薬 剤 学, 81 (2), 182-199 (2021)
〔一 般 論 文〕
湿式粉砕及び滴下凍結乾燥法による
溶解性改善型シクロスポリン含有顆粒剤及び錠剤の設計
小林 正人,近藤 啓太,中田 有紀,船越 映美,丹羽 敏幸*
名城大学薬学部製剤学研究室Design of Cyclosporine Granules and Tablet with Improved Dissolution
Characteristics Using Wet Milling and Drop Freeze-Drying Techniques
MASATO KOBAYASHI, KEITA KONDO, YUKI NAKATA, EMI FUNAKOSHI and TOSHIYUKI NIWA*
Laboratory of Industrial Pharmacy, Faculty of Pharmacy, Meijo University, 150 Yagotoyama, Tempaku-ku, Nagoya, 468–8503, Japan
(Received 9 October 2020; Accepted 21 November 2020)
Summary: Solid pharmaceutical formulations for oral use (granule, tablet) with improved solubility of poorly water-soluble cyclosporine (Cyc) were designed using the combination technique consisting of wet-milling and the drop freeze-drying (DFD) process. The particles of Cyc were dispersed in the aqueous solution composed of hydroxpropyl cellulose as a milling promotor and sodium lauryl sulfate as a dispersion stabilizer, and pulverized into nanometer-sized particles by the wet beads milling technique. Subsequently, the resultant aqueous Cyc nanosuspension with co-dissolving mannitol (MNT) was dropped into liquid nitrogen and freeze-dried to obtain the dried granules. The granules with a wide content range (6.67–47%) could be prepared by adjusting the amount of MNT added. Furthermore, the tablets containing 10 mg of Cyc were manufactured by directly compressing the granules. The kinetic dispersing test indicated that the granules and tablets prepared through these sequential processes had self-dispersion characteristics of Cyc particles. When they were placed in aqueous phase, submicron-sized Cyc particles were spontaneously dispersed. As a result, the granules showed an excellent dissolution behavior of more than 95% at 10 min. On the other hand, the tablets showed fast and high dissolution behavior of more than 80% at 30 min despite no added disintegrant. The outcome of this research would be a new platform technology for improving solubility, which would enable the development of solid formulations with poorly absorbable drugs.
Keywords: wet-milling; drop freeze-drying; self-dispersion; cyclosporine; microsponge; tablet
Cyclosporine(Cyc)は,臓器移植における拒絶 反応の抑制や,乾癬,アトピー性皮膚炎の治療に用 いられる免疫抑制剤である 1).日本では Cyc の経口 投与製剤として液剤,軟カプセル剤及び細粒剤が市 販されているが 2~4),経口投与製剤として汎用される 顆粒剤,錠剤としての製品はない.市場に複数の剤 形を提供することは,患者に多くの選択肢を与え多 様化するニーズに応えることができ,服薬アドヒア ランスの向上につながる.特に錠剤は服用しやすく, 簡便に取り扱えることから医療現場からの要求は高 いものの,先発薬・後発薬を見渡しても市販品が提 供されていないのは,開発の難易度が高いことを物 *〒468–8503 愛知県名古屋市天白区八事山 150 名城大学薬学部製剤学研究室 TEL:052–839–2662,FAX:052–839–2662 E-mail: [email protected]
語っている. Cyc は超難水溶性の薬物であるため,市販医薬品 はいずれも,特別な溶解性改善技術を利用して製剤 化されている 5).すなわち,Cyc の溶解性,ひいては 経口吸収性を効果的に向上させる製剤化技術が, Cycの経口投与製剤を開発する上での鍵となる.Cyc の溶解性向上に関する研究論文は数多くあるもの の,経肺投与製剤を志向した報告が主であり 6~12), 顆粒剤,錠剤などの経口固形製剤化に関する報告は 殆ど見られない. 難水溶性薬物の溶解性改善技術として,湿式粉砕 法が学術領域のみならず,製薬産業においても広く 検討され,溶解性や消化管吸収性の改善を図った報 告が散見される 13~15).当研究室では,湿式ビーズ粉 砕にて得た薬物ナノ懸濁液を噴霧凍結乾燥(Spray freeze-drying: SFD)法にて粉末化し,水相へ投入 した際に薬物ナノ懸濁液を再構築し,溶解速度が大 幅に改善した機能性粒子を設計する製剤化技術につ いて報告してきた 16, 17).本手法による生成物は乾燥 粉末であるため錠剤や顆粒剤などの固形製剤の開発 には適性を有するものの,液滴の噴霧を通じて得ら れた粒子の大きさは 5~20 μmと微粉なため,打錠 やカプセル充填といった製剤化工程において流動性 や充填性などのハンドリング性に課題があった. そこで著者等は,噴霧に代えてノズルを通して薬 液を液体窒素中に自由落下する滴下凍結乾燥(Drop freeze-drying: DFD)法を先に開発した 18).凍結乾 燥後に得られる粒子は,液滴径を反映した 200~500 μmほどの顆粒サイズであり,固形製剤化工程にお ける粉体操作に適した粉末が得られることを示し た.更に,滴下凍結乾燥法を湿式粉砕法と組み合わ せ,湿式粉砕した薬物ナノ懸濁液を滴下凍結乾燥処 理することで,サブミクロン Cyc 粒子を内包した球 形顆粒を調製する製剤技術へと発展させた 19).本工 程にて得られた顆粒は水相に投入後,速やかに包埋 されたナノサイズの薬物一次粒子へと再分散し,難 水溶性 Cyc の溶出性を大きく改善することを明らか にした.本法にて得られる顆粒は多孔質球体を呈し ており,その特徴的構造からマイクロスポンジと命 名した. 日本の医薬品市場にはない汎用剤形(錠剤及び顆 粒剤)を開発することは,医療現場での Cyc による 薬物治療の機会を向上するものと考える.また,本 技術を Cyc 以外の難水溶性薬物へ適用することで, 難水溶性のため開発途中で脱落する約 3 分の 1 の候 補化合物の救済にもつながり 20),製薬産業の更なる 発展に貢献できる.既報 19)ではマイクロスポンジの 設計方法とその物理化学的特性,並びに溶解改善効 果について報告したが,消化管吸収を模した日局溶 出試験第 1 液及び第 2 液での溶出率が水に比べ低値 になる課題があった.加えて,マイクロスポンジ中 の Cyc 含量は 47% と高含量製剤であり,Cyc 製剤 の臨床用量が適応症及び患者の体重に応じて用量調 整されている現状から,市販の細粒剤(17%)と同 様な低含量の顆粒剤,更には現在市販されていない 錠剤の開発が望まれている. そこで筆者等は,マイクロスポンジに配合する添 加剤を処方設計することで,溶解性の向上を図った 顆粒剤や錠剤の開発について試みた.Cyc 顆粒剤の 調製においては,口腔内崩壊錠用賦形剤として汎用 されるマンニトールを添加し,Cyc 含量の調節を試 みた.Cyc 錠の調製では,従来のマイクロスポンジ に賦形剤を混合し,混合末としての打錠を試みたが, マイクロスポンジがかさ高く,混合時の含量均一性 や臼へ充填しきれないといった課題が明らかとなっ た.一般的な改善策として,マイクロスポンジを賦 形剤と造粒し,打錠に適した顆粒を調製することを 発案した.しかしながら,湿式造粒法 21~23)では水と 接触する造粒過程において,乾式造粒法 24)では圧 縮・解砕過程においてマイクロスポンジが保持する 多孔質構造に基づく特有の機能(優れた溶解性改善 性,圧縮成形性)が消失してしまう点が懸念された. そこで,造粒工程を経由せず Cyc 顆粒を更なる添加 剤を配合することなく,直接打錠する簡便かつ効率 的な製法とすることを企図した. 本研究では超難水溶性薬物である Cyc の溶解性の 改善を図った Cyc 顆粒・錠剤を開発することを企図 し,(1)Cyc 顆粒及び錠剤の基本処方となるマイク ロスポンジの処方成分の最適化,(2)含量の異なる Cyc顆粒の設計,及び(3)造粒工程を経由しない直 打法による Cyc 錠の設計について検討を行った.ま た,得られた製剤の物理化学的・薬剤学的物性につ いて評価したので,以下に報告する.
実 験 の 部 1.試 料
難水溶性薬物としてシクロスポリン(Cyc,純度: 99.5%,Teva Czech Industries s.r.o.)を用いた.本 研究で使用した Cyc はレーザー回折・散乱法による メジアン径(D50)が 5.7 μmの粉砕品であり,粉末 X線回折法により非晶質であることを確認した.顆 粒剤及び錠剤の賦形剤として安定結晶形のβ型マン ニトール(MNT)(マンニット P,三菱商事ライフ サイエンス)を採用した.また,湿式粉砕工程にお ける粉砕促進剤として水溶性高分子のヒドロキシプ ロピルセルロース(HPC)(HPC-SL,日本曹達), 分散安定化剤として界面活性剤のラウリル硫酸ナト リウム(SLS)(富士フイルム和光純薬),錠剤製造 の打錠工程の際に使用する滑沢剤としてステアリン 酸マグネシウム(Mg-St)(富士フイルム和光純薬) を用いた.顆粒剤及び錠剤中の MNT の結晶性を確 認する目的で,結晶形帰属の標準品としてδ型 MNT (パーテック デルタ M,メルク)を用いた.また, α型 MNT を Lee 等に報告されている方法に従い調 製した 25).精製水と 99.5% エタノールを体積比 3: 7で混合した溶媒 100 mL にβ型 MNT 15 g を添加 し,加熱して溶解させた.その後,室温で静置し, 成り行きにて放冷して得た析出物をろ過及び乾燥 し,α型 MNT を得た. 2.製剤の調製方法 2.1 Cyc ナノ懸濁液の調製 Cyc 粒子の粉砕には当研究室にて開発した湿式ビ ーズ粉砕法を適用した 15).50 mL 容量のプラスチッ ク製コニカルチューブに粉砕媒体として直径 0.3 mmのジルコニアビーズ(YTZ-03,ニッカトー)60 g及び Cyc 600 mg を仕込み,HPC 150 mg と SLS 30 mg を水に溶解した混合水溶液 15 mL を加えた. 湿式粉砕装置(マルチビーズショッカー,安井器械) を用い,粉砕温度を 0˚C に設定し,2,700 rpm の速 度で 12 分間撹拌して得られた粉砕溶液を目開き 150 μmのふるいに通してビーズを除去し,Cyc ナ ノ懸濁液を調製した.Cyc:HPC:SLS の配合比 は,先の報告にて得られた最適値である 20:5:1 と固定した(粉砕 Cyc 粒子の 50% 累積径は 0.504 μm) 19).湿式粉砕はすべての処方につき,同一成分・
組成比,同一粉砕条件とした(Table I,II の Wet-mill-ing処方欄).
2.2 滴下凍結乾燥法によるマイクロスポンジ及び Cyc 顆粒の調製
湿式粉砕工程により調製した Cyc ナノ懸濁液に, Table I及び Table II の Drop freeze-drying(DFD)
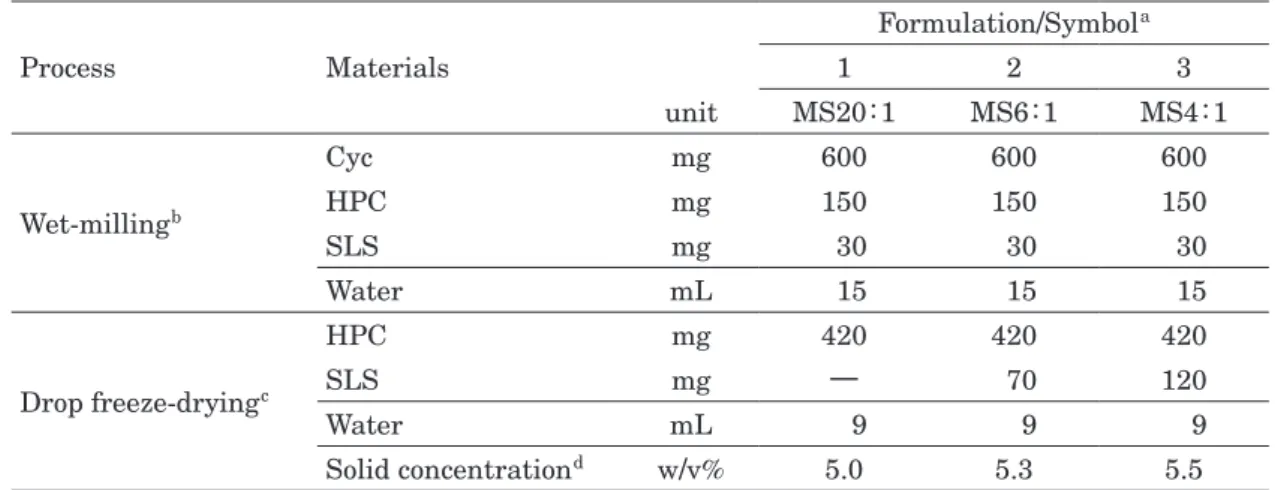
TABLE I Formulation of wet-milling process to produce Cyc nanosuspension and drop freeze-drying
process to produce microsponges with different SLS composition.
Process Materials Formulation/Symbol a 1 2 3 unit MS20:1 MS6:1 MS4:1 Wet-milling b Cyc mg 600 600 600 HPC mg 150 150 150 SLS mg 30 30 30 Water mL 15 15 15 Drop freeze-drying c HPC mg 420 420 420 SLS mg ― 70 120 Water mL 9 9 9 Solid concentration d w/v% 5.0 5.3 5.5
a MS20:1, MS6:1, MS4:1 indicate microsponge (MS) with weight ratio of Cyc: SLS =20:1, 6:1, 4:1 respectively.
b The component/composition in wet-milling process was common in all formulations.
c The composition of SLS was changed to set weight ratio of Cyc: SLS=20:1, 6:1, 4:1 in suspen-sion to be supplied to drop freeze-drying process.
d The solid concentration in nanosuspension to be supplied to drop freeze-drying process was set to around 5 w/v% in all formulations.
処方欄に示した処方量の HPC,SLS 及び MNT を 加え,希釈用の精製水を追加添加し,撹拌混合して HPC,SLS 及び MNT が完全に溶解した滴下溶液を 調製した.この滴下溶液をカプセル化装置(B-390, 日本ビュッヒ)の液滴形成機能を用い,マグネティ ックスターラーによる撹拌の下,液体窒素中に振動 滴下した.適度な大きさの顆粒を得るため,孔径 120 μmのノズルを用いて,1 mL
/
minの速度で送液し, 振動周波数 2,200 Hz,電極電圧 650 V の条件下で滴 下した.液体窒素中で凍結した氷結粒子を凍結乾燥 機(FD-550,東京理化器械)に移して乾燥し,マイ クロスポンジまたは Cyc 顆粒を得た.凍結乾燥は全 ての調製において,-30˚C で 24 時間凍結し,その 後 8 Pa 以下で 30˚C まで徐々に昇温させて行った. 本報告では MNT 未配合の滴下溶液から得られた凍 結乾燥粒子をマイクロスポンジ,MNT 配合凍結乾 燥粒子を顆粒と表記した.Table I には,マイクロス ポンジ中の SLS 配合量の最適化を図った検討処方 を示した(Formulation 1, 2, 3).湿式粉砕法により 調製された Cyc ナノ懸濁液に HPC(一定量)と SLS (可変量)を加え,更に希釈用の精製水を添加し HPC と SLS を完全に溶解させ,Cyc:SLS の配合比が 20:1,6:1 及び 4:1 となるように調製し,それ ぞれの処方名を MS20:1,MS6:1 及び MS4:1 とし た.処方名 MS は Microsponge を略記する. 次に MNT を配合した顆粒の処方を Table II に示 す.①SLS の配合量を最適化した MS6:1(Formu-lation 2)に基づき,Cyc ナノ懸濁液に添加する HPC と SLS の量を 420 mg と 70 mg に固定し,調製す る 顆 粒 中 の Cyc 含 量 が 6.67,10.00,20.00 及 び 30.00%となるように MNT の配合量を調節した (Formulation 4, 5, 6, 7).ここで,滴下溶液の固形 分濃度が 5% となるよう水の添加量を調整した.処 方名を顆粒中の Cyc 含量に対応させ,GR-C6.67%, GR-C10%,GR-C20%,GR-C30%,GR-C47%(MS 6:1 と同一処方)とした(GR は Granule を,C 以 降の表記は Cyc の含量率を示している).続いて, ②Cyc 含 量 率 が 6.67% の Formulation4(GR-TABLE II Formulation of wet milling process and drop freeze-drying process to produce granules with various Cyc
contents.
Process Materials unit
Formulation/Symbol a 4 5 6 7 2 8 9 10 GR-C6.67% (GR-S5%) GR-C10% GR-C20% GR-C30% GR-C47% (MS6:1) GR-S10% GR-S15% GR-S20% Wet-milling b Cyc mg 600 600 600 600 600 600 600 600 HPC mg 150 150 150 150 150 150 150 150 SLS mg 30 30 30 30 30 30 30 30 Water mL 15 15 15 15 15 15 15 15 Drop freeze-drying HPC mg 420 420 420 420 420 420 420 420 SLS mg 70 70 70 70 70 70 70 70 MNT mg 7,730 c 4,730 c 1,730 c 730 c 0 c 7,730 7,730 7,730 Water mL 165 d 105 45 25 9 75 d 45 d 30 d Cyc content in granules w/w% 6.67 10.00 20.00 30.00 47.24 6.67 6.67 6.67 Solid concentration in suspension w/v% 5 5 5 5 5.3 10 15 20
a GR-C6.67%, GR-C10%, GR-C20%, GR-C30%, GR-C47% indicate granule (GR) containing 6.67%, 10%, 20%, 30%, 47% of Cyc (C), respectively. GR-S5%, GR-S10%, GR-S15%, GR-S20% indicate granule (GR) prepared from nanosuspension with 5%, 10%, 15%, 20% of solid concentration (S).
b The component/compositionwas in wet-milling process common in all formulations.
c The amount of MNT in suspension to be supplied to drop freeze-drying process was changed to produce the granules with various Cyc contents.
d The supplemental volume of water was changed to set the various solid concentrations in suspension to be supplied to drop freeze-drying process.
C6.67%)に基づき,Cyc 錠の作製に適した打錠用顆 粒の調製処方を検討した(Formulation 8, 9, 10). Cycナノ懸濁液に添加する HPC,SLS 及び MNT の 量を 420 mg,70 mg 及び 7,730 mg に固定し,滴下 溶液の固形分濃度が 5,10,15 及び 20% となるよ う水の添加量を調整した.処方名を固形分濃度に応 じて GR-S5%(GR-C6.67% と同一処方),GR-S10%, GR-S15%,GR-S20% とした(GR は Granule を,S 以降の表記は滴下溶液の固形分濃度(Solid concen-tration)を示している).打錠用顆粒は GR-C6.67% の処方を用い固形分濃度を調整し検討したため, GR-C6.67%と GR-S5% は同一処方である. 2.3 Cyc 錠の調製 直径 8.0 mm の円形平型杵臼に DFD 法にて調製 した打錠用顆粒 150 mg(Cyc として 10 mg を含有) を手作業により充填し,万能圧縮引張試験機(オー トグラフ AG-X,島津製作所)を用い圧縮速度 10 mm
/
min,25~75 MPa の各種打錠圧にて圧縮成形 して錠剤を得た.GR-S5%,GR-S10%,GR-S15%, GR-S20%より製した錠剤名をそれぞれ TB-S5%, TB-S10%,TB-S15%,TB-S20%とし,TBはTablet, S以降の表記は打錠用顆粒と同様である.なお,杵 臼表面にあらかじめ 1 w/
v%ステアリン酸マグネシ ウムのエタノール懸濁液を塗布・乾燥して滑沢処理 した.一方,錠剤からの Cyc 粒子の分散性を評価す るための試料として,直径 2.8 mm の円形平型杵臼 に顆粒 10 mg(Cyc として 0.67 mg を含有)のミニ 錠剤を作製した(圧縮速度:0.1 mm/
min,打錠圧: 50 MPa).得られた製剤は,シリカゲルを入れたデ シケーター内で 24 時間以上乾燥させ,錠剤物性の 試験に供した. 3.評 価 方 法 3.1 走査電子顕微鏡による表面及び断面の観察 調製した顆粒を導電性粘着両面テープを貼り付け たサンプル台にのせ,プラチナ蒸着(JFC-1600,日 本電子)した後,4~10 kV の加速電圧下で走査型電 子顕微鏡(SEM)(JSM-6060,日本電子)により表 面を観察した.また,錠剤については,ロードセル 式硬度計にて破断した半錠を切断面が天面を向くよ うに設置し,断面を観察した. 3.2 粒度分布測定 調製した顆粒を圧縮空気(0.4 MPa)にて乾式分 散し,粒度分布をレーザー回折・散乱式粒度分布測 定装置(LMS-30,セイシン企業)により測定した. 質量基準の累積粒度分布曲線における 10,50,90% 累積径(D10,D50,D90)を代表値とした. 3.3 粒子密度の測定 DFD 法にて Cyc 顆粒を調製する際の滴下溶液 (GR-S5%,S10%,S15%,S20%,Table II)を, 滴下装置でなく駒込ピペットを用いて大きな液滴状 となるよう液体窒素中に滴下し,続いて凍結乾燥す ることで粒子密度測定用の巨大顆粒を調製した.得 られた巨大顆粒 20 個をデジタルマイクロスコープ (SE-40Z;セルミック)で撮影し,その映像を画像 解析(LUZEX AP,ニレコ)して Heywood 径(d i)を測定し,顆粒を真球と仮定して(1)式より顆粒 20個分の体積(V i)を算出した.画像解析に供した 20個の巨大顆粒の総質量(W)を秤量し,(2)式に て粒子密度(ρ)を算出した. V i=πd i3
/
6 (1) ρ=W/
Σ i20=1V i (2) 3.4 流動性及び充填性の測定 調製した顆粒を目開き 1,000 μmのふるいを通過 させながらロートの孔から質量(M 0)の 5 cm 3のメ スシリンダーに摺り切りいっぱいになるまで充填さ せた.粉体充填時の全質量(M 1)を測定し,式(3) よりかさ密度(最疎かさ密度,ρbulk)を算出した. 厚さ 40 mm のスペーサを用い,顆粒が充填された メスシリンダーをマルチテスター(MT-1001k,セ イシン企業)により 1,000 回(2 回/
秒)タッピング させ,減少後の試料の体積(V 1)をメスシリンダー で読み取り,式(4)よりタップ密度(最密かさ密 度,ρtap)を算出した.また式(5)よりかさ体積の 減少率を表す圧縮度を算出した. ρbulk(g/
cm 3)=(M 1-M 0)/
5 (3) ρtap(g/
cm 3)=(M 1-M 0)/
V 1 (4) Compression ratio(%)=(ρtap-ρbulk)
/
ρtap×100 (5)3.5 比表面積の測定 専用のサンプルセルに試料を適量充填し,室温下 で 6 時間減圧乾燥した.続いて,液体窒素による冷 却下,アルゴンガス吸着装置(Nova-1000,Quan-tachrome)を用い,0.1 から 0.3 までの 5 つの異な る相対圧力でアルゴンガスを吸収させて得られた吸 着等温線を BET 式に当てはめ,比表面積を求めた.
3.6 厚み及び硬度の測定 錠剤の厚みはデジタルリニアゲージ(DG-933, 小野測器)にて計測した.また,錠剤をロードセル 式硬度計(PC-30,岡田精工)により 1.0 mm
/
secの 速度で直径方向に破断した時の硬度を測定した. 3.7 崩壊時間の測定 第 17 改正日本薬局方崩壊試験法に準拠した崩壊 試験装置(NTR-200,富山産業)にて試験器を 30 回/
分の頻度で上下させて錠剤が完全に崩壊する時 間を測定した.試験液として水を用いた. 3.8 粉末 X 線回折装置による結晶性の測定 使用した原料(Cyc 及び各結晶形の MNT),及び 調製した製剤中の結晶状態を粉末 X 線回折装置 (SmartLab,リガク)により評価した.照射 X 線の 条件は Cu Kα線,Ni フィルター,電圧 40 kV,電 流 30 mA,走査範囲は 5~45°/
2θ,走査速度は 5°/
min,ステップは 0.02° とした.錠剤は粉砕するこ となく試料台の中心部に配置し,あらかじめ計測し ておいた錠剤の厚み分だけ照射面を調整して測定し た. 3.9 製剤からの薬物粒子の分散性評価 レーザー回折・散乱式粒度分布測定装置(LMS-30,セイシン企業)に付属の専用セル(容量 15 mL) 内に分散液として温度 25˚C 付近の水 10 mL を注入 し,室温環境下にて顆粒 2 mg またはミニ錠剤 1 錠 を直接投入し,スターラー撹拌下,15 分間連続して 回折強度を測定し,水に投入後の再分散 Cyc 粒子の 粒度分布推移をリアルタイムに測定した.測定時間 に対しその時の D50 値をプロットし,顆粒または錠 剤からの Cyc 粒子の速度論的粒度分布推移(自発的 分散挙動)を評価した.なお同一検体につき測定を 3回繰り返し,平均的な測定結果(n=1)をグラフ 化した. 3.10 製剤中の薬物含有量の定量 マイクロスポンジ及び Cyc 顆粒の場合は Cyc 5 mg 相当の試料を精密に量り取り,メスフラスコに投入 し下記の HPLC 用移動相を加えて溶解し,正確に 100 mL にメスアップした.この液 1 mL を正確に 量り採り,移動相で正確に 10 mL に希釈した.精密 に秤量した Cyc 10 mg 相当の打錠用顆粒,あるいは 錠剤 1 錠をメスフラスコに投入し,移動相を加えて 溶解して 100 mL とした.この液 1 mL を正確に量 り,移動相で正確に 20 mL とした.これらの溶液を 孔径 0.2 μmの親水性セルロースアセテートタイプ のメンブランフィルター(Dismic-13CP,Advantec) にてろ過したろ液を高速液体クロマトグラフィー (LC-10;島津製作所)により製剤中の Cyc 含量を定 量した.測定条件は以下の通り:カラム;ODS 系逆 相カラム(Inertsil ODS-3, 5.0 μm, 4.6×150 mm, GLサイエンス),カラム温度;70˚C,注入量;100 μL,測定波長;210 nm,流速;1.0 mL/
min,移動 相;アセトニトリル:5 mM 酢酸アンモニウム緩衝 液=75:25.分析時間を 10 分とし,Cyc の保持時 間は 5 分付近であった. 3.11 薬物放出性の評価 顆粒及び錠剤からの Cyc の溶出挙動を第 17 改正 日本薬局方溶出試験パドル法に準拠した溶出試験装 置(NTR-3000,富山産業)を用いて測定した.マ イクロスポンジ及び Cyc 顆粒の場合は Cyc 5 mg 相 当の試料を用い,打錠用顆粒及び錠剤の場合は Cyc 10 mg 相当の試料を用いた.試験液には日局溶出試 験第 1 液(pH 1.2),第 2 液(pH 6.8)及び水を 900 mL用いた.試験液温 37±0.5˚C,パドル回転数 50 rpmの条件にて行った.所定時間(2, 5, 10, 15, 20, 30, 45, 60分)ごとに試料溶液を 5 mL 採取し,孔径 0.2 μmの親水性セルロースアセテートタイプのメン ブランフィルター(Dismic-13CP,Advantec)にて ろ過した最初のろ液 3 mL を廃棄し,次の 1 mL の ろ液をメタノールにて適宜希釈し,前項の Cyc 定量 に示した条件を用い,高速液体クロマトグラフィー (LC-10;島津製作所))により測定した.採取され 減少した液量と同量の試験液を補填し,試験液量を 保持した.Cyc の液温 37˚C における各試験液(水, pH 1.2,pH 6.8)への溶解度はそれぞれ 7.3,7.6, 6.2 μg/
mLと報告されており 26),900 mL に溶解され る Cyc の最大溶解量はそれぞれ 6.57,6.84,5.58 mgと計算された. 結 果 と 考 察 1.マイクロスポンジに配合する SLS 量の最適化 著者等はこれまでに,湿式ビーズ粉砕にて得た薬 物ナノ懸濁液を滴下凍結乾燥法(DFD 法)にて造 粒・乾燥することで,難水溶性薬物である Cyc の溶 解性を飛躍的に向上させた多孔質球形顆粒(マイク ロスポンジ)を開発してきた 19).しかしながら,調 製したマイクロスポンジは,試験液に水を用いた溶出試験においては 30 分以内に 90% 以上の高い Cyc 溶出率を達成できたものの,消化管内での溶出挙動 を模擬した日局溶出試験第 1 液及び第 2 液中では, 溶出率が 80% ほどで頭打ちになる結果となった.そ こで本研究では,いかなる試験液中においても優れ た Cyc 溶出挙動を発揮するマイクロスポンジを設計 するため,処方の最適化について検討した. Cyc 粒子の湿式粉砕工程における処方や製法につ いては既報 19)を踏襲し,得られたナノ懸濁液(D 50 =0.504 μm)に配合する SLS を増量させた 2 種類 のマイクロスポンジ(MS6:1, MS4:1)を新たに製 した(Table I).MS20:1 は既報 19)にて最適化した マイクロスポンジであり,MS6:1,MS4:1 ではマ イクロスポンジ中の SLS 量をそれぞれ 3.3 倍,5 倍 増量している.マイクロスポンジを水相に投入後, 再分散した Cyc 粒子の粒度分布を経時的にモニタリ ングした.Fig. 1(a)は経過時間に対して分散液の メジアン径をプロットしたグラフであり,Cyc 以外 の成分は水に可溶なため Cyc 粒子の再分散挙動を表 している.いずれのマイクロスポンジにおいても, 水中に投入後 1 分以内で滴下凍結乾燥前((a)の点 線にて表記)と同程度の粒子径を持つ Cyc サブミク ロン懸濁液を再構築した.Cyc 粒子を包含する支持 体が親水性添加剤から成るスポンジ構造であること から,速やかに水が浸透し,包埋された Cyc ナノ粒 子が自発的分散(Self-dispersion)したものと推察 した.Pongpeerapat 等は,懸濁液中にて粉砕され た難水溶性薬物である Probucol 粒子表面にポリビニ ルピロリドン(PVP)分子鎖が吸着することで,1 次粒子間の凝集が抑制され,微細な粒子状態が維持 されることを提唱している 27).我々の使用した HPC も PVP と同じ水溶性高分子であることから,再分 散後の Cyc ナノ粒子表面に HPC 分子鎖が吸着し, Cyc粒子間の凝集が抑制されナノ懸濁液の再分散・ 安定化に寄与しているものと考察した. 溶出試験の結果,SLS を増量した 2 種類の処方と
Fig. 1. Effect of SLS concentration in dropping suspension on (a) self-dispersion profiles of microsponge in water and dissolution profiles of Cyc from microsponge in (b) water, (c) JP17 1st fluid, pH 1.2, and (d) JP17 2nd fluid, pH 6.8. Key: Microsponges of (□) MS20:1, (○) MS6:1, (△) MS4:1, (×) Cyc bulk. Dotted line in (a) indicates D50 size of original wet-milled nanosuspension. Each point represents the measured value of n=1 in (a). Each point represents the mean ± S.D. (n=3) in (b), (c) and (d).
も開始 10 分時点で 90% 以上の溶出率に達し(Fig. 1(b), (c), (d)),マイクロスポンジからの Cyc 粒子 の自発的ナノ分散特性に連動していた.また,いず れの試験液においてもプラトー域での溶出率が 100%付近へと上昇し,溶出挙動の改善が認められ た.これは SLS の界面活性作用により表面張力が低 下し疎水性薬物である Cyc の試験液への濡れ性が改 善されたためと考察した.SLS の臨界ミセル濃度 (cmc)は 0.236% と報告されている 28).本実験にて 投入したサンプル量に基づき,溶出試験液中の SLS 濃度を計算すると最大でも 1.85×10 -4%であること から,Cyc の溶解性改善に SLS の可溶化能は影響し ていないと推察した.また,一般的に界面活性剤は 経口投与においてその安全性が問題となることがあ り,SLS についても日本国内における経口投与での 1日最大使用量は 300 mg と多くない.今回のマイ クロスポンジ(MS20:1,MS6:1,MS4:1)では, Cycに対しそれぞれ 1
/
20,1/
6,1/
4量の SLS を含 んでいる.Cyc 製剤の製品情報より国内で最も多く 投与される用法用量は,肝移植における拒絶反応の 抑制に対して 1 日 16 mg/
kgである.体重 60 kg の 患者への Cyc の 1 日投与量(960 mg)から換算する と,最も配合量の多いマイクロスポンジ(MS4:1) を服用する場合,SLS の 1 日投与量は 240 mg とな り,日本での使用前例の上限値(300 mg)に近くな る.そこで,SLS 配合量の少ない処方(MS6:1)を マイクロスポンジの最適処方とした(SLS の 1 日投 与量は最大で 160 mg). 2.Cyc 顆粒の設計 Cyc 製剤は用いる適応症や患者の体重により投与 量が調節され 1),日本国内で市販されている用量調整 が可能な製剤には内用液剤と細粒剤があり,それぞ れの製剤の Cyc 含量は内用液剤が 10%,細粒剤は 17%と各剤形 1 濃度の製品が市販されている 29).前 項にて最適処方とした MS6:1 は,Cyc を 47% 含有 する高含量顆粒剤(GR-C47%)であり,市販製品に 比べ Cyc 含量が高く,用量調整が可能かつ市場に無 い新たな剤形と位置付けられる.本項では多様な用 法用量に柔軟に対応するため,Cyc 含量が 47% より も低い複数の含量の顆粒剤の設計を企図した.湿式 粉砕及び滴下凍結時の処方は MS6:1(Formulation 2)を基本とし,用量調整するため口腔内崩壊錠用 賦形剤として汎用されている MNT を滴下溶液に新 たに添加することとした.なお,MNT を選択した 理由は水溶性であることから,①滴下溶液のノズル 閉塞のリスクを回避でき,②再分散試験においてCyc のみの粒子径が評価できるためである.顆粒剤中の Cyc含量が 6.67%,10%,20% 及び 30% となるよ う MNT 配合量を調整し(Table II,GR-C6.67%, GR-C10%, GR-C20%, GR-C30%),Cyc 含量の異な る顆粒剤を製した.前項までは調製した球形粒子を 既報 19)に合わせてマイクロスポンジと称してきた が,市販製剤としての適用を考慮し,以降は調製物 を顆粒(Granule)と呼ぶこととする.Table II の 処方名 GR は Granule を,C 以降の表記は Cyc の含 量率を示している(C20% は Cyc を 20 w/
w%含有 する).SEM 観察(Fig. 2)及び粒度分布測定の結 果(Fig. 3),いずれの顆粒も D50 値が 300~400 μm の粒度の揃った球形を呈していた.著者等は,湿式 粉砕して得た薬物懸濁液を噴霧凍結乾燥(Spray freeze-drying: SFD)工程にて処理すると,噴霧さ れた液滴が液体窒素中で瞬時に凍結し,生成した氷 粒は自身の形状を保ったまま水分が蒸発するため, 液滴のサイズ・形状と同等な微細な球形粒子が生成 すると報告した 17).本研究では,顆粒サイズの粒子 を製するため,液滴の形成方法を噴霧様式からノズ ル先端部からの重力落下方式へと変更し,滴下凍結 乾燥(Drop freeze-drying: DFD)技術の開発へと至 った.本装置の液滴形成部は,ノズル内径の 2~3 倍 程度の液滴が高速かつ連続的に形成するよう設計さ れており,いずれの処方においても使用したノズル 径(120 μm)の 3 倍程度のサイズを有する球形粒子 が形成された(Fig. 3).本研究では検討しなかった が,DFD 法では使用するノズル径を変更することに より,滴下液滴径,ひいては生成粒子の粒子サイズ を自由に調整できることを以前に報告した 30).得ら れた顆粒の表面を観察すると,MNT を配合してい ない顆粒では,先に報告 19)したように凍結乾燥時に 水分が揮発することで形成される多孔質な顆粒であ るが(Fig. 2 e-2),MNT の配合量が増えるほど表面 の微細な亀裂模様が減少していく様子が観察された (Fig. 2 e-2→a-2).Table II の処方に示した通り, MNTの添加量に応じて滴下液の水量を調整して固 形分濃度を 5% 付近に統一しているため,粒子密度 は異ならないと推測している(後述).表面状態に相 違が生じたのは,MNT の結晶化が液滴表面で推進Fig. 2. Scanning electron microphotographs of granules prepared from dropping suspension with various Cyc concentrations. Key: (a) GR-C6.67% (GR-S5%), (b) GR-C10%, (c) GR-C20%, (d) GR-C30%, (e) GR-C47% (MS6:1). Appearance under 1) low and 2) high magnification.
した結果,HPC からなるネットワーク構造(骨組 み)を緻密に穴埋めする壁土効果をもたらしたため と推察した. 次に,調製した Cyc 含量の異なる顆粒からの Cyc 粒子の再分散性を評価した(Fig. 4(a),挿入図は 10分以降のメジアン径を標準軸に拡大プロットした ものである).いずれの顆粒も投入後 5 分以内にサ ブミクロン懸濁液を再構築し,水相中での優れた自 発的再分散性を呈した.これは水相へ投入後,直ち に顆粒を構成する水溶性基剤が溶解し,HPC
/
MNT マトリックスに包埋されている Cyc ナノ粒子が凝集 することなく一次粒子の状態で水相中に分散したも のと考察した.これら顆粒の Cyc 溶出性を評価した 結果,いずれの顆粒も 10 分時点で 95% 以上の溶出 性を示し,初期溶出速度に優れ,極めて高い Cyc 溶 出性を有する顆粒剤であることが確認された(Fig. 4(b)).6.67~47% の Cyc 含量の異なる顆粒からの Cycの速く高い溶出挙動は,Fig. 4(a)に示した水 相中における優れた自発的再分散性に基づくと推察 した.本研究では,湿式粉砕法と滴下凍結乾燥法を 組み合わせることにより Cyc の溶解性改善型顆粒剤 の調製が可能となった.また,幅広い含量(6.67~ 47%)の顆粒剤が調製可能であることが示された. 3.Cyc 錠の設計 3.1 打錠用顆粒の調製 日本では Cyc 含量が異なる 3 用量(10 mg,25 mg 及び 50 mg)の軟カプセル剤が市販されている.前 述したように Cyc 製剤は用いる適応症や患者の体重 により投与量が調節されている 1).そのため,本研究 では 10 mg, 25 mg, 50 mg の中で細やかな投与量調 整に対応可能な 10 mg 含量を研究の端緒として選択 し,Cyc 錠剤を設計することとした.経口服用性を 考慮し,錠剤径を Cyc 10 mg 軟カプセル剤の長径と 同じく 8 mm とし,試打検討を踏まえて錠剤質量を 適度な厚みとなる 150 mg に設定した.Cyc 10 mg を含有し質量 150 mg の錠剤とするため,第 1 項 にて SLS 配合量を最適化したマイクロスポンジ (MS6:1)に賦形剤として MNT を混合し,成錠化 を試みた.しかしマイクロスポンジのかさ密度が MNTと比べてかなり小さく,両者の混合均一性や 錠剤毎の含量均一性に課題を生じた.また,混合末 のかさ体積が大きくなり,1 錠分の混合末が打錠臼 へ入りきらないという問題点も露呈した.マイクロ スポンジを更に他の賦形剤とともに造粒することもFig. 3. Effect of Cyc content in dropping suspension on particle size distri-bution of granules measured by laser diffraction scattering method. Key: (◇) GR-C6.67% (GR-S5%), (◆) GR-C10%, (△) GR-C20%, (▲) GR-C30%, (○) GR-C47% (MS6:1). Each point represents the mean ±S.D. (n=3).
考えられたが,その特徴的な多孔質構造を破壊して しまい,優れた溶解性が損なわれることが危惧され た.そこで,マイクロスポンジの設計段階で賦形剤 をあらかじめ配合しておく方が得策であるとの結論 に至り,第 2 項で設計した MNT 配合 Cyc 顆粒を直 打することで錠剤を製することとした.即ち,本技 術にて得られた顆粒をそのまま打錠用顆粒として用 い,他の添加剤(結合剤,崩壊剤)との混合工程を 経由しない簡便かつ効率的な調製法とすることを発 案 し た.GR-C6.67% 処 方(Formulation 4)で は 150 mg あたりの Cyc 含量が 10 mg となり,目標と する錠剤処方に合致したことから,Cyc 顆粒(GR-C6.67%)を 10 mg 含有 Cyc 錠の打錠用末に選定し た.しかしながら,MNT を配合した本顆粒におい ても依然としてかさ高く,打錠臼へ 1 錠分の打錠用 末を充填するのが困難であった.そこで,打錠用顆 粒としての適性を付与するため,顆粒の重質化を試 みた.滴下液滴中の固形分濃度を上げることで粒子 密度が高い重質な顆粒が得られるものと考え,GR-C6.67%処方の成分・組成は固定し,希釈水量を減 ずることで滴下溶液中の固形分濃度を 5% から 20% まで順次増加させ,打錠用顆粒を調製した(Table II,GR-S5%,GR-S10%,GR-S15%,GR-S20%). すべての処方で滴下液中の MNT は完全に溶解して いることを付記する.ここで処方名の GR は前述と 同様に Granule を,S 以降の表記は滴下溶液の固形 分濃度(Solid concentration)を示している(S20% は滴下溶液の固形分濃度 20%). 得られた顆粒はいずれの処方でも,第 2 項で調製 された固形分濃度 5% の顆粒(Fig. 2-a,GR-S 5%) と同じく MNT による緻密な表面状態を有した球形 状を呈していた(写真は省略).次に,各種顆粒の密 度に関する物性値を Table III に一覧化した.なお, 滴下液を滴下装置の代わりに駒込ピペットの先端か ら液体窒素中に落下させ,Fig. 5 の拡大写真に示す ような 4 mm 程度の球形顆粒を調製し,画像解析よ り算出した 20 個分の総体積(真球と仮定のもと)と 総質量から粒子密度を算出した.得られた顆粒の粒 子密度は滴下溶液の固形分濃度とほぼ同じ値となり (例えば 5 w
/
v%の場合,0.047 g/
cm 3),滴下時の固 形分濃度がそのまま乾燥後の粒子密度となることが 検証された.これらの結果は,DFD 法では目標値通 りの粒子密度を持つ顆粒を調製することができるこ とを示している.こうして固形分濃度の変更にて, 調製した顆粒による粉体層の密度(かさ密度,タッ プ密度)は増大し,GR-S15% と GR-S20% 顆粒で は臼への充填の問題が解決した.また Carr による 流動性の尺度によれば,圧縮度が 20% 未満の場合に ハンドリング性が良好であると提唱されている 31). 測定に用いたサンプル量が 0.5 g 未満と少量であっFig. 4. Effect of Cyc content on (a) self-dispersion profiles of granules and (b) dissolution profiles of Cyc from granules prepared with various Cyc con-tents in water. Insert graphs in (a) represents the median diameter in normal enlarged vertical axis. Key: (◇) GR-C6.67%, (◆) GR-C10%, (△) GR-C20%, (▲) GR-C30%, (○) GR-C47% (MS 6:1), (×) Cyc bulk. Dotted line in (a) indicates D50 size of original wet-milled nanosuspension. Each point represents the measured value of n=1 in (a). Each point represents the mean±S.D. (n=3) in (b).
たことに影響し,GR-S15%,GR-S20% で標準偏差 が大きくなったものの,滴下溶液が高濃度になるに 従い圧縮度が小さくなる傾向が認められ,粒子密度 の重質化による流動性の改善が示された.一方,顆 粒の比表面積値は,Cyc 原末(3.24 m 2
/
g)と比較し 顕著に増大し,その多孔質構造や速やかな粒子分散 性(後述)を裏付ける結果であった.顆粒間で比較 すると,液滴中の固形分濃度が高くなることで顆粒 内部の細孔体積が減少して比表面積は低下するもの の,GR-S20% でも 10.96 m 2/
gと大きな値を持つこ とが示された. 3.2 打錠用顆粒の錠剤化 このようにして調製した同一組成で粒子密度の異 なる打錠用顆粒(GR-S5%, GR-S10%, GR-S15%, GR-S20%)150 mg を量り取り,直径 8 mm の打錠 用臼に手充填し,万能圧縮引張試験機にて錠剤を製 した.顆粒以外の添加剤(結合剤,崩壊剤)は追加 配合していない.HPLC を用いた定量により錠剤中 の Cyc 含量は,1 錠当たり 9.5~10.6 mg と目標値 10 mgに対し±6.0%以内の偏差となっていることを 確認した.錠剤の処方名をそれぞれ S5%, TB-S10%, TB-S15%, TB-S20%とした(TB は Tablet, S以降の表記は打錠用顆粒と同様).粒子密度の異な る顆粒を用い,3 水準の圧力(25 MPa,50 MPa 及 び 75 MPa)にて圧縮成形した際に得られた錠剤の 厚み,硬度の打錠圧に対する関係を Fig. 6 に示した. いずれの打錠用顆粒も打錠圧が高くなるに従い錠厚 は薄くなり,錠剤硬度が高くなる傾向が認められた. 打錠用顆粒に新たに結合剤を追加混合することなく 直打したが,50 MPa 以上の打錠圧では 70 N 以上と 実用上十分な硬度(≥50 N 32))を示し,良好な圧縮 成形を有していた.そこで以降の検討では打錠圧を 50 MPa に固定した.湿式粉砕工程では粉砕促進剤 として,その後の固形化工程では賦形剤として配合 した HPC が結合剤として機能したためと考える. また,特異のスポンジ構造が塑性変形を助長し,優 れた圧縮成形性に寄与したことも一因であると推察 した.打錠用顆粒間で錠剤の硬度を比較した時,低 粒子密度の顆粒ほど錠剤硬度が増大すると予測した が,実際にはそうならなかった(15%>10%>5%> 20%の順に錠剤硬度は高い).原因として,粒子密 度が低い打錠用顆粒(GR-S5%,GR-S10%)では打 錠用臼への充填時に,1 度に全量を充填することが できず,GR-S5% では 3 回,GR-S10% では 2 回に 分けて充填しては手作業で軽く圧縮する操作を繰り 返したため,これらの錠剤では内部に強度偏析が生 じ,硬度が見かけ上低くなったものと考察した. 3.3 錠剤の薬剤学的特性 薬剤的挙動として,錠剤の水相中での崩壊挙動を 評価した.50 MPa にて打錠した錠剤の崩壊時間は, 日局に準拠した試験法において 10 分程度であったFig. 5. Pictures of big granules for particle density measurement.
TABLE III Particle, bulk and tapping density and specific surface area of granules prepared from dropping
suspension with various solid concentrations.
Symbol unit GR-S5% GR-S10% GR-S15% GR-S20%
Particle density g/cm 3 0.047 0.112 0.168 0.197
Bulk density a g/cm 3 0.045±0.001 0.058±0.000 0.070±0.003 0.102±0.002 Tapping density a g/cm 3 0.054±0.001 0.069±0.001 0.089±0.000 0.112±0.008 Compression ratio a % 16.93±0.95 15.47±0.81 20.87±2.91 8.33±5.10
Specific surface area m 2/g 16.74 13.40 11.91 10.96
(TB-S5%:542±164 S10%:635±24 s,TB-S15%:577±26 s, TB-S20%:633±19 s, n=3).な お,本錠剤の崩壊は接水した基剤が溶解する浸食型 であることを崩壊試験での観察から確認している. 崩壊剤を配合せずとも水相投入後 10 分程度で完全 に崩壊した.錠剤(50 MPa にて打錠)の破断面を SEM観察すると,いずれの錠剤においても打錠用 顆粒の形状は失われ,微細な空隙を残した錠剤の内 部構造が観察された(Fig. 7).著者等は,滴下凍結 乾燥法で調製したマイクロスポンジの内部は粉砕さ れた Cyc 粒子が支持層内に均一に分散したマトリッ クス構造を有していることを前報にて報告した 19). 本研究では MNT を追加添加し,同一の手法により 顆粒を調製していることから,打錠用顆粒において も Cyc 粉砕粒子が均一に分散した構造を成し,さら にそれを直打して得た錠剤においても Cyc 粒子が MNT結晶間に点在したマトリックス構造を形成し ていると推察した.積層構造の主成分が親水性の MNTと HPC であり,微小空隙を伝わって錠剤内部 へ急速に導水し,接水する錠剤表面のみならず,内 部からの基剤の浸食により錠剤の崩壊が促進された ものと考察した.
Fig. 6. Effect of particle density of granules on (a) thickness and (b) hardness of tablets compressed at various compression pressures. Key: ( ) S5%, ( ) TB-S10%, ( ) TB-S15%, ( ) TB-S20%. Each point represents the mean ± S.D. (n=3) in (b).
Fig. 7. Scanning electron microphotographs of cross-section of tablet prepared by compression (50 MPa) of granules with various particle densities. Key: (A) S5%, (B) S10%, (C) TB-S15%, (D) TB-S20%. Cross-section under high magnification.
次に,調製した各粒子密度を有する打錠用顆粒, 及びそれより製した錠剤の再分散性と Cyc 溶出性を 評価した(Fig. 8(a), (b)が顆粒,(c), (d)が錠 剤).なお(a), (c)の挿入図は,平衡粒子径を明確 に表示するため,10 分以降のメジアン径を標準軸に プロットした拡大図である.打錠用顆粒では,粒子 密度に依らずいずれも水相投入後 5 分以内にサブミ クロン懸濁液を再構築し,15 分時点で 90% 以上と 優れた Cyc 溶出性能を示した.一方,これらの顆粒 を圧縮成形し製した錠剤のリアルタイムでの分散挙 動を検出するため,レーザー回折・散乱装置に付属 する測定セルの少ない分散液量に応じたミニ錠剤を 新たに調製した.錠剤形状が普通錠と相似形となる ようミニ錠剤の直径を 2.8 mm に設定した.このミ ニ錠剤を精製水に投入した後の分散挙動を Fig. 8 (c)に示した.水相に投入後,分散粒子の大きさが 数百μmから数μm域に小さくなる推移は錠剤への 水の浸食に伴う崩壊過程を表しており,分散時間は 併記した顆粒のそれと比べ,5~7 分程度延長した. また錠剤が完全に崩壊した 10 分以降でも分散した 粒子はシングルミクロン域に留まり,1 μm未満のサ ブミクロンサイズに到達しない錠剤が認められた. これは打錠により Cyc ナノ粒子同士が強く圧着し, サブミクロンサイズの 1 次粒子への再分散性機能を 発揮できなかったためと推察した.錠剤からの溶出 挙動(Fig. 8(d))は,打錠用顆粒(Fig. 8(b))に 比べて初期溶出がやや緩慢で,30 分時点で到達した 平衡溶出率は 80% ほどとやや低い結果となった.顆 粒と対比して考察すると,錠剤の崩壊に時間を要す るため初期溶出がその分遅延し,またミニ錠剤の再 分散性試験の結果(Fig. 8(c))から推察されるよう に,圧縮工程による薬物粒子の圧着により Cyc 分散
Fig. 8. Effect of particle density on (a, c) self-dispersion profiles and (b, d) dis-solution profiles of Cyc in water from (a, b) granules and (c, d) tablets prepared from suspension with various solid concentrations. Insert graphs in (a) and (c) represents the median diameter in normal enlarged vertical axis. Key: ( ) S5%, ( ) S10%, ( ) S15%, ( ) GR-S20%, ( ) TB-S5%, ( ) TB-S10%, ( ) TB-S15%, ( ) TB-GR-S20%, (×) Cyc bulk. Dotted line in (a, c) indicates D50 size of original wet-milled nanosuspension. Each point represents the measured value of n=1 in (a) and (c). Each point represents the mean±S.D. (n=3) in (b) and (d).
粒子が若干肥大化したため,平衡溶出率が低下した ものと考察した.溶出試験の際に浸食により錠剤が 完全に崩壊するまでの時間は,ミニ錠剤の分散挙動 で示された時間や崩壊試験で計測された崩壊時間よ りも長く,20 分程度要することが観察された.これ はミニ錠剤の分散試験では撹拌子を用いており,崩 壊試験では上下に試験器を動かすことにより錠剤へ 水流の負荷が加わり,溶出試験の緩やかなパドル撹 拌(50 rpm)に比べ水による浸食を促しているもの と考えられた.また通常錠はミニ錠剤と比べ錠厚が 厚く(3.4 mm vs 1.2 mm),溶出に関わる導水距離 が長い(肉厚な)通常錠では溶出時間が遅延する一 因となったことも考えられる.つまり浸食時間が異 なることから錠剤からの Cyc 粒子の再分散性も低下 しているものと推察した.加えて,この水流の負荷 は溶出試験中で再分散した Cyc 粒子サイズにも影響 し,撹拌子を用いて分散したサイズよりも大きな粒 子で分散しているものと考えられ,この点も顆粒と 錠剤の平衡溶出率に差が生じた要因であると考察し た.更に,Fig. 4 に示した GR-C6.67%(◇)と Fig. 8に示した GR-S5%(〇)は同一製剤であるが,Fig. 4に比べ Fig. 8 では平衡溶出率が低下し,Fig. 8 の 溶出試験では 60 分以降も溶出率 100% には到達し なかった.Fig. 8 に示した打錠用顆粒及び錠剤の溶 出試験は Cyc 10 mg 相当の試料を投入しており,前 述した溶出試験(Fig. 1, 4)の 2 倍量である.これ は Cyc の溶解度から勘案すると,Fig. 8 では過飽和 量(900 mL 中 6.6 mg 程度)を越えており,この点 も平衡溶出率が低下した一因であると推察した.い ずれにしても,錠剤においても Cyc の溶出挙動は原 末と比較し,劇的に改善した.グラフは示さないが, Cyc顆粒及び Cyc 錠の局方第 1 液,第 2 液中におけ る溶出挙動は,Fig. 8 に示した精製水中での挙動と ほぼ同等であった.Liu 等は,水に対する Cyc の溶 解性が高い製剤は in vivo における吸収性も高いこ とを報告しており 33),本研究で開発した Cyc 含量の 異なる複数の顆粒剤及び Cyc10 mg 含有錠剤は消化 管内でも高い溶解性を発揮することが暗示され,消 化管吸収性の高い製剤を開発できることが示唆され た. 4.錠剤中の結晶性の評価 Cyc は分子量が 1,202 と大きく環状の分子構造を 有しており,結晶を形成し難いと言われている.ま た,顆粒剤と錠剤を賦形した MNT には安定結晶形 であるβ型に加え,準安定形のα 型,δ型の合計 3 種類の結晶形が存在することが知られており 34),吉 成等は湿潤条件において MNT の結晶転移が起きる こと,そして,各結晶形はそれぞれ特異的な粒子構 造を持ち,塑性変形性が異なるため圧縮特性に違い が生じることを報告している 35).そこで,本研究に て調製した製剤の物理的安定性を確認するため,調 製工程中及び調製した錠剤の保存期間中での結晶状 態の変化について粉末 X 線回折法にて精査した.検 出した試料の粉末 X 線回折像をまとめて Fig. 9 に示 す(A は全回折角範囲,B,C は低回折角範囲のみ を拡大).(A)は原料の MNT と Cyc,並びに調製 したマイクロスポンジ(MS6:1),打錠用顆粒(GR-S20%)及び錠剤(TB-S20%)の回折チャートであ る.また MNT の結晶形を同定するため,α,β及び δ型 MNT 純品の回折チャートを(B)に示す.原料 に用いた MNT(a)はβ型に特異の回折ピーク (14.6°,▼)を持ち,β型であることが確認された. Cyc原料(b)の回折像はハローパターンを示し,非 晶質であることを確認した.MNT を配合していな いマイクロスポンジ(MS6:1,c)も同様にハロー パターンを示し,凍結乾燥後でも Cyc は非晶質状態 を維持していることを確認した.処方に MNT を配 合した打錠用顆粒(GR-S20%,d)及びこれを圧縮 成形した錠剤(TB-S20%,e)にはδ型 MNT に特 徴的な回折ピーク(9.7°,*)が認められ,凍結乾燥 にて再結晶化した際に MNT がβ型からδ型に結晶 転移したことが明らかとなった.冷却下で結晶化し た際のδ型への結晶転移は吉成等も報告しており 35), これに符合する結果となった.また圧縮成形工程中 には,結晶形が転移しないことも分かった.次に, 準 安 定 形 MNT 結 晶(δ型)を 含 有 す る 製 剤 の 保 存 安 定 性 を 予 見 す る た め, 加 速 試 験 条 件 (40˚C
/
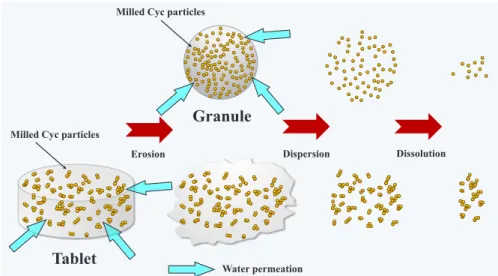
75%RH)下で保存した錠剤(TB-S20%)の 結晶測定を行った(C).その結果,開始時のδ型 MNTに特異の回折ピークは 28 日間後も不変であ り,短期間ではあるが加速条件下でδ 型の結晶形を 維持し,比較的結晶安定性が高い錠剤であることが 示された.この結果に呼応して,保存後の Cyc 分散 挙動,溶出挙動には開始時の挙動から変動はなかっ た(データは省略).更に長期間の保存安定性を評価 する必要はあるものの,本研究にて開発された顆粒剤・錠剤は,市販流通後において MNT の結晶転移 による物性や薬剤特性の変化が生じ難い錠剤である と推測した. 結 論 湿式粉砕と滴下凍結乾燥を組み合わせた製剤化技 術にて,溶解性の向上を図った Cyc 顆粒及び Cyc 錠 を開発した.凍結乾燥工程において滴下した液滴が 液体窒素中で瞬時に凝固することで,真球状の顆粒 を調製した.本研究では,(1)滴下溶液中の SLS の 配合量を最適化することで溶解性を改善したマイク ロスポンジ処方を確立した.また,(2)処方成分中 に MNT を配合し,6.67~47% の Cyc 含量を有する ハンドリング性に富んだ顆粒剤を製した.更に,(3) 設計した顆粒に新たな添加剤を配合することなく直 打することで,効率的な錠剤の設計法を確立した. 得られた顆粒及び錠剤は親水性マトリックス内に湿 式粉砕された Cyc ナノ粒子が包埋された構造を有し ており,難溶性 Cyc の溶解改善が達成されることを 示した.これら製剤からの溶出機構を Fig. 10 に模 式化した.顆粒を水相に投入すると,多孔性顆粒内 への急速な導水に伴いマトリックス基剤が溶解し, 微細 Cyc 粒子が水相中に速やかに分散してサブミク ロン懸濁液を形成する.水相中に分散した Cyc 微粒 子は膨大な表面積を有しているため,急速な初期溶 出と高い溶出量を達成した(Fig. 10 上図).一方 Cyc 錠の場合,錠剤自身が浸食・崩壊するのに時間を要 するため顆粒剤に比べ初期溶出速度が低下するもの の,浸食による崩壊後は速やかに溶解し,水相中に Cyc微粒子を分散する.ただし,圧縮過程中に一次 粒子間で圧着された凝集 Cyc 粒子となって分散する ことにより,溶出量が顆粒と比べて低くなると推察 した(Fig. 10 下図).錠剤で認められたこうした課 題に対しては,添加剤の配合により改善できるので はないかと考える.例えば,Cyc 顆粒にクロスカル メロースナトリウムに代表されるスーパー崩壊剤 36) を配合し,錠剤自身に速やかな崩壊性を付与するこ とで初期溶出速度を向上する,あるいは平衡溶出量 の低下に対しては,結晶セルロースに代表される圧 縮成形剤 37)を処方成分に添加し,低打錠圧にて成形 し,Cyc 粒子間の圧着を抑制するなどの対処策が思 案される.本研究では,Cyc 粒子の速度論的分散挙 動を検出する都合上,水に不溶な添加剤をあえて配 合しなかった側面があることを補足する. 以上,湿式粉砕法に新規な乾燥工程を組み合わせ た技術にて,シクロスポリン顆粒剤及び錠剤を開発 した.滴下凍結乾燥工程では設備投資を含めたスケ ールアップ適性など実生産化に向けた検討が必要と
Fig. 9. X-ray powder diffraction patterns of (A) raw materials, mi-crosponge, granule and tablet, (B) MNT polymorphs and (C) TB-S20% under accelerated test condition (40˚C/75%RH). Key: (a) MNT (β-form), (b) Cyc bulk, (c) MS6:1, (d) GR-S20%, (e) TB-S20%. ▼:specific peak in β-form of MNT. *: specific peak in δ-form of MNT.
なるが,得られた顆粒剤を単味で直打することで錠 剤の製造が可能なことから,生産の効率化において 極めて有用な製法であると考える.また,本研究で 開発した顆粒剤及び錠剤は平衡溶出率が 100% に達 しない課題があるため,市販製剤(液剤,軟カプセ ル,細粒剤)と生物学的同等な製剤の製品化を考え た場合,溶出率の更なる向上と市販製剤の溶出挙動 との一致を目指した検討が必要と考えられるもの の,市場にはない汎用剤形(顆粒剤,錠剤)の開発 への足掛かりになるものと期待される.今後,処方 成分の種類や配合量及び顆粒の調製条件について詳 細に検討し最適化を図ることで,本研究で開発した 含量(10 mg)よりも高含量で,国内で市販されて いる軟カプセル剤と同一含量(25 mg 及び 50 mg) の Cyc 錠剤の開発へと発展していきたい.また,凍 結乾燥工程を経由したマンニトール処方の錠剤は, 薬液と MNT を含む添加剤の懸濁液を凍結乾燥によ り固形製剤化し口腔内崩壊錠(OD 錠)を製する Zydis技術とも共通しており 38, 39),OD 錠の開発に も発展できる可能性を秘めている.加えて,本法は シクロスポリン以外の難水溶性薬物に対しても適用 可能と考えられ,難経口吸収性の問題により製品化 が困難となっている薬物へ向けた溶解性改善のため の新たなプラットフォーム技術となることが期待さ れる. 引 用 文 献
1) L. Dehesa, A. Abuchar, A. Nuno-Gonzalez, M.
Viti-ello, FA. Kerdel, The use of cyclosporine in derma-tology, J. Drugs Dermatol., 11 (8), 979–987 (2012). 2) D. I. Min, Neoral: a microemulsion cyclosporine, J.
Transpl Coord., 6 (1), 5–8 (1996).
3) WA. Ritschel, Microemulsion technology in the re-formulation of cyclosporine: the reason behind the pharmacokinetic properties of Neoral, Clin. Trans-plant., 10 (4), 364–373 (1996).
4) A. Husek, High-perfomance liquid chromatograph-ic analysis of cyclosporine A and its oral solutions, J. Chromatogr. A, 759 (1), 217–224 (1997).
5) P. R. Beauchesne, N. S. C. Chung, K. M. Wasan, Cy-closporine A: A review of current oral and intrave-nous delivery systems, Drug Dev. Ind. Pharm., 33 (3), 211–220 (2007).
6) J. M. Tam, J. T. McConville, R. O. Williams. III, K. P. Johnston, Amorphous cyclosporin nanodispersions for enhanced pulmonary deposition and dissolution, J. Pharm. Sci., 97 (11) , 4915–4933 (2008).
7) S. Onoue, H. Suzuk, Y. Kojo, S. Matsunaga, H. Sato, T. Mizumoto, K. Yuminoki, N. Hashimoto, S. Yama-da, Self-micellizing solid dispersion of cyclosporine A with improved dissolution and oral bioavailabil-ity, Eur. J. Pharm. Sci., 62, 16–22 (2014).
8) H. Suzuki, K. Ueno, T. Mizumoto, Y. Seto, H. Sato, S. Onoue, Self-micellizing solid dispersion of cyclo-sporine A for pulmonary delivery: Physicochemical, pharmacokinetic and safety assessments, Eur. J. Pharm. Sci., 96, 107–114 (2017).
9) H. Sato, Y. Kawabata, K. Yuminoki, N. Hashimoto, Y. Yamauchi, K. Ogawa, T. Mizumoto, S. Yamada, S. Onoue, Comparative studies on physicochemical stability of cyclosporine A-loaded amorphous solid dispersions, Int. J. Pharm., 426, 302–306 (2012). 10) H. Suzuki, T. Moritani, T. Morinaga, Y. Seto, H. Sato,
S. Onoue, Amorphous solid dispersion of cyclospor-ine A prepared with fine droplet drying process: Physicochemical and pharmacokinetic characteri-zation, Int. J. Pharm., 519, 213–219 (2017). Fig. 10. Schematic diagram of redispersing process of granule and tablet in
11) T. Niwa, H. Shimabara, K. Danjo, Novel spray freeze-drying technique using four-fluid nozzle de-velopment of organic solvent system to expand Its application to poorly water soluble drug, Chem. Pharm. Bull., 58 (2), 195–200 (2010).
12) T. Niwa, D. Mizutani, K. Danjo, Spray freeze-dried microparticles of a poorly water-soluble drug for respiratory delivery, Chem. Pharm. Bull., 60 (7), 870–876 (2012).
13) Y. Tanaka, M. Inkyo, R. Yumoto, J. Nagai, M. Takano, S. Nagata, Nanoparticulation of poorly water-solu-ble drugs using a wet-mill process and physico-chemical properties of the nanopowders, Chem. Pharm. Bull., 57 (10), 1050–1057 (2009).
14) A. Afolawemi, O. Akinlabi, E. Bilgili, Impact of pro-cess parameters on the breakage kinetics of poorly water-soluble drugs during wet stirred media mill-ing: A microhydrodynamic view, Eur. J. Pharm. Sci., 51, 75–86 (2014).
15) T. Niwa, S. Miura, K. Danjo, Universal wet-milling technique to prepare oral nanosuspension focused on discovery and preclinical animal studies―De-velopment of particle design method, Int. J. Pharm., 405, 218–227 (2011).
16) T. Niwa, H. Shimabara, M. Kondo, K. Danjo, Design of porous microparticles with single-micron size by novel spray freeze-drying technique using four-flu-id nozzle, Int. J. Pharm., 382, 88–97 (2009). 17) T. Niwa, K. Danjo, Design of self-dispersible dry
nanosuspension through wet milling and spray freeze-drying for poorly water-soluble drug, Eur. J. Pharm. Sci., 50, 272–281 (2013).
18) A. Nakashima, T. Izumi, K. Ohya, K. Kondo, T. Niwa, Design of highly dispersible PLGA microparticles in aqueous fluid for the development of long-acting release injectables, Chem. Pharm. Bull., 65 (2), 157–165 (2017).
19) M. Kobayashi, K. Kondo, C. Nakashima, Y. Kida, T. Niwa, Design of self-dispersible microsponge con-taining cyclosporine through wet milling and drop freeze-drying processes to improve dissolution be-havior, J. Drug Deliv. Sci. Technol., 57 , 101577 (2020).
20) S. T. Buckley, K. J. Frank, G. Fricker, M. Brandl, Biopharmaceutical classification of poorly soluble drugs with respect to “enabling formulations”, Eur. J. Pharm. Sci., 50, 8–16 (2013).
21) H. Sunada, M. Hasegawa, T. Makino, H. Sakamoto, T. Tanino, H. Kokubo, T. Kawaguchi, Study of stan-dard tablet formulation based on fluidized-bed granulation, Drug Dev. Ind. Pharm., 24 (3), 225– 233 (1998).
22) J. Z. H. Gao, A. Jain, R. Motheram, D. B. Gray, M. A. Hussain, Fluid bed granulation of a poorly water soluble, low density, micronized drug: comparison with high shear granulation, Int. J. Pharm., 237, 1–14 (2002).
23) S. Gupta, P. Thool, S. Meruva, J. Li, J. Patel, A. Agrawal, S. Karki, W. Browen, Development of low dose micro-tablet by high shear wet granulation process, Int. J. Pharm., 587 , 119571 –119582
(2020).
24) M. G. Herting, P. Kleinebudde, Roll compaction/dry granulation: Effect of raw material particle size on granule and tablet properties, Int. J. Pharm., 338, 110–118 (2007).
25) Y. Y. Lee, J. X. Wu, M. Yang, P. M. Young, F. van den Berg, J. Rantanen, Particle size dependence of poly-morphism in spray-dried mannitol, Eur. J. Pharm. Sci., 44, 41–48 (2011).
26) H. Ismailos, C. Reppas, J. B. Dressman, P. Macheras, Unusual solubility behaviour of cyclosporine A in aqueous media, J. Pharm. Pharmacol., 43, 287–289 (1991).
27) A. Pongpeerapat, C. Wanawongthai, Y. Tozuka, K. Moribe, K. Yamamoto, Formulation mechanism of colloidal nanoparticles obtained from probucol/PVP/
SDS ternary ground mixture, Int. J. Pharm., 352, 309–316 (2008).
28) M. Vogt, K. Kunath, J. B. Dressman, Dissolution improvement of four poorly water soluble drugs by cogrinding with commonly used excipients, Eur. J. Pharm. Biopharm., 68, 330–337 (2008).
29) G. C. Yee, Dosage form of cyclosporine, Pharmaco-therapy, 11 (6), 149S–152S (1991).
30) I. Funahashi, K. Kondo, Y. Ito, M. Yamada, T. Niwa, Novel contamination-free wet milling technique using ice beads for poorly water-soluble compounds, Int. J. Pharm., 563, 413–425 (2019).
31) Carr, R. L, Evaluating flow properties of solids, Chem. Eng., 72, 163–168 (1965).
32) Y. Okuda, Y. Okamoto, Y. Irisawa, K. Okimoto, T. Osawa, S. Yamashita, Formulation design for oral-ly disintegrating tablets containing enteric-coated particles, Chem. Pharma. Bull., 62 (5), 407–414 (2014).
33) C. Liu, J. Wu, B. Shi, Y. Zhang, T. Gao, Y. Pei, En-hancing the bioavailability of cyclosporine a using solid dispersion containing polyoxyethylene (40) stearate, Drug Dev. Ind. Pharm., 32 (1), 115–123 (2006).
34) M. Mehta, S. P. Bhardwaj, R. Suryanarayanan, Con-trolling the physical form of mannitol in freeze-dried systems, Eur. J. Pharm. Biopharm., 85, 207– 213 (2013).
35) T. Yoshinari, R. T. Forbes, P. York, Y. Kawashima, The improved compaction properties of mannitol after a moisture-induced polymorphic transition, Int. J. Pharm., 258, 121–131 (2003).
36) N. Zhao, L. L. Augsburger, The influence of granu-lation on super disintegrant performance, Pharm. Dev. Technol., 11 (1), 47–53 (2006).
37) G. Thoorens, F. Krier, B. Leclercq, B. Carlin, B. Ev-rard, Microcrystalline cellulose, a direct compres-sion binder in a quality by design environment―A review, Int. J. Pharm., 473, 64–72 (2014).
38) H. Seager, Drug-delivery products and the zydis fast-dissolving dosage form, J. Pharm. Pharmacol., 50, 375–382 (1998).
39) B. P. Badgujar, A. S. Mundada, The technologies used for developing orally disintegrating tablets: A review, Acta Pharm., 61, 117–139 (2011).