Fabrication of α-elastin and elastic model
polypeptide cross-linked nanoparticles by
gamma irradiation
著者
Fujimoto Mari
内容記述
学位授与大学: Osaka Prefecture University(大阪
府立大学), 学位の種類: 博士(理学), 学位記番号:
論理第89号, 学位授与年月日: 2010-03-31, 指導教
員: 古田雅一.
0
Fabrication of
α
-elastin and elastic model polypeptide
cross-linked nanoparticles by gamma irradiation
(ガンマ線照射によるα-エラスチン
およびエラスチンモデルポリペプチドの架橋ナノ粒子化)
Mari Fujimoto
藤本 真理
Osaka Prefecture University
2010
0
Table of contents
Chapter 1. Introduction 1
Chapter 2. Preparation of α-elastin nanoparticles by gamma irradiation 15
Chapter 3. Preparation of nanoparticles using the elastic model polypeptide by gamma irradiation 32
Chapter 4. Analysis of the aggregation process of (GVGVP)251 (polypeptideI): Optimization of nanoparticle formation by gamma-ray cross-linking 52
Chapter 5. Loading and timed release of drugs by using the cross-linked polypeptide nanoparticles 81
Chapter 6. Summary 94
Acknowledgements 101
1 Chapter 1 Introduction
Elastin, a highly insoluble extracellular matrix (ECM) protein, is the core protein
of elastic fibers that contribute to the maintenance of elasticity of soft tissues, such as
the skin, lungs, arteries, and ligaments. The breakdown of elastic tissue causes damage
and leads to diseases such as supravalvular aortic stenosis and cutis laxa. Elastin is
secreted from smooth muscle cells, fibroblasts, and chondrocytes as an approximately
70-kDa soluble protein referred to as tropoelastin, which has alternating hydrophobic
and cross-linking domains. The assembly of tropoelastin into a fibrillar matrix is
believed to be a complex stepwise process. In the first step, the secreted tropoelastin
molecules are thought to self-aggregate via coacervation, a process that concentrates
and aligns the tropoelastin molecules for cross-linking. Tropoelastin aggregates are then
deposited onto preformed microfibrillar templates that act as a molecular scaffold.
Finally, lysyl oxidase (LOX) catalyzes the oxidative deamination of the peptidyl lysine
residues in tropoelastin to α-aminoadipic semialdehyde (allysine), which can
spontaneously condense with the neighboring amino groups or other peptidyl aldehydes
to form covalent cross-links such as desmosine or isodesmosine. After cross-linking,
2
the elasticity of tissues such as the skin, aorta, and lungs (Fig. 1-1) [1–7].
Fig. 1-1 Formation of elastic fibers [3]
α-Elastin can be fractionated from soluble elastin by coacervation as described by
Partrige et al., [8] in which the coacervated α-elastin is separated from the insoluble
β-elastin using oxalic acid at pH 4.7 and 50°C. Coacervation is repeated 2 more times in
order to improve the purity of the α-elastin fractions. α-Elastin has a molecular weight
of 10 to 60 kDa and runs as smear bands on SDS-PAGE. α-Elastin has hydrophobic
amino acid residues, such as GVGVP or GVGVAP, and hydrophilic domains
3
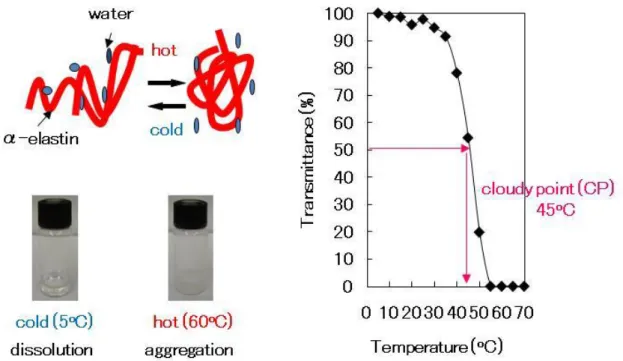
aggregation under specific concentration and temperature conditions referred to as the
cloudy point (CP) (Fig. 1-2).
On increasing the temperature of the α-elastin solution above the CP, the solution
becomes turbid due to the formation of aggregates. This process is readily reversible,
i.e., on lowering the temperature, the cloudiness clears, and the aggregates readily
redissolve. This phenomenon results from the hydrophobic amino acid residues, such as
GVGVP, within α-elastin. At temperatures above the CP, α-elastin can form aggregates
at a concentration of 0.11 mg/ml and a temperature of 21.5°C. This process is called
microcoacervation [9]. This is based on solvent entropy change, i.e., ΔG = ΔH – TΔS
(ΔG: Gibbs free energy, ΔH: enthalpy, T: temperature, and ΔS: entropy). Generally, in
polymers such as α-elastin, the solvent entropy is nearly zero. Further, in order to cause
dissolution of polymers such as α-elastin (the Gibbs free energy change becomes
negative), enthalpy has to become negative. Weis-Fogh and Andersen proposed that the
solvent entropy decreases on extension of the α-elastin, as a result of the exposure of the
hydrophobic groups to water [10]. On stretching, there occurs an exothermic hydration
of the hydrophobic groups, with a decrease in the entropy of water [11]. The water of
hydrophobic hydration can be quantified and characterized as a function of the variables
4
[12].
Fig. 1-2 Aggregation above the specific temperature, CP. CP in the figure is 10 mg/ml α-elastin.
Thermosensitive elastin model polypeptides such as (GVGVP)n, synthesized by
Escherichia coli recombinant DNA technology, have specific characteristics that mimic
α-elastin. The thermosensitivity of polypeptides has been utilized to develop micro- and
nanoparticles [13–15]. However, the thermosensitivity of polypeptides poses a serious
problem in the fabrication of stable particles by chemical cross-linking. The major
demerit of chemical cross-linking is the residual chemical reagents, including toxic
cross-linkers. However, irradiation-induced cross-linking by ionizing radiations does
5
materials, which are also effectively sterilized at the same time [16].
In recent years, gamma-rays have been used as cross-linkers to form hydrogels and
sheets using proteins and polysaccharides. Generally, proteins and polysaccharides are
destroyed by gamma irradiation. However, at high concentrations, chitosan, starch, and
cellulose have been shown to form cross-linked hydrogels on gamma irradiation
[17–19]. Collagen forms cross-linked hydrogels by gamma irradiation [20]. Elastin
model polypeptides, such as those containing GVGVP, have been used to develop
cross-linked sheets by gamma irradiation [21].
Therefore, I thought of obtaining nano-sized aggregates of α-elastin and
subsequently, nanoparticles of α-elastin cross-linked by gamma irradiation.
In recent years, nanoparticles have become an important area of research in the
field of drug delivery because they have the ability to deliver a wide range of drugs.
Nanoparticles have another advantage over larger microparticles in that they are better
suited for intravenous drug delivery. The smallest capillaries in the body are 5–6 μm in
diameter. The size of the particles being dispersed in the bloodstream must be
significantly smaller than 5 μm. Thus, a wide variety of drugs can be delivered via a
number of routes using nanoparticle carriers. Nanoparticles can be used to deliver
6
etc. They can be formulated for targeted delivery to the lymphatic system, brain, arterial
walls, lungs, liver, and spleen or for long-term systemic circulation [22–26]. Moreover,
approximately 100-nm particles can be used for tumor targeting owing to their enhanced
permeability and retention effect (EPR) [27]. These 100-nm particles are able to reach
the tumors because the blood vessels of tumors are larger in diameter than microvessels.
Thermosensitive block copolymers such as poly(ethylene glycol) conjugated with
poly(N-isopropylacrylamide) have specific characteristics that mimic α-elastin. These
polymers also aggregate at temperatures above their respective CPs and form
nanoparticles, and the particle size differs with the heating process [28].
Therefore, I focused on the establishment of α-elastin and elastic model
polypeptide aggregation procedures and characterization of the aggregation process by
using different heating processes in order to obtain stable nanoparticles by gamma-ray
cross-linking. As a result, I successfully obtained stable cross-linked α-elastin and
elastic model polypeptide nanoparticles. I also studied the factors necessary to obtain
stable cross-linked nanoparticles and their use for loading methotrexate and cisplatin to
check their applicability to drug delivery.
In chapter 2, I have shown the effect of the heating process on cross-linked
7
irradiation. I also studied the effect of the concentration and dose of gamma irradiation
on the formation of cross-linked α-elastin nanoparticles. As a result, I successfully
obtained stable cross-linked nanoparticles at a temperature below the CP (Fig. 1-3).
Fig. 1-3 Principle of cross-linked nanoparticle synthesis
α-Elastin has hydrophobic amino acid residues, such as GVGVP, that aggregate on
heating as well as hydrophilic domains containing charged amino acids.
In chapter 3, I employed 3 types of elastic model polypeptides to study the details
of the aggregation procedure and to obtain cross-linked elastic model polypeptide
nanoparticles by gamma irradiation. I used the following elastic model polypeptides: (1)
8
repeats of such hydrophobic amino acids are responsible for aggregation as observed in
the α-elastin model polypeptide); (2) Polypeptide II:(GVGVP GVGFP GEGFP GVGVP
GVGFP GFGFP)17(GVGVP) (containing Glu); and (3) Polypeptide III:(GVGVP
GVGFP GKGFP GVGVP GVGVP GVGVP)22(GVGVP) (containing Lys). I
successfully obtained stable cross-linked nanoparticles of Polypeptide I and II,
respectively, at a temperature below the CP, reconfirming that repeats of hydrophobic
domains such as GVGVP were important for the aggregation and formation of the
cross-linked structure.
In chapter 4, I focused on the heating process for optimizing nanoparticle
formation using polypeptide I solution in order to obtain information on the efficiency
of the nanopariticle formation by gamma-ray cross-linking. I changed the heating rate
and monitored the aggregation of polypeptide I by circular dichroism (CD)
spectrometry. Moreover, I used molecular dynamics (MD) simulation as well as simple
amino acids, such as Val, Pro, and Gly, to guess the point of cross-linking.
In chapter 5, I have described an attempt to load methotrexate and cisplatin on the
cross-linked elastic model polypeptide I and II nanoparticles to explore the possibility
of efficient drug delivery.
9
polypeptide aggregation procedures and (2) characterization of the aggregation to obtain
stable cross-linked nanoparticles by gamma-ray cross-linking.
References
[1] Vrhovski, B., Jensen, S., Weiss, A. S., Coacervation characteristics of recombinant
human tropoelastin, European Journal of Biochemistry 1997, 250, 92–98.
[2] Ross, R., Fialkow, R. J., Altman, L. K., The morphogenesis of elastic fibers,
Advances in Experimental Medicine and Biology 1977, 79, 7–17.
[3] Sato, F., Wachi, H., Ishida, M., Nonaka, R., Onoue, S., Urban, Z., Starcher, B. C. S.,
Seyama, Y., Distinct steps of cross-linking, self-association, and maturation of
tropoelastin are necessary for elastin fiber formation, Journal of Molecular Biology
2007, 369, 841–851.
[4] Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., Walter, P., In: The
Molecular Biology of the Cell. 4th edition. Garland, New York, 2002, pp. 151–152.
[5] Urban, Z., Riazi, S., Seidl, T. L., Katahira, J., Smoot, L. B., Chitayat, D., Boyd, C.
D., Hinek, A., Connection between elastin haploinsufficiency and increased cell
proliferation in patients with supravalvular aortic stenosis and Williams-Beuren
10
[6] Urban, Z., Michels, V. V., Thibodeau, S. N., Donis-Keller, H., Csiszar, K., Boyd, C.
D., Supravalvular aortic stenosis : a splice site mutation within the elastin gene results
in reduced expression of two aberrantly spliced transcripts, Human Genetics 1999,
104, 135–142.
[7] Kitano, Y., Nishida, K., Okada, N., Mimaki, T., Yabuuchi, H., Cutis laxa with
ultrastructural abnormalities of elastic fiber, Journal of the American Academy of
Dermatology 1989, 21, 378–380.
[8] Partrige, S. M., Davis, H. F., Adair, G. S., The chemistry of connective tissue 2.
Soluble protein derived from partial hydrolysis of elastin, Biochemical Journal 1955
61, 11–30.
[9] Kaibara, K., Watanabe, T., Miyakawa, K., Characterization of critical processes in
liquid-liquid phase separation of the elastomeric protein-water system: Microscopic
observations and light scattering measurements, Biopolymers 2000, 53, 369–379.
[10] Weis-Fogh, T., Andersen, S. O., New molecular model for the long-range elasticity
of elastin, Nature 1970, 227, 718–721.
[11] Urry, D. W., Woods, C. T., Hayes, L. C., Xu, J., McPherson, D. T., Parker, T. M.,
Iwama, M., Furuta, M., Hayashi, T., Murata, M., Elastic protein-based biomaterials:
11
J., Wiss, D. L. (eds.) Tissue Engineering and Novel Delivery System. CRC Press,
New York, 2004, pp.31–54.
[12] Urry, D. W., Physical chemistry of biological free energy transduction as
demonstrated by elastic protein-based polymers, The Journal of Physical Chemistry
B, 1997, 101, 11007–11028.
[13] Fujita, Y., Mie, M., Kobatake, E., Construction of nanoscale protein particle using
temperature-sensitive elastin-like peptide and polyaspartic acid chain, Biomaterials
2009, 30, 3450–3457.
[14] Osborne, J. L., Farmer, R., Woodhouse, K. A., Self-assembled elastin-like
polypeptide particles, Acta Biomaterialia 2008, 4, 49–57.
[15] Herreo-Vanrell, R., Rincon, A. C., Alonso, M., Reboto, V., Molina-Martinez, I. T.,
Rodriguez-Cabello, J. C., Self-assembled particles of an elastin-like polymer as
vehicles for controlled drug release, Journal of Controlled Release 2005, 102,
113–122.
[16] Bessho, M., Furuta, M., Kojima, T., Okuda, S., Hara, M., Gelatin hydrogels
cross-linked by gamma-rays irradiation: materials for absorption and release of dye,
Journal of Biomaterials Science, Polymer Edition 2005, 16, 715–724.
12
processing, Radiation Physics and Chemistry 2002, 63, 625–627.
[18] Kume, T., Matsuhashi, S., Shimazu, M., Ito, H., Fujimura, T., Adachi, K., Uchida,
H., Shigeta, N., Matsuoka, H., Osa, A., Sekine, T., Uptake and transport of
positron-emitting tracer (18F) in plants, Applied Radiation and Isotopes 1997, 48,
1035–1043.
[19] Yoshii, F., Zhao, L., Wach, R. A., Nagasawa, N., Mitomo, H., Kume, T.,Hydrogels
of polysaccharide derivatives crosslinked with irradiation at paste-like condition,
Nuclear Instruments and Methods in Physics Research B 2003, 208, 320–324.
[20] Koshimizu, N., Bessho, M., Suzuki, S., Yuguchi, Y., Kitamura, S., Hara, M.,
Gamma-crosslinked collagen gel without fibrils: analysis of structure and heat
stability, Bioscience, Biotechnology, and Biochemistry2009, 73, 1915–1921.
[21] Urry, D. W., Pattanaik, A., Accavitti, M. A., Luan, C. X., Mcpherson, D. T, Xu, J.,
Gowda, D. C., Parker, T. M., Harris, C. M., Jing, N., Transductional elastic and
plastic protein-based polymers as potential medical devices, Handbook of
biodegradable polymers, Domb, A. J., Kost, J., Wiseman, D. M., (eds), Harwood
Academic Publishers, Chur, Switzerland, 1997, pp. 367-386.
[22] Hua, W., Zhang, X-Z., Han, C., Chen, W-Q., Cheng, S-X., Zuo,R-X.,
13
acids-b-N-isopropylacrylamide) for drug delivery, Journal of Controlled Release
2006, 116, 266–274.
[23] Herrero-Vanrell, R., Rincon, A. C., Alonso, M., Reboto, V., Molina-Martinez, I.T.,
Rodriguez-Cabello, J. C., Self-assembled particles of an elastin-like polymer as
vehicles for controlled drug release, Journal of Controlled Release 2005, 102,
113–122.
[24] Scholes, P. D., Coombes, A. G. A., Illum, L., Davis, S. S., Vett, M., Davies, M. C.,
The preparation of sub-200 nm poly(lactide-co-glycolide) microspheres for
site-specific drug delivery, Journal of Controlled Release 1993, 25, 145–153.
[25] Zhang, Y., Zhuo, R., Synthesis and drug release behavior of poly(trimethlene
carbonate)-poly(ethylene glycol)-poly(trimethylene carbonate) nanoparticles,
Biomaterials 2005, 26, 2089–2094.
[26] Hans, M. L., Lowman, A. M., Biodegradable nanoparticles for drug delivery and
targeting, Current Opinion Solid State Materials Science 2002, 6 , 319–327.
[27] Duncan, R., Vincent, M. J., Greco, F., Nicholson, R. I., Polymer-drug conjugates:
Towards a novel approach for the treatment of endocrine-related cancer, Endocrine-
Related Cancer 2005, 12, 189–199.
14
processing and formulation parameters on the size of nanoparticles based on block
copolymers of poly(ethyleneglycol) and poly(N-isopropylacrylamide) with and
15
Chapter 2 Preparation of α-elastin nanoparticles by gamma irradiation
Introduction
α-Elastin is a component of soft tissue such as skin, lung, arteries, and ligaments.
To purify α-elastin, bovine ligament elastin is treated with mild acid hydrolysis to yield
a high molecular weight digest that retains the amino acid composition of native elastin.
Despite structural heterogeneities resulting from the hydrolysis, α-elastin retains several
key physicochemical properties of elastin. Recently, films of α-elastin from bovine
aortic smooth muscle cells have been developed for vascular tissue [1]. However,
α-elastin–based biomaterials are in the very early stages of development [2].
α-Elastin (from bovine neck ligament) is unique in that it undergoes aggregation at
a specific concentration and temperature called the cloudy point (CP). When the
temperature of the solution is raised above the CP, α-elastin starts a complex,
self-assembly process that leads to aggregation. At a concentration of 0.11 mg/ml and a
temperature of 21.5ºC, α-elastin can aggregate and form micro- or nano-sized particles
[3]. Therefore, I tested whether stabilized α-elastin nanoparticles, which could be useful
as a career for drug delivery, might be obtained by using gamma irradiation to
16
α-elastin concentration on nanoparticle formation, the formation of nanoparticles in
solution above the CP using various heating protocols, and the effect of gamma
irradiation dose on the stabilization of cross-linked nanoparticles.
Materials and Methods
Materials
α-Elastin (extracted from bovine neck ligaments) was obtained from Elastin
Product Company, Inc. The coacervated α-elastin is separated from insoluble β-elastin
with oxalic acid at pH 4.7 and 50ºC. Coacervation is repeated two more times to
improve the purity of the α-elastin fractions. α-Elastin has a molecular weight of
10–60 kDa and runs as a smear on polyacrylamide gel electrophoresis (PAGE) [4].
Evaluation of the CP of α-elastin solutions
Transmittance was measured with a HITACHI U-3210 UV-Vis spectrophotometer
at various temperatures using a water bath (EYELA SB-350) with a magnetic stirrer/hot
plate (CORNING). Turbidity measurement was based on the change in absorbance at
300 nm [5]. At a given temperature, the disposable plastic cuvette containing the sample
17
considered the actual turbidity for the sample at the given temperature.
Preparation of α-elastin aggregates in aqueous solution heated above the CP
Particle formation of α-elastin in aqueous solution was examined by transmittance
after heating from 4ºC to 60ºC at various heating rates (1–6ºC/min) [6]. In brief, 50 mg
of α-elastin was dissolved in 5 ml of deionized water (10 mg/ml) in a stoppered, Pyrex
test tube and cooled on ice. The α-elastin solution was heated from 4ºC to 60ºC while
stirring in a water bath (EYELA SB-350) with a magnetic stirrer (EYELA RCN-3D).
Measurement of the size distribution of the aggaregated α-elastin
Particles sizes were measured by dynamic light scattering (DLS) (Particle Sizing
Systems; Nicomp 370) at a 90º scattering angle. The solution was placed into a
disposable plastic cuvette (Kartell ART.01961-00) and incubated at 60ºC in the cuvette
holder of the Nicomp 370.
Gamma irradiation of aggregated α-elastin
The heated samples containing aggregated α-elastin were placed in thermos bottles
18
doses of 1–30 kGy using the 60Co gamma irradiation pool at Osaka Prefecture
University (dose rate, 8.8 kGy/h). The temperature of the sample was maintained at
60ºC during irradiation by replacing the hot water in the thermos bottle every 1 h.
The size distributions of the aggregated α-elastin after irradiation at temperatures
from 4ºC to 60ºC were measured by DLS.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Aggregated α-elastin was recovered by centrifugation at 10,000 rpm at 4°C for 5
min (HITACHI himac CR21, rotor no. 26) and dissolved in deionized water to a final
concentration of 10 mg/ml. The solution (0.1 ml) was mixed with sample buffer (0.1
ml) and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE). SDS-PAGE was performed using a 10% polyacrylamide gel (thickness: 1
mm); the sample (10 l/well) was run on a Mini-PROTEAN 3 cell (BIO-RAD) at a constant voltage (200 V) for 45 min. Separated polypeptides were visualized with the
19 Transmission electron microscopy
Samples for transmission electron microscopy (TEM) were dissolved in deionized
water to obtain a final concentration of 10 mg/ml. The sample was added to
carbon-coated, 400-mesh copper grids (VECO) on ice. Excess sample was absorbed on
filter paper, and the grid was dried at room temperature before observing under TEM.
The prepared samples were viewed using a HITACHI, EF2000 (HITACHI Ltd.)
operating at 200 keV.
Atomic force microscopy
Samples for atomic force microscopy (AFM) were dissolved in deionized water to
a final concentartion of 10 mg/ml. The samples were deposited on mica on ice and dried
at room temperature. AFM analysis was performed in air with a Digital Instruments
MultiMode (SII Nano Technology Inc., SPI 3800N, SPI 4000 NanoNavi) using
SI-DF20 cantilevers in the tapping mode with a resonant frequency of less than 200
20 Results and Discussion
Effect of the concentration of α-elastin on particle formation
The optical transmittance of 0.1, 0.5, 1.0, 5.0, and 10 mg/ml aqueous solutions of
α-elastin as a function of temperature was examined. The CP of the α-elastin solution
was when the transmittance of the solution was 50%. α-Elastin underwent aggregation
at temperatures of 45ºC for 10 mg/ml, 32ºC for 1.0 mg/ml, and 30ºC for 5.0 mg/ml. The
CP was observed at concentrations of more than 0.5 mg/ml as shown in Table 2-1.
Table 2-1. Cloudy point of aggregated α-elastin solution
Concentration (mg/ml) Cloudy point (CP) (oC)
0.1 Not detected
0.5 Not detected
1.0 30
5.0 32
21
Table 2-2. Effect of α-elastin concentration on particle size. Mean size of α-elastin was measured by DLS analysis at 60ºC Concentration (mg/ml) Size (nm) 0.1 No stable particles 0.5 No stable particles 1.0 No stable particles 5.0 No stable particles 10.0 240 (±50)a a standard deviation
The effect of α-elastin concentration (0.1–10 mg/ml) on the formation of
nanoparticles at a heating rate of 1.9ºC/min was also investigated (Table 2-2). A
concentration of 10 mg/ml of α-elastin was optimal for nanoparticle formation. All the
aggregates redissolved when the temperature was brought back to below the CP.
Effect of heating protocol on α-elastin particle formation
The effects of various heating rates on particle formation of 10 mg/ml α-elastin in
aqueous solutions were examined. When the solution temperature increased from 4ºC to
60ºC (above CP), α-elastin formed aggregates in aqueous solution at 60ºC (above CP)
22
particles were within the range of 200–300 nm for both “Slow heating (1.9ºC/min)” and
“Fast heating (5.6ºC/min)” protocols.
After a radiation dose of 30 kGy, the particles could not be dissolved by reducing
the temperature to 5ºC (below CP), and transmittance of the solution was almost zero at
all temperatures examined. These results indicate that gamma-ray cross-linking
stabilized the aggregates, which then lost their thermosensititvity. After irradiation and
collection of aggregates by centrifugation at 4ºC, particle size distributions were
analyzed by DLS. The mean of the size distributions shifted to less than 200 nm
(Table 2-3), and only the “Slow heating” protocol yielded a narrow size distribution
containing a single peak, while the other condition yielded broader and unstable size
distributions containing several peaks. Based on these obserevation, gamma-ray
cross-linking under “Slow heating” conditions was the optimal protocol to produce
23
Table 2-3. Mean size of aggregated α-elastin by heating after 30-kGy gamma irradiation as measured by DLS at 5ºC and 60ºC
Heating process Size (nm) 60oC Size (nm) 5oC Slow heating 180 (±40)a 160 (±30)a Fast heating 100(±20)a, 530(±60)a 99.8% 0.2% No stable particles a standard deviation
Effect of gamma irradiation doses onα-elastin particle formation
The effects of gamma irradiation doses (1–30 kGy) on particle formation of
α-elastin in aqueous solutions was also examined. After gamma irradiation, aggegated
α-elastin could not be dissolved by reducing the temperature to 5ºC (below CP), and
transmittance of the solution was almost zero at all temperatures examined when the
gamma irradiation dose was more than 15 kGy (Table 2-4). These results indicated that
the aggregates were stabilized by cross-linking. The aggregates were collected by
centrifugation at 4ºC after irradiation and analyzed by SDS-PAGE (Fig. 2-1). At doses
of more than 7.5 kGy, the molecular weight of α-elastin was greater than that before
irradiation (Fig. 2-1). New higher molecular weight bands with a narrow distribution
24 209000 124000 80000 49100 34800 28900 20600 ( Da ) m a rk e
r
0 k G y 1 k G y 3 k G y 7 .5 k G y 1 5 k G y 3 0 k G yFig. 2-1 Changes in the molecular weight of cross-linked α-elastin after gamma irradiation
Table 2-4. Effect of the gamma irradiation doses on α-elastin particle size Radiation dose (kGy) Cloudy point (oC) Size (nm) 60oC Size (nm) 5oC Yield (%) 1.0 45 No stable particles No stable particles < 1 3.0 45 No stable particles No stable particles <1 5.0 45 240(±50)a No stable particles <1 7.5 30 190(±20)a 60(±10)a 10 15 Not detected 150(±30)a 70(±10)a 10 30 Not detected 180(±40)a 160(±30)a 10 a standard deviation
25
α-elastin were approximately 200 nm after irradiation with more than 5 kGy at 60ºC.
When the temperature of the aggregated α-elastin solution decreased, the mean of the
size distributions of aggregated α-elastin shifted to approximately 60, 70, and 160 nm
after irradiation with 7.5, 15, and 30 kGy, respectively, at 5ºC (Table 2-4). Images of
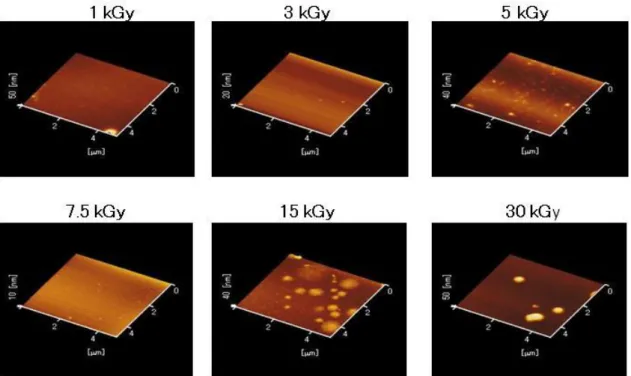
aggregated α-elastin nanoparticles were obtained by AFM (Fig. 2-2). Analysis of the
images showed that almost all of the aggregated α-elastin nanoparticles were spherical,
and their size increased with increasing radiation doses.
26
Fig. 2-3 TEM image of α-elastin nanoparticles after gamma irradiation
In addition, images of the cross-linked nanoparticles (irradiated at 30 kGy) were
obtained by TEM (Fig. 2-3). This image shows that almost all the cross-linked
nanoparticles were spherical. The size distribution obtained from TEM imaging was
consistent with the DLS results (irradiated at 30 kGy). The yield of the aggregated
α-elastin nanoparticles was 10% at radiation doses of more than 7.5 kGy (Table 2-4).
From these results, I conclude that doses of 30-kGy gamma irradiation form aggregates
by cross linking α-elastin.
The optimal condition to produce stable nanoparticles was irradiation of 10 mg/ml
27
other within the aggregated nanoparticles above the CP (60ºC). This molecular
assembly would facilitate cross-linking by gamma-rays. The high concentration of
α-elastin (10 mg/ml) would contribute to cross-linking efficiency in forming stable
cross-linked nanoparticles. Generally, polysaccharides are destroyed by gamma
irradiation. However, high concentration of chitosan, starch, and cellulose were also
cross-linked to form hydrogels by gamma irradiation [7-9].
Moreover, the present results clearly indicated that the heating protocol may be
crucial for forming stable nanoparticles of aggregated α-elastin by gamma irradiation.
“Slow heating” might be effective for efficient aggregation to facilitate cross-linking.
Generally, it is necessary for polymer chains to be close for efficient cross-linking.
α-Elastin has hydrophobic amino acids such as Val, Pro, and Ala (Table 2-5). Elastin
fiber assembly reportedly requires a GVGVP or GVGVAP amino acid sequence for
aggregation. Hydrophobic amino acids may cause aggregation, so these amino acids
might contribute to aggregation and cross-linking by gamma irradiation. In contrast,
“Fast heating” might not allow enough hydrophobic side-chain interactions to form
stable, cross-linked structures. Hydrophobic side-chains need time to interact because of
repulsive charges within the hydropholic domain of α-elastin.
28
analyze the aggregation process of α-elastin under different heating rates. However, data
for stable, cross-linked nanoparticles from the aggregates produced by the “Slow
heating” protocol could not be obtained. It was difficult to analyze the aggregation and
cross-linking procedures for proteins such as elastin, because the proteins have various
amino acids with polar side-chains (hydrophobic or hydrophilic amino acid and plus- or
minus-charged amino acids)
In the next chapter, to further analyze the aggregation procedure, the effect of
charge on the aggregation processes was studied using three types of elastic model
polypeptides containing charged amino acids. I tried to make cross-linked elastic model
polypeptide nanoparticles by gamma irradiation.
Conclusions
Nanoparticles of α-elastin were successfully obtained from only a 10 mg/ml
solution by gamma-ray cross-linking, raising the temperature above the CP with the
“Slow heating” protocol, and maintaining the temperature during irradiation. The size of
the resulting nanoparticles was distributed within the range of approximately 140 nm
under 5ºC (below CP) with a narrow size distribution. Yield of the cross-linked
29
the particles increased with the radiation doses and α-elastin concentration.
Table 2-5. Amino acid analyses for α-elastin. Values are expressed as residues per thousand residues [10]
Amino acid Value
Asp 6.6 Glu 16.1 Hyp 13.4 Ser 8.4 Gly 344.1 His 0.7 Arg 5.9 Thr 7.5 Ala 206.7 Pro 119.9 Tyr 8.7 Val 137.5 Met 2.1 Cys 0.8 Ilu 25.5 Leu 62.9 Phe 30.2 Lys 2.8
30 References
[1] Jennie, B. L., Jesse, B. W., Phillip, J. S., Joyce, Y. W., Cross-linked α-elastin
biomaterials: towords a processable elasin mimetic scaffold, Acta Biomaterialia,
2005, 1, 155-164.
[2] Gomes, M., Azevedo, H., Malafaya, P., Silvia, S., Oliveira, J., Silva S., Sausa, R.,
Mano, J., Reis, R., Natural polymers in tissue engineering application (chapter 6). In:
Blitterswijk, C., et al. (eds), Tissue engineering, Academic Press, San Diego, 2008,
pp. 146-192.
[3] Kaibara, K., Watanabe, T., Miyakawa, K., Characterization of critical processes in
liquid-liquid phase separation of the elastomeric protein-water system: Microscopic
observations and light scattering measurements, Biopolymers 2000, 53, 369-379.
[4] Technical data sheet of the Elastin Product Co. Inc. for alpha-elastin (AE17)
[5] Urry, D. W., Nichol, A., McPherson, D. T., Harris, C. M., Parker, T. M., Xu, J,
Gowda, D C., Peter, R. Shewry, Properties, preparations, and applications of
bioelastic materials. In: Weseman, D. L., Trantolo, D, J., Altobelli, D. E., Yaszemski,
M, J., Gresser, J. D., Schwartz, E. R. (eds) Encyclopedic handbook of biomaterials
and bioengineering Part A: Materials, Volume 2. Marcel-Dekker, New York, 1995,
31
[6] Neradovic, D., Soga, O., Van Nostrum, C. F., Hennink, W. E., The effect of the
processing and formulation parameters on the size of nanoparticles based on block
copolymers of poly(ethyleneglycol) and poly(N-isopropylacrylamide) with and
without hydrolytically sensitive groups, Biomaterials 2004, 25, 2409-2418.
[7] Kume, T., Nagasawa, N., Yoshii, F., Utilization of carbohydrates by radiation
processing. Radiation Physics and Chemistry 2002, 63, 625-627.
[8] Kume, T., Matsuhashi, S., Shimazu, M., Ito, H., Fujimura, T., Adachi, K., Uchida,
H., Shigeta, N., Matsuoka, H., Osa, A., Sekine, T., Uptake and transport of
positron-emitting tracer(18F) in plants, Applied Radiation and Isotopes 1997, 48,
1035-1043.
[9] Yoshii, F., Zhao, L., Wach, R. A., Nagasawa, N., Mitomo, H., Kume, T., Hydrogels
of polysaccharide derivatives crosslinked with irradiation at paste-like condition,
Nuclear Instruments and Methods in Physics Research B 2003, 208, 320-324.
[10] Vyavahare, N., Ogle, M., Schoen, F. J. Levy, R. S., Elastin calcification and its
prevention with aluminum chloride pretreatment, American Journal of Pathology
32
Chapter 3 Preparation of nanoparticles using the elastic model polypeptide by gamma irradiation
Introduction
As described in Chapter 2, I obtained α-elastin cross-linked nanoparticles.
However, I could not obtain any information about the difference in efficiency to obtain
stable cross-linked nanoparticle size with a narrow size distribution-specific heating rate.
It is difficult to analyze the aggregation and cross-linking procedure for α-elastin
through different heating processes, because the protein has various types of amino
acids (hydrophobic or hydrophilic amino acids and positively or negatively charged
amino acids). To obtain the details of the aggregation and cross-linking procedure, I
used 3 types of the elastic model polypeptide, namely, polypeptide I: [(GVGVP)251]
(containing GVGVP whose repeat caused aggregation), polypeptide II: [(GVGVP
GVGFP GEGFP GVGVP GVGFP GFGFP)17(GVGVP)] (containing Glu), and
polypeptide III: [(GVGVP GVGFP GKGFP GVGVP GVGVP GVGVP)22(GVGVP)]
(containing Lys). These polypeptides were synthesized with Escherichia coli
recombinant DNA technology described by Urry et al. Polypeptide II and III were
synthesized as a controlled release delivery system for amphiphilic drugs and
33
hydrophobicity by replacing Val with a much more hydrophobic residue, namely, Phe.
As a result, there is a systematic shift in pKa values of the polypeptides. This is based
on the polar-polar repulsive free energy of hydration [1]. I attempted to prepare
aggregated polypeptide I, II, and III in an aqueous solution above their CP and to form
nanoparticles of the polypeptides by various heating methods.
Materials and Methods
Materials
Elastic model polypeptide I: (GVGVP)251, II: (GVGVP GVGFP GEGFP GVGVP
GVGFP GFGFP)17(GVGVP), and III: (GVGVP GVGFP GKGFP GVGVP GVGVP
GVGVP)22(GVGVP) were supplied from Prof. Urry (Minnesota University) and BRL
(Bioelastic Research Ltd.) through Bioelastic Japan Co. [1]. Sodium phosphate, dibasic,
anhydrous (NaHPO4); sodium phosphate, monobasic (NaH2PO4 ・ 2H2O); and
hydrochloric acid were obtained from Wako Pure Chemicals Industries Ltd.
Evaluation of CP of polypeptide solution
Transmittances were measured with a HITACHI U-3210 UV-Vis
34
SB-350) with a magnetic stirrer/hot plate (CORNING). Turbidity was assessed by the
change in absorbance at 300 nm for a 1 mg/ml solution [2]. At a given temperature, the
disposable plastic cuvette containing the sample was allowed to stand until a constant
turbidity value was obtained. This constant value is considered to be the actual turbidity
of the sample at the given temperature.
Preparation of polypeptide I, II, and III aggregates in an aqueous solution heated above CP
The aggregates of Polypeptide I, II, and III were prepared by heating the aqueous
solutions from 4ºC to 60ºC at various heating rates (1–6ºC/min) as described in Chapter
2. Moreover, “Heat shock,” procedure was employed as an additional heating process,
in which the polymer solution at a high concentration was dropped into hot water
(above CP) with constant stirring in a water bath (EYELA SB-350) by using a magnetic
stirrer (EYELA RCN-3D) at 600 rpm.
Measurement of size distribution of aggregated polypeptide I, II, and III
Particles size was measured by dynamic light scattering (DLS) (Particle Sizing
Systems, Nicomp 370) at a 90° scattering angle. The polymer solution described above
35
plastic cuvette (Kartell ART.01961-00) and incubated at 42°C or 50°C in the cuvette
holder of Nicomp 370.
Gamma irradiation of aggregated polypeptide I, II, and III
The heated polymer samples were placed in thermos bottles (TIGER MWE-C350)
and irradiated with 60Co gamma-rays at doses of 8.5–30 kGy by using 60Co gamma
irradiation pool at Osaka Prefecture University (dose rate: 10.5 kGy/h). The temperature
of the sample was maintained at 42°C or 50°C during irradiation.
Transmission electron microscopy
Samples for transmission electron microscopy (TEM) were dissolved in deionized
water to a final concentration of 10 mg/ml. The sample solution was added to
carbon-coated 400-mesh copper grids (VECO) on ice, excess sample was absorbed with
a filter paper, and the grid was dried at room temperature before observation by TEM.
The prepared samples were viewed using a HITACHI EF2000 (HITACHI Ltd.)
36 Atomic force microscopy
Samples for Atomic force microscopy (AFM) were dissolved in deionized water to
a final concentration of 1 mg/ml. The samples were deposited on mica on ice and dried
at room temperature. AFM analysis was performed in air by using a digital instrument
multimode (SII Nano Technology Inc., SPI 3800N, SPI 4000 NanoNavi) using SI-DF20
cantilevers working in the tapping mode with a resonant frequency of less than 200
kHz.
Results and Discussion
Preparation of polypeptide I nanoparticles
Effect of the heating process on aggregation of polypeptide I
I examined the effect of the heating process on particle formation of polypeptide I
in aqueous solution. The polypeptides in the solution aggregated at a temperature
greater than the CP (30°C) irrespective of the heating rate from 1.3 to 3.8°C/min. I
attempted to obtain aggregated polypeptide I at a concentration of 5 mg/ml and
temperature of 42°C. This concentration and temperature were the optimum for forming
aggregated polypeptide I particles. The average size of the aggregated particles was
within the range of 380–440 nm for all the heating processes before irradiation
37
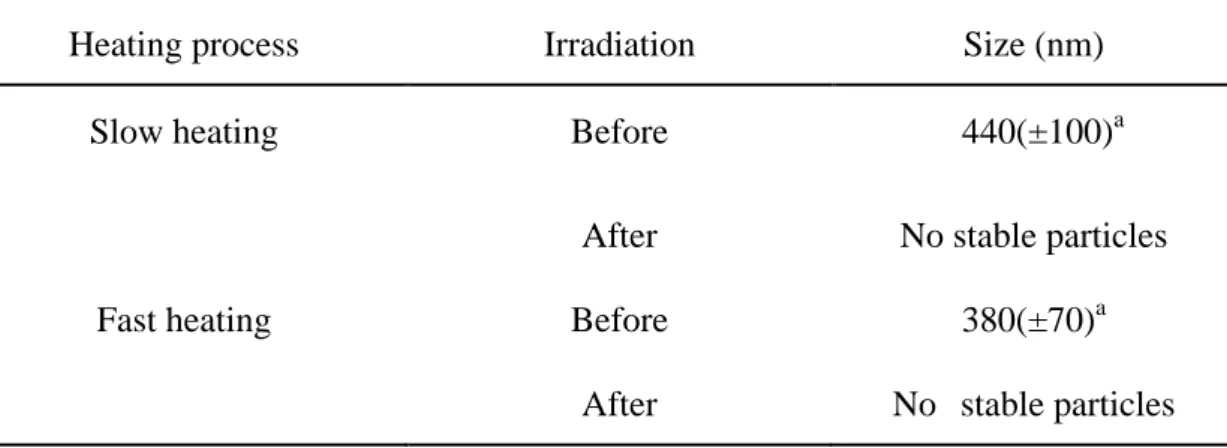
Table 3-1. Effect of the heating process on polypeptide I particle size. Mean size of the polypeptide I measured by DLS analysis at 42°C.
Heating process Irradiation Size (nm)
Slow heating Before 440(±100)a
After No stable particles Fast heating Before 380(±70)a
After No stable particles
a
standard deviation
I also investigated the effect of gamma irradiation on the aggregated polypeptides
prepared by each heating process at a temperature greater than the CP. After irradiation,
“Slow heating” (1.3°C/min) yielded a gel of the aggregated polypeptides, while “Fast
heating” (3.8o
C/min) yielded sediments of the aggregated polypeptides (aggregated
polypeptide could not form nanoparticles), which did not dissolve again at temperatures
lower than the CP.
Therefore, I employed the “Heat shock (40oC/min)” heating process and attempted
to obtain cross-linked nanoparticles. In “Heat shock,” 25 mg/ml of the polypeptide I
solution was dropped into the water at 42oC. This concentration and temperature were
the optimum to form polypeptide I aggregates. The size of aggregated polypeptide I was
38
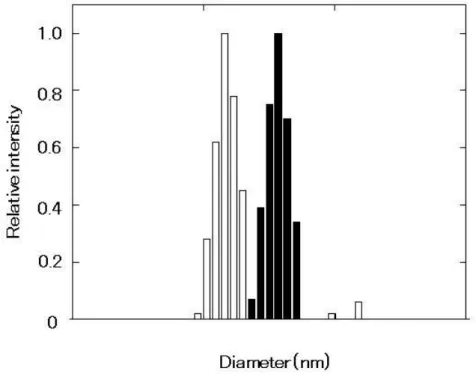
suspension of nanoparticles (ca. 150 nm). The results of the DLS measurements are
shown in Fig. 1. The nanoparticles did not dissolve at temperatures lower than the CP.
Fig. 3-1 Mean size of the polypeptide I particles measured by DLS analysis. The sample is prepared using the “Heat shock” process at 42°C in deionized water. The shaded square and blank square in the figure are before irradiation and after irradiation, respectively.
These results strongly suggest that the nanoparticles were cross-linked by irradiation
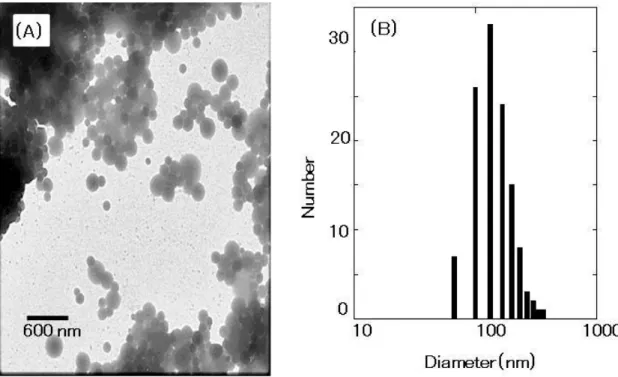
with gamma-rays. Fig. 3-2(A) shows the TEM image of cross-linked nanoparticles. As
seen in the figure, almost all the cross-linked nanoparticles were spherical. I measured
the size of the 120 particles that appeared in the TEM image by using the scale bar. The
39
(Fig. 3-2(B)) was consistent with the DLS result (average size and standard deviation:
150 ± 30 nm). The AFM image was also consistent with the DLS result (Fig. 3-3). The
yield of the cross-linked nanoparticles increased with an increase in radiation doses, as
shown in Table 3-2. The size of the cross-linked nanoparticles decreased slightly with
an increase in radiation doses, as shown in Table 3-3.
Fig. 3-2 TEM image of polypeptide I particles cross-linked by gamma irradiation (A) and the size distribution histogram obtained from the TEM image (B). The scale bar in the micrograph represents a length of 600 nm. The size distribution histogram was obtained by measuring the diameter of 120 particles in the TEM image by using the scale bar.
40
Fig. 3-3 AFM image of the polypeptide I particles cross-linked by gamma irradiation.
Table 3-2. Radiation dose response of the yield of the polypeptide I particles. Deposit of the particles obtained by centrifugation of the particle solution at 19,000 rpm at 4°C for 1 hour (HITACHI himac CR21, rotor no. 46) and dissolving in deionized water. Yield is determined by measuring the dry weight of the deposit.
Radiation doses (kGy) Yield (%)
8.5 40
17 50
41
Table 3-3. Effect of the dose rate of gamma-rays. Mean sizes of polypeptide Iparticles were determined by DLS analysis.
Radiation doses (kGy) Size (nm)
42oC Size (nm) 15oC 8.5 380(±50)a 200(±30)a 15 200(±20)a 120(±10)a 30 180(±30)a 170(±50)a a standard deviation
During the “Heat shock” process, because the polypeptide concentration was 25
mg/ml, the initial polypeptide concentration might be higher than that during any other
heating process during particle formation of the polypeptides when the polypeptides
were dropped into hot deionized water at a temperature greater than the CP, although
the final concentration of the polypeptide after particle formation was 5 mg/ml. In
contrast, the polypeptide concentration was maintained at 5 mg/ml all the time during
“Slow heating” and “Fast heating.” Therefore, I compared particle formation by “Slow
heating” and “Fast heating” in aqueous solutions with different concentrations of
42
yielded clouded suspensions after “Slow heating” and “Fast heating,” and no significant
difference was observed before irradiation. After irradiation, nanoparticles were not
formed in the aggregated polypeptide solutions in both 25 mg/ml solution and 5 mg/ml
solution, as described above. These results suggest that the “Heat shock” process was
the most important process with respect to the formation of nanoparticles. I also
attempted to increase the sample volume of the droplet of the polypeptide solution
during the “Heat shock” process by using needles of different gauge sizes. The cooled
polypeptide I solution (25 mg/ml cooled at 4°C) was dropped into hot deionized water
at 42°C with a tuberculin syringe using 18-G (droplet volume, 20 μl) and 27-G (droplet
volume, 5 μl) needles, and the final concentration after aggregation of polypeptide I was
5 mg/ml. A stable particle was obtained when the 27-G needle was used but not
obtained when the 18-G needle was used. The average size of the particles was 150 nm
when the 27-G needle was used. Thus, the 27-G needle was more effective than the
18-G needle in forming stable cross-linked nanoparticles distributed within a narrow
size range.
These results suggest that in cases of “Slow heating” and “Fast heating,” the
hydrophobic side-chain interaction of the polypeptides might be poor and might not be
43
yielded gels and sediments, respectively. It was thought that the inter-molecular contact
between hydrophobic side chains was important to form cross-linked structures.
Moreover, I reconfirmed that repeat of hydrophobic domains such as (GVGVP) was
important to form α-elastin aggregation and cross-linked structure.
Preparation of polypeptide II and III nanoparticles
Effect of the heating process on the aggregation of polypeptide II and III
To study the effect of charge within α-elastin, I used 2 types of elastic model
polypeptide II and III. I attempted to dissolve polypeptide II and III in deionized water.
However, solubility of both polypeptide II and III were low in the deionized water at
4oC and any CP could not be observed at any temperature in deionized water. Therefore,
polypeptide II and III were prepared in 10 mM phosphate buffer (PB). In the solution of
polypeptide II in PB (7 mg/ml), polypeptide II underwent aggregation at a temperature
corresponding to 30oC with an increase in temperature. However, polypeptide III did
not show CP in deionized water and PB at any temperature.
I examined the effects of the three heating processes on the particle formation of
polypeptide II in PB, in which the time of the heating was varied from 4oC to 50oC. I
attempted to obtain aggregated polypeptide II at a concentration of 7 mg/ml and
44
aggregated polypeptide II. As shown in Table 3-4, polypeptide II was aggregated in PB
(7 mg/ml) with heating at 50oC (above CP) by any of the heating processes, but the
main peak size distribution of the aggregates varied among “Slow heating (1.5oC/min)”
(70 ± 15 nm), “Fast heating (4.6oC/min)” (60 ± 10 nm), and “Heat shock (50oC/min)”
Table 3-4. Effect of the heating process on polypeptide II particle sizes. Mean size of polypeptide II particles measured by DLS analysis at 50°C.
Heating process Irradiation Size (nm)
Slow heating Before 20(±3)a 12% ,70(±15)a 88% After 60(±10)a
Fast heating Before 10(±2)a 4% ,60(±10)a 96% After 55(±10)a
Heat shock Before 80(±20)a
After 40(±10)a 56% ,90(±20)a 44%
a
standard deviation
(80 ± 20 nm), as shown in Fig. 3-5. All the aggregates were dissolved again by reducing
the temperature below CP.
On the other hand, after 30-kGy irradiation, the particles could not be dissolved by
reducing the temperature to 15oC (below CP), and transmittance of the solution was
45
stabilized and lost their thermosensitivity by gamma-ray irradiation cross-linking. I
collected the aggregates by centrifugation at 4oC after the irradiation and analyzed their
size distributions by DLS (Table 3-4). As shown in Fig. 3-6, the mean of the size
distributions was shifted to lower than 100 nm for the preparation conditions “Slow
heating” and “Fast heating”; both these conditions yielded narrow size distribution
containing a single peak around 60 nm, while the other conditions yielded broader and
unstable size distributions containing several peaks. However, the yield rate of particles
for “Slow heating” was more than that of “Fast heating” after gamma irradiation (Table
3-5). Figure 3-7 shows the cross-linked nanoparticle image obtained from AFM. On the
basis of these observations, I assume that nanoparticles were formed from the
aggregated polypeptide II particles by gamma-ray irradiation cross-linking and that
“Slow heating” was the optimum process to obtain crosslinked nanoparticles with a
46
Fig. 3-5 Mean size of the aggregated polypeptide II particles by the three heating processes measured by DLS before gamma irradiation ((A): Slow heating (B): Fast heating (C): Heat shock).
Fig. 3-6 Mean size of the aggregated polypeptide II particles by the three heating processes measured by DLS after 30 kGy gamma irradiation ((A): Slow heating (B): Fast heating (C): Heat shock).
47
Table 3-5. Yield rate of polypeptide II nanoparticles after gamma irradiation by using different heating methods. Supernatant obtained by centrifugation of the suspension at 19,000 rpm at 4°C for 1 hour (HITACHI himac CR21, rotor no.46) was dried. The yield was determined by measuring its dry weight.
Heating process Yield (%)
Slow heating 60
Fast heating 30
Fig. 3-7AFM image of polypeptide II nanoparticles cross-linked by gamma irradiation (Preparation by “Slow heating”).
48
Table 3-6. Radiation dose response of the yield of polypeptide II nanoparticles. Deposit of the particles was obtained by centrifugation of the particle solution at 19,000 rpm at 4°C for 1 hour (HITACHI himac CR21, rotor no.46) and dissolved in deionized water. The yield was determined by measuring the dry weight of the deposit.
Radiation doses (kGy) Yield (%)
8 60
32 70
Table 3-7. Effect of the dose rate of gamma-rays. Mean sizes of polypeptide II particles were determined by DLS analysis.
Radiation doses (kGy) Size (nm) 50oC Size (nm) 15oC 8.5 50(±10)a 40(±10)a 15 40(±10)a 35(±10)a 30 60(±10)a 70(±20)a a standard deviation
I also investigated the effect of gamma irradiation dose on the formation of stable
nanoparticles by using the “Slow heating” condition. At 8 and 32 kGy irradiation doses,
the yield of the cross-linked nanoparticles collected at 4oC after irradiation was
49
distributions of the particles were within the order of less than 100 nm at 15oC (Table
3-7).
From these results, it is suggested that the heating process might be crucial to form
stable nanoparticles of aggregated polypeptide II by gamma irradiation. Generally,
polymer chains should be close to each other to obtain efficient cross-linking. “Slow
heating” might contribute to efficient aggregation to help cross-linking. In cases of
“Heat shock” and “Fast heating,” the hydrophobic side-chain interaction of the
polypeptides might be poor and might not be effective to form the cross-linked structure.
The inclusion of charged amino acids within the polypeptides would take a longer time
for interaction of hydrophobic side chains because of repulsion of charges.
I also attempted to prepare aggregated polypeptide III by each heating process at a
temperature greater than the CP. However, polypeptide III did not show CP in deionized
water and PB at any temperature. Therefore, aggregated polypeptide III could not be
prepared like aggregated polypeptide II. Thus, I could not obtain polypeptide III
nanoparticles.
From these results, it is suggested that the “Slow heating” process is the optimum
process to aggregate polypeptides containing charged amino acids, because it causes
50
cross-linked nanoparticles could not be obtained without aggregation of the
polypeptides. To efficiently obtain cross-linked nanoparticles of α-elastin, “Slow
heating" is required because α-elastin contains charged amino acids.
In the next chapter, to obtain information about aggregation of the hydrophobic
domain (GVGVP) within α-elastin, I focus on the heating process for optimizing
nanoparticle formation. I changed the heating rate and monitored the aggregation
process of polypeptide I by circular dichroism (CD) spectrometry in order to yield the
nanoparticles more efficiently. Moreover, I used molecular dynamics (MD) simulation
to analyze the mechanism of aggregate formation. I also used simple amino acids such
as Val, Pro, and Gly for analyzing cross-linked points.
Conclusions
Stable nanoparticles of polypeptide I were successfully obtained by gamma-ray
cross-linking on increasing the temperature to a value greater than the CP with the
“Heat shock” process and by using a needle with a small droplet volume before
irradiation and maintaining the temperature during gamma irradiation. The size of the
nanoparticles was ca. 150 nm.
51
irradiation through choosing the appropriate heating rate before irradiation and
maintaining the temperature during irradiation. Although stable cross-linked
nanoparticles of polypeptide II were formed by gamma irradiation with the “Slow
heating” process, the size of the nanoparticles was approximately less than 100 nm.
References
[1] Urry, D. W., Biorefinery (34), In: Kamm, B., Gruber, P. R., Kamm, M. (eds)
Biorefineries- industrial process and products, Status quo and future directions, 2
volumes. Wiley VCH, Weinheim, 2006, pp. 217-253.
[2] Urry, D. W., Nichol., A., Mcpherson, D. T, Harris, C. M., Parker, T. M., Xu, J.,
Gowda, D. C., Shewry, P. R., Properties, preparations, and applications of bioelastic
materials. In: Wiseman, D. M.., Trantolo, D. J., Altobelli, D. E., Yaszemski, M. J.,
Gresser, J. D., Schwartz, E. R., (eds), Encyclopedic handbook of biomaterials and
bioengineering-Part A-Materials, Volume 2. Marcel Dekker, New York, 1995,
52
Chapter 4 Analysis of the aggregation process of (GVGVP)251 (polypeptide I):
Optimization of nanoparticle formation by gamma-ray cross-linking
Introduction
Stable nanoparticles of polypeptide I was successfully obtained by gamma-ray
cross-linking. The temperature was increased to a value higher than the CP using a heat
shock process. A needle with a small droplet volume was used before irradiation and the
temperature was maintained during the gamma irradiation as described in Chapter 3.
Aggregation above the CP was found to be essential in order to obtain the cross-linked
nanoparticles using the heat shock process. This suggests that the repeating hydrophobic
GVGVP motif of polypeptide I play a crucial role in the formation of the cross-linked
nanoparticles. Moreover, heat shock might help to facilitate the process of formation of
the nanoparticles.
Murasato et al., developed a novel C3 symmetric peptide conjugate known as
“Wheel-FKFE.” This conjugate consists of three β-sheet-forming peptides formed in a
wheel-like arrangement. It was demonstrated that the nano-fibers are formed by
stacking and hydrogen bonding interactions between the molecules. Each fiber has an
extended β-sheet structure and a hydrophobic cavity as determined by circular
dichroism (CD) spectroscopy and fluorescence spectroscopy in combination with
53
of polypeptides [1]. This study indicates that the specific conformation produced by
hydrophobic interactions of the polypeptide sequence might be important for the
cross-linking process. The aggregation process of polypeptide I was investigated under
the three different heating processes described in previous chapters (“Slow heating”,
“Fast heating” and “Heat shock”) using circular dichroism (CD) spectroscopy and the
hydrophobic characteristics were monitored using a fluorescent reagent (ANS).
Li et al., simulated the molecular basis for the extensibility of the (GVGVP)18
elastin model polypeptide by molecular dynamics (MD). MD is a form of computer
simulation in which atoms and molecules are allowed to interact for a specified period
of time by approximations of physical parameters. This provides a view of the motion
of the atoms and molecules [2-4]. The principles of MD have also been described as
“statistical mechanics by numbers” and “Laplace's vision of Newtonian mechanics.”
MD is used to predict biomolecular interactions and provide insights into molecular
motion at the atomic scale [7-8]. Therefore, in order to characterize the aggregation
process, the aggregation of three molecules of (GVGVP)18 in water was simulated by
MD to identify the amino acid residues involved in hydrophobic interactions among the
three molecules. The reactivity of single amino acid mixtures containing a combination
54
to identify the cross-linking points.
Materials and Methods Materials
Elastin protein-based polypeptide I: (GVGVP)251 was supplied from Prof. Urry
(Minnesota University) and BRL (Bioelastic Research, Ltd.) through Bioelastic Japan
Co. [5]. 8-Anilino-1-naphthalenesulfonic acid magnesium salt (ANS-Mg)
((C6H5NHC10H6SO3)2Mg ・H2O) was obtained from Nacalai Tesque, Inc. Sodium
chloride (NaCl), potassium chloride (KCl), disodium hydrogen phosphate (Na2HPO4)
potassium dihydrogen phosphate (KH2PO4), valine, glycine, and proline were obtained
from Wako Pure Chemical Industries Ltd.
Circular dichroism spectroscopy of aggregated polypeptide I
The polypeptide structure was analyzed using circular dichroism (CD)
spectroscopy after heating to 42ºC using three different heating processes, “Slow
heating”, “Fast heating”, and “Heat shock”.
Aqueous solutions of the polypeptide I were prepared at 4ºC and heated to 42ºC
Slow heating: 25 mg of the polymer was dissolved in 5 ml of deionized water in a
55
heated from 4ºC to 42ºC at a rate of 1.3°C/min while stirring in a water bath (EYELA
SB-350) with a magnetic stirrer (EYELA RCN-3D) at a speed of 600 rpm.
Fast heating: 25 mg of the polymer was dissolved in 5 ml of deionized water at
room temperature (5 mg/ml) in a stoppered Pyrex glass test tube and cooled to 4ºC. The
polymer solution was quickly heated from 4ºC to 42ºC for 10 min. while stirring in a
water bath (EYELA SB-350) with a magnetic stirrer (EYELA RCN-3D) at a speed of
600 rpm.
Heat shock: 25 mg of the polymer was dissolved in 1 ml of deionized water at
room temperature and cooled to 4ºC. Drops of the solution at 4ºC were added to a 4-fold
volume of water at 42ºC in a stoppered Pyrex glass test tube using a 1-ml tuberculin
syringe with a 27-G needle (0.4 × 19 mm) (TERUMO) while stirring in a water bath
(EYELA SB-350) with a magnetic stirrer (EYELA RCN-3D) at a speed of 600 rpm.
The heated samples were diluted to 0.5 mg/ml in deionized water to bring the
absorbance within the measurable range (<1.0) for CD spectroscopy.
CD spectra were measured using a JASCO J-720 spectropolarimeter (JASCO
Corporation, Tokyo, Japan) with a standard analysis program. The temperature was
controlled using a recirculating water bath (42ºC), and the spectra were recorded over
56
scanning speed of 50 nm/min. The spectral bandwidth was 1.0 nm, and the integration
time was 5 s. Data are represented as molar ellipticities ([θ] deg cm2/d mol).
Gamma irradiation
The heated samples were placed in thermos bottles (TIGER MWE-C350) and
irradiated with 60Co gamma-rays at doses of 8.5–30 kGy using the 60Co-gamma
irradiation pool at Osaka Prefecture University (dose rate: 10.5 kGy/h). The temperature
of the sample was maintained at 42ºC during irradiation. The gamma irradiated samples
were subjected to CD spectroscopy in the same manner as described above.
Fluorescence spectroscopy of polypeptidemixed with 8-anilino-1-naphthalenesulfonic
acid magnesium salt
The aggregates of polypeptide Iprepared by each heating process were mixed with
8-anilino-1-naphthalenesulfonic acid magnesium salt (ANS-Mg) in 10 mM PBS. The
concentrations of the polypeptide and the ANS-Mg were 5 mg/ml and 0.1 mM,
respectively. Fluorescence spectroscopy measurements were performed using a
Shimadzu spectrofluorophotometer RF-5000 (Shimadzu Co. Ltd.). The emission spectra
57
Analyzing the aggregation of the (GVGVP)18 with molecular dynamics simulation
Molecular dynamics simulations were performed using an energy refinement
(AMBER) force field [9] with the Groningen Machine for Chemical Simulations
(GROMACS 3.3.0) software package [10] using the GROMACS 96 force field [11] and
the TIP3P water model. Each molecular system simulated was described using an
all-atoms representation of both the polypeptide and water. The idealized β-spiral
structure proposed by Urry was used as the starting structure for all simulations [2, 12].
The initial structure was immersed in a periodic water box with a truncated octahedral
shape. Electrostatic energy was calculated using the particle mesh Ewald method [13].
Cutoff distances for the calculation of the Coulomb and van der Waals interactions were
set at 0.9 nm. After energy minimization using a steepest descent method, the system
was equilibrated at 315 K and normal pressure for 20 picoseconds (ps) under the LINCS
constraints [14] for all bonds. The system was coupled to the external bath by the
Berendsen pressure and v-rescale temperature coupling [15]. The final MD calculations
were performed under the same conditions used for the 10000 ps calculations. The
58
Amino acid analysis of irradiated single amino acid mixtures
Single amino acid mixtures combining single amino acids such as Val-Val,
Pro-Pro, Gly-Gly, Val-Pro, Val-Gly, Pro-Gly, and Val-Pro-Gly were prepared. Each
mixture was dissolved in deionized water (concentration:1720 nmol/ml) and irradiated
with 60Co gamma-rays at a dose of 30 kGy as described above. After gamma irradiation,
these samples were freeze dried and redissolved in 0.1 N HCl (concentration: 200
nmol/ml). Amino acid analyses were performed with a HITACHI Model L-8500
analyzer using MCl buffer and ninhydrin.
Results and Discussion
CD spectroscopy of polypeptide I
Polypeptide I prepared by the three different heating processes (“Slow heating”,
“Fast heating” and “Heat shock”) was characterized by CD spectroscopy (0.5 mg/ml in
deionized water) at 42ºC (above CP). The CD spectra of the samples are shown in Fig.
4-1. The absorbance of the aggregated polypeptides in an aqueous solution was under
OD 1.0, and the intensities of bands of the CD spectra were within the measurable range.
As shown in Fig. 4-1, the CD spectrum of the sample prepared by “Heat shock” has a




![Table 2-5. Amino acid analyses for α-elastin. Values are expressed as residues per thousand residues [10]](https://thumb-ap.123doks.com/thumbv2/123deta/8513415.1805675/32.892.122.779.259.1066/table-analyses-elastin-values-expressed-residues-thousand-residues.webp)