−
37
−*
Department of Chemistry, Faculty of Science, Fukuoka University,
Nanakuma 8-19-1, Jonan-ku, Fukuoka 814-0180 Japan.
福岡大学理学集報
37
⑵37
〜44
(2007
)High-Level and Functional Expression of a Recombinant Protein of Onion Trypsin Inhibitor
Mika F
URUKI, Keiko S
UEMATSU, Masanobu D
ESHIMARU*, and Shigeyuki T
ERADA( Received June 30 , 2007 )
Abstract
We previously reported purification, characterization, and cDNA cloning of a onion ( Allium cepa L.) trypsin inhibitor (OTI). To investigate its inhibitory mechanism, we have developed bacterial expression system of recombinant OTI. Among three different bacterial strains exam- ined, Rosetta-gami B strain, which has mutations in genes for thioredoxin reductase and gluta- thione reductase, showed a prominent result in the functional expression of recombinant OTI.
Using this bacterial strain with expression plasmid for thioredoxin (TrxA)-OTI fusion protein, the recombinant protein in soluble form was yielded up to 10 mg per 100 -ml bacterial culture.
This recombinant OTI showed trypsin inhibition to a similar extent as inhibitor purified from onion tissue. Utilizing present expression system, several mutant proteins of OTI can be pro- duced in order to elucidate its inhibitory mechanism.
INTRODUCTION
Onion trypsin inhibitor (OTI) is a mem- ber of Bowman-Birk type inhibitor (BBI) family originally found in seeds of legumi- nae plants [1]. Similar to leguminae BBIs, OTI is a monomeric protein with a molecu- lar weight of 7 kDa and relatively many cysteine residues. The rigid conformation and the high stability against extreme temperature and pH may be due to the for- mation of many disulfide bonds. Applying the 2-D model for soybean BBI proposed by Odani and Ikenaka [2], the first and the second reactive loops of OTI seemed topologically similar to those of soybean.
As compared with soybean BBI,
17Arg and
46
Leu in OTI are predicted to be responsible for the inhibition of trypsin/plasmin and
chymotrypsin, respectively [1]. To confirm such a structure-function relationship of OTI, recombinant expression system has needed to be developed.
Bacterial cells are frequently used for pro- duction of recombinant proteins because of their simplicity in handling and the rapid growth [3]. However, they often fail to pro- duce the recombinant proteins with proper and functional conformations, especially when eukaryotic genes are introduced. In such a case, most of the recombinant pro- teins form insoluble aggregates, so-called inclusion bodies, through inappropriate as- sociation or scrambled disulfide bonds [4].
Considering the relatively high cysteine
content, it had seemed difficult to produce
recombinant OTI using bacterial expression
system. Thus, in the present study, several
−
38
−bacterial strains and plasmid vectors which have been reported to enhance the solubi- lization of recombinant proteins [5, 6] were examined for production of recombinant OTI in functional form.
MATERIALS AND METHODS To construct all the expression plasmids, OTI-coding DNA fragments were prepared by PCR using cDNA clone, pOTI-2.1, as template [1]. Oligonucleotides used for PCR were designed upon 5 ' - and 3 ' -terminal nucleotide sequences of mature OTI-coding region, and appropriate restriction sites were added to their 5 ' -temini, if necessary.
PCR-amplified DNA fragments were gel- purified, digested by restriction enzymes, and ligated to compatible restriction sites of expression plasmid, pGEX-2T (GE Healthcare Bioscience) or pET-32a (Nova- gen). Nucleotide sequences of completed expression vectors were analyzed by ABI Prism 377 DNA sequencing system (Applied Biosystems).
Coding region of thioredoxin gene ( TrxA ) was amplified from genomic DNA of E.
coli strain JM109 by PCR. It was once sub- cloned into a cloning vector, pTV118N (Takara Biotechnology), and its nucleotide sequence was determined to be identical to that reported previously [7]. TrxA frag- ment was then removed together with Lac operator/promoter sequence of pTV118N by Eco RI/P vu II cleavage, and transferred into corresponding site of pACYC184 (New England Biolabs).
Bacterial cells were transformed with re- spective expression plasmids by modified Hanahan method [8], and single colonies with appropriate drug resistance were iso- lated. 100 ml of fresh 2xYT medium was inoculated with 1 ml of bacterial overnight culture, and recombinant expression was
induced by addition of isopropyl- β -D- thiogalactopyranoside (IPTG) when the OD
600reached 0.6. After 6 hours of induc- tion period, a 200-µl portion of the culture was removed and bacterial cells were col- lected by centrifugation and lysed using Laemmli ' s sample buffer for SDS-PAGE.
Residual bacterial sample in which the re- combinant expression was observed was suspended in 2.5 ml of phosphate-buffered saline, followed by sonication and centrifu- gation to separate the soluble and insoluble fractions. GST- and histidine hexamer (His
6)-tagged proteins were purified using glutathione sepharose 4B resin (GE Health- care Bioscience) and Ni-NTA His-Bind resin (Novagen), respectively, according to manufacturers ' instructions. Proteins in eluates were isolated from low molecular impurities by gel filtration using Sephadex G-25 (GE Healthcare Bioscience).
Molecular mass and N-terminal amino acid sequence of recombinant protein were determined by MALDI-TOF/MS using Voy- ager-DE STR (Applied Biosystems) and Ed- man degradation using protein sequencer, PPQS-21 (Shimadzu), respectively. Purified recombinant OTIs were tested for trypsin inhibitory activity [9]. Following to prein- cubation of TPCK-treated bovine trypsin (25 µg/ml) with various amount of recombinant proteins in 50 mM Tris-HCl (pH 8.0) con- taining 10 mM CaCl
2, benzoyl-L-arginine p -nitroanilide (Bz-L-Arg- p NA) was added.
Hydrolysis of Bz-L-Arg- p NA was estimated from the measurement of OD
410at 25 ℃ .
RESULTS
First, the glutathione- S -transferase (GST)- fusion expression system using the plas- mid pGEX-2T was tested for recombinant expression of OTI. Mature protein-coding region for OTI consisting of 213 bp was
−
38
−−
39
−High-Level and Functional Expression of a Recombinant Protein of Onion Trypsin Inhibitor
(
M. Furuki et al.
) −39
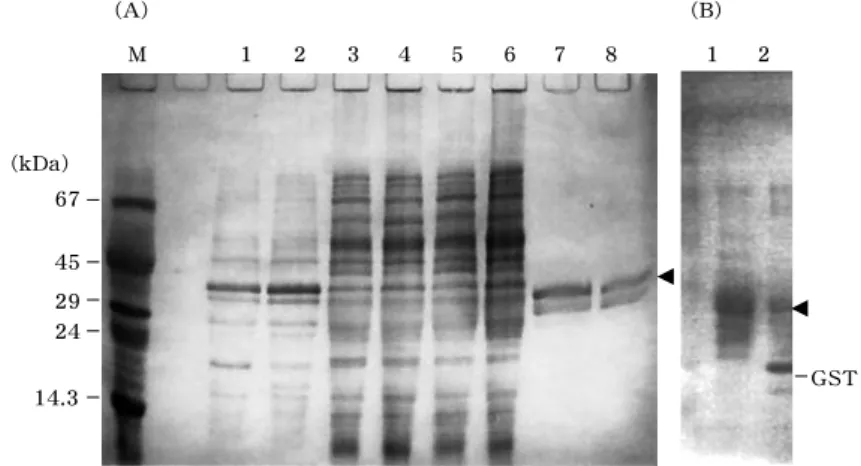
−PCR-amplified and ligated to Bam HI- Sal I restriction site of pGEX-2T. The nucleo- tide sequence of resulted plasmid clone, pGST-OTI, was confirmed to be in frame with GST-coding sequence. This expres- sion construct was then used to transform a bacterial strain JM109 or ER2267 and ampicillin-resistant bacterial transformants were isolated. Expression of recombinant OTI was induced by addition of IPTG to a 10-ml culture of bacterial transformant to a final concentration of 1 mM at 37 ℃ . 6 hours after IPTG addition, whole cell sam- ples were subjected to SDS-PAGE analysis and high-level expression of GST-OTI were observed in either bacterial strain.
To determine whether GST-OTI was expressed in soluble or insoluble form, bac- terial cells were disrupted by sonication, and soluble and insoluble fractions were isolated by centrifugation. As the result of SDS-PAGE analysis, distinct band cor-
responding to GST-OTI, expected to be 〜
32 kDa, was found in insoluble fraction (Figure 1A, lanes 1-4). To isolate GST-OTI from the soluble fraction, in which various host-derived protein constituents could pre- vent the detection of recombinant, affinity chromatography using glutathione-sepha- rose was employed. As a result, a protein corresponding to GST-OTI was recovered in glutathione-bound fraction (Figure 1A, lanes 7 and 8), but showed no activity to inhibit hydrolysis of Bz-L-Arg- p NA by trypsin even with considerably high con- centration (data not shown), suggesting that GST-OTI produced by this expression system has a misfolded but soluble con- formation. Modification in the incubation temperature (25 ℃ instead of 37 ℃ ) and/or IPTG concentration (0.1 mM instead of 1.0 mM) did not confer the inhibitory function to GST-OTI, suggesting difficulty in the production of recombinant OTI in func-
GST (kDa)
67 45 29 24 14.3
(A) (B)
M 1 2 3 4 5 6 7 8 1 2
Fig. 1 . Expression profile of GST-OTI analysized by SDS-PAGE. (A) Insoluble (lanes 1 and 2 ) and soluble fractions (lanes 3 and 4 ) of bacterial cells, and unbound (lanes 5 and 6 ) and bound fractions (lanes 7 and 8 ) from glutathione-affinity chromatography were analyzed on 10 % polyacrylamide gel. Bacterial strain, JM 109 (Lanes 1 , 3 , 5 , and 7 ) or ER 2267 (lanes 2 , 4 , 6 , and 8 ) was used as host cell. Protein bands corresponding to GST-OTI are indicated by arrowhead. (B) Limited proteolysis of GST-OTI by thrombin.
Untreated (lane 1 ) and thrombin-treated (lane 2 ). OTI isolated was not
detected in this analysis because of its low molecular weight. Bands for
GST is indicated.
−
40
−tional form using this expression system.
Considering that GST interfered the activ- ity of fused OTI moiety, these two moieties were separated by limited proteolysis using thrombin (Figure 1B). However, the prod- uct showed any inhibition against trypsin (data not shown).
According to the report that co-expres- sion of TrxA affects the folding of several recombinant proteins [10], we next made attempt to construct a co-expression sys- tem of thioredoxin and recombinant OTI.
Bacterial TrxA gene sequence was PCR- amplified and subcloned into pACYC184 plasmid together with a Lac operator/pro- moter element. Resulted TrxA expression plasmid, which bares p15A-type replication origin ( ori ) sequence, is compatible in a bacterial cell with pGST-OTI with ColE1 ori [11]. E . coli JM109 was once transformed with pTrxA and tetracycline-resistant clone was further transformed with pGST- OTI. Tetracycline/ampicillin double resis- tant clone, which corresponds to a double- transformant, was isolated and subjected to the expression experiment. Expression of TrxA and GST-OTI was induced by ad- dition of IPTG (0.1 mM) to the bacterial culture. The results of SDS-PAGE analy- sis showed that GST-OTI, as well as co- expressed TrxA, was produced partially as soluble form, similar to the case of GST- OTI only (data not shown). This soluble GST-OTI was then purified and used to examine the activity. However, no trypsin- inhibiting activity was observed again (data not shown).
When TrxA is co-expressed or fused with a recombinant protein with multiple disulfide bonds, its folding assisting ability is strongly exerted by use of the bacterial strain which has mutant alleles of thiore- doxin reductase ( trx B) and/or glutathione reductase ( gor ) genes [12]. Accordingly, a
trxB/gor E. coli host, Rosetta-gami B, was used to construct a new expression system for recombinant OTI. In this case, OTI- coding DNA was subcloned into pET-32a in order to produce the recombinant OTI as a TrxA-fused, His
6-tagged form (TrxA-OTI).
This expression construct, referred to pTrx- OTI, was used to transform E . coli Rosetta- gami B and the recombinant expression was induced. When IPTG concentration was varied 0.1 to 1.0 mM, dose-dependent increase of recombinant expression was observed and sufficient expression of Trx- OTI was achieved with 0.4 mM IPTG (Fig.
2). As soluble and insoluble fractions were analyzed by SDS-PAGE, most of the re- combinant protein was detected as soluble (data not shown). Whole bacterial lysate from 100-ml culture with 0.4 mM IPTG was applied to an affinity chromatography us- ing Ni
2+-chelating resin, which can bind His
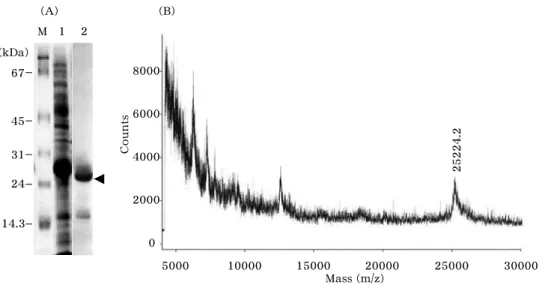
6-tagged recombinant protein, and 10 mg of bound protein was recovered. As analyzed by SDS-PAGE, single protein band of ~17 kDa corresponding to TrxA- OTI was observed (Fig. 3A). Its molecular mass was measured to be 25224.2 (M + H
+), which was in good accordance with the value expected for the recombinant protein with a deleted N-terminal methionine resi- due (25224.01) (Fig. 3B). The truncation at N-terminal was confirmed by peptide se- quence analysis for intact recombinant pro- tein, and it seemed to be caused by post- translational proteolysis found commonly in bacteria [13].
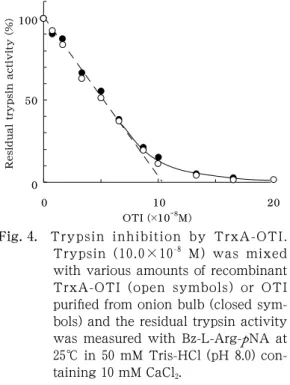
A potent inhibitory activity of TrxA-OTI
was observed using an assay system with
trypsin and Bz-L-Arg- p NA. As shown in
Fig. 4, this inhibition was dose dependent
and the result suggested 1:1 association of
inhibitor with trypsin. Such a inhibition
profile was highly similar to that of native
OTI protein, suggesting that TrxA-OTI
−
41
−High-Level and Functional Expression of a Recombinant Protein of Onion Trypsin Inhibitor
(
M. Furuki et al.
)(kDa) 67
45
31
24
14.3
IPTG (mM) 0 0.25 0.1 0.4 1.0
Fig. 2 . IPTG concentration dependency of TrxA-OTI expression. pTrx-OTI- transformed Rosetta-gami B bacterial cells were pre-cultured to a OD
600of 0 . 6 , and the recombinant expression was induced for 6 hours after addition of IPTG. Arrowhead indicates the bands corresponding to TrxA-OTI.
(kDa) 67
45 31 24
14.3
M 1 2
5000 10000 15000 20000 25000 30000 Mass (m/z)
8000
6000
4000
2000
0
25224.2
Counts
(A) (B)
Fig. 3 . Purification and identification of TrxA-OTI. (A) Total proteins in
Bacterial soluble fraction (lane 1 ) and proteins purified by Ni
2+-affinity
chromatography (lane 2 ) were analyzed by SDS-PAGE. Protein bands
corresponding to TrxA-OTI are indicated by arrowhead. (B) Molecular
mass determination for purified TrxA-OTI by MALDI-TOF/MS.
−
42
−could reproduce accurately the molecular behavior of OTI although it retained the extra moiety of ~17 kDa fused to its N-ter- minal.
0 50 100
0 10 20
OTI (
×10-
8M)
Residual trypsin activity (%)
Fig. 4 . Trypsin inhibition by TrxA-OTI.
Trypsin ( 10 . 0 × 10
-8M) was mixed with various amounts of recombinant TrxA-OTI (open symbols) or OTI purified from onion bulb (closed sym- bols) and the residual trypsin activity was measured with Bz-L-Arg- p NA at 25 ℃ in 50 mM Tris-HCl (pH 8 . 0 ) con- taining 10 mM CaCl
2.
DISCUSSION
OTI is the first BBI found in liliceae plant and has a primary structure similar to that of leguminae BBIs [1]. However, it is sug- gested that the conformation of OTI is somewhat different from those of legumi- nae BBIs, because OTI lacks two cysteine residues in its sequence corresponding to those found at the the second reactive loop of leguminae BBIs [2]. Although its inhibi- tory activities against trypsin, chymotryp- sin, and plasmin were demonstrated, it has been unknown what structural elements are responsible for respective activities. If desirable amino acid substitutions can be introduced to OTI by protein engineering, its inhibitory mechanism will be solved readily. In present study, we tested three
different expression systems for produc- tion of recombinant OTI in functional form; GST-fusion system, co-expression with TrxA, and TrxA-fusion system with a trxB/gor -mutant bacterial strain.
GST- and TrxA-fusion tags are com- monly used for recombinant expression of proteins with multiple disulfide bonds, and their effectiveness in improvement of the solubility of recombinant proteins have been reported in many cases [5, 6]. In the present case, despite of relatively low amount, GST-OTI was purified as a soluble form and tested for its activity. However, no trypsin inhibition was detected with GST-OTI and even the co-expression of TrxA protein had no effect on the function of recombinant protein. Since GST moiety could be removed easily from recombinant protein by proteolysis using thrombin, OTI moiety seemed to be folded independently;
i.e. , GST did not form any covalent or non- covalent association with OTI to interfere its activity. After cleavage by thrombin, some precipitation was formed in the solu- tion of GST-OTI, implying that removal of the GST moiety triggers aggregation of OTI. Thus, it seems likely that, under spe- cific conditions, recombinant OTI tends to take an conformation which is soluble but inert and unstable.
In contrast, TrxA-fusion system with bacterial strain Rosetta-gami B produced much more amount of recombinant OTI in soluble form. Approximately 10 mg of purified TrxA-OTI was obtained per 100-ml bacterial culture, which contains around 0.5 g wet weight of bacterial cells. The purity of TrxA-OTI after affinity chromatography and gel filtration was estimated to be ~94%
using BioAnalyzer (Agilent). Such a promi-
nency in the quantity and quality of TrxA-
OTI seemed to be achieved by an oxida-
tive condition of bacterial cytosol resulted
−
43
−High-Level and Functional Expression of a Recombinant Protein of Onion Trypsin Inhibitor
(
M. Furuki et al.
)from trxB mutation, which acts coopera- tively with enhancement of solubility of TrxA. Purified recombinant protein showed trypsin inhibition at quite a similar level as OTI protein purified from onion bulb. This activity of TrxA-OTI was derived from OTI moiety itself, since TrxA protein produced in the same system showed no trypsin in- hibition (data not shown).
In conclusion, recombinant OTI was pro- duced in soluble and functional form by use of TrxA-fused system with a bacterial strain with trxB/gor mutations. Although it is believed that cysteine-rich proteins are generally difficult to be produced as soluble and functional recombinants, the expres- sion system in the present study might improve the problems, such as insoluble aggregates or dysfunctions of recombinant proteins. Utilizing this expression system, production of several mutant proteins of OTI is in preparation. Systematic inves- tigation that determines which mutation abolish the inhibitory activity of OTI will elucidate the structural elements essential for its function.
References
1 . Deshimaru, M., Watanabe, A., Sue- matsu, K., Hatano, M., and Terada, S. Purification, amino acid sequence, and cDNA cloning of trypsin inhibi- tors from onion ( Allium cepa L.) bulbs.
Biosci. Biotechnol. Biochem . 67, 1653-1659 (2003).
2 . Odani, S. and Ikenaka, T. Studies on soybean trypsin inhibitors. 8. Disulfide bridges in soybean Bowman-Birk pro- teinase inhibitor. J Biochem ( Tokyo ) 74, 697-715 (1973).
3 . Terpe, K. Overview of bacterial expres- sion systems for heterologous protein production: from molecular and bio-
chemical fundamentals to commercial- systems. Appl. Microbiol. Biotechnol. 72, 211-222 (2006).
4 . Fahnert, B., Lilie, H., and Neubauer, P.
Inclusion bodies: formation and utilisa- tion. Adv. Biochem. Eng. Biotechnol. 89, 93-142 (2004).
5 . Esposito, D. and Chatterjee, D. K. En- hancement of soluble protein expres- sion through the use of fusion tags.
Curr. Opin. Biotechnol . 17, 353-358 (2006).
6 . Sheibani, N. Prokaryotic gene fusion expression systems and their use in structural and functional studies of proteins. Prep. Biochem. Biotechnol. 29, 77-90 (1999).
7 . Lim, C. J., Geraghty, D., Fuchs, J. A.
Cloning and nucleotide sequence of the trxA gene of Escherichia coli K-12. J.
Bacteriol . 163, 311-316 (1985).
8 . Hanahan, D., Jessee, J., and Bloom, F. R. Plasmid transformation of Esch- erichia coli and other bacteria. Methods Enzymol . 204, 63-113 (1991).
9 . Terada, S., Fujimura, S., Kino, S., and Kimoto, E. Purification and character- ization of three proteinase inhibitors from Canavalia lineata seeds. Biosci.
Biotechnol. Biochem . 58, 371-375 (1994).
10 . Yasukawa, T., Kanei-Ishii, C., Maeka- wa, T., Fujimoto, J., Yamamoto, T., and Ishii, S. Increase of solubility of foreign proteins in Escherichia coli by coproduction of the bacterial thiore- doxin. J. Biol. Chem. 270, 25328-25331 (1995).
11 . Martinez, E., Bartolome, B., de la Cruz, F. pACYC184-derived cloning vectors containing the multiple cloning site and lacZ α reporter gene of pUC8/9 and pUC18/19 plasmids. Gene 68, 159-162 (1988).
12 . Bessette, P. H., Aslund, F., Beckwith,
J., and Georgiou, G. Efficient folding of
−