メサドン塩酸塩
CTD 第 2 部
2.6 非臨床試験の概要文及び概要表
2.6.1 緒言
メサドン塩酸塩 2.6.1 緒言 Page 1
目 次
2.6.1 緒言 ... 2 2.6.1.1 医薬品の構造及び薬理的特性に関する簡潔な情報 ... 2
メサドン塩酸塩 2.6.1 緒言 Page 2
2.6.1 緒言
2.6.1.1 医薬品の構造及び薬理的特性に関する簡潔な情報
(1) 医薬品の構造 1) 化学構造式: 2) 一般的名称: [日本名] メサドン塩酸塩 [英 名] Methadone Hydrochloride 3) 化 学 名: [日本名] (6RS)-6-(ジメチルアミノ)-4,4-ジフェニルヘプタン-3-オン 一塩酸塩 [英 名] (6RS)-6-(Dimethylamino)-4,4-diphenylheptan-3-one monohydrochloride 4) 分 子 式:C21H27NO · HCl 5) CAS 番号:1095-90-5 (2) 薬理学的特性 モルヒネをはじめとするオピオイドは,強い鎮痛作用から多くの国において疼痛治療の中心的 な役割を担っている。これらは,鎮痛作用に関わると考えられている異なる3 種のオピオイド受 容体,μ,κ 及び δ 受容体のうち,主には μ 受容体を活性化させることで鎮痛作用を発揮させるこ とが知られている。 メサドンは,1937 年に合成されたジフェニルヘプタン誘導体オピオイドアゴニストでありモ ルヒネなどのオピオイド系アルカロイドとは構造的に関連しない。しかしながら,メサドンは, モルヒネと同様にμ オピオイド受容体へ作用し,鎮痛効果を発揮する。また,オピオイド受容体 アゴニストに見られる多様な作用をもち,中枢神経系や平滑筋に対する作用が特に顕著である。 更に,オピオイド系アルカロイドには見られない NMDA 受容体を介した鎮痛作用も報告されて いる。 一般に使用されるメサドンは,左旋性(l-体)と右旋性(d-体)のラセミ体混合物(dl-体)で あり,親水性塩のメサドン塩酸塩の形態で扱われる。 塩基性,脂溶性の物質であり,経口,経直腸,静注,筋注など,種々の経路にて投与可能だが, 経口投与において高い生物学的利用率を持つことから,多くは経口経路にて投与される。 他のオピオイドと同様に身体依存性を有するが,退薬症候の発現はモルヒネなどの半減期の短 いオピオイドに比べて遅く,また症状も比較的軽い。 メサドンはこの特性を利用して,世界各国で既に承認・臨床使用されており,麻薬依存症治療メサドン塩酸塩 2.6.1 緒言 Page 3 及びがん性疼痛をはじめとする慢性疼痛の軽減を目的として,広く使用されている(1.6.1 参照)。 (3) 承認申請における効能・効果及び用法・用量 【効能又は効果】 他の強オピオイド鎮痛剤で治療困難な下記疾患における鎮痛 中等度から高度の疼痛を伴う各種癌 〈効能又は効果に関連する使用上の注意〉 本剤は,他の強オピオイド鎮痛剤の投与では十分な鎮痛効果が得られない患者で,かつオピオイ ド鎮痛剤の継続的な投与を必要とするがん性疼痛の管理にのみ使用すること。 【用法及び用量】 本剤は,他の強オピオイド鎮痛剤から切り替えて使用する。 通常,成人に対し初回投与量は本剤投与前に使用していた強オピオイド鎮痛剤の用法・用 量を勘案して,メサドン塩酸塩として1回5~15 mgを1日3回経口投与する。 その後の投与量は患者の症状や状態により適宜増減する。 〈用法及び用量に関連する使用上の注意〉 1. 初回投与量 (1)本剤の薬物動態は個人差が大きく,他のオピオイド鎮痛剤との交差耐性が不完全で あるため,本剤と他のオピオイド鎮痛剤の等鎮痛比は確立していない。(【警告】 の項参照) (2)初回投与量を選択する下記換算表は目安であり,換算比は本剤投与前に使用してい たオピオイド鎮痛剤の投与量により大幅に異なる。患者の症状や状態,オピオイド 耐性の程度,併用薬剤を考慮して適切な用量を選択し,過量投与にならないよう注 意すること。(【警告】,「慎重投与」,「相互作用」の項参照) (3)経口モルヒネ量60 mg/日未満のオピオイド鎮痛剤からの切り替えは推奨されない。 換算表(本剤1日投与量の目安) メサドン塩酸塩(mg/日) 15 mg/日 (5 mg/回×3回) 30 mg/日 (10 mg/回×3回) 45 mg/日 (15 mg/回×5回) ↑ ↑ ↑ モルヒネ経口剤(mg/日) 60≦~≦160 160<~≦390 390< 2. 初回投与時 (1)本剤投与後少なくとも7日間は増量を行わないこと。[本剤の血中濃度が定常状態に 達するまでに時間を要することから,7日未満の増量は過量投与となる可能性があ る。(【警告】,【薬物動態】の項参照)] (2)フェンタニル貼付剤から本剤へ変更する場合には,フェンタニル貼付剤剥離後にフ ェンタニルの血中濃度が50%に減少するまで17時間以上かかることから,剥離直後
メサドン塩酸塩 2.6.1 緒言 Page 4 の本剤の使用は避け,本剤の使用を開始するまでに,フェンタニルの血中濃度が適 切な濃度に低下するまでの時間をあけるとともに,本剤の低用量から投与すること を考慮すること。 3. 疼痛増強時 本剤服用中に疼痛が増強した場合や鎮痛効果が得られている患者で突発性の疼痛が発現 した場合は,直ちに速放性のオピオイド製剤の追加投与(レスキュードーズ)を行い鎮痛 を図ること。 4. 増量 (1)本剤初回投与後及び増量後少なくとも7日間は増量を行わないこと。[呼吸抑制を発 現するおそれがある。(【警告】,「薬物動態」の項参照)] (2)鎮痛効果が得られるまで患者毎に用量調整を行うこと。鎮痛効果が得られない場合 は,1日あたり本剤1日投与量の50%,1回あたり5 mgを上限に増量すること。 (3)本剤を増量する場合には,副作用に十分注意すること。(【警告】の項参照) 5. 減量 連用中における急激な減量は,退薬症候があらわれることがあるので行わないこと。副 作用等により減量する場合は,患者の状態を観察しながら慎重に行うこと。 6. 投与の中止 本剤の投与を中止する場合には,退薬症候の発現を防ぐために徐々に減量すること。副 作用等により直ちに投与を中止する場合は,退薬症候の発現に注意すること。
メサドン塩酸塩
CTD 第 2 部
2.6 非臨床試験の概要文及び概要表
2.6.2 薬理試験の概要文
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 1
用語及び略語一覧
略語 定義 定義(日本語)
[35S]GTPγS Guanosine 5’-O-(3-[35S] thio)triphosphate グアノシン5'-O-(3-[35S]チオ三リン酸)
5-HT 5-hydroxytryptamine セロトニン
CHO 細胞 Chinese hamster ovary 細胞 チャイニーズハムスター卵巣細胞
DADL [D-Ala2-D-Leu5]enkephalin D-アラニン(2)-D-ロイシン(5)-エンケファリン
DAMGO(DAGO) [D-Ala2-N-MePhe4-Gly-ol5]enkephalin D-アラニン(2)-N-メチルフェニルアラニン(4)-
グリシンオール(5)-エンケファリン DPDPE [D-Pen2,D-Pen5]enkephalin D-ペニシラミン(2)-D-ペニシラミン(5)-
エンケファリン ED50 50% effective dose 50%効果量
EDDP 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine 2-エチリデン-1,5-ジメチル-3,3-ジフェニル ピロリジン
EMDP 2-ethyl-5-methyl-3,3-diphenyl-1-pyrroline 2-エチル-5-メチル-3,3-ジフェニル-1-ピロリン HERG Human ether-a-go-go related gene ヒト遅延整流性カリウムイオンチャネル遺伝子
IC50 50% inhibitory concentration 50%阻害濃度
LD50 Lethal dose, 50% 半数致死量 Ki Inhibition constant 阻害定数 %MPE Percent maximum possible effect
(処置後測定値 - 処置前測定値) × 100 (cut-off 値 - 処置前測定値) NA noradrenaline ノルアドレナリン NMDA N-methyl-D-aspartate N-メチル-D-アスパラギン酸 QT(間隔) - 心電図上のQ 波と T 波の間隔 SD Standard deviation 標準偏差
SEM Standard error of the mean 標本平均の標準誤差 SNL モデル Spinal nerve ligation モデル 脊髄神経部分結紮モデル
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 2
目 次
2.6.2 薬理試験の概要文 ... 3 2.6.2.1 まとめ ... 3 2.6.2.2 効力を裏付ける試験 ... 5 2.6.2.2.1 オピオイド受容体への結合親和性 ... 5 2.6.2.2.2 鎮痛作用 ... 9 2.6.2.2.3 モルヒネ耐性に対する作用 ... 12 2.6.2.3 副次的薬理試験 ... 15 2.6.2.3.1 NMDA 受容体を介する作用 ... 15 2.6.2.3.2 5-HT 及び NA 再取り込み阻害作用 ... 17 2.6.2.3.3 催吐作用及び制吐作用 ... 21 2.6.2.4 安全性薬理試験 ... 22 2.6.2.4.1 中枢神経系に及ぼす影響 ... 22 2.6.2.4.2 循環器系に及ぼす影響 ... 22 2.6.2.4.3 呼吸器系に及ぼす影響 ... 23 2.6.2.4.4 消化器系に及ぼす影響 ... 24 2.6.2.5 薬力学的薬物相互作用試験 ... 24 2.6.2.6 考察及び結論 ... 24 2.6.2.7 図表 ... 26 2.6.2.8 参考文献 ... 26メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 3
2.6.2 薬理試験の概要文
2.6.2.1 まとめ
メサドンの鎮痛薬としての主な作用機序は,オピオイド受容体を介するものである。 オピオイド受容体に対する結合親和性は,l-メサドンはμ1 及びμ2のμオピオイド受容体及びδ受 容体に対してモルヒネとほぼ同等の親和性を示す一方で,κオピオイド受容体に対しては低い親和 性を示した。dl-メサドンのμ1及びμ2オピオイド受容体への親和性は,l-メサドンの約半分であっ た。更に,メサドンはμオピオイド受容体の選択性が高いことが示されている。 また,ラットに髄腔内投与したdl-メサドンが,脊髄後角ニューロンの電気的刺激による侵害誘 発活動に対しナロキソンにより可逆的に拮抗されたことから,抗侵害受容作用は主にオピオイド 受容体を介したものであると考えられた。鎮痛作用として,ラットでの疼痛モデル(Tail flickテスト,Hot plateテスト及びPaw pressureテ スト)において,l-及びdl-メサドンは有意な疼痛反応抑制作用を示した。また,SNLモデルを用 いた機械刺激誘発性アロディニアと冷感刺激性アロディニアにおいてl-及びdl-メサドンは用量依 存性の抗アロディニア作用を示した。更に,Tail flickテスト及び神経障害性疼痛モデルでの痛覚 過敏に対し,モルヒネの鎮痛効果に対して耐性が見られた後も,メサドンは鎮痛効果を示した。 副次的薬理作用として,メサドンはNMDA受容体拮抗作用を有する。ラット前脳及び脊髄を用 いた結合親和性試験において,dl-,d-及びl-メサドンはNMDA受容体のMK-801 結合部位に対し親 和性を示したが,CGS-19755 結合部位に対しては親和性を示さなかった。in vitroの試験の結果, メサドンには5-HT及びNA再取り込み阻害作用が示され,メサドンをラットに皮下投与したとき, ED50量では5-HTの再取り込み作用は影響されないが,ED90量の投与では約50%阻害された。な お,ラットに鎮痛用量を超えるメサドンを腹腔内に投与しても,脳での5-HTの代謝は変化しない ことが示されている。また,イヌを用い,メサドンの催吐及び制吐作用について評価した結果, メサドン投与群ではモルヒネに見られる催吐作用は見られず,薬物誘発性の嘔吐を抑制した。 一方,安全性薬理に関連する試験では,メサドンの中枢神経系,循環器系,呼吸器系及び消化 器系に対する以下の報告がある。 中枢神経系に及ぼす作用として,ラットにおいて自発運動量を変化させ,筋硬直,カタレプシ ー,協調運動の障害,接触及び聴覚刺激に対する過敏反応及び体温変動などが見られ,これらは モルヒネにも見られた症状変化であった。中枢神経系に及ぼす作用は,l-メサドンに強く見られ, オピオイド受容体を介したものと考えられた。 循環器系に対しては,覚醒下では昇圧作用を持ち,心拍数を低下させた。ウサギの摘出心臓標 本に対してQT 間隔の有意な増加を示し,HERG 阻害においてもフェンタニルやブプレノルフィ ンと同じく薬物誘発性の QT 間隔延長作用を有することが確認された。HERG 阻害作用は,d-及 び l-メサドンのいずれにおいても認められている。 呼吸器系に対しては,ラット及びイヌを用いた試験の結果,呼吸抑制作用が見られ,血液中の 平均pO2とpH の有意な低下と,平均 pCO2の有意な増加が観察された。pCO2の増加は血清中の メサドン濃度に依存することが示唆され,l-メサドンのみが抑制作用を示した。
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 4
消化器系に対しては,モルヒネと同様にラットの小腸輸送能を抑制し,摘出モルモット回腸の 薬物誘発収縮反応の抑制及びウサギに対する糞便排泄抑制作用は,モルヒネよりも強いことが示 唆された。消化器系に及ぼす作用として,平滑筋の弛緩作用に光学異性体間で顕著な違いは見ら れておらず,大きな差はないと考えられた。
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 5
2.6.2.2 効力を裏付ける試験
2.6.2.2.1 オピオイド受容体への結合親和性 2.6.2.2.1.1 ウシの脳・尾状核のオピオイド受容体に対する結合親和性の検討 (2.6.3.2.1,資料番号 4.2.1.1.1,参考資料) ウシの脳・尾状核を用い,放射性リガンドと競合させ,l-,d-及び dl-メサドン並びにモルヒネ のオピオイド受容体への結合親和性を検討した。 [方法] ウシの脳より尾状核を摘出し,試験に用いるホモジネートを調製した。それぞれの放射性リガ ンド{[3H]DADL(μ1),[3H]DAGO(μ2),[3H]DPDPE(δ)及び[3H]U69593(κ)}に対し,l-,d-及び dl-メサドン並びにモルヒネの IC50を求めた。 [結果] 試験結果を表2.6.2.2.1.1-1 に示す。l-,d-及び dl-メサドンのうち,l-メサドンは μ1オピオイド受 容体に対する親和性が一番高かった(IC50=3.01 ± 0.18 nM)。また,l-メサドンの μ1,μ2オピオイ ド受容体に対する親和性は,d-メサドンの約 10 倍高く,dl-メサドンの約 2 倍高かった。 モルヒネと比較し,l-メサドンは μ1,μ2,δ オピオイド受容体においてモルヒネと同程度の親和 性を示す一方で,κ オピオイド受容体に対してはモルヒネの 213 nM に対し 1332 nM と低い親和 性を示した。 表 2.6.2.2.1.1-1 オピオイド受容体への結合親和性(ウシの脳・尾状核) 薬物 IC50 (nM)[平均遊離塩基濃度(ng/mL)] μ1 μ2 δ κ l-メサドン塩酸塩 3.01 ± 0.18 6.94 ± 1.3 371 ± 75 1332 ± 280 [0.9] [2.1] [114.8] [412.2] d-メサドン 26.4 ± 3.7 87.5 ± 9.0 9532 ± 854 2137 ± 881 [8.2] [27.1] [2949.7] [661.3] dl-メサドン塩酸塩 5.73 ± 1.5 10.0 ± 3.1 752 ± 686 1817 ± 573 [1.8] [3.1] [232.7] [562.3] モルヒネ塩酸塩 2.55 ± 0.37 6.59 ± 1.73 365 ± 97 213 ± 102 [0.7] [1.9] [104.1] [60.8] 平均 ± SD(n = 3 - 4) [4.2.1.1.1 Table 2 を改変引用]
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 6 2.6.2.2.1.2 オピオイド受容体の形質移入細胞及びラット脳スライスを用いた結合親和性の比較 (2.6.3.2.1,資料番号 4.2.1.1.2,参考資料) オピオイド受容体の形質移入細胞を用いて,メサドンと他のオピオイドの結合力価を評価した。 また,μ オピオイド受容体への GTP 結合作用を DAMGO と比較した。 [方法] C6 細胞に μ 又は δ,CHO 細胞に κ を形質移入し調製した細胞膜について,メサドンを含むオ ピオイド作動薬(0.3 nM~1 μM)と[3H]diprenorphine(0.2 nM)との拮抗試験から結合親和性を評 価した。また,各作動薬(0.001~10 μM)を C6μ 細胞に 60 分間作用させ,[35S]GTPγS 結合活性 をDAMGO と比較した。脳スライスについては,20 μm 厚の視床切片に[35S]GTPγS(40 pM)と 作動薬を2 時間作用させた後,放射活性を調べた。 [結果] 試験結果を表2.6.2.2.1.2-1 に示す。細胞膜での結合試験において,メサドンの Ki 値は C6μ,CHOκ 及びC6δ に対してそれぞれ 2.90 ± 0.90,1427 ± 249 及び 358 ± 119 nM を示し,メサドンの選択性 指数は,κ/μ が 493,δ/μ が 124 と算出され,フェンタニル,モルヒネ及びオキシコドンと比べて もμ オピオイド受容体に対する高い選択性を示した。μ オピオイド受容体への GTP 結合試験とし て,C6μ 細胞での DAMGO と[35S]GTPγS の結合比を算出したとき,メサドンは DAMGO に対し, 100 ± 7.0%の活性を示した。これは,モルヒネ,フェンタニル及びオキシコドンよりも高く,更 にラット視床スライス標本での[35S]GTPγS の結合も,対 DAMGO 最大結合活性比でフェンタニル, モルヒネ及びオキシコドンより高い値を示した。なお,メサドンの結合活性に性差は認めなかっ た。 表 2.6.2.2.1.2-1 ラットにおける各オピオイドの力価比較 薬物 Ki(nM) [平均遊離塩基濃度(ng/mL)] 選択性指数 [35S] GTPγS 結合活性 - 対 DAMGO 最大結合活性(%) C6μ CHOκ C6δ κ/μ δ/μ C6μ 雄 - 視床 雌 - 視床 メサドン 2.90 ± 0.90 1427 ± 249 358 ± 119 493 124 100 ± 7.0 58.5 ± 5.6 45.2 ± 9.6 [0.9] [441.6] [110.8] オキシモルフォン 0.98 ± 0.06 80.0 ± 25.0 84.20 ± 26.19 82 86 86.6 ± 2.4 37.1 ± 6.4 35.4 ± 6.1 [0.3] [24.1] [25.4] モルヒネ 1.70 ± 0.50 65.5 ± 22.6 104.57 ± 27.18 38 61 83.8 ± 1.0 42.8 ± 5.3 41.7 ± 8.1 [0.5] [18.7] [29.8] ヘロイン 158 ± 22.0 5634 ± 790 3895 ± 871 37 25 83.4 ± 6.4 39.2 ± 5.9 42.8 ± 6.4 [58.4] [2081.3] [1438.9] フェンタニル 0.71 ± 0.27 86.0 ± 24.0 51.16 ± 12.98 121 72 77.1 ± 6.0 50.9 ± 7.3 59.4 ± 10.7 [0.2] [28.9] [17.2] コデイン 727 ± 128 25411 ± 10015 52207 ± 25421 35 72 67.1 ± 5.9 31.4 ± 5.2 25.5 ± 6.0 [217.6] [7607.0] [15628.7] オキシコドン 43.9 ± 7.04 5943 ± 671 2160 ± 397 135 49 66.1 ± 3.6 36.6 ± 4.9 37.4 ± 4.5 [13.8] [1874.2] [681.2] ハイドロモルフォン 0.50 ± 0.03 12.9 ± 1.40 9.08 ± 1.95 26 18 65.1 ± 2.2 25.6 ± 4.9 34.3 ± 5.2 [0.2] [4.2] [2.9] ハイドロコドン 32.6 ± 2.19 2212 ± 502 1238 ± 332 68 38 59.5 ± 4.1 17.8 ± 5.8 20.3 ± 3.7 [9.8] [662.2] [370.6] 平均 ± SEM (n = 3) [4.2.1.1.2 Table 2~4 を改変]
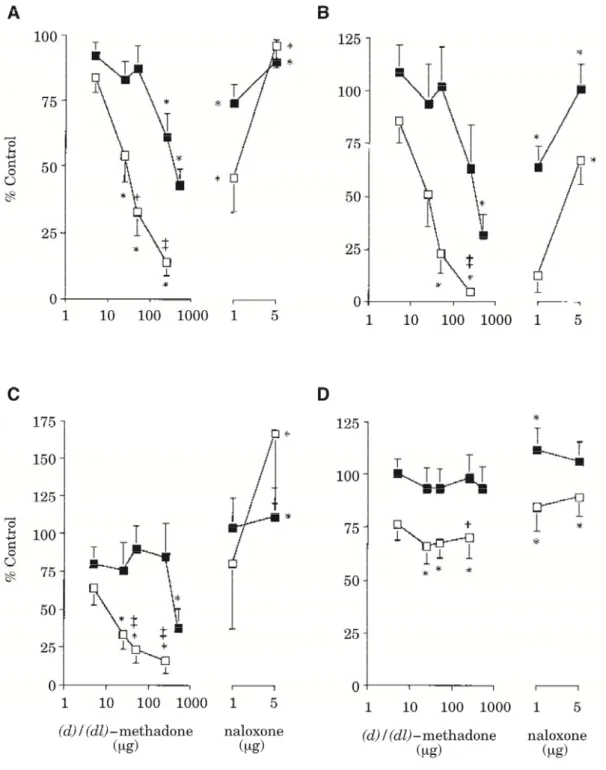
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 7 2.6.2.2.1.3 ラットの脊髄オピオイド受容体を介した侵害神経活動へ及ぼす作用 (2.6.3.2.1,資料番号 4.2.1.1.3,参考資料) ラット髄腔内に d-及び dl-メサドンを投与し,電気的刺激による脊髄後角ニューロンの侵害神経 活動に対する抑制作用を検討し,ナロキソンによる拮抗作用についても検討した。 [方法] ラットの脊髄(L4 - L5)をハロセン麻酔下で露出させ,脊髄後角に細胞外記録電極を挿入し, 後肢電気刺激後に得られる C-及び A-線維を経由した脊髄後角ニューロンの単一細胞活動を記録 した。刺激は,C-線維閾値の 3 倍の電気刺激(2 ms 幅,0.5 Hz,16 回)の入力を行った。d-メサ ドン(5, 25, 50, 250 及び 500 μg/50 μL)及び dl-メサドン(5, 25, 50 及び 250 μg/50 μL)を露出さ せた脊髄に投与し,神経活動の変化を測定した。また,dl-メサドン(250 μg/50 μL)及び d-メサ ドン(500 μg/50 μL)投与状態におけるナロキソンの拮抗作用を検討した。 [結果] 試験結果を図2.6.2.2.1.3-1 に示す。dl-メサドンは後肢への電気的な侵害刺激に対して,25 μg/50 μL 以上の濃度で C-線維誘発による反応(90~300 ms),Aβ-線維誘発による反応(0~20 ms),Aδ-線維誘発による反応(20~90 ms),50 μg/50 μL 以上では input(初回刺激の反応)を,250 μg/50 μL では後発射(300~800 ms)を有意に抑制した。また,C-線維誘発による反応及び後発射の 16 回 の累積回数とinput の比較から算出されたワインドアップは,25 μg/50 μL の投与により有意に減 少した。 dl-メサドン(250 μg/50 μL)による C-線維誘発による反応,Aβ-線維誘発による反応及び Aδ-線維誘発による反応の抑制作用は1 μg のナロキソンで,input,後発射及びワインドアップの抑 制作用は5 μg のナロキソンで打ち消された。また,d-メサドンは 250 μg/50 μL 以上の濃度で C-線維誘発による反応を抑制し,500 μg/50 μL で input,後発射,ワインドアップ及び Aδ-線維誘発 による反応を有意に抑制したが,これらの効力は dl-メサドンに比べて弱く Aβ-線維誘発による 反応については抑制しなかった。d-及び dl-メサドンの阻害作用は,ナロキソンにより可逆的に 拮抗されたことから,この抗侵害受容作用は主にオピオイド受容体を介したものであると考え られた。
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 8
図 2.6.2.2.1.3-1 電気的な侵害刺激に対する d- 及び dl-メサドンの脊髄神経への作用 (A)C-線維誘発による反応,(B)input,(C)ワインドアップ及び(D)Aβ-線維誘発による反応に対する d-(■)
及び dl-メサドン(□)の抑制作用とその抑制作用に対するナロキソンの拮抗作用(平均 ± SEM, n = 6 - 14)
*:p < 0.05 vs コントロール (multiple paired t-test)
+:p ≤ 0.05,‡:p ≤ 0.01 は,d- 及び dl-群間の比較(unpaired t-test)
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 9 2.6.2.2.2 鎮痛作用 2.6.2.2.2.1 ラットを用いた鎮痛作用 - 他オピオイド薬との比較 – (2.6.3.2.2,資料番号 4.2.1.1.4,参考資料) ラットを用いて鎮痛試験を行い,モルヒネ,オキシコドン,dl-,l-及び d-メサドンの疼痛反応 抑制作用を評価した。 [方法]
疼痛反応は,Tail flick テスト,Hot plate テスト,Paw pressure テストを行い下記の指標について 評価した。各薬物を皮下投与し,投与前,投与30, 60, 90, 120, 180 及び 240 分後に評価を実施した。 用量依存性(メサドン:1.12, 2.24, 4.47 mg/kg,モルヒネ:2.22, 4.43, 8.87 mg/kg,オキシコドン: 1.12, 2.24, 4.48 mg/kg,遊離塩基換算,表 2.6.2.2.2.1-1 参照)については,投与 30 分後に評価を実 施した。結果は,それぞれの塩酸塩投与量を記載した。
・Tail flick テスト(Ugo Basile 製,回避時間を測定,cut-off:8 秒)
・Hot plate テスト(Harvard Apparatus Ltd. 製,52.5 ± 0.3°C の熱板上における肢を舐める, 振る又は飛び上がるまでの時間を測定,cut-off:60 秒)
・Paw pressure テスト(paw pressure device,Ugo Basile 製,回避圧力を測定)
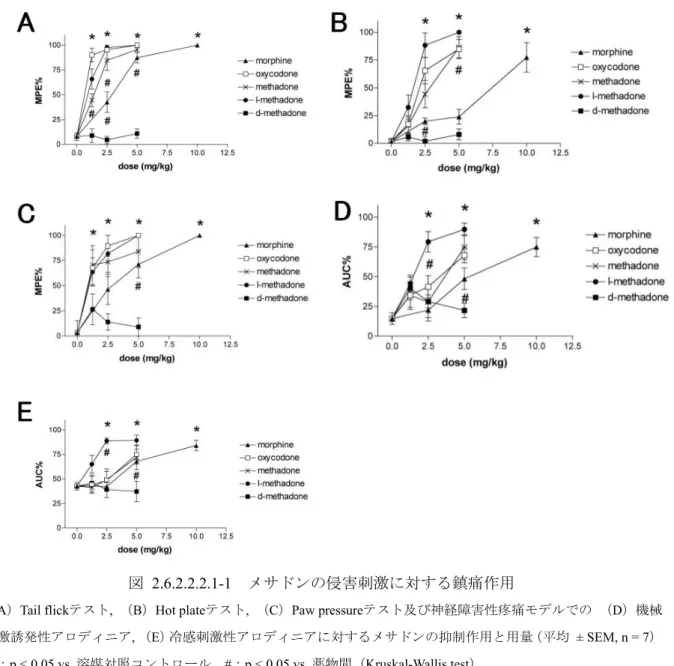
神経障害性疼痛モデルとしては,SNL モデルについて von Frey filaments を用いた機械刺激及び アセトンによる冷感刺激に対する回避反応の評価を実施した。 表 2.6.2.2.2.1-1 薬物の塩酸塩表記における投与量 薬物 投与物質 投与量(塩:mg/kg) 投与量(遊離塩基:mg/kg) メサドン メサドン塩酸塩 1.25, 2.5, 5 1.12, 2.24, 4.47 モルヒネ モルヒネ塩酸塩 2.5, 5, 10 2.22, 4.43, 8.87 オキシコドン オキシコドン塩酸塩 1.25, 2.5, 5 1.12, 2.24, 4.48 [結果] 試験結果を図2.6.2.2.2.1-1 に示す。溶媒を投与したコントロール群に対し,モルヒネ,オキシ コドン,dl-及び l-メサドンは Tail flick テスト,Hot plate テスト及び Paw pressure テストにおいて, 用量依存性の疼痛反応抑制作用を投与30 分後から示した。Tail flick テストでは,100%MPE を示 した最小用量は,オキシコドン2.5 mg/kg,l-メサドン 2.5 mg/kg,dl-メサドン 5.0 mg/kg 及びモル ヒネ10.0 mg/kg であった。Hot plate テストでは,l-メサドンが最も効果的で 2.5 mg/kg で 100%MPE を示した。Paw pressure テストでは,100%MPE を示した用量はオキシコドン 5.0 mg/kg,l-メサド ン5.0 mg/kg,モルヒネ 10.0 mg/kg であった。d-メサドンはどのテストにおいても疼痛反応に対す る抑制作用を示さなかった(図2.6.2.2.2.1-1, A~C)。
また,SNL モデル動物において機械刺激誘発性アロディニアと冷感刺激性アロディニアに対す る評価を行ったとき,dl-及び l-メサドン,モルヒネ及びオキシコドンは用量依存性の抗アロディ ニア作用を示し,いずれのアロディニアに対しても l-メサドンの効果が最も強かった
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 10
(図2.6.2.2.2.1-1, D, E)。d-メサドンは抗アロディニア作用を示さなかった。なお,モルヒネ(5.0 mg/kg),オキシコドン(2.5 mg/kg)及び l-メサドン(1.25 mg/kg)は抗アロディニア作用を示し た濃度で自発運動を減少させ,鎮静作用を示した。
図 2.6.2.2.2.1-1 メサドンの侵害刺激に対する鎮痛作用
(A)Tail flickテスト, (B)Hot plateテスト, (C)Paw pressureテスト及び神経障害性疼痛モデルでの (D)機械 刺激誘発性アロディニア, (E)冷感刺激性アロディニアに対するメサドンの抑制作用と用量(平均 ± SEM, n = 7)
*:p < 0.05 vs 溶媒対照コントロール,#:p < 0.05 vs 薬物間(Kruskal-Wallis test)
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 11
2.6.2.2.2.2 ラットを用いた鎮痛作用-雌雄の比較(2.6.3.2.2,資料番号 4.2.1.1.2,参考資料) 雌雄SD ラットを用いて,メサドンと他のオピオイドの皮下投与による鎮痛力価を評価した。
[方法]
各薬物を皮下投与し,30 分後に 50℃の温水に尾部を浸漬した。浸漬後の回避時間を計測した後 (Warm-Water Tail-Withdrawal Assay),%MPE を算出し ED50を求めた。
[結果] 試験結果を表2.6.2.2.2.2-1 に示す。メサドンの ED50は雄で1.49 ± 0.20 mg/kg,雌では 1.81 ± 0.37 mg/kg であり,オキシコドンと同程度で,モルヒネ,ハイドロコドンよりも強いことが示された。 また,メサドンにおいて性差は確認されなかった。 表 2.6.2.2.2.2-1 ラットにおける各オピオイドの鎮痛力価比較 薬物 ED50(mg/kg) 性差(雌/雄) 雄 雌 フェンタニル 0.06 ± 0.002 0.06 ± 0.0004 1.0 オキシモルフォン 0.18 ± 0.02 0.29 ± 0.06 1.6 ハイドロモルフォン * 0.27 ± 0.03 0.42 ± 0.05 1.6 ヘロイン 0.62 ± 0.11 0.58 ± 0.06 0.9 メサドン 1.49 ± 0.20 1.81 ± 0.37 1.2 オキシコドン 1.53 ± 0.29 1.71 ± 0.30 1.1 モルヒネ *** 2.17 ± 0.25 6.08 ± 0.62 2.8 ハイドロコドン ** 2.23 ± 0.21 3.70 ± 0.37 1.7 コデイン 46.3 ± 10.4 46.6 ± 5.9 1.0 *:P < 0.05,**:P < 0.01,***:P < 0.001 雌雄比較(二元配置分散分析,Student’s t test), 平均 ± SEM (n = 6) [4.2.1.1.2 Table 1 を引用] 2.6.2.2.2.3 ラットを用いた主要代謝物の鎮痛作用(2.6.3.2.2,資料番号 4.2.1.1.5,参考資料) ラットを用いて,EDDP 及び EMDP について皮下あるいは経口投与による鎮痛効果を評価した。 [方法] 各薬物を皮下(100 mg/kg)あるいは経口(200 mg/kg)投与し,30 及び 60 分後に熱刺激に対す る回避時間をtail-jerk assay により評価した。 [結果] EDDP 及び EMDP のいずれも,皮下あるいは経口投与によって鎮痛作用を示さなかった。更に, EDDP(100 mg/kg 皮下あるいは 200 mg/kg 経口投与)は,モルヒネ(1.0 mg/kg 皮下投与)による 鎮痛効果に拮抗しなかった。また,EMDP(100 mg/kg 皮下あるいは経口投与)は,dl-メサドン(0.05 mg/kg 皮下投与)による鎮痛作用に拮抗せず,EDDP 及び EMDP は鎮痛作用もオピオイドに対す る拮抗作用も持たないことが示唆された。
メサドン塩 2.6.2.2.3 2.6.2.2.3 レーザ サドンの [方法 ラット 幅0.3 m す用量の 遊離塩基 た機械刺 与に対す 物を交差 [結果 試験結 モルヒネ った(図 鎮痛作用 日目から (A) (B)投 塩酸塩 3 モルヒネ 3.1 ラット (2.6. ザー照射によ の交差耐性に 法] トの坐骨神経 mm, 2 分)の のメサドン塩 基として7.59 刺激に対する するメサドン 差投与し評価 果] 結果を図2.6. ネでは6 日及 図 2.6.2.2.3.1-用は減弱しラ ら完全な鎮痛 図2.6 投薬前,†: p 投薬前後の差, ネ耐性に対す ト神経障害性 3.2.3,資料 より坐骨神経 について検討 経を麻酔下で の照射により 塩酸塩(5 mg/ 9 mg/kg)を皮 る回避反応を ン及びメサド 価した。 .2.2.3.1-1 及び 及び生理食塩 -1 A)。メサド ラットに耐性 痛作用が見ら 6.2.2.3.1-1 < 0.05 及び † *: p < 0.05 vs 2.6.2 する作用 性疼痛モデル 料番号 4.2.1.1 経を傷害した 討を行った。 で露出させ, 神経障害を /kg,遊離塩基 皮下投与し, を測定するこ ドン 21 日間 び図2.6.2.2.3 塩水では11 日 ドンにより, 性形成が認め られなくなっ 1 日 2 回 反 ††:p < 0.01 vs 1 日目(Wilcoxo □ メサド ○ モルヒ ◆生理食塩 2 薬理試験の概 ルに対する作 1.6,参考資 たラット神経 514 nm アル を誘発した。 基として4.47 ,午前の投与 ことで,耐性形 間投与に対す 3.1-2 に示す 日目にいずれ 完全な鎮痛 められた(図 た。 反復投与後の s モルヒネ及び on signed-rank te ドン ネ 塩水 概要文 用 料) 経障害性疼痛モ ルゴンイオン 誘発7 日後 7 mg/kg)もし 与前及び30 分 形成を評価し するモルヒネ す。薬物投与前 れの群におい 痛作用の発現 図 2.6.2.2.3.1-の閾値の変化 びメサドン est)及び †:p < モデルを用い ンレーザー(波 から1 日 2 回 しくはモルヒ 分後にvon F した。更に, の交差耐性を 前の閾値は,メ いても初期の 現は9 日間継 -1 B)。一方 化(中央値 ± 絶 0.05 vs モルヒ [4. いて,モルヒ 波長514 nm 回,完全な鎮 ヒネ塩酸塩(1 Frey filament モルヒネ14 を最終投与翌 メサドンでは の値に対し,小 続したが,そ 方,モルヒネ 絶対偏差,n = 8) ヒネ(Mann–Whit .2.1.1.6 Fig. 3 ■ ● Page 12 ヒネとメ m, 0.16 W, 鎮痛を示 10 mg/kg, ts を用い 4 日間投 翌日に薬 は7 日, 小さくな その後, ネでは,3 ) tney U-test) 3 を引用] ■メサドン ●モルヒネ
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 13 耐性形成後に交差投与を行った結果,モルヒネ耐性形成を示したラットに対し,メサドンは鎮 痛効果を示したが,メサドン耐性形成を示したラットに対し,モルヒネは無効であった(図 2.6.2.2.3.1-2)。 図2.6.2.2.3.1-2 耐性形成後の交差投与による閾値の変化(中央値 ± 絶対偏差,n = 8) (A)14 日間モルヒネ処理に対するメサドン,(B)21 日間メサドン処理に対するモルヒネ
*: p < 0.05 vs 最終投与日(Wilcoxon signed-rank test)及び ††:p < 0.01 vs モルヒネ 14 日目(Mann–Whitney U-test)
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 14 2.6.2.2.3.2 持続投与後の疼痛反応に対する作用(2.6.3.2.3,資料番号 4.2.1.1.7,参考資料) ラットにモルヒネ,フェンタニル及びメペリジンを1 週間持続投与し,その前後における各オ ピオイドの疼痛反応抑制作用を評価した。 [方法] 疼痛反応は,Tail flick テストにより評価した。各オピオイドを皮下投与し,投与 15 または 25 分後にTail flick テスト及び次の用量の投与を行い,%MPE 及び ED50を求めた。モルヒネ,フェ
ンタニル及びメペリジンに対する交差耐性は,浸透圧ポンプを用いてそれぞれを(1/4 ED50)/時
間で1 週間皮下投与し,これらの処理前及びポンプ摘出 24 時間後に各オピオイドの ED50を算出
し,ED50を比較したRelative Potency(RP)を求めた。
[結果] モルヒネ及びフェンタニルに対する交差耐性の試験結果を表 2.6.2.2.3.2-1 に示す。モルヒネ処 理に対するRP は,モルヒネ及びフェンタニルが 0.54 及び 0.70 となり,ED50が有意に増加したの に対し,メサドンは 0.78 と ED50の有意な増加を示さなかった。フェンタニル処理に対しては, モルヒネ,フェンタニル及びメサドンのRP が,0.99,1.27 及び 0.98 を示し,いずれの薬剤も ED50 の有意な増加を認めなかった。メペリジン処理に対しては,モルヒネ,フェンタニル及びメサド ンのRP が,0.40,0.49 及び 0.51 を示し,いずれの薬剤も ED50が有意に増加した。 表 2.6.2.2.3.2-1 モルヒネあるいはフェンタニル前処理に対する各オピオイドの鎮痛力価比較 薬物 モルヒネ(0.8 mg/kg/hr 7 日処理) フェンタニル(0.01 mg/kg/hr 7 日処理) ED50 (遊離塩基 mg/kg, n = 7 - 9) 処理前 処理後 RP 処理前 処理後 RP モルヒネ硫酸塩 3.3 6.6** 0.54 2.5 3.8 0.99 フェンタニルクエン酸塩 0.037 0.055 ** 0.70 0.037 0.032 1.27 メペリジン塩酸塩 20.9 53.7 ** 0.49 28.9 46.3 0.75 メサドン塩酸塩 1.5 2.2 0.78 2.0 2.2 0.98 エトルフィン塩酸塩 0.0023 0.0028 0.85 0.0021 0.0018 1.20 レボルファノール酒石酸塩 0.45 1.12 ** 0.44 0.54 0.78 0.81 **:p < 0.01 vs 処理前値 [4.2.1.1.7 Table 1 及び 3 を引用]
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 15
2.6.2.3 副次的薬理試験
2.6.2.3.1 NMDA 受容体を介する作用 2.6.2.3.1.1 NMDA 受容体への結合親和性(2.6.3.3.1,資料番号 4.2.1.2.1,参考資料) ラットの前脳及び脊髄を用いて,NMDA 受容体に対するメサドンの結合作用について評価した。 [方法] ラットの前脳(全脳より小脳と脳幹を除く部分)及び脊髄(腰椎から仙椎)から神経膜を調製 した。放射性リガンド{[3H]MK-801(非競合性 NMDA 受容体拮抗薬)及び[3H]CGS-19755(競合 性NMDA 受容体拮抗薬)}と競合させ,dl-,d-及び l-メサドン,デキストロメトルファン(非競 合性NMDA 受容体拮抗薬),モルヒネ,ハイドロモルフォン及びナルトレキソンのグルタミン酸 受容体の一種であるNMDA 受容体への結合親和性を検討した。[3H]MK-801(5 nM)は,薬物と 前脳では4 時間,脊髄では 2 時間室温でインキュベーションした。[3H]CGS-19755(10 nM)は, 薬物と50 分間 4℃でインキュベーションし,いずれもフィルター濾過の後,結合量を計測し,IC50 値を求めた後,Ki 値の算出を行った。 [結果] 試験結果を表2.6.2.3.1.1-1 に示す。dl-,d-及び l-メサドン及びデキストロメトルファンは,前脳 及び脊髄において[3H]MK-801 を置換し,NMDA 受容体に対し親和性を示した。同様に検討を行 ったモルヒネ, ハイドロモルフォン及びナルトレキソンは MK-801 を置換せず,親和性を示さな かった。 一方,[3H]CGS-19755 はいずれの薬物によっても置換されなかったことから,メサドンは NMDA 受容体のグルタミン酸結合部位に対しては親和性を示さないことが示唆された。このことから, メサドンは非競合性のNMDA 受容体拮抗作用を有することが示唆された。 表 2.6.2.3.1.1-1 NMDA 受容体への結合親和性(ラット前脳・脊髄) 薬物 CNS region Ki(μM) [平均遊離塩基濃度(ng/mL)] [3H]MK-801 [3H]CGS-19755 前脳 脊髄 前脳 デキストロメトルファン 5.0 ± 0.3 [1357.0] 0.8 ± 0.6 [217.1] > 100 [> 27140.0] dl-メサドン 8.3 ± 1.2 [2568.4] 2.5 ± 0.0 [773.6] > 100 [> 30945.0] d-メサドン塩酸塩 7.4 ± 1.2 [2289.9] 2.6 ± 1.4 [804.6] > 100 [> 30945.0] l-メサドン塩酸塩 3.4 ± 0.3 [1052.1] 2.8 ± 0.9 [866.5] > 100 [> 30945.0] モルヒネ > 100 [> 28534.0] - - ハイドロモルフォン > 100 [> 32180.0] - - ナルトレキソン > 100 [> 34140.0] - - 平均 ± SEM(n = 2 - 4) [4.2.1.2.1 Table 1 を改変引用]メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 16
2.6.2.3.1.2 d-メサドンの NMDA 受容体を介した抗侵害受容作用 (2.6.3.3.1,資料番号 4.2.1.2.2,参考資料)
ラットを用いて,Tail flick テスト及びホルマリン誘発性並びに NMDA 誘発性の侵害反応を指標 に,d-メサドンの NMDA 受容体を介した抗侵害受容作用を評価した。
[方法]
ラジエントヒーターの熱を尾部に照射し潜時を測定(Tail flick テスト)した後,ラットの脊髄 に,塩酸を用いて溶解させた d-又は l-メサドンを髄腔内投与(IT)し,15 分後に再度 Tail flick テ ストを実施し潜時が2 倍以上の伸びを示したものについて鎮痛効果有りと判断した。また,d-メ サドンを脊髄に投与し,15 分後に右足蹠にホルマリンを投与した後,60 分間の侵害反応(右肢の 振り回数)を計測した。同様に,ナロキソンを d-メサドンと同時投与を行った場合についても侵 害反応を計測した。また,髄腔内に NMDA を投与して起こる侵害反応(尾部の引っ掻き,噛み 及び舐め)に対する d-メサドンの作用を検討した。 [結果] 試験結果を図2.6.2.3.1.2-1 及び表 2.6.2.3.1.2-1 に示す。Tail flick テストでは,l-メサドンは用量 依存的な鎮痛作用(ED50:15.6 μg /ラット)を示したが,d-メサドンでは 460 μg /ラットでも鎮痛 作用を示さなかった(図 2.6.2.3.1.2-1, A)。l-メサドンの鎮痛作用はナロキソンで拮抗された。ホ ルマリン誘発性の侵害反応については,d-メサドンはホルマリン投与 10~60 分後に起こる 2 相目 の反応を用量依存的に抑制し(図2.6.2.3.1.2-1, B),これはナロキソンにより拮抗されなかった。 更に,d-メサドンの 250 μg /ラット投与は,侵害反応を示すための NMDA の ED50を3 倍に増加さ せ(表2.6.2.3.1.2-1),d-メサドンは NMDA 受容体拮抗作用による抗侵害受容作用を持つことが示 唆された。 図 2.6.2.3.1.2-1 d-メサドンの鎮痛作用
(A)d-, l-メサドンのTail flickテスト及び(B)d-メサドンのホルマリン誘発性の疼痛反応(第2相)に対する抑制 作用(平均 ± SEM, n = 9)
*p < 0.05 vs 生理食塩水コントロール(一元配置分散分析,Student’s t test)
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 17 表 2.6.2.3.1.2-1 NMDA 誘発侵害反応に対する d-メサドン(250 μg /ラット)の作用 処置 NMDA ED50 a(nmol /ラット) [ng/ラット] 95%信頼区間 a 効力比 生理食塩水+ NMDA 1.3 [191.3] 1.0 - 1.6 1.00 d-メサドン+ NMDA 4.3 * [632.7] 3.4 - 5.0 0.30 *:p < 0.05 vs 生理食塩水 + NMDA (n = 7)
a:BLISS-21 computer program を用いて算出
[4.2.1.2.2 Table 1を改変引用] 2.6.2.3.2 5-HT 及び NA 再取り込み阻害作用 2.6.2.3.2.1 5-HT あるいは NA 再取り込み阻害作用(2.6.3.3.2,資料番号 4.2.1.2.3,参考資料) ラットの大脳皮質あるいは延髄-橋のホモジネートからシナプトソームを調製し,メサドンと他の オピオイドの5-HT 及び NA 再取り込み量を測定し,各オピオイドの阻害定数(Ki)を求めた。 [方法] 大脳皮質あるいは延髄-橋のホモジネートからシナプトソームを調製した。メサドンを含む各オ ピオイド作動薬(それぞれ 5~10 濃度)及びリガンドとして,10 nM [3H]5-HT あるいは 10 nM [3H]NA を作用させ,フィルター濾過の後,それぞれのリガンドの再取り込み量を測定し,Ki を 求めた。また,ラット前脳のホモジネートを用いて,オピオイド受容体リガンドに対する Ki を求 めた。試験条件を表2.6.2.3.2.1-1 に示した。 表 2.6.2.3.2.1-1 オピオイド受容体結合並びに 5-HT 及び NA 再取り込み阻害の試験条件 試験条件 オピオイド受容体への結合阻害 μ δ κ 容量(mL) 1 1 2
3H-リガンド DAMGO DPDPE U-69,593
リガンド濃度(nM) 0.33 2.2 0.49
反応温度(℃) 25 25 25
反応時間(hr) 1 1 1.5
フィルター予浸 0.5%PEI 0.5%PEI 0.1%BSA 5-HT,NA 再取り込み阻害 5-HT NA 脳部位 大脳皮質 延髄-橋 反応時間(min) 5 6 3H-リガンド 5-HT NA リガンド濃度(nM) 10 10 [結果] 試験結果を表 2.6.2.3.2.1-2 に示す。メサドン及び(±)-トラマドールについては,遊離塩基濃度 を併記した。d-及び l-メサドンは,いずれも 5-HT 再取り込み阻害作用を示し,それぞれの Ki は 992 及び 14.1 nM であった。更に d-及び l-メサドンは,いずれも NA 再取り込み阻害作用を示し, それぞれの Ki は 12700 及び 702 nM であった。同様に比較を行ったモルヒネ,オキシコドン,ブ プレノルフィン,ナロキソンなどのテトラヒドロフラン環を有しC4-C5 に橋状酸素を持つオピオ イドは,5-HT あるいは NA 再取り込み阻害作用を示さなかった。メサドンと同様に非フェナント レンとして評価を行ったトラマドールについては,(±)-,(+)-及び(-)-トラマドールのいずれも 5-HT あるいは NA 再取り込み阻害作用を示し,Ki はそれぞれ 992, 528 及び 2350 あるいは 785, 2510 及び432 nM であった。
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 18 表 2.6.2.3.2.1-2 オピオイド受容体結合並びに 5-HT 及び NA 再取り込み阻害作用の比較 薬物 Ki (nM) [遊離塩基濃度(ng/mL)] μ δ κ NA 5-HT DAMGO 0.365 85 210 > 100,000 > 100,000 DPDPE 380 4.45 11700 > 100,000 > 100,000 U-69,593 1000 3750 6.65 > 100,000 > 100,000 フェナントレン構造(+橋状酸素) コデイン 160 5130 5970 > 100,000 > 100,000 オキシコドン 8.69 901 1350 > 100,000 > 100,000 ハイドロコドン 11.1 962 501 > 300,000 > 300,000 (- )-モルヒネ 1.24 145 23.4 > 100,000 > 100,000 (+)-モルヒネ > 100,000 > 100,000 > 300,000 > 300,000 > 300,000 ブプレノルフィン 4.18 25.8 12.9 > 100,000 > 100,000 (- )-ナロキソン 0.559 36.5 4.91 > 100,000 > 100,000 (+)-ナロキソン 3550 122000 8950 > 100,000 > 100,000 ナルトレキソン 0.0825 8.02 0.509 > 100,000 > 100,000 フェナントレン構造(- 橋状酸素) レボルファノール 0.42 3.61 4.2 1210 86.3 デキストロルファン 420 34700 5950 340 401 レボメトルファン 11.2 249 225 179 35.8 デキストロメトルファン 1280 11500 7000 240 23 レバロルファン 0.213 2.18 1100 19100 7950 デキストラロルファン 1140 2660 34.6 804 27700 非フェナントレン構造 dl-メサドン [0.5] [134.6] [125.3] 1.7 435 405 - - l-メサドン 0.945 371 1860 702 14.1 [0.3] [114.8] [575.6] [217.2] [4.4] d-メサドン 19.7 960 1370 12700 992 [6.1] [297.1] [423.9] [3930.0] [307.0] LAAM 9.86 169 1020 67400 1440 d-プロポキシフェン 34.5 380 1220 10000 30000 (±)-トラマドール 2120 57700 42700 785 992 [558.4] [15197.0] [11246.3] [206.8] [261.3] (+)-トラマドール 1330 62400 54000 2510 528 (- )-トラマドール 24800 213000 53500 432 2350 各5~10濃度を用いて評価(2~3試行)。2~3回の結果の平均 [4.2.1.2.3 Table 3を引用]
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 19 2.6.2.3.2.2 in vitro 及び ex vivo 投与による 5-HT 再取り込み阻害作用 (2.6.3.3.2,資料番号 4.2.1.2.4,参考資料) ラットの大脳皮質からシナプトソームを調製し,メサドンの5-HT 再取り込み量を測定し,阻害定 数(Ki)を求めた。更に,メサドン皮下投与後に脳を摘出し,5-HT 再取り込みの評価を行った。 [方法] 大脳皮質のホモジネートからシナプトソームを調製し,メサドン及びリガンドとして 5 nM [3H]5-HT を 6 分間作用させ,リガンドの再取り込み量を測定し,Ki を求めた。ex vivo の試験とし て,急性投与試験ではホットプレート法から算出したメサドンのED50量もしくはED90量を皮下 投与し,30 分後にシナプトソームを調製し,リガンドの再取り込み量を測定した。慢性投与試験 では,メサドンを15 日間皮下投与した。最終投与 72 時間後にシナプトソームを調製し,リガン ドの再取り込み量を測定した。更に,最終投与24 時間後に ED90量を投与し,30 分後に調製した シナプトソームを用いた評価を実施した。 [結果] 試験結果を表2.6.2.3.2.2-1 に示す。メサドン及びトラマドールの 5-HT 再取り込み阻害作用にお ける Ki はそれぞれ 270 及び 760 nM であり,遊離塩基濃度に換算した場合 83.6 及び 200.2 ng/mL であった。一方,モルヒネは再取り込み阻害作用を示さなかった。モルヒネ,メサドン及びトラ マドールについて各塩酸塩を皮下投与した時のホットプレート法でのED50が求められ,この用量 を皮下投与した時に,トラマドール処理ラットでは5-HT の取り込みが 63%まで阻害されたが, モルヒネ及びメサドンでは認められなかった。一方,ED90を投与した際に,メサドンは52%まで 5-HT 取り込み量を阻害したが,モルヒネでは認められなかった。メサドン慢性投与の結果,休薬 72 時間後に 5-HT 取り込みが 90%増加したが,トラマドールには認められなかった。一方,24 時間後にED90量を作用させた場合,メサドンによる5-HT 再取り込み阻害作用は認められなかっ た。 表 2.6.2.3.2.2-1 メサドン,モルヒネ及びトラマドールの 5-HT 再取り込み阻害作用 生理食塩水 モルヒネ塩酸塩 メサドン塩酸塩 トラマドール塩酸塩 [3H]-5HT 取り込み阻害 Ki(μM), [遊離塩基濃度(ng/mL)] - > 300,000 [> 85600000] 0.27 ± 0.038 [83.6] 0.76 ± 0.045 [200.2] 鎮痛作用/ED50 [遊離塩基 mg/kg] - [4.3] [3.5] [31] [3H]-5HT 取り込み (%) a 投与量 [遊離塩基 mg/kg] 急性投与 ED50 - [4.3]: ≈100 [3.5]: ≈100 [31]: 63 ± 3.4 ED90 - [40]: ≈100 [35]: 52 ± 3.8 - 慢 性 投 与 投与量 1mL/kg - 初期→最終:[5→40] 増加量/日:[2.5] 初期→最終:[15→120] 増加量/日:[7.5] 72 時間後の 再取り込み量 100 ± 6.6 - 190 ± 15 b 110 ± 4.8 24 時間後に再投与, 再取り込み量測定 - - [35]:102 ± 4.4 [31]:58 ± 4.2 b a:平均 ± SEM(n = 4)
b:p < 0.01 vs 生理食塩水処理72時間後(one way ANOVA followed by REGWF test.)
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 20
2.6.2.3.2.3 メサドン投与による脳内 5-HT 代謝の検討 (2.6.3.3.2,資料番号 4.2.1.2.5,参考資料)
ラットを用いてメサドンを腹腔内投与し,5-HT 及び 5-hydroxyindole acetic acid(5-HIAA)の脳内濃 度を測定した。 [方法] メサドンを含む各薬物を生理食塩水に溶解し腹腔内投与し,1 時間後に脳を摘出し,5-HT 及び 5-HIAA の濃度を測定した。さらに,プロベネシド及びパルギリンを併用し,5-HIAA 及び 5-HT の変化について評価を行い,5-HT 代謝の指標とした。 [結果] 試験結果を表2.6.2.3.2.3-1 に示す。メサドン(10 mg/kg)の腹腔内投与により,脳内の 5-HT, 5-HIAA 濃度に変化は認められなかった。モルヒネ(10 及び 20 mg/kg)は,5-HT 濃度に変化を及 ぼさなかったが,5-HIAA 濃度は 20 mg/kg の腹腔内投与により有意に増加した。同様に評価を行 ったペチジン(50 mg/kg)及びクロルイミプラミン(15 mg/kg)では,5-HIAA 濃度の減少が認め られた。 表 2.6.2.3.2.3-1 メサドンを含む鎮痛薬投与後 1 時間の脳内 5-HT 及び 5-HIAA 変化 薬物
遊離塩基換算 5-HT(μg/g ± SEM) 5-HIAA(μg/g ± SEM) 投与量 コントロール 処置 コントロール 処置 モルヒネ塩酸塩 10 0.46 ± 0.02 0.47 ± 0.01 0.36 ± 0.02 0.36 ± 0.02 20 0.41 ± 0.02 0.41 ± 0.02 0.34 ± 0.01 0.42 ± 0.01 ** ペンタゾシン乳酸塩 60 0.47 ± 0.01 0.46 ± 0.02 0.30 ± 0.02 0.32 ± 0.01 メサドン塩酸塩 10 0.45 ± 0.01 0.45 ± 0.01 0.29 ± 0.02 0.29 ± 0.02 ペチジン塩酸塩 50 0.49 ± 0.02 0.53 ± 0.01 0.34 ± 0.01 0.28 ± 0.01 ** クロルイミプラミン塩酸塩 15 0.52 ± 0.02 0.57 ± 0.02 0.31 ± 0.01 0.28 ± 0.01 * ナロキソン塩酸塩 5 0.48 ± 0.02 0.47 ± 0.02 0.35 ± 0.02 0.35 ± 0.02 *: p<0.05, **: p<0.001 対コントロール(n = 10~15) [4.2.1.2.5 Table 1を引用] また,プロベネシド(200 mg/kg)及びパルギリン(75 mg/kg)投与による脳内の 5-HIAA 及び 5-HT の経時的な増加がモルヒネ(20 mg/kg)では亢進されたが,メサドンには認められなかった。
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 21 2.6.2.3.3 催吐作用及び制吐作用(2.6.3.3.3,資料番号 4.2.1.2.6,参考資料) 各オピオイドのイヌにおける覚醒下での催吐及び制吐作用に対する効果を評価した。 [方法] 雑種犬を用いてアポモルフィン(静脈内投与:0.03 mg/kg)又は硫酸銅(胃内投与:16 mg/kg) を投与し,催吐作用を確認した。次に,オピオイドを静脈内投与した後,15 分後にアポモルフィ ン単独又はアポモルフィンと硫酸銅を投与した後,15 分後までの嘔吐の有無を観察した。 [結果] 試験結果を表2.6.2.3.3-1 に示す。メサドン(0.2, 0.5, 1 mg/kg),フェンタニル(0.005, 0.01 mg/kg) 及びモルヒネ(0.3, 1, 2 mg/kg)を投与した結果,メサドン及びフェンタニルではいずれの投与量 においても嘔吐は見られなかったが,モルヒネは0.3 mg/kg 投与で 6 例中 6 例,1 mg/kg 投与で 5 例中1 例に嘔吐が確認された。以上のことより,メサドンの催吐作用は本条件下では見られず, モルヒネよりも低いものと考えられた。 また,アポモルフィン単独又は硫酸銅併用による催吐作用は,オピオイドの高用量投与によっ て抑制された。 表2.6.2.3.3-1 イヌにおける各オピオイドの催吐作用及び制吐作用 オピオイド 投与量(mg/kg) 嘔吐動物数/使用動物数 薬物 オピオイド +アポモルフィン +アポモルフィン +硫酸銅 0 - 5/5 5/5 メサドン 0.2 0/5 5/5 - 0.5 0/5 4/5 - 1 0/10 0/5 0/5 0 - 6/6 5/5 モルヒネ 0.3 6/6 6/6 - 1 1/5 3/5 - 2 0/28 0/23 0/5 0 - 7/7 5/5 フェンタニル 0.005 0/6 5/6 - 0.01 0/12 3/7 1/5 [4.2.1.2.6 Table 1 を改変引用]
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 22
2.6.2.4 安全性薬理試験
2.6.2.4.1 中枢神経系に及ぼす影響 (2.6.3.4.1,2.6.3.4.5,資料番号 4.2.1.3.1~4.2.1.3.10,参考資料) ラットの運動量に対するメサドン塩酸塩の作用は,腹腔内への投与量がメサドンとして5 mg/kg であれば,運動量が低下した後に運動量の増加が認められる二相性の反応を示し,10 mg/kgの投 与では運動量を低下させることが示唆された(4.2.1.3.1 参照)。この作用はメサドン特有のもので はなく,モルヒネでも同様な作用が認められ,投与経路にかかわらずメサドンは,モルヒネと同 程度又はやや強い作用を有することが示唆された(4.2.1.3.2 参照)。ラット腹腔内投与において, 単回投与後の投与1 時間後までの行動量は,l-メサドンが 2.68 mg/kgで 50%程度の抑制が見られた のに対し,d-メサドン 2.68 mg/kg投与では行動量に変化は見られず,8.9 及び 26.8 mg/kg投与でそ れぞれ50 及び 0%の抑制が見られた(4.2.1.3.3 参照)。1 時間ごとの行動量は,l-メサドン(2.68 mg/kg) が投与1 時間後まで行動抑制を示すが,3 及び 4 時間目までは行動量の亢進を示したのに対し, d-メサドン(15.7 mg/kg)には投与 1 時間後までの行動抑制が観察されたのみであった。 また,ラットにおける行動変化について,メサドン10 mg/kg程度を腹腔内又は皮下投与するこ とにより,筋硬直やカタレプシーが引き起こされることが示唆された(4.2.1.3.4,4.2.1.3.5 参照)。 モルヒネでも同様な作用が見られるが,メサドンの方が筋硬直やカタレプシーの発症が早く,ま た,同一投与量での比較においてはメサドンの方が強い筋硬直が認められており,メサドンの作 用はモルヒネよりも強いことが考えられた(4.2.1.3.5,4.2.1.3.6 参照)。マウスにおける協調運動 への影響を検討した結果,メサドンはモルヒネと同様にマウスの協調運動に対し障害作用を有し ていることが示唆された(4.2.1.3.7 参照)。 感覚及び運動反射反応に対する影響については,メサドンの投与によりラットにおいて用量依 存的に正向反射の消失が認められた(4.2.1.3.6 参照)。モルヒネでも同様に正向反射の消失が認め られたが,同一投与量での比較では,メサドンの作用のほうが強いことが示唆された。なお,触 覚及び聴覚刺激に対する過敏反応についてもメサドン,モルヒネ双方に認められており,メサド ン特有の作用ではないものと考えられ,脳波については,l-メサドンが影響を与えることが示さ れた(4.2.1.3.4,4.2.1.3.6,4.2.1.3.8 参照)。 体温に与える影響については,メサドンを皮下投与することにより体温に変化が認められるが, その変化は環境の温度により大きく異なり,マウスでは20℃から 25℃程度の環境下では直腸温を 低下,30℃程度の環境下では直腸温を上昇させることが示唆された(4.2.1.3.9 参照)。なお,モル ヒネやフェンタニルにおいてもメサドンと同様の作用が認められ,メサドンの体温に与える影響 はラット及びマウスへの腹腔内投与でも同様の結果が得られている(4.2.1.3.4,4.2.1.3.7 参照)。 この作用は,l-メサドンに由来すると考えられた(4.2.1.3.10 参照)。 2.6.2.4.2 循環器系に及ぼす影響(2.6.3.4.2,2.6.3.4.5,資料番号 4.2.1.3.10~4.2.1.3.17,参 考資料) ラットにおけるテレメトリー試験(4.2.1.3.11 参照),覚醒下のイヌ(4.2.1.3.12)及びウマ (4.2.1.3.14)における投与試験から,メサドンは昇圧作用を持っていることが示された。一方, これらの試験において,メサドンはウマ以外の試験では心拍数を低下させた。また,麻酔下のイメサドン塩酸塩 2.6.2 薬理試験の概要文 Page 23 ヌ及びネコで得られた結果では,d-及びl-メサドンのいずれも血圧の低下作用を示したが,静脈内 投与においては一過性であり,摘出心臓を用いた結果では,心拍などに光学異性の影響は見られ ていない(4.2.1.3.10 参照)。心電図への影響は,ウサギの摘出心臓標本を用いて灌流下にメサド ン投与の影響を評価した結果,30 μMにてQT間隔の有意な延長が認められ,%QT変化量は,1 及 び3 μMで 10~12%の増加,10 及び 30 μMで 21 及び 44%の有意な増加を示した(4.2.1.3.15 参照)。 また,メサドンをHERG発現系(HEK293 細胞)において評価した結果,メサドンのHERG阻害は, 1, 3, 10, 30 及び 100 μMでそれぞれ 3.7, 16.9, 42.7, 64.5 及び 79.8%(グラフ読み取り値)を示し,IC50 は9.8 μMであり,フェンタニルやブプレノルフィン(それぞれIC50=1.8 及び 7.5 μM)と同じく 薬物誘発性のQT間隔延長作用を有することが確認された(4.2.1.3.16 参照)。同様に作用させたメ サドンの主要代謝物であるEDDP(50 μM)にはQT間隔延長作用が確認されておらず,メサドン の未変化体特有の作用であることが確認された。なお,モルヒネ(1 mM)にQT間隔延長作用は 確認されなかった。メサドンのHERGに対する作用については,アフリカツメガエル卵母細胞に HERGを発現させたin vitroの試験において,d-及びl-メサドンのいずれにおいても阻害作用が認め られ,d-メサドンがより低濃度でQT間隔延長作用を示す可能性も示唆されている(4.2.1.3.17 参照)。 2.6.2.4.3 呼吸器系に及ぼす影響 (2.6.3.4.3,2.6.3.4.5,資料番号 4.2.1.3.10~4.2.1.3.14,4.2.1.3.18~4.2.1.3.22,参 考資料) ラットに腹腔内投与を行ったところ,呼吸パラメータと用量の関係から,メサドンの過量投与 は呼気時間の延長を伴った呼吸抑制を招きやすいことが示唆された(4.2.1.3.18 参照)。メサドン をモルヒネ,フェンタニル及びブプレノルフィンと過量投与状態で比較した際に,メサドンの投 与30 分前から 240 分後までの分時換気量,呼吸頻度,呼吸時間及び呼気時間の各曲線下面積で 示されるパラメータはフェンタニルと同様のプロファイルを示し,フェンタニル,メサドン,モ ルヒネの順に投与の影響が見られた。吸気時間の延長がすべての薬剤で同様に見られたが,1 回 換気量には影響が認められなかった(4.2.1.3.19 参照)。更に,ラットでのテレメトリー試験にお いて,皮下への持続注入により呼吸数の減少が示された(4.2.1.3.11 参照)。また,イヌに皮下投 与を行った試験においても肺換気量の低下を示す呼吸抑制が観察された(4.2.1.3.20 参照)。モル モット及びウサギを用いた光学異性体別の評価の結果,l-メサドンのみが抑制作用を示し,dl-メ サドンの持つ呼吸抑制作用は中枢神経系を介したl-メサドンの作用に負うことが大きいと考えら れた(4.2.1.3.21,4.2.1.3.10 参照)。血液ガスの変化は,ラットに腹腔内投与を行った試験におい ては,平均pO2とpHの有意な低下と平均pCO2の有意な増加が観察されている(4.2.1.3.22 参照)。 メサドンをモルヒネ,フェンタニル及びブプレノルフィンと過量投与状態で比較した際に,メサ ドンとモルヒネの変化は類似のプロファイルを示し,フェンタニルのPaO2,pH及び重炭酸の変化 に及ぼす影響が最も大きかった(4.2.1.3.19 参照)。pHの低下とpCO2の増加は,イヌに静脈内投与 を行った試験においても観察されている(4.2.1.3.12,4.2.1.3.13 参照)。これらのことから,メサ ドンは他のオピオイドと同じく呼吸機能に作用し,投与量の増加に伴って呼吸抑制作用を示すと 考えられた。
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 24 2.6.2.4.4 消化器系に及ぼす影響 (2.6.3.4.4,2.6.3.4.5,資料番号 4.2.1.3.10,4.2.1.3.23~4.2.1.3.27,参考資料) 炭末輸送能を調べた結果,メサドンにラットの小腸輸送能を抑制する作用が認められた。その 用量・作用比較を行った場合,モルヒネと同程度かやや強く,フェンタニルより弱いものである と考えられた(4.2.1.3.23,4.2.1.3.24 参照)。メサドンは,摘出モルモット回腸の薬物による収縮 反応を抑制し,その作用はモルヒネよりも強いことが示唆されている(4.2.1.3.25 参照)。この摘 出平滑筋の薬物誘発性収縮反応に対する弛緩作用は,d-及び l-メサドンのいずれにも見られ,そ の作用強度に鎮痛作用に見られるほどの違いは見られていない(4.2.1.3.10,4.2.1.3.26 参照)。 また,ウサギを用いて糞便排泄抑制作用を検討した結果,メサドンの作用はモルヒネよりも強 いことが示唆されている(4.2.1.3.27 参照)。
2.6.2.5 薬力学的薬物相互作用試験
該当資料なし2.6.2.6 考察及び結論
メサドンの鎮痛薬としての主な作用機序は,オピオイド受容体を介するものである。 効力を裏付ける試験として,オピオイド受容体に対する結合親和性を検討した。ウシの脳・尾 状核を用いた放射性リガンドによる評価では,ラセミ体であるdl-メサドンのμオピオイド受容体 への親和性は,l-メサドンの約半分であった。l-メサドンはμ1,μ2のμオピオイド受容体において モルヒネとほぼ同等の親和性を示し,κオピオイド受容体に対しては低い親和性を示した (2.6.2.2.1.1 参照)。更に,受容体を形質移入させた細胞の試験結果からは,dl-メサドンは,フェ ンタニル,モルヒネ及びオキシコドンよりも高いμオピオイド受容体への選択性及びμオピオイド 受容体のGTP結合活性化が示唆されている(2.6.2.2.1.2 参照)。In vivoの試験でも,ラットに髄腔 内投与したdl-メサドンが,脊髄後角ニューロンの電気的刺激による侵害誘発活動に対し,ナロキ ソンにより可逆的に拮抗された(2.6.2.2.1.3 参照)。このことからも,メサドンの抗侵害受容作用 は主にオピオイド受容体を介したものであることが示された。種々の疼痛モデルにおいても,dl-及びl-メサドンは有意な疼痛反応抑制作用を示し,モルヒネと同様に強力な鎮痛作用を持つこと が示されている(2.6.2.2.2.1 参照)。また,Tail flickテスト及び神経障害性疼痛モデルでの痛覚過 敏に対し,モルヒネの鎮痛効果に対して耐性が見られた後も,メサドンは鎮痛効果を示したこと から,モルヒネとの交差耐性は不完全であり,がん性疼痛においてもモルヒネで効果が見られな くなった際にメサドンを使用することで除痛効果が期待できると考えられる(2.6.2.2.3.1, 2.6.2.2.3.2 参照)。 以上の結果から,dl-メサドンは,モルヒネよりも少ない量でμオピオイド受容体を介した鎮痛 作用を示し,特にその作用は,l-メサドンに負うところが大きいと結論される。 副次的な薬理試験として,NMDA受容体を介した作用,5-HT及びNA再取り込み阻害作用及び 嘔吐に対する作用を検討した。メサドンは,モルヒネやフェンタニルには見られないNMDA受容 体拮抗作用を有し,この受容体を介した鎮痛作用も有していることが示され,これにはd-メサド ンも関与すると考えられている(2.6.2.3.1 参照)。一方,モルヒネ,オキシコドン及びブプレノルメサドン塩酸塩 2.6.2 薬理試験の概要文 Page 25 フィンといったテトラヒドロフラン環を持つオピオイドには見られない5-HT及びNA再取り込み の阻害作用も確認されているものの,鎮痛に関わる直接的な結果は得られていない。さらに,ラ ットにおいては鎮痛用量のメサドン投与によって脳内5-HTの代謝に変化は見られておらず,各 Kiの比較からも,メサドンの持つ鎮痛効果はμオピオイド受容体を介したものが主であると考えら れる(2.6.2.3.2 参照)。オピオイド投与において,しばしば問題とされる嘔吐については,オピオ イド誘発性の催吐作用を確認した結果,メサドンによる嘔吐はイヌでは見られなかった(2.6.2.3.3 参照)。このことから,メサドンはモルヒネと比較して嘔吐を引き起こす可能性が低いと考えられ る。 安全性薬理試験の結果から,メサドンは,中枢神経系に対しモルヒネなどと同様にオピオイド に見られる抑制作用を有することが示されている(2.6.2.4.1 参照)。メサドンの高用量投与でラ ットにおいて見られた運動量の低下は,モルヒネと同程度かそれ以上の効力を示し,また,筋硬 直やカタレプシーといった行動変化も,モルヒネよりも早期に強く発現することが示されている。 メサドンは協調運動を障害し,用量依存的に正向反射を消失させ,その作用がモルヒネよりも強 いことも示されている。聴覚及び触覚刺激に対しての過敏反応については,モルヒネと同様の反 応を有することが示されている。更に,モルヒネやフェンタニルと同様に周囲の温度に影響を受 ける体温変化を示した。これらは,主にl-メサドンのオピオイドとしての作用に由来すると考え られた。 循環器系に対しては,覚醒下の試験の結果,メサドンは昇圧作用を持ち,心拍数を低下させる 傾向が示唆された。これは,メサドン投与によるアルギニンバソプレッシンの遊離が関与したも のと考えられ,μオピオイド受容体作動薬に一般的に認められる作用と同じものであった。一方, 麻酔下では,d-及びl-メサドンのいずれにおいても降圧作用が確認されている。心室筋に及ぼす作 用として,ウサギの摘出心臓標本やHERG発現系を用いた評価の結果,フェンタニルやブプレノ ルフィンと同様にメサドンにはQT間隔延長作用を持つことが示唆された(2.6.2.4.2,4.2.1.3.13 参 照)。また,HERG発現系を用いた評価では,いずれの光学異性体も阻害効果を有するが,d-メサ ドンがやや強い阻害作用を持つことが示唆されている。臨床における最高血漿中濃度とIC50の比 較から,dl-メサドンの安全域が 2.7 と求められ,この安全域は,モルヒネ(> 400),ブプレノル フィン(208)及びフェンタニル(60)と比べると狭いことから,心臓伝達系疾患の合併や催不整 脈作用のある薬剤の併用等,不整脈発現素因を有する患者にメサドンを使用する場合には,心機 能や血中電解質の評価を行うなど,慎重な管理が必要と考える。 呼吸器系に対しては,ラットにおいて呼吸数の減少が示され,また,血液中の平均pO2とpHの 有意な低下と平均pCO2の有意な増加が観察されている。これらの作用は,l-メサドンに由来し, メサドンは他のオピオイドと同様に呼吸機能を抑制する作用を有すると考えられた。 消化器系に対しては,メサドンにラットの小腸輸送能を抑制する作用が認められ,その効力は モルヒネと同程度かやや強く,フェンタニルより弱いものであると考えられた。メサドンは,摘 出モルモット回腸の各種薬物誘発の収縮反応を抑制し,その作用はモルヒネよりも強いことが示 唆された。また,ウサギを用いて糞便排泄抑制作用を検討した結果,メサドンの効力はモルヒネ よりも強いことが示唆された。 安全性薬理試験の結果から,光学異性体間で見られる急性毒性反応が鎮痛用量に見られるほど の比を示さない理由として,呼吸器系及び中枢性の抑制には l-メサドンの関与が大きく,循環器
メサドン塩酸塩 2.6.2 薬理試験の概要文 Page 26 系に対しては d-メサドンが l-メサドンと同程度かやや強く作用するためと考えられた。 以上の薬理試験の検討結果より,メサドンは他のμ オピオイド作動性の鎮痛薬と同様に鎮痛作 用を有し,臨床使用においてその鎮痛効果を期待することが確認された。また,d-メサドンには NMDA 誘発侵害反応の抑制効果が確認され,ほかのオピオイドに見られない鎮痛作用,例えば, がん性の神経障害性疼痛に対する鎮痛作用を持つことが期待される。 一方,安全性薬理で見られた結果も,他のμ オピオイド作動性の鎮痛薬と同様のものであった。 メサドンの不整脈誘発のリスクに関しては,モルヒネ及びフェンタニルといった従来臨床で使用 されるオピオイド系鎮痛薬よりも高いことが示唆されたが,十分な管理のもとで使用される場合, メサドンは鎮痛薬として臨床的有用性が期待できるものと考える。