目次
略号一覧表 ... 3 2.4 非臨床試験の概括評価 ... 5 2.4.1 非臨床試験計画概略 ... 5 2.4.2 薬理試験 ... 9 2.4.2.1 効力を裏付ける試験 ... 9 2.4.2.2 副次的薬理試験 ... 10 2.4.2.3 安全性薬理試験 ... 11 2.4.2.4 薬力学的薬物相互作用試験 ... 11 2.4.3 薬物動態試験 ... 11 2.4.3.1 吸収 ... 12 2.4.3.2 薬物動態学的薬物相互作用試験 ... 14 2.4.3.3 その他の薬物動態試験 ... 14 2.4.4 毒性試験 ... 14 2.4.4.1 単回投与毒性試験 ... 14 2.4.4.2 反復投与毒性試験 ... 15 2.4.4.3 遺伝毒性試験 ... 17 2.4.4.4 がん原性試験 ... 17 2.4.4.5 生殖発生毒性試験 ... 17 2.4.4.6 局所刺激性試験 ... 18 2.4.4.7 その他の毒性試験 ... 18 2.4.4.8 安全域 ... 26 2.4.5 総括及び結論 ... 27 2.4.6 図表 ... 28 参考文献一覧 ... 28表
表 2.4-1 Overview of preclinical studies for BAX 855 ... 6表 2.4-2 Overview of preclinical studies for PEG2ru20KCOOH ... 8
表 2.4-3 Comparative immunogenicity studies conducted with BAX 855 in mouse models in the study IMM_R&D_012_11 ... 20
表 2.4-4 BAX 855: NOAELs from the toxicity studies... 26
表 2.4-5 BAX 855: AUC comparison from the Phase I clinical study and cynomplgus monkeys after first dosing (Based on FVIII activity) ... 26
略号一覧表
略号 英語 日本語
ADME absorption, distribution, metabolism and excretion 吸収,分布,代謝及び排泄
ANOVA analysis of variance 分散分析
APC antigen-presenting cell 抗原提示細胞
aPTT activated partial thromboplastin time 活性化部分トロンボプラスチン時間
AST aspartate aminotransferase アスパラギン酸アミノ基転移酵素
AUC area under the curve 曲線下面積
AUC0-tlast area under the curve from 0 to the last quantifiable sampling time point
時間 0 から測定可能な最終採血時点までの
曲線下面積
CHO Chinese hamster ovary チャイニーズハムスター卵巣
Cmax maximum concentration 最高濃度
DNA deoxyribonucleic acid デオキシリボ核酸
ECG electrocardiogram 心電図
ELISA enzyme-linked immunosorbent assay 酵素標識免疫吸着測定法 FACS fluorescence-activated cell sorter 蛍光活性化セルソーター
FDA Food and Drug Administration 米国食品医薬品局
FEIBA Factor Eight Inhibitor Bypassing Activity 凝固因子抗体迂回活性複合体
FVIII factor VIII 血液凝固第VIII 因子
GLDH glutamate dehydrogenase グルタミン酸脱水素酵素
GLP Good Laboratory Practice 医薬品の安全性に関する非臨床試験の実施の基準
HCT hematocrit ヘマトクリット
HGB haemoglobin ヘモグロビン
ICH International Conference on Harmonisation 日米EU 医薬品規制調和国際会議
IgG immunoglobulin G 免疫グロブリンG IL interleukin インターロイキン IU international unit 国際単位 kDa kilodalton キロダルトン ko knock out ノックアウト LPS lipopolysccharide リポポリサッカライド
MHC major histocompatibility complex 主要組織適合性複合体
MRT mean residence time 平均滞留時間
NCE normochromatic erythrocyte 正染性赤血球
NOAEL no observed adverse effect level 無毒性量
PCE polychromatic erythrocyte 多染性赤血球
RBC red blood cell 赤血球
RETI reticulocyte 網状赤血球
rFVIII recombinant factor VIII 遺伝子組換え型ヒト血液凝固第VIII 因子
t1/2 half-life 半減期
t1/2terminal terminal half-life 終末相半減期
TAT thrombin-antithrombin complex トロンビン-アンチトロンビン複合体
TK toxicokinetic(s) トキシコキネティクス
TNF-α tumor necrosis factor-α 腫瘍壊死因子-α
2.4 非臨床試験の概括評価
2.4.1
非臨床試験計画概略
FVIII は内因系血液凝固経路の重要な因子である.血友病 A は FVIII の欠損又は障害により引き 起こされるX 連鎖劣性先天性出血性障害であり,関節及び組織に出血症状をもたらす.血友病 A 患者には,出血症状の治療及び予防に十分な止血効果を示すFVIII 濃度を達成するため,FVIII 濃 縮製剤(血漿由来又は遺伝子組換え型)が使用される. 定期補充療法は,FVIII 濃度を正常値の 1%以上に維持し,自然出血症状を効果的に予防又は減 少させることを目的とする.現在のところ,FVIII を目標レベルに維持するためには,薬物動態プ ロファイル,出血タイプ及びライフスタイルに応じてFVIII を繰り返し静脈内投与するしかない. 半減期を延長させたFVIII 濃縮製剤は,有効性の向上又は投与頻度の減少若しくはその両方につながると考えられる.FVIII 分子を PEG で修飾することにより,FVIII を含む治療用たん白質の循
環時間を延長可能である.
バクスター社は,CHO 細胞からプラズマ/アルブミンフリー細胞培養法及びウイルス不活化工
程により得られたrFVIII を用いて PEG 化遺伝子組換え型ヒト FVIII である BAX 855(以下,「本
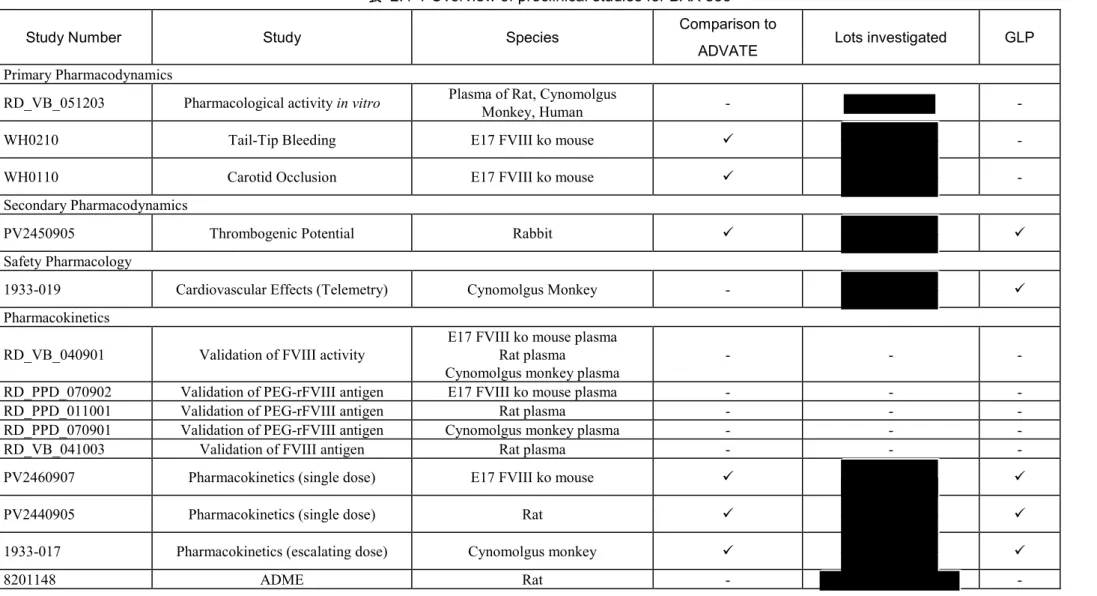
剤」)を開発した.この製造に用いるrFVIII はバクスター社の既承認製剤アドベイトのバルク原 薬である. 本剤の非臨床開発計画にはin vitro 及び in vivo での効力を裏付ける試験,副次的薬理試験,安全 性薬理試験,薬物動態試験及び毒性試験が含まれる.In vitro 薬理活性試験,本剤の標識体を用い たラットADME 試験,ウサギ局所刺激性試験,ヒト組織の交差反応性試験及び本剤の不純物とし て同定されたPEG2ru20KCOOH を用いた試験を除くすべての試験で本剤 2 ロットを用いて検討し た.一部の試験では,下記の表 2.4-1 にまとめるように,アドベイトを対照物質とした.本剤の 特性データ(3.2.P.5.4 項を参照)から,非臨床試験に使用した本剤は構造及び機能において臨床 試験用及び市販予定の本剤と同等であることが示されている. 本剤の非臨床有効性及び安全性プロファイルをE17 FVIII ko マウス,並びに正常ラット及びカ ニクイザルにおいて検討した.E17 FVIII ko マウス(エクソン 17 ノックアウトマウス, 系統, )は,患者集団と臨床的に類似する血友病マウスモデルとして十分に確立され,広 く用いられていることから選択した[4].試験に用いた E17 FVIII ko マウスは,FVIII 遺伝子のエク ソン17 に neo 発現カセットを挿入して作製した.エクソン 17 の変異が重症血友病 A(FVIII の残 存活性1%未満)を引き起こす.ラットは広範な動物試験に用いられていることから選択した.ラ ットは一般に規制当局により適切な動物モデルとして認識されている(ICH M3(R2)「医薬品の 臨床試験及び製造販売承認申請のための非臨床安全性試験実施についてのガイダンス」).カニ クイザルは霊長類とヒトの血液凝固系が類似しているため,ヒト血液凝固たん白質の試験に適す る動物種であると考えて選択した.本剤の薬理活性は全動物種においてin vitro 及び/又は in vivo で示された. 非臨床試験の概要を各試験のGLP 適合状況と共に表 2.4-1 及び表 2.4-2 に示す.
表 2.4-1 Overview of preclinical studies for BAX 855
Study Number Study Species Comparison to
ADVATE Lots investigated GLP
Primary Pharmacodynamics
RD_VB_051203 Pharmacological activity in vitro Plasma of Rat, Cynomolgus Monkey, Human - -
WH0210 Tail-Tip Bleeding E17 FVIII ko mouse -
WH0110 Carotid Occlusion E17 FVIII ko mouse -
Secondary Pharmacodynamics
PV2450905 Thrombogenic Potential Rabbit
Safety Pharmacology
1933-019 Cardiovascular Effects (Telemetry) Cynomolgus Monkey -
Pharmacokinetics
RD_VB_040901 Validation of FVIII activity E17 FVIII ko mouse plasma Rat plasma
Cynomolgus monkey plasma - - -
RD_PPD_070902 Validation of PEG-rFVIII antigen E17 FVIII ko mouse plasma - - -
RD_PPD_011001 Validation of PEG-rFVIII antigen Rat plasma - - -
RD_PPD_070901 Validation of PEG-rFVIII antigen Cynomolgus monkey plasma - - -
RD_VB_041003 Validation of FVIII antigen Rat plasma - - -
PV2460907 Pharmacokinetics (single dose) E17 FVIII ko mouse
PV2440905 Pharmacokinetics (single dose) Rat
1933-017 Pharmacokinetics (escalating dose) Cynomolgus monkey
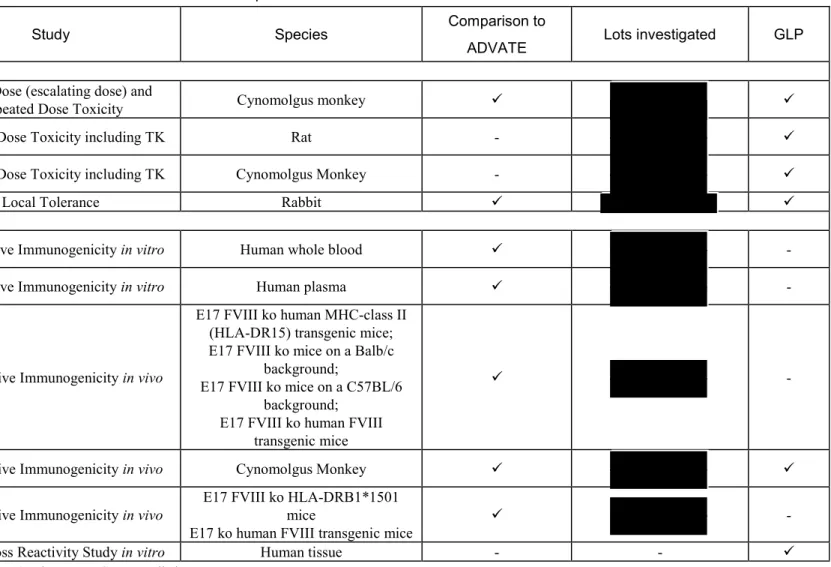
表 2.4-1 Overview of preclinical studies for BAX 855(続き)
Study Number Study Species Comparison to
ADVATE Lots investigated GLP
Toxicology
1933-017 Single Dose (escalating dose) and Repeated Dose Toxicity Cynomolgus monkey
8202366 Repeated Dose Toxicity including TK Rat -
1933-018 Repeated Dose Toxicity including TK Cynomolgus Monkey -
PV2651201a Local Tolerance Rabbit
Other Toxicity Studies
IVFS_001_10 Comparative Immunogenicity in vitro Human whole blood -
IVFS_002_10 Comparative Immunogenicity in vitro Human plasma -
IMM_R&D_012_11 Comparative Immunogenicity in vivo
E17 FVIII ko human MHC-class II (HLA-DR15) transgenic mice; E17 FVIII ko mice on a Balb/c
background;
E17 FVIII ko mice on a C57BL/6 background;
E17 FVIII ko human FVIII transgenic mice
-
8220805 Comparative Immunogenicity in vivo Cynomolgus Monkey
IMM_R&D_025_12b Comparative Immunogenicity in vivo E17 FVIII ko HLA-DRB1*1501 mice
E17 ko human FVIII transgenic mice -
GLP2572 Tissue Cross Reactivity Study in vitro Human tissue - -
-: not compared to ADVATE, not used BAX 855 lots or not GLP complied a: Investigation of BAX 855 with a nominal potency of 2000 IU FVIII/5 mL vial
表 2.4-2 Overview of preclinical studies for PEG2ru20KCOOH
Study Number Study Species Test item GLP
8298494 Repeated Dose Toxicity including TK Rat PEG2ru20KCOOHa
6291-259 Bacterial Reverse Mutation Assay TA100, TA1535 and TA1537, and Salmonella typhimurium TA98,
Escherichia coli WP2uvrA PEG2ru20KCOOH
a -
6291-260 Chromosomal Aberrations Assay Human Peripheral Blood Lymphocytes PEG2ru20KCOOHa -
6291-261 Bone Marrow Micronucleus Assay Mouse PEG2ru20KCOOHa -
-: not GLP complied
2.4.2
薬理試験
2.4.2.1
効力を裏付ける試験
本剤の効力を裏付ける試験をin vitro で 1 試験及び in vivo で 2 試験実施した.In vitro 試験にお
いて,本剤とラット及びカニクイザルの血液凝固系との相互作用をaPTT 測定により示した.In
vivo 試験において,2 種類の異なる動物モデル(E17 FVIII ko マウスの尾端出血モデル及び頸動脈
閉塞モデル)を用いて本剤の有効性を評価した.バクスター社の既承認rFVIII 製剤であるアドベ イトを対照物質とした. 2.4.2.1.1 ラット及びサルにおける本剤のin vitro薬理活性評価 本剤の毒性プロファイルをラット及びカニクイザルで評価した.そこで,本剤とラット及びカ ニクイザルの血液凝固系との相互作用をaPTT 測定により検討した(RD_VB_051203 試験).ラ ット,カニクイザル及びヒトの希釈血漿に本剤を添加すると,aPTT は濃度依存的に短縮した.こ れは本剤がラット,カニクイザル及びヒトの血液凝固系と相互作用することを示している.した がって,ラット及びカニクイザルは本剤の非臨床安全性評価に適する動物種であると考えられた.
2.4.2.1.2 E17 FVIII koマウスの尾端出血モデルにおける有効性
E17 FVIII ko マウスの尾端出血モデルを用いて本剤又はアドベイト投与後の失血量を評価した (WH0210 試験).本試験では各群 16 匹(雌雄各 8 匹)の E17 FVIII ko マウスを使用した.本剤 又はアドベイトの200 IU FVIII/kg を尾端切断の 18~48 時間前に単回静脈内投与した.Baumgartner ら[3]が報告した社内試験の結果に基づいて,E17 FVIII ko マウスにおいて有効性が示された 200 IU FVIII/kg の用量を選択した.本剤は尾端切断の 30,40 又は 48 時間前に E17 FVIII ko マウスに投与
した.対照物質であるアドベイトは尾端切断の18,24,30 又は 40 時間前に投与した.処方緩衝 液(10 mL/kg)は尾端切断の 5 分前に投与した.尾端切断後 60 分にわたり血液を採取した.有効 性は60 分後の明らかな失血量の減少と定義した. アドベイトの有効性は尾端切断の18 時間前に投与した動物で示されたが,それ以前(24,30 及び40 時間前)では認められなかった.本剤の有効性は尾端切断の 30 時間前に投与した動物で 示されたが,40 時間前では明確ではなく,48 時間前では示されなかった.臨床的意義のある有効 性は,本剤では尾端切断の30 時間前,アドベイトでは 18 時間前に投与した時に認められた.結 果の統計学的評価から,本剤はアドベイトと比較して1.5 倍以上の投与間隔で投与した時,アド ベイトと同程度に有効であることが示された.また,すべての投与群のいずれの動物においても 本剤の静脈内投与の忍容性は良好であり,急性毒性の徴候は認められなかった.
2.4.2.1.3 E17 FVIII koマウスの頸動脈閉塞モデルにおける有効性
FeCl3誘発動脈血栓症モデルである頸動脈閉塞モデルを用いてE17 FVIII ko マウスにおける本剤
及びアドベイトの有効性を評価した(WH0110 試験).本試験では各群 12 匹(雌雄各 6 匹)の
E17 FVIII ko マウスを使用した.本剤又はアドベイトの 200 IU FVIII/kg を内皮剥離の 12~64 時間 前にE17 FVIII ko マウスに予防的に静脈内投与した.Baumgartner ら[3]が報告した社内試験の結果
に基づいて,200 IU FVIII/kg の用量を選択した.本剤は内皮剥離の 24,30,40,48,54 又は 64 時間前に,アドベイトは内皮剥離の12,18,24,30 又は 40 時間前に E17 FVIII ko マウスに投与 した.処方緩衝液(10 mL/kg)は内皮剥離の 15 分前に投与した.超音波血流プローブを用いて傷 害前後のFeCl3誘発動脈血栓をモニターした.永続的血管閉塞までの時間を測定し,30 分間の観 察時間内に処方緩衝液を投与した動物と比較して閉塞までの時間の中央値の明確な短縮が認めら れた場合,有効性ありと定義した. 処方緩衝液を投与した動物では30 分間の観察時間内に血管閉塞は認められなかった.アドベイ トは12 又は 18 時間前に投与した時有効であると考えられたが,より長時間の時点では有効性の 低下が示された.アドベイトの有効性は,処方緩衝液と比較して,投与後30 時間まで有意であっ たが,より長時間(投与後24 及び 30 時間)における有効性はわずかであり,臨床的に意義があ るとは考えられなかった.本剤は内皮剥離の24 時間前に投与した時,臨床的に意義のある有効性 を有すると考えられた.30 時間前に投与した時には,処方緩衝液と比較して統計学的に有意であ ったものの有効性はわずかであった.これを上回る時点(40~64 時間後)では,有効性を確認で きなかった.本試験の結果に基づいて,本剤はFVIII ko マウスの頸動脈閉塞モデルにおいて有効 であると結論できる.本剤は投与後24 時間の時点で依然として有効であったが,アドベイトは有 効ではなかったことから,アドベイトと比較して臨床的に意義のある有効性の持続が示された. 2.4.2.1.4 効力を裏付ける試験の概要 ラット,カニクイザル及びヒト血漿を用いたin vitro 試験において,本剤の活性と血液凝固系と の相互作用を示す濃度依存的なaPTT の短縮が認められた. 効力を裏付ける試験に用いた両モデルにおいて,アドベイトと比較して本剤の臨床的に意義の ある有効性の延長が認められた.
2.4.2.2
副次的薬理試験
本剤の非臨床試験において,副次的薬理作用は認められなかった.本剤の血栓形成性をウサギ 静脈血停滞モデルにおいて別途評価した. 2.4.2.2.1 ウサギにおける血栓形成性 本剤及びアドベイトの単回静脈内(ボーラス)投与後の血栓形成性をWessler ら[18]が開発した ウサギ静脈血停滞モデルにおいて評価した(PV2450905 試験).予定臨床最高用量である 100 IU FVIII/kg と比較して広い安全域(9 倍)を設定し,表示用量 900 IU FVIII/kg を投与した.各群 6 匹(雌雄各3 匹)から成る 3 群及び陽性対照物質投与用の 2 匹(活性型プロトロンビン複合体製 剤FEIBA の 20 U/kg を静脈内投与)を用いた.本剤及びアドベイト投与後の単離頸静脈切片にお ける血栓形成を半定量的に評価した. 被験物質である本剤又は対照物質であるアドベイトのいずれを静脈内投与した場合でも血栓形 成性は認められなかった.陽性対照物質であるFEIBA を投与したすべての動物では最大スコアで あったことから,試験モデルの妥当性が確認された.2.4.2.2.2 副次的薬理試験の概要 本剤の非臨床試験において副次的薬理作用は認められなかった.ウサギ静脈血停滞モデルにお いて本剤の血栓形成性はアドベイトと同程度であった.
2.4.2.3
安全性薬理試験
2.4.2.3.1 テレメトリー装着カニクイザルにおける心血管系及び呼吸器系安全性 統合ラジオテレメトリーシステムを用いて,無麻酔・非拘束サルにおいて,体温,心血管系(大 動脈血圧及び単極誘導ECG)及び呼吸器系(胸腔内圧)パラメータを投与前 24 時間から投与後 24 時間までの 48 時間にわたり連続的にモニターした(1933-019 試験).本剤 2 ロットを 8 匹の 雄カニクイザルに単回静脈内投与した(150 又は 600 IU FVIII/kg).いずれの用量でも本剤投与の 忍容性は良好であり,臨床的,心血管系又は呼吸器系有害作用を誘起しなかった. 2.4.2.3.2 本剤の中枢神経系に及ぼす影響 カニクイザルの安全性薬理試験(1933-019 試験),カニクイザルの用量漸増及び予備的反復投 与毒性試験(1933-017 試験),並びにラット(8202366 試験)及びカニクイザル(1933-018 試験) の反復投与毒性試験において中枢神経系に対する有害作用は認められなかった. 2.4.2.3.3 安全性薬理試験の概要 以上のように,テレメトリー装着無麻酔カニクイザルにおいて本剤の両ロットの単回静脈内投 与後の忍容性は良好であり,予定臨床最高用量100 IU FVIII/kg の 6 倍に相当する 600 IU FVIII/kg の用量まで臨床的,心血管系又は呼吸器系有害作用を誘起しなかった.本剤の安全性薬理試験及 び毒性試験において中枢神経系に対する影響は認められなかった.2.4.2.4
薬力学的薬物相互作用試験
本剤は単独投与するため薬力学的薬物相互作用試験は実施しなかった.2.4.3
薬物動態試験
E17 FVIII ko マウス,並びに正常ラット及びカニクイザルに本剤又はアドベイトを静脈内投与し て本剤2 ロットの薬物動態プロファイルを評価した.ラットでは,放射性標識した本剤の ADMEに関する更なる情報が得られた.FVIII 活性発色測定法,PEG-rFVIII 抗原測定法及び FVIII 抗原測
定法を用いて薬物動態試験3 試験の試料を分析した.
FVIII 活性発色測定法及び FVIII 抗原測定法では非修飾型 rFVIII(アドベイト)及び PEG 化 rFVIII
(本剤)の両方が検出された.PEG-rFVIII 抗原の PEG 化に基づく測定法では本剤の測定可能シグ
ナルのみが検出された.主要評価項目は用量補正AUC0-tlast(E17 FVIII ko マウス及びラット)又 はAUC0-tlast(カニクイザル)及びMRT とし,副次的評価項目は t1/2terminalとした.AUC0-tlastは循 環血液中の薬物量に比例する代表的な指標であることから,バイオアベイラビリティに関する情
報が得られる.MRT は薬物が循環血液中に滞留する平均時間と定義され,修飾した FVIII 製剤の 循環時間の延長を示す最適なパラメータと考えられる.t1/2terminalは消失相において血漿中薬物濃度 が半減するまでに要する時間と定義され,循環時間の延長を示す重要なパラメータと考えられる.
2.4.3.1
吸収
2.4.3.1.1 E17 FVIII koマウスにおける薬物動態試験
E17 FVIII ko マウスに本剤(2 ロット)又はアドベイトの 200 IU FVIII/kg を投与した(PV2460907 試験).FVIII 活性について,本剤群の用量補正 AUC0-tlastの点推定値はアドベイト群の1.9 倍であ った(それぞれ0.0797 及び 0.0421 h·IU FVIII/mL /IU FVIII/kg).本剤群の MRT の点推定値はア ドベイト群の1.6 倍であった(それぞれ 7.9 及び 4.9 時間).本剤群の t1/2terminalの点推定値はアド
ベイト群の1.4 倍であった(それぞれ 5.9 及び 4.3 時間).本傾向は,両投与群における他のすべ
ての薬物動態変数で同様であった.したがって,ヒトrFVIII の PEG 化により E17 FVIII ko マウス において循環時間が延長し,その結果,曝露時間が全体として増大した.
2.4.3.1.2 ラットにおける薬物動態試験
ラットにおいても本剤2 ロットの薬物動態プロファイルをアドベイトと比較評価した
(PV2440905 試験).200 IU FVIII/kg を単回静脈内ボーラス投与した.また,本剤のより高用量 (350 及び 700 IU FVIII/kg)も検討した.ヒト FVIII 活性とラット FVIII 活性ベースライン値との
相互作用を回避するため,ヒトFVIII に対して選択性を有しラット FVIII 抗原との交差反応を示さ
ないFVIII 抗原を測定した.すべての血漿試料について FVIII 抗原及び rFVIII 結合 PEG を分析し た.
FVIII 抗原について,200 IU FVIII/kg では,本剤群の用量補正 AUC0-tlastの幾何平均値はアドベイ
ト群の1.4 倍であった(それぞれ 0.071 及び 0.050 h·IU FVIII/mL /IU FVIII/kg).本剤群の MRT の 幾何平均値はアドベイト群の1.2 倍であった(それぞれ 7.5 及び 6.2 時間).本剤群の t1/2terminalの
幾何平均値はアドベイト群の1.1 倍であった(それぞれ 6.1 及び 5.5 時間).本傾向は,これら投
与群における他のすべての薬物動態変数で同様であり,PEG 化により rFVIII の循環時間が改善す
ることが本モデルで示された.本剤の中用量群では,用量補正AUC0-tlastの幾何平均値は,低用量
群の1.2 倍,高用量群の 1.4 倍であった(それぞれ 0.071[低用量],0.084[中用量]及び 0.060 [高用量]h·IU FVIII/mL /IU FVIII/kg).予想通り,アドベイトの投与により rFVIII 結合 PEG は 検出されなかった.ラットにおける用量比例性を本剤の低用量(200 IU FVIII/kg),中用量(350 IU FVIII/kg)及び高用量(700 IU FVIII/kg)を比較して評価した.rFVIII 結合 PEG について,本剤の
低用量群の用量補正AUC0-tlastの幾何平均値は中用量群及び高用量群の約80%であった(それぞれ
0.051[低用量],0.065[中用量]及び 0.064[高用量]h·ng rFVIII 結合 PEG/mL /ng rFVIII 結合 PEG/kg).本剤の用量比例性は,統計学的に検出されなかったが,本動物モデルでは起こり得る 範囲内であった.本剤の低用量投与後,rFVIII 結合 PEG の薬物動態プロファイルは,FVIII 抗原
の薬物動態プロファイルと類似するパターンを示した.これはPEG 化によりラットにおける曝露
また,放射性標識した本剤の2,088 IU FVIII/kg を排泄物及び血液採取用ラットに単回静脈内投 与し,ADME を検討した.全身オートラジオグラフィ用の動物には 4,176 IU FVIII/kg を単回静脈 内投与した.両用量共に,用いた検出方法(液体シンチレーション計測法,オートラジオグラフ
ィ)で6 週にわたり十分に高い放射活性が測定可能であった(8201148 試験).
[3H] PEG-rFVIII はトリチウム化した[3H] PEG 試薬の結合により合成し,rFVIII に結合した PEG
骨格ではいくつかの水素[1H]がトリチウム原子[3H]により交換された.本剤の標識体は,他の非臨 床試験で用いた本剤の非標識体と同等の有効成分を産生する,類似の工程で調製された.[3H] PEG-rFVIII の比活性は,他の非臨床 in vivo 試験で用いた非臨床用 4 ロットの比活性の範囲(3,691.6 ~4,355.7 IU FVIII/mg たん白質)を下回っていた.予測される放射性分解による経時的な活性の漸 減を制限するために,本試験のin vivo 部分は標識体合成後可能な限り速やかに開始するようにデ ザインした.したがって,放射性標識体は標準的設備を備えていない別施設( )で調製し なければならなかった.調製の違いにもかかわらず,保持された放射活性は小規模なバッチとし ては妥当な高さであると考えられた.したがって,FVIII 活性に認められた差異は[3H] PEG-rFVIII の分布に影響を及ぼさないと判断された. 血液,血漿,組織,尿及び糞を,投与後1,008 時間までの計画した時間に採取した.薬物動態 パラメータにはわずかに性差が認められ,雄のt1/2が長かったが,[3H] PEG-rFVIII 由来放射活性へ の曝露量は同程度であった.[3H] PEG-rFVIII 由来放射活性の分布は雌雄共に広範であり,血液及 び血漿,並びにすべての分析マトリックスにおいて放射活性が定量可能であった.放射活性の最 高濃度は,雌雄共に血漿,血液,腸間膜リンパ節,脾臓,肝臓,副腎及び腎臓で認められた.放 射活性は,血液及び血漿から雄ではそれぞれ827 及び 655 時間,雌ではそれぞれ 306 及び 276 時 間のt1/2を以て,主に尿から排泄された.組織における放射活性のCmaxは1,8,24 及び 168 時間 後の採取時に観察された.雄及び雌での総回収率の平均値はそれぞれ97.4 及び 107.0%であり,単 回投与後1,008 時間で完全に排泄されることが示された.以上の試験結果から,[3H] PEG-rFVIII は分析した組織に広範に分布し,放射活性は主に尿を介して6 週以内に定量的に排泄されること が示された. 2.4.3.1.3 カニクイザルにおける薬物動態試験 カニクイザルに本剤2 ロットを 350,700 又は 1,500 IU FVIII/kg の用量で,アドベイトを 350 IU FVIII/kg の用量で試験 1 及び 8 日目に投与した(1933-017 試験).FVIII 活性及び rFVIII 結合 PEG
を分析した.FVIII 活性測定では,ヒトとサルの FVIII 活性は区別されないため,投与前にすべて
のサルにおいてベースラインFVIII 活性が検出された.
FVIII 活性については,アドベイト 350 IU FVIII/kg と比較して,本剤のロット 1 及び 2 の AUC0-tlast
の幾何平均値はそれぞれ1.3 及び 1.6 倍大きく(それぞれ 61,77 及び 47 h·IU FVIII/mL),MRT の幾何平均値はそれぞれ1.4 及び 1.6 倍長く(それぞれ 10.7,12.3 及び 7.5 時間),t1/2terminalの幾 何平均値はそれぞれ1.6 及び 1.7 倍長かった(それぞれ 9.0,9.8 及び 5.7 時間).カニクイザルに おける本剤の用量比例性を350,700,及び 1500 IU FVIII/kg で評価した.本剤の用量を 2 倍にし た時のFVIII 活性に基づく AUC0-tlastの増加は,ロット1 で 1.51,ロット 2 で 1.85 と推定された.
これらの結果から,検討した用量範囲において,ロット2 では用量に比例した AUC0-tlastの増加が,
ロット1 では用量比例性を下回る増加が示された.本剤の用量を 2 倍にした時の rFVIII 結合 PEG
に基づくAUC0-tlastの増加は,ロット1 で 1.47,ロット 2 で 2.00 と推定され,FVIII 活性の結果と
類似していた.結果のばらつきは内因性FVIII 活性レベルに関しカニクイザルで想定されるもの
であった.
2.4.3.1.4 薬物動態試験の概要
E17 FVIII ko マウス,並びに正常ラット及びカニクイザルにおける薬物動態試験から,アドベイ トと比較して本剤ではFVIII 活性(E17 FVIII ko マウス及びカニクイザル)又は FVIII 抗原(ラッ
ト)の循環時間が長く,結果として,FVIII への曝露時間が増大することが示された.放射性標識 した本剤をラットに単回静脈内投与し,その動態を6 週まで追及した.[3H] PEG-rFVIII は分析し た組織に分布し,放射活性は糞中排泄を伴いながら主に尿を介して排泄され,投与した総放射活 性は6 週以内に消失した.
2.4.3.2 薬物動態学的薬物相互作用試験
本剤は単独投与するため薬物動態学的薬物相互作用試験は実施しなかった.2.4.3.3
その他の薬物動態試験
その他の薬物動態試験は実施しなかった.2.4.4
毒性試験
2.4.4.1
単回投与毒性試験
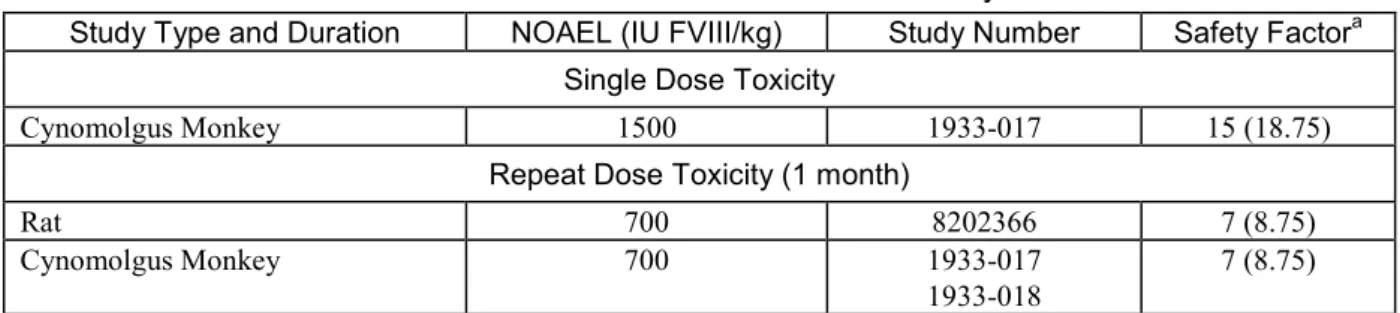
単回投与毒性試験は実施しなかった.ICH M3(R2)ガイドライン「医薬品の臨床試験及び製造 販売承認申請のための非臨床安全性試験実施についてのガイダンス」の4. 急性毒性試験に「いず れかの試験から急性毒性に関する情報が得られる場合には,別途に単回投与試験を実施すること は推奨されない.」と記載されていることから,単回投与毒性はカニクイザルにおける用量漸増 試験データに基づいて評価した. 2.4.4.1.1 カニクイザルにおける用量漸増試験 試験1 及び 8 日目に,雌雄各 2 匹のカニクイザルに本剤 2 ロットの 350,700 及び 1,500 IU FVIII/kg を静脈内に単回緩徐ボーラス投与した(1933-017 試験).アドベイトは試験 1 日目に 350 IU FVIII/kg を静脈内投与した.毒性評価は,一般状態観察,心血管検査,血液学的検査及び血液生化学的検 査,体重測定,器官重量,並びに肝臓の病理組織学的評価に基づいて行った.いずれの用量レベ ルにおいても毒性徴候及び死亡例はなかった.本剤の低,中及び高用量群の個別動物に毒性学的 に意義のある所見を伴わないGLDH 及び AST の軽度上昇が,また,用量漸増フェーズでは薬理学 的作用に起因し,被験物質投与に関連すると考えられたaPTT の短縮,並びに TAT 及び D-ダイマ ーの上昇がそれぞれ個別動物で散見されたが,その他の評価項目に本剤又はアドベイトに関連した変化はなかった.したがって,本剤の異なる2 ロットの投与は忍容性が良好であり,NOAEL は1500 IU FVIII/kg であった.
2.4.4.2
反復投与毒性試験
反復投与毒性試験を以下の2 動物種で実施した. ラット(8202366 試験) カニクイザル(1933-017 及び 1933-018 試験) 2.4.4.2.1 ラットにおける反復投与毒性試験 ラットに本剤(2 ロット)の 350 又は 700 IU FVIII/kg を 29 日間隔日投与(15 回)し,2 週間の 回復期間を設定した(8202366 試験).毒性評価は,死亡,一般状態観察,眼科学的検査,臨床 病理学的検査(血液学的検査,血液凝固検査,血液生化学的検査及び尿検査),器官重量,並び に病理学的検査(雄生殖能を含む肉眼的及び顕微鏡的評価)に基づいて行った.全身的TK プロ ファイルを求めるため,TK コホートから 1 及び 29 日目に一連の血液試料を採取した. 高用量の700 IU FVIII/kg まで,本剤に関連した臨床徴候,並びに体重,摂餌量,眼科学的検査, 臨床病理検査,尿検査,精液検査及び器官重量に本剤に関連した変化は認められなかった.試験 27 日目の 350 IU FVIII/kg 投与直後に 1 匹が死亡したが,剖検所見は認められなかったため,本剤 との関連性はないと考えられた.本剤の作用による肉眼的所見は認められなかった.顕微鏡的に は,700 IU FVIII/kg を投与した動物の肺での泡沫化マクロファージの発生率/重症度が対照群と 比較して軽微に増加していた.この軽微な増加は炎症又は組織傷害を伴わず,2 週間の無投与期 間後に回復が認められた.したがって,これらの所見は有害なものとは考えられなかった.脳及 び脊髄にPEG に起因する空胞形成が認められるかどうかを詳細に評価した.本剤を投与した動物 の脳又は脊髄には病理組織学的変化を示す所見はなく,これらの器官に空胞形成がないことが確 認された.雄生殖器に病理組織学的所見はなかった. 1 日目の TK 評価の結果から適正に投与されていることが示された.29 日目に低レベルの FVIII 活性及びrFVIII 結合 PEG が検出され,中和抗体の存在と相関していた.以上より,ラットに 29 日間,15 回隔日投与した時の本剤の NOAEL は 700 IU FVIII/kg であった. 2.4.4.2.2 カニクイザルにおける反復投与毒性試験 カニクイザルにおける予備的反復投与毒性試験(1933-017 試験)において,本剤を雌雄各 2 匹 に700 IU FVIII/kg の用量で 5 日毎に計 26 日間(計 6 回)静脈内投与し,更に,雌雄各 1 匹に処方 緩衝液を投与した.毒性評価は,一般状態観察,血液学的検査及び血液生化学的検査,体重測定, 器官重量及び抗体形成検査に基づいて行った. 試験期間を通じて,反復投与フェーズ中の毒性徴候,死亡例,あるいは本剤又は処方緩衝液に 関連した体重変動はなかった.反復投与フェーズの本剤初回投与後にaPTT の短縮,また,反復 投与によりTAT の増加が認められ,これらは薬理学的作用に起因した,本剤の投与に関連するものと考えられた.反復投与フェーズ中にaPTT が延長し,これは抗 FVIII 中和抗体の形成に関連す る可能性があった. 反復投与フェーズの本剤を投与したすべての動物は,ヒトFVIII 及び PEG-rFVIII に特異的な抗 体を発現した.また,4 匹中 3 匹に抗 PEG 抗体が発現した.対照の処方緩衝液を投与した動物は, ヒトFVIII,PEG-rFVIII 及び PEG に特異的な抗体を発現しなかった.本剤を投与した 4 匹中 2 匹 ではヒトFVIII 活性に対する中和抗体が発現し,4 匹中 3 匹では PEG-rFVIII 活性に対する中和抗 体が発現した.他のすべての動物における抗体力価は定量限界未満であった.本剤に対する抗体 形成は,カニクイザルへの異種たん白質の反復投与後に予測される免疫反応であり,これは非PEG 化FVIII 製剤においてもよく知られている[7].抗 PEG 抗体の形成による有害作用はなかった.以 上のように,本剤の反復投与の忍容性は良好であり,NOAEL は 700 IU FVIII/kg であった. カニクイザルにおける主要反復投与毒性試験(1933-018 試験)において,本剤 2 ロットを 150, 350 及び 700 IU FVIII/kg の用量で 5 日毎に 26 日間(計 6 回)投与した.毒性評価は,体重測定, 一般状態観察,眼科学的検査,血液学的検査,血液凝固検査,血液生化学的検査,尿検査,剖検 時の肉眼的所見及び組織病理学的所見に基づいて行った.また,TK 評価及び抗体評価も実施した. 試験期間を通じていずれの用量においても毒性徴候及び死亡例はなかった.投与26 日目に赤血 球パラメータ(RBC,HGB,HCT)の軽微な低下所見(高用量群の 3 匹及び低用量群の 1 匹), 吐血(中用量群の1 匹及び高用量群の 2 匹)又は血腫(低用量群の 1 匹,中用量群の 1 匹及び高 用量群の1 匹)を伴う RETI の増加及び aPTT の延長(すべての動物)が認められた.これらの所 見は,カニクイザルのFVIII 活性に部分的低下をもたらす,内因性 FVIII に対する交差反応性を有 する抗FVIII 中和抗体の発現に起因する可能性が高い.回復期の 12 日目までに aPTT の部分的回 復が認められた.本剤の作用による肉眼的及び顕微鏡的所見は認められなかった.脳及び脊髄の 詳細な病理組織学的検査から,これらの器官に空胞形成がないことが確認された. 本剤を投与したのすべての動物は,投与に関連したヒトFVIII,PEG-rFVIII 及び PEG に特異的 な結合抗体を発現した.本剤投与群の30 匹中 28 匹は,31 日目にヒト FVIII に対する結合及び中 和抗体を発現した.投与前の試料に中和抗体は認められなかった.結合及び中和抗体の形成は, TK 解析において本剤を 6 回投与した動物の大半で曝露量が統計学的に有意に減少(AUC0-tlast及び Cmax共にp<0.0001,ANOVA,両側,有意水準 5%)したことにも反映された.1 日目と比較した
29 日目の FVIII 活性及び rFVIII 結合 PEG レベルの低下は本剤に対する中和抗体の存在とよく相関 していた.
本剤に対する抗体形成はカニクイザルへの異種たん白質の反復投与後に予想される免疫反応で あり,これは非PEG 化 FVIII 製剤においてもよく知られている[7].抗 PEG 抗体の形成による有 害作用はなかった.したがって,本試験のNOAEL は高用量の 700 IU FVIII/kg であった.
2.4.4.2.3 慢性毒性試験
ラット及びカニクイザルの反復投与毒性試験の結果,ラット及びサルにおいてヒトFVIII に特 異的な抗体が誘導されることが示された.抗FVIII 抗体は FVIII 活性及び PEG-rFVIII 活性に対し て中和特性を示した.結果として,本剤の全身曝露量は,性別及び投与用量に関係なく,反復投 与後(29 日目)に単回投与後(1 日目)と比較して著明に低下していた. バクスター社は,ヒトFVIII に対する特異的抗体が本剤の反復投与後にラット及びサルで誘導 されることから,本剤の更なる長期投与毒性試験を実施してもヒトにおける本剤の安全性に関す る追加情報は得られないと考える. 一方,有効性観察期間の平均値(標準偏差)が定期補充療法群(120 例)で 5.94(1.14)ヵ月, 出血時補充療法群(17 例)で 6.31(0.41)ヵ月である本剤の臨床試験(261201 試験)において (M2.7.3.2.1),本剤の投与を受けた全 137 例(本剤の累積実投与日数 6,717 日,総投与量 21,803,981 IU FVIII)の忍容性は良好であった(M2.5.5.14).
2.4.4.3
遺伝毒性試験
ICH S6(R1)ガイドライン「バイオテクノロジー応用医薬品の非臨床における安全性評価」に基 づき,遺伝毒性試験はPEG 化 FVIII 等のバイオテクノロジー応用医薬品の安全性評価には適用さ れない.本剤の作用機序及び特性から本剤とDNA や他の染色体成分との直接相互作用は示唆され ていないため,本剤に遺伝毒性はないと考える.したがって,遺伝毒性試験は適用されず,標準 的な組み合わせのin vitro 及び in vivo 試験は実施していない.2.4.4.4
がん原性試験
本剤は遺伝子組換え型たん白質であり,その薬理作用に基づく突然変異原性,染色体異常誘発 性又はがん原性の可能性(免疫抑制又は増殖作用等)はないと考えられる.ICH S6(R1)ガイドラ イン「バイオテクノロジー応用医薬品の非臨床における安全性評価」に記載されているように, がん原性に対する懸念がない限り,生物学的製剤のがん原性試験は不要である.また,本剤の150, 350 及び 700 IU FVIII/kg を 26 日間にわたり投与したサル組織の病理組織学的検査で増殖作用は認 められなかった(1933-018 試験).したがって,本剤にがん原性の懸念はないため,がん原性試 験は実施しておらず,また実施計画もない.2.4.4.5
生殖発生毒性試験
本剤については生殖発生毒性試験を実施していないが,雌雄のラット及びサルを用いた反復投 与毒性試験において生殖器への影響は認められず,ラット反復投与毒性試験の精液検査において 本剤に関連する影響はなかった.また,PEG 化ヒト rFVIII である本剤を生殖発生毒性試験の試験 期間にわたり異種動物へ投与する試験の実施は,抗体形成のリスクがあるため困難である.ラッ ト及びカニクイザルを用いた反復投与毒性試験において本剤に対する中和抗体が産生され,rFVIII の全身曝露量は,本剤の反復投与(6~15 回投与)後に単回投与(1 日目)後と比較して著明に低 下していた.したがって,抗原抗体反応に基づく両立し難い反応の発現リスクにより,生殖発生 毒性試験の結果はヒトにおける状況を表さないと考えられる.更に,血液凝固第XII 因子[17]等の 低下による凝固亢進状態が不育症のリスク要因となることが知られており,本剤の過量投与による凝固亢進により個体発生に関係する毒性が発現するリスクは想定可能と考える.これらのこと から,バクスター社は動物を用いた生殖発生毒性試験の実施を省略できると判断し,妊婦又は妊 娠している可能性のある女性患者には治療上の有益性が危険性を上回ると判断される場合にのみ 投与すべきであることを添付文書で注意喚起する. 成人と比較して小児集団での本剤の有害事象のリスクは高くないと推定される. 一般的に,止血については生後6 ヵ月で完全な成熟に達し,小児と成人でほぼ同等であると推 測される.健康人では,FVIII は出生時及び小児期に成人の値の範囲内にあることが示された [11][10][2].また,胎児及び新生児の凝固系たん白質は分布容積が大きく血漿クリアランスが速い ため,成人と比較して曝露の増加がないことが示唆されている[11]. FVIII 製剤の薬効分類別の薬理作用として,非臨床試験の結果から本剤はアドベイトと類似した 安全性プロファイルを有することが示唆された.ラット及びカニクイザルにおける本剤の反復投 与毒性試験において有害事象は発現しなかったため,小児においてまだ発達過程にある標的器官 に対する懸念は提起されなかった.したがって,本剤を小児に投与した時の安全性プロファイル はアドベイトと類似していると考えられる. この評価に基づき,バクスター社は幼若動物における非臨床試験の実施を計画していない.成 人と小児集団との間で本剤の薬理作用,薬物動態及び安全性に関連した又は予期できない差異は 予想されない[1][5].本剤の投与に関連した有害事象は未成熟もしくは成熟動物の試験で検出でき るか,又は成人の安全性データから外挿可能である.
2.4.4.6
局所刺激性試験
ラット(8202366 試験)及びカニクイザル(1933-018 試験)における反復投与毒性試験中に局 所刺激性を評価した.本剤を投与した動物の注射部位での顕微鏡的所見は対照動物と同等であり, 静脈内投与後に予想される正常反応と一致していた. ウサギにおける追加試験を1 試験実施し,2,000 IU FVIII/5 mL バイアルの力価における本剤の 局所刺激性を検討した.表示力価2000 IU FVIII/5 mL バイアルの本剤は,静脈内(臨床投与経路), 動脈内及び静脈周囲投与後の局所忍容性が良好であった(PV2651201 試験).2.4.4.7
その他の毒性試験
2.4.4.7.1 免疫原性試験 各種in vitro 及び in vivo モデルを用いて,本剤とアドベイトの免疫原性を比較評価した.モデル にはヒト全血又は血漿を用いた2 種類の in vitro モデル,非臨床免疫原性評価のために特別に開発した3 種類の新規マウスモデル(E17 FVIII ko マウス,E17 FVIII ko ヒト MHC-クラス II(HLA DR-15)トランスジェニックマウス及び E17 FVIII ko ヒト FVIII トランスジェニックマウス),並
また,本剤中に存在するたん白質凝集物によるリスクを評価するため,凝集物含量の異なる本 剤2 ロットの比較免疫原性を特定の血友病 A 患者集団を反映する 2 種類の非臨床血友病マウスモ デルににおいて評価した. 2.4.4.7.1.1 In vitro試験 健康供血者8 例(男女各 4 例)から得たヒト全血を用いて,in vitro サイトカイン放出アッセイ を実施し,本剤2 ロット及びアドベイト 1 ロットがヒトの自然免疫系を活性化する可能性を比較 評価した(IVFS_001_10 試験).アドベイトの処方緩衝液を陰性対照とした.トール様受容体 4 を介して自然免疫細胞を活性化することが知られているLPS を陽性対照とした.全血試料を本剤, アドベイト,処方緩衝液又はLPS のいずれかと 37°C で 20~22 時間インキュベートし,培養液上
清中に放出されたサイトカインIL-1β,IL-6,IL-8 及び TNF-α を測定した.In vitro にてヒト全血 とのインキュベーション後,本試験で検討したいずれの本剤ロットにおいても,一貫してきわめ て低レベルのサイトカインが放出されるのみであった.IL-1β,IL-6,IL-8 及び TNF-α の放出量は, 処方緩衝液対照群で認められたそれぞれの放出量と同程度であった.また,サイトカイン放出量 は,全血とアドベイトをインキュベートした時と同程度であった.本試験において,本剤の両ロ ットから同様の結果が得られた.健康供血者8 例(男女各 4 例)から得たヒト血漿を用いて in vitro 補体活性化アッセイを実施し,本剤2 ロット及びアドベイト 1 ロットが補体活性化を誘発する可 能性を比較評価した(IVFS_002_10 試験).アドベイトの処方緩衝液を陰性対照とした. Saccharomyces cerevisiae の細胞壁から抽出した多糖類であり,代替経路を介して補体系を活性化 することが知られているザイモサンを陽性対照とした.補体系は代替経路,古典経路及びレクチ ン経路の3 種類の経路で活性化される.3 経路共に補体因子 C5 を C5a と C5b に分解する.C5a は多機能の炎症性メディエータであり,C5b は補体因子 C6,C7,C8 及び C9 と複合体を形成し, 膜侵襲複合体となる.補体活性化のマーカーとしてC5a の産生を ELISA 法で測定した.ヒト血漿 試料を本剤,アドベイト,処方緩衝液又はザイモサンのいずれかと1 時間インキュベートし,産
生したC5a を測定した.本試験で検討した本剤のいずれのロットにおいても,in vitro でヒト血漿
とのインキュベーション後に低レベルのC5a が一貫して誘発された.C5a レベルは,処方緩衝液 対照物質又は標準物質アドベイトとのインキュベーション後と同程度であった.また,本試験で 用いた本剤2 ロットは,in vitro でヒト血漿において補体系を活性化する能力に差異がなかった. 以上のように,本剤及びアドベイトは,in vitro でヒト全血とのインキュベーションによりサイ トカイン放出を誘発せず,ヒト血漿とのインキュベーションにより補体活性化も誘発しなかった. 2.4.4.7.1.2 In vivo試験
本剤2 ロット及びアドベイト 1 ロットの免疫原性を,E17 FVIII ko マウス,E17 FVIII ko ヒト主 要組織適合性複合体(MHC)-クラス II(HLA-DR15)トランスジェニックマウス及び E17 FVIII ko
ヒトFVIII トランスジェニックマウスの 3 種類の血友病マウスモデルにおいて比較評価した
(IMM_R&D_012_11 試験).
E17 FVIII ko マウスは,Bi ら[4]が述べているように,Balb/c 又は C57BL/6 を背景とする,マウ
E17 FVIII ko ヒト MHC-クラス II(HLA-DR15)トランスジェニックマウスは,マウス FVIII を
ノックアウト,マウスMHC-クラス II 複合体を完全ノックアウト及びヒト MHC-クラス II
HLA-DRB1*1501 をノックインした血友病マウスである[14][15].APC に発現した MHC-クラス II
たん白質は,CD4+ T 細胞にペプチドを提示することによりたん白質(この場合,FVIII)に対する
抗体反応の開始に重要な役割を果たす.この新規トランスジェニック血友病マウスモデルでは, ヒトFVIII に対する抗体反応の誘導はヒト MHC-クラスハプロタイプ HLA-DR15 による FVIII ペプ チドの提示に依存する.ヒトMHC-クラスハプロタイプ HLA-DR15 は,血友病患者での FVIII イ
ンヒビターの発現リスクの増大に関連することが示されている[12][13].
E17 FVIII ko ヒト FVIII トランスジェニックマウスは,マウス FVIII をノックアウトし,ヒト FVIII
を遺伝子導入により発現させた血友病マウスである[15].これらのマウスは生得のヒト FVIII には
免疫学的に耐性であるが,免疫原性の高いヒトPEG-rFVIII 製剤を投与すると免疫反応を発現する.
このモデルを用いて,アドベイトと比較して本剤がヒトFVIII に対する免疫寛容を維持するかど
うかを検討した.
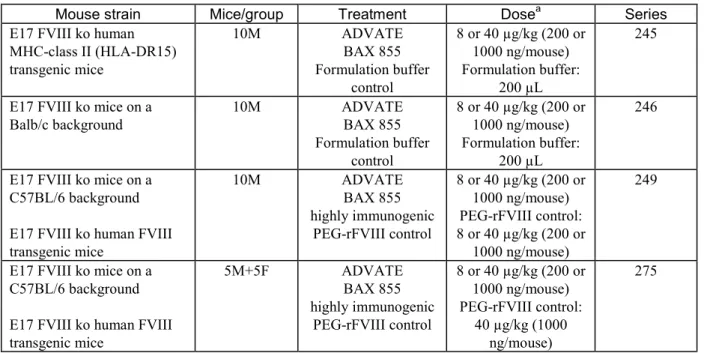
マウスで実施した比較免疫原性試験(シリーズ)の概要を表 2.4-3 にまとめる.
表 2.4-3 Comparative immunogenicity studies conducted with BAX 855 in mouse models in the study IMM_R&D_012_11
Mouse strain Mice/group Treatment Dosea Series
E17 FVIII ko human MHC-class II (HLA-DR15) transgenic mice 10M ADVATE BAX 855 Formulation buffer control 8 or 40 µg/kg (200 or 1000 ng/mouse) Formulation buffer: 200 µL 245
E17 FVIII ko mice on a
Balb/c background 10M ADVATE BAX 855 Formulation buffer control 8 or 40 µg/kg (200 or 1000 ng/mouse) Formulation buffer: 200 µL 246
E17 FVIII ko mice on a C57BL/6 background E17 FVIII ko human FVIII transgenic mice 10M ADVATE BAX 855 highly immunogenic PEG-rFVIII control 8 or 40 µg/kg (200 or 1000 ng/mouse) PEG-rFVIII control: 8 or 40 µg/kg (200 or 1000 ng/mouse) 249
E17 FVIII ko mice on a C57BL/6 background E17 FVIII ko human FVIII transgenic mice 5M+5F ADVATE BAX 855 highly immunogenic PEG-rFVIII control 8 or 40 µg/kg (200 or 1000 ng/mouse) PEG-rFVIII control: 40 µg/kg (1000 ng/mouse) 275
a: Eight doses in weekly intervals. Doses in ng or µg refer to protein doses. A protein dose of 8 and 40 µg/kg correlates to an activity-based dose of ca 50 and 250 IU FVIII/kg, respectively.
各マウスモデルにおいて本剤2 ロットとアドベイト 1 ロットとを比較評価した.シリーズ 249
及び275 では,E17 FVIII ko ヒト FVIII トランスジェニックマウスにおいてヒト FVIII に対する免
対照とした.初回投与前,4 回目投与後及び最終投与 1 週後に血液試料を採取し,ヒト FVIII 及び PEG-rFVIII に対する結合抗体を ELISA 法で測定した.また,PEG に対する結合抗体を FACS によ
り分析した.各マウスモデルにおいて,本試験で検討した本剤の両ロットは,ヒトFVIII 及び
PEG-rFVIII に対する抗体を同程度に誘導し,これはアドベイトとも同程度であった.
更に,本剤の両ロットは,E17 FVIII ko マウス(35 匹中 20 匹;シリーズ 246 及び 275)におい てPEG に特異的な抗体を誘導したが,E17 FVIII ko ヒト FVIII トランスジェニックマウスでは誘 導しなかった.上述のように,E17 FVIII ko ヒト FVIII トランスジェニックマウスはヒト FVIII に
は免疫学的に耐性であるが,免疫原性の高いヒトPEG-rFVIII 製剤を投与するとヒト FVIII に対し て免疫反応を発現する.マウスに本剤を投与した時PEG に対する抗体が発現しなかったことから, PEG に対する抗体の誘導は,免疫原性たん白質のエピトープの存在に依存することが示された. PEG 自体はポリエーテル化合物であり,FVIII のようなたん白質に対する抗体反応の開始に必要な CD4+ T 細胞が認識するたん白質エピトープを含まない.これらの試験で用いたマウスモデルの免 疫系が本剤のヒトFVIII 部分を異種たん白質と認識した場合にのみ本剤投与により PEG に対する
抗体が誘導された.一方,E17 FVIII ko ヒト FVIII トランスジェニックマウスにおいて免疫寛容を
破壊することが知られている免疫原性の高い陽性対照PEG-rFVIII は,これらの試験で用いたすべ てのマウスモデルにおいてPEG に対する抗体を誘導した.この場合,免疫原性の高い PEG-rFVIII 製剤のFVIII 部分が免疫原性のたん白質エピトープとなる. 以上のように,本剤及びアドベイトは,比較免疫原性試験で検討したすべての血友病マウスモ デルにおいて類似したFVIII 免疫原性プロファイルを示した. カニクイザルにおいて本剤2 ロット及びアドベイト 1 ロットの免疫原性を比較評価した (8220805 試験).本剤及びアドベイトは共に 8 又は 40 µg/kg の用量で週 1 回,8 週間(計 8 回) 静脈内投与した.活性用量としてそれぞれ50 及び 250 IU FVIII/kg に相当する.毒性及び抗体反応 の評価は,一般状態観察,体重測定,免疫原性解析及び肉眼的観察に基づいて行った.抗体評価 用の血液試料は,初回投与前,5 回目投与直前及び最終投与 1 週後(9 週目の 1 日目)に採取した. いずれの用量においても毒性徴候は認められなかった.処方試料から動物に適正に投与されたこ とを確認した.雄1 匹(40 μg/kg,本剤)は試験 29 日目に瀕死状態に陥り,安楽死させた.本動 物の健康状態不良及び損傷は,ケージ内の同居動物に起因するものであり,本剤の投与と関連が あるとは考えられなかった.本剤又はアドベイトに関連した体重変動又は臨床徴候は観察されな かった.本剤又はアドベイトを投与したすべての動物にヒトFVIII 及び PEG-rFVIII に特異的な抗 体が発現した.試験群間での抗体価の著明な差異は,4 及び 8 回目投与後に認められなかった. また,1 匹を除いて本剤投与群のすべての動物に PEG に特異的な抗体が発現した.CHO たん白質 に特異的な抗体はいずれの動物においても検出されなかった.8 回目投与後に,FVIII に対する中 和抗体がアドベイトを投与した8 匹すべて及び本剤を投与した 8 匹中 7 匹の動物で検出された.8 μg/kg の本剤を投与した 1 匹には FVIII 活性に対する中和抗体は発現しなかった. 以上のように,カニクイザルにおいて本剤及びアドベイトの忍容性は良好であり,類似した免 疫原性プロファイルを示した.
本剤中に存在するたん白質凝集物によるリスクを評価するため,一方は凝集物含有量が低く,
もう一方は凝集物含有量が高い本剤2 ロットの免疫原性を比較評価した.特定の血友病 A 患者集
団を反映する2 種類の血友病マウスモデルである,E17 FVIII ko ヒト MHC-クラス II(HLA DR-15) トランスジェニックマウス及びE17 FVIII ko ヒト FVIII トランスジェニックマウスを用いた (IMM_R&D_025_12 試験).
本剤の凝集物含有量 %のロット 1 と %のロット 2 とを比較し( で分析),アド
ベイトを対照とした.本剤の各ロット(8 及び 40 µg/kg)又はアドベイト(40 µg/kg)を週 1 回,
計8 回投与した.初回投与前,4 回目投与後及び最終投与 1 週後に血液試料を採取し,ヒト FVIII
及びPEG-rFVIII に対する結合抗体を ELISA 法で測定した.PEG に対する結合抗体を FACS によ り分析した.
本剤2 ロットは,E17 FVIII ko ヒト MHC-クラス II(HLA-DR15)トランスジェニックマウスに
おいて,ヒトFVIII 及び PEG-rFVIII に対して類似した範囲内の結合抗体力価を示した.重要な点
として,アドベイトを投与したE17 FVIII ko ヒト FVIII トランスジェニックマウスの 10 匹中 8 匹,
アドベイトと同用量の本剤ロット1(凝集物含有量 %)を投与した 10 匹中 10 匹及びロット 2 (凝集物含有量 %)を投与した 10 匹中 9 匹で FVIII に対する免疫寛容が維持された. 以上のように,凝集物含有量の異なる本剤2 ロットは,2 種類の血友病マウスモデルにおいて 類似した免疫原性プロファイルを示し,アドベイトのFVIII 免疫原性プロファイルと異ならなか った. 2.4.4.7.1.3 毒性試験及び免疫原性試験の概要 要約すると,ラット及びカニクイザルにおいて本剤の忍容性は良好であり,単回投与時の NOAEL はサルで 1500 IU FVIII/kg,1 ヵ月にわたる反復投与時の NOAEL はラット及びカニクイザ
ル共に700 IU FVIII/kg であった.本剤又はアドベイトを反復投与した動物の大半に結合抗体及び 中和抗体の形成が認められ,これは異種たん白質の反復投与後に予想される免疫反応であり,非 PEG 化 FVIII 製剤においてもよく知られている[7].比較免疫原性試験で得られた結果から,本剤 及びアドベイトは類似したFVIII 免疫原性プロファイルを有することが示された.凝集物含量の 異なる本剤2 ロットは 2 種類の血友病マウスモデルにおいてアドベイトと類似した FVIII 免疫原 性プロファイルを示した.アドベイトの患者における安全性プロファイルは確立している.本剤 は非臨床動物モデルにおいてアドベイトと同様のFVIII 免疫原性プロファイルを示したことから, 患者においても同様の安全性プロファイルを示すと考えられる. 2.4.4.7.2 その他の試験 2.4.4.7.2.1 ヒト組織パネルを用いた本剤の交差反応性の評価 組織交差反応性試験において,3 例の異なるドナーから選択した広範な正常ヒト組織に対する 抗PEG ウサギモノクローナル抗体(本剤に特異的に結合することが示された抗 PEG- 抗体)と
Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use」[8]に従い組 織を選択した. 免疫組織化学的解析から,抗PEG ウサギモノクローナル抗体と正常ヒト組織との特異的な交差 反応性は生じないことが示された.陽性対照組織( を添加した肝臓)は抗PEG 抗体に より特異的に染色され,染色法の頑健性が確認された. 以上のように,ヒト組織に対する高親和性抗PEG 抗体の反応は生じ難い. 2.4.4.7.2.2 ラットにおけるPEG2ru20KCOOHの反復投与毒性試験 PEG の評価に適した動物種であるラットに週 2 回,28 日間反復投与し,PEG に関連する毒性を 評価した.生涯にわたり本剤を投与した場合の複数倍に相当するPEG 投与量を設定した(8298494 試験). PEG2ru20KCOOH は,本剤の代謝によるたん白質部分の異化後に遊離すると考えられるため, 非結合型20 kDa モデル PEG として用いた.また,PEG2ru20KCOOH は最終製剤の不純物として 特定されている(3.2.S.3.2 項を参照.同項では「PEG2ru20KCOOH」を「PEG2ruButanoic acid20K (PEG-acid)」と記載.). PEG2ru20KCOOH を 0.65,6.5 及び 65 mg/kg の用量で週 2 回,28 日間(計 8 回)静脈内(ボー ラス)投与し,4 及び 13 週間の無投与期間中,遅発性毒性又は毒性の可逆性を評価した.また, 被験物質のTK プロファイル及び抗薬物抗体の発現も評価した. 用量は,本剤で通常想定される臨床最高用量(80 IU rFVIII/kg/日,週 2 回投与)の生涯投与量 に基づいて設定した.生涯投与期間を70 年と仮定した.rFVIII 1 U 当たりの PEG 量は 0.095 µg で あることから,長期間の定期補充療法で予測される最高用量は7.6 µg/kg となる.したがって,PEG 投与用量は,単回投与量の約100~10,000 倍,平均生涯投与量の約 10 分の 1~10 倍に相当する. PEG2ru20KCOOH を 0.65,6.5 及び 65 mg/kg の用量で静脈内投与した時の忍容性は良好であり, 一般状態,体重,摂餌量,眼科学的検査,器官重量,臨床又は解剖病理学的検査に及ぼす有害所 見はなかった.最高用量の65 mg/kg においてもマクロファージ又はその他の細胞の空胞化は認め られなかった.PEG に対する結合 IgG 抗体は検出されなかった. PEG2ru20KCOOH を投与したすべての動物の曝露は,投与 1 及び 22 日目の TK 評価で確認され た. 以上のように,PEG2ru20KCOOH をラットに 4 週間静脈内投与した時の忍容性は良好であり, NOAEL は予定臨床最大用量に基づく単回投与量の約 10,000 倍及び平均生涯投与量の約 10 倍に相 当する,最高用量の65 mg/kg であった. 2.4.4.7.2.3 PEG2ru20KCOOHの遺伝毒性試験 復帰突然変異試験においては,S9 mix の存在及び非存在下でネズミチフス菌株 TA98,TA100,
TA1535 及び TA1537,並びに大腸菌株 WP2uvrA を試験菌株として用い,適切な溶媒対照及び陽 性対照と共に突然変異誘発性を評価した.試験したPEG2ru20KCOOH の用量は 100,333,1000,
3330 及び 5000 μg/プレートとし,用量当たり 3 枚のプレートを用いた.初回試験の結果は独立 した別試験により確認された.本試験結果から,本試験条件下でのPEG2ru20KCOOH は,AroclorTM 誘導ラット肝臓(S9)から調製したミクロゾーム酵素の有無にかかわらず,いずれの試験菌株で もプレート当たりの平均復帰突然変異体数において陽性の増加を誘発しないことが示された. 染色体異常試験においては,培養ヒト末梢血液リンパ球をあらかじめ設定した濃度の PEG2ru20KCOOH,溶媒及び陽性対照と共に,S9 mix 非存在下では 3 時間(初回試験)又は約 22 時間(確認試験),S9 mix 存在下では 3 時間(初回及び確認試験),37 ± 2oC でインキュベート し,コルセミドをインキュベーション終了前2 ± 0.5 時間に添加した.培養細胞は PEG2ru20KCOOH の添加後約22 時間で採取した.試験結果から,初回及び確認試験のいずれにおいても,S9 mix の存在及び非存在下でのPEG2ru20KCOOH には,染色体異常,倍数体又は核内倍加の有意な上昇 は観察されなかった.以上のように,PEG2ru20KCOOH は,培養ヒト末梢血液リンパ球において, 代謝活性化系の有無にかかわらず染色体異常の誘発に関して陰性であった. 小核試験においては,各群雄5 匹のマウスに PEG2ru20KCOOH の 500,1000 及び 2000 mg/kg(そ れぞれ,250,500 及び 1000 mg/kg を約 1~2 時間の間隔で 20 mL/kg/回の容量にて 2 回投与),生 理食塩液(溶媒対照)及びシクロホスファミド(陽性対照)を緩徐に静脈内投与し,毒性徴候及 び/又は死亡について,最終投与後24 時間(すべての群)又は 48 時間(PEG2ru20KCOOH の 2000 mg/kg 及び生理食塩液投与群)観察した.骨髄は後肢骨から採取した.試験結果から,被験物質 であるPEG2ru20KCOOH は,2000 mg/kg の用量レベルで 10 匹中 1 匹の自発運動をわずかに抑制 した以外,2000 mg/kg まで投与した動物において臨床毒性徴候を引き起こさなかった. PEG2ru20KCOOH は,試験したいずれの用量においても小核を伴う PCEs の有意な増加を誘発し なかった.また,PEG2ru20KCOOH は,試験したいずれの用量及び時間(24 又は 48 時間)にお いてもPCE:NCE 比を統計学的に有意に減少せず,骨髄に対して細胞毒性を示さなかった.以上 から,被験物質であるPEG2ru20KCOOH は,本試験条件下でのマウス骨髄小核試験において陰性 と評価された. 2.4.4.7.3 添加物及び不純物 2.4.4.7.3.1 最終製剤に用いた添加物の毒性評価 本剤の処方はアドベイトと同一である(2.3.P 項を参照).したがって,製剤に用いる添加物は 安全であることが示されている.ポリソルベート80 に関連する過敏性反応を除き,添加物に起因 する既知の又は想定される毒性はない.そのため,ポリソルベート80 に対する過敏性反応が知ら れている患者への本剤の使用は避ける必要がある. 2.4.4.7.3.2 最終製剤のPEG関連不純物の毒性評価 本剤の最終製剤に含まれる可能性のあるPEG 関連不純物の毒性を PEG の主な代謝物であり, 本剤の主な不純物であるPEG2ru20KCOOH のラットにおける 28 日間反復投与毒性試験及び遺伝 毒性試験において評価した(2.6.6.8.2.2 項及び 2.6.6.8.2.3 項を参照).すべての動物において PEG2ru20KCOOH の忍容性は良好であり,試験した最高用量の 65 mg/kg でも有害作用は認められ
ず,また,遺伝毒性も示されなかった.更に,本剤の最終製剤に含まれる可能性のあるPEG 関連 不純物の毒性の詳細な評価結果をバクスター社のExpert Report(RA13RS22R2)に記載する. 2.4.4.7.4 PEG及びPEG化生物製剤の安全性 PEG は非イオン性ポリエーテルの化合物に属し,消費生活用製品に,また多くの医薬品の添加 物として使用されている.5~40 kDa の PEG は極めて水に溶けやすく,通常,毒性及び免疫原性 が軽微であると考えられている.過去数十年間,このようなPEG による生物製剤の化学的修飾は, 薬理学的,薬物動態学的及び安全性プロファイルを向上させることが示されている. 同様のサイズのPEG 単独又はたん白質との複合体,また,本剤の予定投与量よりも高い PEG 投与量の安全性データから,臨床用量でPEG に起因するあるいは関連する安全上の懸念は示唆さ れない.文献に報告されているPEG に関連する唯一の所見は,高用量の PEG を反復投与した動
物試験で認められた特定の種類の細胞における空胞化である[16][9][6](Baxter Expert Report RA13RS22R2).EMA の CHMP Safety Working Party は最近,0.4 μmol/kg/月以上の PEG(主に 40 kDa
超)の投与により発現する上衣細胞の空胞化リスクについて考察した.本剤の場合,1 日当たり のPEG 投与量 7.6 µg/kg は閾値である 0.4 μmol/kg/月の 105 分の 1 である. このような所見の確率,本質及び機能的意義はまだ完全には理解されていないが,本剤の動物 試験ではこのような空胞化は発現しなかったことを注目することが重要である.本剤に適用され ているPEG サイズ及び PEG 用量は空胞化の発現が想定されない範囲内にある.マウス,ラット, ウサギ及びカニクイザルにおける本剤の非臨床試験では,rFVIII の PEG 化に関連する有害作用は 発現しなかった.ADME 試験では,標識した本剤の極めて高用量をラットに単回静脈内投与して も,投与後6 週以内に完全に排泄されることが示された.動物及びヒトにおける PEG の in vivo 最終主要分解産物であり,本剤の不純物であるPEG2ru20KCOOH のラットでの 28 日間反復投与 毒性試験及び遺伝毒性試験も実施した.PEG2ru20KCOOH は遺伝毒性がなく,マクロファージ又 は他の細胞の空胞形成を含むいかなる有害作用又は非有害作用も誘起しなかった. 上記から,1)本剤投与時の 1 ヵ月当たりの PEG 投与量は他の既承認 PEG 化製剤よりも少ない, 2)PEG 投与量は動物試験で認められた上衣細胞の空胞化の閾値(0.4 µmol/kg/月以上)よりも低 い,3)本剤に結合する PEG のサイズ(20 kDa)は蓄積のリスクを伴うことなく,生体からの完 全な消失を可能にする,と結論できる.したがって,長期投与時のPEG 量は現時点で安全上の懸 念をもたらすことは少ないか,全くないと考えられる. PEG 及び他の PEG 化生物製剤の安全性プロファイルに基づくと,本剤による臨床治療において 発現する可能性のある有害作用は特定されなかった.安全性データの包括的な概要は2.6.6.8.4 項
に記載されている.PEG 及び本剤の安全性はバクスター社の Expert Report(報告書 RA13RS22R2) 中で更に詳細に評価されている.