアロ抗原感作を行った 白 血 球 を 移 植 す る こ と に よ り GVHD が 起 こ る こ と が 報 告 さ れ て い る (Nakanishi and Ototake, 1999; Qin et al., 2002)。ギンブナにおけるGVHR誘 導方法は前述のマウスの実験モデルと同様に、三倍体クローン系統である S3N 系統のギンブナをドナー、S3N 系統に近縁種であるキンギョを掛け合わせて作 出した四倍体である S4Nをレシピエントとして移植を行うものである。このモ デルではドナーS3N 細胞にとって、キンギョ由来の組織抗原を持つレシピエン トは非自己と認識され、攻撃される。
しかし、魚類ではこれまで T 細胞サブセットに対するモノクローナル抗体
(mAb)が存在しなかったため、実際にどの細胞がGVHRの誘導や組織傷害に 関わっているかを明らかにすることはできなかった。当研究室では、ギンブナ において、魚類ではじめてCD8α鎖およびCD4に対するmAbの作成に成功し、
T細胞サブセットについての解析が可能となった(Toda et al., 2009; Toda et al.,
2011b)。そこで、本章では魚類GVHRにおけるT細胞の関与を明らかにするた
め、GVHR 誘導に伴う T細胞サブセットの動態を解析した。また、魚類におけ るGVHDによる組織傷害の担い手を明らかにするため、MACS法を用いてCD8 陽性Tリンパ球を除いた細胞の移植によるGVHDの誘導について検討した。
10
2.2 材料及び方法
2.2.1 供試魚
諏訪湖産3倍体クローンギンブナ(Carassius auratus langusdorfii)S3N系統(体 重約20 g)とS3Nにキンギョ(Carassius auratus)をかけ合わせた4倍体雑種(S4N、 体重約20 g)を用いた。いずれも水温25°Cの循環濾過式アクアトロンで飼育し、
1日二回給餌を行った。
2.2.2 アロ抗原感作
血球の移植に先立ち、鱗移植によるアロ抗原感作を行った。ベンゾカイン
(Sigma)による麻酔後、アロ抗原感作群には1回につきS4Nの12枚の鱗をS3N
の体側に移植し、初回鱗移植から14日後に、再度同様な手順で移植をおこなっ た。なお、対照群として鱗移植によるアロ抗原感作を行わない群を用意した。
2.2.3 移植細胞の採取および調整、移植法
最終感作から 7 日後にドナーS3N より頭腎及び体腎を摘出し、スチールメッ シュ(#80)上でピンセットを用いてこれらの組織を磨砕し、0.5% FBS加Hank's Balanced Salt Solution(HBSS)液に遊離細胞を浮遊させた。細胞浮遊液をパーコ ール液(1.080 g/ml)に重層し、遠心(650×g、30分、4°C)後、パーコール液 と細胞浮遊液層の間に形成された単核白血球層を回収した。
上記の方法により回収した細胞を1×108個/mlの濃度でPBSに浮遊させた。調 整した細胞浮遊液0.1mlを、レシピエントの尾部血管より注入し血球移植を行っ
11
た。以上の作業はすべて氷上で行った。
2.2.4 病理組織標本の作製
ドナー細胞移植後7日目(体腎)および14日目(体腎、腸管)のレシピエン トより摘出した体腎および腸管を 4% パラホルムアルデヒド(PFA)加 PBS に より4°Cで24時間固定した。その後、組織をTween-20加PBS(PBST)で洗浄 後自動包埋機にて包埋した。包埋終了後、パラフィンブロックを作成しマイク ロミクロトームを用いて、厚さ4 μmの組織切片を作成した。作成した組織切片 はヘマトキシリン・エオジン(HE)染色法にて染色を行った。
2.2.5 フローサイトメトリー(FACS)解析
ドナーのS3NとレシピエントのS4Nの細胞は、そのDNA含量の違いからDNA 染色色素である Hoechst33342 を用いて染め分けることができる。このことを利 用して、移入したドナー細胞の動態及びレシピエントの細胞の反応をフローサ イトメトリーにより解析した。
・細胞の調整
移植3、7、14日後のレシピエントより、頭腎、体腎、末梢血、脾臓、肝臓及
び腸管から細胞を回収した。摘出した肝臓及び腸管を 1 mM DTT 及び 1 mM EDTA加PBSに入ったシャーレに入れ、4°C、15分間インキュベートした。洗浄、
細切後、5% FBS、0.1 g/ml Collagenase、0.1 g/ml DNase加カルシウムマグネシウ
ム不含HBSS(CMF-HBSS)に入れ、25°C、90分間振とうした。その後、スチ
ールメッシュ上でピンセットを用いて組織を磨砕し、血球細胞を得た。混入し
12
た赤血球を溶血処理により除き白血球を得た。
・Hoechstを用いた蛍光染色および免疫染色
回収した白血球を、7.5 μM Hoechst33342、5% FBS加HBSS液中に浮遊させ25°C で90分染色した。Hoechst染色後、mAb 2C3(抗ギンブナCD8 ラットmAb)ま たは6D1(抗ギンブナ CD4 ラットmAb)、抗ラットビオチン標識抗体、ストレ プトアビジンPE標識体を用いて免疫染色を行った。この後に2.5 μg/ml Propidium Iodide(PI) 加HBSS上に細胞を浮遊し、FACS解析を行った。
・FACS解析
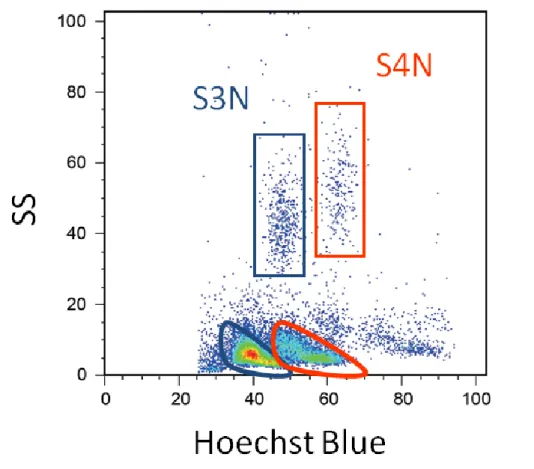
FACS解析には、EPICS Altra(Beckman Coulter)を用いた。Hoechst励起のた めに UV レーザー、PI、PE 励起および前方散乱光(FS)、側方散乱光(SS)の 解析のために488 nmアルゴンレーザーを使用した。Hoechst Blueの検出には410 band pass(BP)filter、PIの検出には675 BP filter、PEの検出には 575BP filterを 用いた。FACS解析では、リンパ球分画のうち、PI-、PE+の細胞集団をHoechst Blue vs. SSのpseudo-color plot上に展開し、ドナー(S3N)およびレシピエント(S4N) 細胞にそれぞれゲートを設定し(Fig. 2-1)、ドナーTリンパ球の割合を解析した。
細胞数については、細胞浮遊液中に蛍光ビーズを添加し、検出されるビーズ の割合から絶対数を求めた。
2.2.6 MACS法を用いたCD8陰性細胞群の分取及び移植
前述の感作と同様に14 日間隔で二回のアロ抗原感作を行い、最終感作から 7 日後のドナーS3Nの頭腎及び体腎を摘出し、2.2.3で述べた方法で細胞を分離し、
0.5% FBSを加えたOPTI-MEM(OPTI-0.5)に浮遊した。1.08 g/mlのパーコール
13
を重層し密度勾配遠心(650×g、4°C、30分)により白血球を分離した。2回遠 心洗浄(450 ×g、4°C、5分)後、1 × 107個/mlに単核白血球を浮遊させ、mAb 2C3
(腹水)を添加し4°Cで45分間反応させた。3回遠心洗浄後、1×108個/mlにな るように調整した細胞浮遊液に対し、5倍希釈になるようにマイクロビーズ標識 ヤギ抗ラットIgG抗体(Miltenyi Biotec GmbH, Germany)を添加し4°Cで20分 間反応させた。2回遠心洗浄後、magnetic activated cell sorting (MACS; Mini Macs, Miltenyi Biotec)によりCD8α-細胞を分離した。
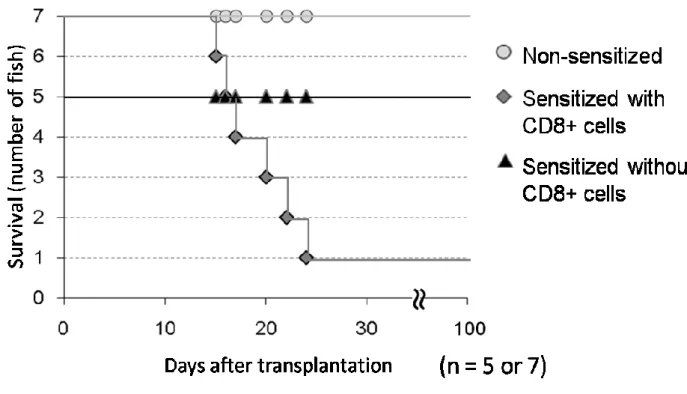
この CD8α-細胞を、感作に用いたレシピエントへ前述の方法を用いて 1×107 個移植し、その後生存率の検討を行った。その後、感作群および未感作と比較 した。
2.2.7 統計解析
生存率の解析では、Fisher's exact probability test (two-tail test)を用いて両群間の 有意差を検定した。フローサイトメトリーにおける有意差検定は、two-way
ANOVAによる分散分析の後、Bonferroniの多重比較検定によって行った。
14
2.3 結果
2.3.1 GVHRの誘導と組織傷害
GVHD の発症を確認するため、感作ドナー細胞投与後のレシピエントの生存 率について検討した。鱗移植によるアロ抗原感作を行った個体より採取したリ ンパ球を移植した群(感作群)にのみ立鱗、腹水の貯留、貧血等の典型的なGVHD の症状が認められ、移植後 15 日目から 21 日目に死亡する個体が認められた。
未感作個体より採取したリンパ球を移植した群(未感作群)においては移植後 100日を経過しても死亡する個体は認められなかった(Fig. 2-2)。Fisherの正確 確立検定により、感作群と未感作群の死亡率には有意差が認められた。また、
GVHD を発症し死亡した個体には、脾臓の腫張、腸の発赤および脆弱化、肝臓 の緑色変化等が認められた。
続いて、GVHD における組織傷害を検討するため、病理組織学的検討を行っ た。感作群、未感作群及び移植を行わなかった正常組織を比較したところ、感 作群の体腎において、移植後 7 日目から単核球の浸潤および増加が認められ、
14日目では尿細管の崩壊像や細胞の増殖が認められた(Fig. 2-3)。肝臓、腸管、
皮膚における移植後14日目の組織像を観察した。その結果、肝臓においては中 心静脈における囲管性の細胞浸潤や肝臓組織の崩壊(Fig. 2-4)、腸管では単核球 の浸潤、粘膜下組織の肥厚や単核球の浸潤(Fig. 2-4)、皮膚においては皮下の筋 肉層の崩壊と単核球の浸潤が認められた。未感作群においては、いずれの組織 についても病理組織学的変化は認められなかった。
15
2.3.2 GVHR誘導時におけるT細胞サブセットの動態
レシピエントの体腎から回収したCD4およびCD8陽性T細胞のうち、ドナー 由来の細胞の占める割合の経時的な変化について解析した。感作群は未感作群 に比べ、時間の経過に伴いCD4、CD8陽性T細胞ともに、ドナー由来細胞がよ り多くの割合を占めた(Fig. 2-5)。
次に、GVHRの誘導に伴うT細胞サブセットの動態を明らかにするために、
移植3、7、14日後に、哺乳類においてGVHDの標的器官として知られている脾 臓および肝臓、並びに硬骨魚類の代表的なリンパ・造血器官である腎臓および 末梢血からそれぞれ白血球を採取した。ドナー細胞由来細胞とレシピエント細 胞を識別するため、生体染色色素であるHoechst33342によるDNA染色を行う とともに、ギンブナCD8α鎖およびCD4に対するモノクローナル抗体を用いた 免疫染色を行い、フローサイトメーターを用いて、レシピエントにおけるドナ ーT細胞サブセットの動態について解析を行った。その結果、感作群において、
ドナー由来CD4陽性T細胞は移植7日後には著しい増加が認められた。一方、
CD8陽性Tリンパ球数はCD4陽性T細胞に遅れて増加し、組織傷害が顕著とな る移植後14日目に最も高い値を示した。一方、未感作群においては、ドナー由 来CD4陽性T細胞、CD8陽性T細胞いずれも有意な増加は認められなかった(Fig.
2-6)。
感作群においては、特に体腎で多くのドナー由来リンパ球が認められ、移植 後14日目の体腎から得られたCD4陽性Tリンパ球数(132.2±60.1×104個)は、
移植した単核白血球集団に含まれていたCD4陽性T細胞数(75.9±16.1×104個)
より多かった。
16
2.3.3 GVHD誘導におけるドナー細胞からのCD8陽性T細胞除去の影響
哺乳類においては、GVHDの組織傷害にCD8陽性T細胞が関わっていること が明らかとなっているが、魚類においては不明である。そこで、ギンブナにお けるGVHD と CD8 陽性T 細胞の関係を調べるため、MACS 法により、感作し たドナーリンパ球より CD8 陽性 T 細胞を除いた白血球(CD8-細胞群)を移植 し、GVHDの発症の有無について検討した。その結果、CD8-細胞群を移植した 群では、移植後 100 日を経過しても、GVHD の特徴的な症状を呈する個体や、
死亡する個体は認められなかった(Fig. 2-7)。
17
2.4 考察
哺乳類では急性GVHDの発症時に、まずTh1細胞の増加が認められ、次いで Th1細胞により産生されるIL-2やIFN-γなどのサイトカインの働きによって CTLの活性化が起こり、組織傷害が起こることが知られている(Yi et al., 2009)。
今回のギンブナにおける研究においてもGVH反応の進行に伴い、CD4陽性T 細胞がまず増加し、その後CD8陽性T細胞の増加が認められた。CD4陽性T細 胞は、ドナー細胞移植後いずれの時点においてもCD8陽性T細胞より多くの細 胞が認められた。特に、移植後14日目の体腎においては、感作群では移植した 細胞に含まれるCD4陽性T細胞数よりも、レシピエントの体腎より採取したド ナー由来CD4陽性T細胞数のほうが多くなっており、CD4陽性T細胞が特に活 発に増殖していることが明らかとなった。未感作群では感作群に認められたよ うな細胞の増加は認められず、移植前の感作により、CD4陽性T細胞の増殖が 促進されたと考えられる。
これまでの哺乳類における知見および今回の研究の結果から、今回増殖の認 められたCD4陽性細胞はTh1細胞と考えられる。ギンブナにおいてTh1系のサ イトカイン遺伝子がいくつか単離されていることから、これらTh1系サイトカ イン遺伝子のメッセンジャーRNAの発現解析により増殖した細胞がTh1細胞か どうかを確認することが可能であり、GVH反応の際のT細胞の相互作用の解明 につながると思われる。
組織学的観察により感作群の腸管、肝臓、皮膚などの組織に組織傷害が認め られた。これらの組織においては、未感作群と比較し、顕著なリンパ球の浸潤
18
が認められた。移植後7日目の体腎では顕著な組織傷害は認められなかったが、
移植後14日目では顕著な組織傷害が認められた。FACS解析より、CD8陽性T 細胞は体腎において移植後7日目から細胞数の増加が認められ、移植後14日目 には顕著に増加が認められた。CD8陽性T細胞の細胞数の変化と組織傷害の進 行には相関が認められたこと、ならびにCD8陽性T細胞を除いた細胞を移植し た際にはGVHDの発症及び死亡が認められなかったことから、GVHDによる組 織傷害においてCD8陽性T細胞が関与していることが示唆された。
これまでのギンブナにおける研究から、CD8陽性T細胞がアロ抗原特異的細 胞傷害性を示すこと(Toda et al., 2009)、哺乳類の細胞傷害経路と相同な
Perforin/Granzyme経路(Kurobe et al., 2007; Toda et al. 2011a, 2011c)により傷害す ることが明らかとなっている。
以上より、ギンブナにおけるGVHRの誘導にはCD4陽性T細胞が関与すると ともに、GVHRに伴う組織傷害には、CD8陽性T細胞が主要な役割を担ってい ると考えられる。
今回、移植後のレシピエントにおけるドナー細胞の動態に着目したが、ドナ ー由来CD4及びCD8陽性T細胞の活性がレシピエント内で高まっていることが 考えられる。今後、細胞数だけでなくCD4及びCD8陽性T細胞の活性化に注目 して検討する必要があると思われる。
19
Fig. 2-1. Ploidy analysis of leukocytes by flow cytometry. Leukocytes were obtained from a S4N recipient fish and stained with Hoechst 33342. Four distinct cell populations were observed, and the two upper gated regions show neutrophils and the two lower gated regions show lymphocytes. Lymphocytes could be divided into two populations, donor-derived (triploid) and recipient (tetraploid) cells by Hoechst blue intensity. The two populations on the left with lower Hoechst blue staining and the two populations on the right with greater Hoechst blue intensity represent S3N donor- and S4N recipient-derived leukocytes, respectively.
20
Fig.2-2 Comparison of the survival rate of S4N recipients injected with leukocytes.
Sensitized ( ) or non-sensitized ( ) S3N donor cells (0.1 ml of 1×108 cells/ml) were injected into S4N recipients via the caudal blood vessels 7 days after the last sensitization.
Asterisks indicate statistical significance (p < 0.05) compared to non-sensitized control experiment using fisher’s exact probability test.
21
Fig.2-3 Histopathological changes in recipient trunk kidney during GVHR. HE staining of trunk kidney sections from one or two weeks after the transfer of sensitized donor cells, two weeks after the transfer of non-sensitized donor cells or non-treated healthy fish.
Asterisks indicate renal tubules. Arrow indicates collapsed renal tubules. Bars: 50μm.
22
Fig. 2-4 Histopathological changes in recipient target organs. Liver of recipient receiving sensitized (A) and non-sensitized (B) donor cells. Second segment of intestine of recipient receiving sensitized (C) and non-sensitized (D) donor cells. Skin of recipient receiving sensitized (E) and non-sensitized (F) donor cells. All tissues were examined 2 weeks after donor cell injection. Arrows indicate infiltrated mononuclear cells. Abbreviations used are: bd, bile duct; cv, central vein; ed, epidermis; ep, epithelium; lp, lamina propria; mm, muscularis mucosae; mu, muscle; sc, scale. Bars: 100μm.
23
CD4 CD8
Fig. 2-5 FACS analysis of CD4- and CD8α-positive T cells in the recipient trunk kidney.
Leukocytes were obtained from the trunk kidney of S4N recipients at day 3 (d3), 7 (d7) and 14 (d14) after donor cell transplantation. Lymphocytes were gated as SS low fraction on a pseudo-color plot of Hoechst blue vs. SS plot shown in Fig. 2-1 and ploidy analysis was conducted. Numbers show the percentages of donor (S3N)-derived and recipient (S4N) cells.
24
Fig. 2-6 Kinetics of donor-derived CD4- and CD8α-positive T cells in lymphoid tissues of recipients. Lymphocytes were obtained from recipient fish at day 3, 7 and 14 after the donor cell transplantation. (A) trunk kidney; (B) liver; (C) spleen; (D) PBL. Dark and grey histograms represent recipients injected with sensitized and non-sensitized donor cells, respectively. Error bars indicate the standard deviation of three independent experiments. Asterisks indikate statistically significant using the two-way ANOVA and succeeding Bonferroni’s multiple tests (p < 0.05).
Days after transplantation Days after transplantation
25
Fig. 2-7 Comparison of the survival rate of S4N recipients injected with leukocytes containing or lacking CD8α-positive T cells. S3N donor cells (0.1 ml of 1×108 cells/ml) with CD8α ( ; n
= 7) or without CD8α ( ; n = 5) were injected into S4N recipients via the caudal blood vessels 7 days after the last sensitization. Control fish ( ; n = 7) received the same number of non-sensitized S3N donor cells.
26
第 3 章
魚類特有のインターフェロン、 IFNγrel の同定
27
3.1 序論
インターフェロンは最も古くから知られるサイトカインであり(Isaacs and Lindenmann, 1957)、抗ウイルス活性、抗増殖活性、免疫調節能を示すサイトカイ ンとして知られている(Stark et al., 1998; Platanias, 2005)。脊椎動物におけるイン ターフェロンは、リガンドの構造、相互作用する受容体、および関与する転写 因子(JAK-STAT経路)によって、I型、II型およびIII型の3種類に分類されて いる(Fig. 3-1)(Samuel, 2007)。I型とIII型は単量体であり、II型はホモ二量体 を形成する。I型インターフェロンは、いずれもヘテロ受容体IFNAR1/IFNAR2 と相互作用し、非受容体型チロシンキナーゼであるJak1とTyk2により活性化(リ ン酸化)された転写因子STAT1、STAT2とIRF-9が会合し、ISGF3と呼ばれる転 写複合体を形成、核内移行しISREエレメントに結合することで遺伝子発現調節 を行う。また、III型インターフェロンであるIFNλは、ヘテロ受容体
IFNLR1/IL10R2と相互作用し、I型インターフェロンと同様の細胞内シグナル伝
達経路を用いて作用する。一方、II型インターフェロンであるIFNγは、ヘテロ
受容体IFNGR1/IFNGR2をと相互作用し、Jak1およびJak2を介して転写因子
STAT1のホモ二量体が細胞質より核内に移行してプロモーター上のGASエレメ
ントに結合し、転写調節を行うことが知られている。
哺乳類のI型インターフェロンは、これまでに9種類(IFNα、IFNβ、IFNε、 IFNκ、IFNω、IFNδ、IFNτ、IFNν、IFNζ)が知られており(Pestka, 2007; Pestka et al.,
2004)、そのうち、IFNδはブタ、IFNτはウシ、IFNζはマウスといった様に、種
特有のインターフェロンも見つかっている(Lefevre et al., 1998; Oritani et al.,
28
2000; Roberts et al., 1991)。
魚類においてもインターフェロン様の抗ウイルス活性をもつ物質の存在はヒ メハヤ(Pimephales promelas)において1965年にはじめて報告され(Gravell and Malsberger, 1965)、1975年にはニジマス(Oncorhynchus mykiss)由来のRTG-2細 胞株を用いて、リガンドの部分精製が試みられた(de Sena and Rio, 1975)。その後、
I型インターフェロンのホモログがゼブラフィッシュ(Danio rerio)において2003 年に単離された(Altmann et al., 2003)。II型インターフェロンは2004年にフグ
(Takifugu rubripes)で遺伝子が単離され(Zou et al., 2004)、これまでにギンブナ を含め、8種の魚種から単離されている(Zou and Secombes, 2011)。魚類以外の脊 椎動物ではII型インターフェロンとしてIFNγのみが知られており、それぞれの 種が1つのIFNγを持つことが知られている。しかし、前述のように一部の魚種 ではIFNγに加えて魚類特有のIFNγであるIFNγrelの存在が報告されている (Grayfer et al., 2010; Igawa et al., 2006)。さらに、クローンギンブナを含む複数の 魚種においては、IFNγについても2種類存在することが明らかとなっている (Yabu et al., 2011;Zou and Secombes, 2011)。
魚類IFNγは生物活性と作用機序に関する研究が進んでおり、哺乳類と同様に、
ホモ二量体を形成してIFNγ受容体IFNGRに結合し、転写因子STAT1を介した 抗ウイルス活性やマクロファージの貪食活性を促進することがギンブナやキン ギョで明らかとなっている(Grayfer et al., 2010)。一方、IFNγrelの機能については、
キンギョにおいてマクロファージの貪食能を増強することが報告されているが (Grayfer et al., 2010, 2011)、その作用機序やインターフェロンの定義として重要な 抗ウイルス活性については不明である。
29
さらに、これまでに報告されているコイ科魚類のゼブラフィッシュ、キンギ ョ、コイのIFNγrelのアミノ酸配列を比較すると、C末端側において塩基性アミ ノ酸に富んだ核移行シグナル(NLS)様配列を有するものと欠くものがあり、
IFNγrel遺伝子が複数存在することが推察された。
そこで本研究では、まずギンブナよりIFNγrel遺伝子の単離を試みた。その結 果、NLS配列の有無により2種類のIFNγrelが得られ、それぞれIFNγrel 1、IFNγrel 2と名付けた。さらに、それぞれのIFNγrelについて、機能や特性を明らかにす るために、抗ウイルス活性やリガンドの構造および作用機序をインターフェロ ンの分類に沿って解析した。
30
3.2 材料及び方法
3.2.1 細胞培養
ギンブナ胸腺由来細胞株GTS9はLeibovitz's L-15 (Life Technologies)に10% 牛 胎児血清(FBS)を添加した培地を用い 25°C で維持した。ヒト胎児腎細胞株 HEK293細胞はRPMI1640 (Life Technologies)に10% FBSを添加した培地を用 い37°Cで維持した。
3.2.2 ギンブナIFNγrel 1、IFNγrel 2およびキンギョIFNγrel 1のcDNAの単離 ギンブナまたはキンギョの脾臓よりTRIzol(Invitrogen)を用いて全RNAを回 収し、その後High-Capacity cDNA Reverse Transcription Kit(Life Technologies)を 用いて cDNA を合成した。既報のキンギョやゼブラフィッシュの塩基配列の情 報をもとに設計したプライマー(Table 3-1)および上記の cDNA を用いて、
PrimeSTAR HS DNA Polymerase(Takara)にてRT-PCRを行った。反応液は説明 書に従って調整し、最初に98°Cで 30秒の反応を行った後、98°Cで10秒、55°C で5秒、72°Cで60秒の反応を35サイクル行った。増幅産物をillustra GFX PCR DNA and Gel Band Purification Kits(GE ヘルスケア)を用いて精製し、GoTaq Master Mix(Promega)を用いて、72°Cで15分間の反応を行い、3’末端側へのA 付加を行った。反応産物をillustra GFX PCR DNA and Gel Band Purification Kitsを 用いて精製し、pGEM-T Easyベクター(Promega)にLigation high Ver.2(東洋紡)
を用いて16°C で 30 分間ライゲーションを行った。その後、大腸菌DH5α に形 質転換を行った。得られたコロニーをGoTaq Master Mix(Promega)を用いて、
31
インサートチェックPCRとして、最初に94°Cで60秒の反応を行った後、94°C で10秒、50°Cで5秒、72°Cで30 秒の反応を25サイクル行った。その後、イ ンサートが確認された菌をLB液体培地にて増菌し、QIAprep Spin Miniprep Kit
(QIAGEN)を用いてプラスミドを回収した。そのプラスミドを用いて、BigDye Terminator v3.1 Cycle Sequencing Kit(Life Technologies)とApplied Biosystems 3130 ジェネティックアナライザ(Life Technologies)によりDNAシークエンスを行い、
配列を確認した。
3.2.3哺乳類細胞発現系を用いた組み換えIFNγrelの作製
ギンブナIFNγrel 1およびIFNγrel 2のcDNAを鋳型として制限酵素認識配列を 付加した哺乳類発現ベクター作製用プライマー(表 1)を用いて、3.2.2 と同様 にPrimeSTAR HS DNA PolymeraseにてPCRを行い、pGEM-T Easyベクターにサ ブクローニングした。プラスミドを回収後、制限酵素HindIII およびBamHI を 用いてDNAを消化・精製し、インサートを得た。同様に哺乳類細胞発現ベクタ ーであるp3xFLAG-CMV-14(Sigma)を制限酵素 HindIII および BamHI を用い て消化した。得られた両DNAとLigation high Ver.2を用いてライゲーションし、
大腸菌 DH5α に形質転換を行った。作製したベクターのシークエンスを確認し た後、QIAGEN Plasmid Midi Kit(QIAGEN)を用いて精製し、発現ベクターを得 た。作製した発現ベクターCMV-IFNγrel 1 およびCMV-IFNγrel 2 をFuGENE 6 Transfection Reagent(Promega)を用いて、2 × 106に播種したHEK293細胞に遺 伝子導入した。
32
3.2.4大腸菌発現系を用いた組み換えIFNγrelの発現および精製
<IFNγrel発現大腸菌株の作製>
ギンブナIFNγrel 1およびIFNγrel 2のcDNAを鋳型として制限酵素認識配列を 付加した大腸菌発現ベクター作製用プライマー(表 1)を用いて、3.2.2 と同様 にPrimeSTAR HS DNA PolymeraseにてPCRを行い、pGEM-T Easyベクターにサ ブクローニングした。上記のプラスミドと大腸菌発現ベクターである pET-16b
(Novagen)を共に制限酵素 NdeI および BamHI を用いて消化し、精製後、
Ligation high Ver.2を用いてライゲーションし、大腸菌DH5αに形質転換を行っ た。その後、作製したベクターのシークエンスを確認した。作製したベクター pET-16b-IFNγrel 1 またはpET-16b-IFNγrel 2をそれぞれタンパク発現用大腸菌株 BL21(DE3)PlysSまたはBL21(DE3)(Novagen)に形質転換した。
<組換えIFNγrel 1およびIFNγrel 2の発現および精製>
作製したタンパク発現用大腸菌株をLB培地で液体培養し、OD600=0.5程度に 増殖した後、0.1 mMの IPTGを加え、20°Cで16 時間振騰培養し、タンパク発 現を誘導した。大腸菌をPBSにて3回洗浄した後、培養液1 L当たり50 mLの 精製バッファー(20 mM リン酸ナトリウム pH7.4, 80 mM イミダゾール, 500 mM NaCl, 1×Protease Inhibitor Cocktail(Roche))に懸濁し、超音波破砕機にて破 砕した。その後 0.1%となる様に Triton X-100 を加え、遠心(9000×g、4°C、90 分)分離した後、上清を0.45 μmのディスクフィルターにてろ過し、組換えIFNγrel を 含 む 可 溶 性 画 分 を 得 た 。 精 製 バ ッ フ ァ ー で 平 衡 化 し た Ni-NTA Agarose
(QIAGEN)を組換え体1 mgあたり200 μl程度加え、4°Cで4時間から一晩反
33
応させた。洗浄バッファー(20 mM リン酸ナトリウム pH7.4, 80 mM イミダゾ ール, 500 mM NaCl)で洗浄した後、溶出バッファー(20 mM リン酸ナトリウム pH 7.4, 500 mM イミダゾール, 500 mM NaCl)に組換え体を溶出した。
<ゲルろ過クロマトグラフィー>
溶出した組み換え体をHiPrep 16/60 Sephacryl S-100 HRカラム(GEヘルスケ ア)を用いて精製した。精製にはリン酸ナトリウムバッファー(50 mMリン酸 ナトリウムpH 7.4, 300 mM NaCl)を用いた。精製後、サンプルをフィルターろ
過し(0.22 μm)、以下の試験に用いた。
3.2.5免疫沈降法によるIFNγrel分泌性の確認
<免疫沈降>
発現ベクターp3xFLAG-CMV-14、CMV-IFNγrel 1 および CMV-IFNγrel 2 を
HEK293 細胞に導入後、48 時間後に培養上清を回収し、上清 1ml と 1μg の
anti-FLAG M2 mAb (Sigma)と4°Cで2時間反応させた後、Protein G Sepharose
(GEヘルスケア)30μlを加え、4°Cで2時間反応させた。その後、免疫沈降の 産物にSDS サンプルバッファーを加え、ウェスタンブロッティング解析に供し た。
<ウェスタンブロッティング>
免疫沈降の産物を2μlずつ各レーンに添加し、SDS-PAGE を行った後、PVDF 膜に転写した。ブロッキングを行い、3,000倍に希釈したHRP標識抗FLAG M2
34
抗体(Sigma)を添加し、4°Cにて一晩反応させた。洗浄後、Chemi-Lumi One L
(ナカライ)にて発色し、Hyperfilm ECL(GEヘルスケア)に感光させ、シグナ ルを検出した。
3.2.7 ギンブナ細胞を用いた抗ウイルスアッセイ
抗ウイルスアッセイには、遺伝子導入後の HEK293 細胞の培養上清に含まれ
る組換えIFNγrelまたは大腸菌発現系によって作製し精製した組換え IFNγrel を
用いた。GTS9細胞を1 × 104個/wellなる様に96穴プレートに播種し細胞がフラ スコの底面に付着しコンフルエントになった後、精製した組換えIFNγrelを添加 し、25°C にて24 時間培養した。その後、PBS にて一回洗浄し、48 時間培養後 に 1 TCID50 となる感染価に調整したフナ造血器壊死症ウイルス(crucian carp hematopoietic necrosis virus; CHNV)を加え、48時間培養した。PBSによる洗浄 後、4% PFAにて15分固定後、0.1%クリスタルバイオレット染色を行い、プレ ートリーダーにて595nmの吸光度を測定した。
3.2.8 eGFPコンストラクトの作製および発現
pEGFP-N2ベクターにIFNγrel 1のC末端側に存在するNLS様配列、KHHHR を付加したpEGFP-KHHHR、およびアラニンを付加したpEGFP-AAAAAを作成 した。pEGFP-N2ベクター、pEGFP-KHHHRおよびpEGFP-AAAAA をFuGENE 6 Transfection Reagent (Promega)を用いて、1 × 106に播種したGTS9細胞へ遺伝子 導入した。導入24時間後に細胞をカバーガラスに播種しなおし、生着後4% PFA にて固定を行い、共焦点レーザー顕微鏡FV 1000(オリンパス)にてそれぞれの
35
GFPの細胞内局在を観察した。
3.2.9 IFNγrel処理後のリン酸化解析
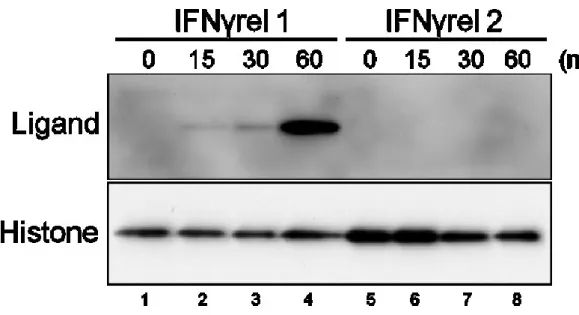
<細胞のIFNγrel処理>
IFNγrel 1またはIFNγrel 2処理後のGTS9細胞におけるSTATのリン酸化を調 べた。100 ng/mLの組換えIFNγrel 1またはIFNγrel 2をGTS9細胞に加え、0、15、 30、60分間、25°Cで培養後、それぞれPBSで3回洗浄し、溶解バッファー(50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% TritonX-100, 1×Protease Inhibitor Cocktail, 1×Phos STOP(Roche))に懸濁し、27G針付きシリンジを用いて20~30回吐出 及び吸引を繰り返し、細胞を破砕した。細胞溶解液を遠心沈殿(12,000×g、4°C、 10分)後、その上清をサンプルとした。
<ウェスタンブロッティングによる検出>
ブラッドフォード法によって蛋白濃度を測定しサンプル濃度を調整した後、
SDSサンプルバッファーとサンプルを混合し、96°Cで2分間煮沸した。STAT お
よびtubulin検出用にそれぞれ12 μg/レーンまたは2 μg/レーンとなる様にレーン
に分注し、それぞれ7.5% または10% SDS-PAGE ゲルを用いて電気泳動後、セ ミドライ式でPVDF膜に転写した。膜をブロッキング処理後、1,000倍に希釈し たPhospho-Stat抗体およびtotal-Stat抗体(Cell Signaling Technology)、ならびに 3,000倍に希釈した抗α tubulin抗体を添加し、4°Cにて一晩反応させた。洗浄後、
10,000倍に希釈したHRP標識ヤギ抗ウサギIgGまたは抗マウスIgG抗体(DAKO)
を添加し、1時間室温にて反応させた。洗浄後、Western Lightning ECL Proまた
36
はWestern Lightning ECL Ultra(PerkinElmer)を用いて発色し、Hyperfilm ECL(GE ヘルスケア)に感光させ、シグナルを検出した。
3.2.10 ルシフェラーゼレポーターアッセイ
STAT3 結合性プロモーター領域を含んだルシフェラーゼレポーターベクター
pGL3-APRE は、慶応大学医学部 吉村昭彦教授より分与されたものを用いた。
STAT6 結合性プロモーター領域を含んだルシフェラーゼレポーターベクター
pGL4-STAT6はpGL4.15ベクター(Promega)に制限酵素SacIまたは XhoIを用 いてサブクローニングし作製した。それぞれのレポーターベクターをコントロ ール測定用レポーターベクターであるpRL-TK(Promega)と共に、GTS9細胞に 遺伝子導入後、96穴プレートに1 × 104/wellになる様に播種した。生着後、IFNγrel 1 または IFNγrel 2 を加え、24 時間 25°C にて培養した。その後、Dual-Glo™
Luciferase Assay Systemを用いて転写活性を測定した。蛍光の測定にはCentro LB 960(BERTHOLD)を用いた。
3.2.11 統計解析
抗ウイルスアッセイの有意差検定は one-way ANOVA による分散分析の後、
Turkeyの多重比較検定によって行った。