神経変性疾患に対する新規治療薬開発を目的とした
レチノイン酸受容体アゴニストおよびムスカリン M
1受容体ポジティブ アロステリックモジュレーターに関する研究
2019
栗本 恵実
Study on retinoic acid receptor agonists and muscarinic M
1receptor positive allosteric modulators as novel therapeutic approaches
for neurodegenerative diseases Emi Kurimoto
Neurodegenerative disorders are characterized by chronic and progressive neuronal dysfunction and the loss of neurons in the specified regions of brain and/or spinal cord. Alzheimer disease (AD) and Parkinson disease (PD) are the most common age-related neurodegenerative disorders, affecting several millions of aged people worldwide. Both genetic susceptibility and environmental factors are believed to be responsible for these diseases. Although symptomatic treatments for both AD and PD are currently available, unfortunately, there are neither treatments that slow the neurodegenerative process nor sufficient treatments without severe side effects. A better understanding on the molecular mechanisms of these neurodegenerative diseases will help us to obtain effective drugs for treatment of AD and PD in future. Thus, the strategy for the development of potential neuroprotective drugs and effective drugs for symptomatic relief without severe adverse effects in patients with PD and AD, offers great challenge for basic science and clinical development. In this study, I investigated the following three issues to develop fundamental treatments for PD and beneficial treatments for AD.
1. Neuroprotective effects of retinoic acid receptor agonist on midbrain dopaminergic neurons as a potential therapeutic approach for treatment of PD.
Retinoids are reported to inhibit inflammatory responses in activated microglia, which is associated with the pathogenic processes of PD, although the roles of retinoic acid receptor (RAR) in regulation of dopaminergic neuronal death have not been investigated. In this study, I examined the effects of RAR agonists against interferon-γ/lipopolysaccharide (LPS)-induced dopaminergic neuronal death in midbrain slice culture. An RAR agonist Am80 protected dopaminergic neurons from microglia-mediated injury without affecting microglial activity. In addition, oral administration of Am80 could suppress LPS-induced degeneration of nigral dopaminergic neurons in mice. Next, I investigated the mechanisms of the neuroprotective effect of RAR agonist, and found that the neuroprotective effect of Am80 was mediated by up-regulation of brain-derived neurotrophic factor (BDNF) and its downstream signaling. These results suggest that activation of RAR is a promising approach to prevent dopaminergic neuronal death mediated by inflammatory responses.
2. An approach to discovering novel muscarinic M1 receptor positive allosteric modulators (M1
PAMs) with minimized gastrointestinal (GI) dysfunction as promising therapeutic agents for the treatment of AD.
Activation of muscarinic M1 receptor (M1R) is a promising approach for improving cognitive
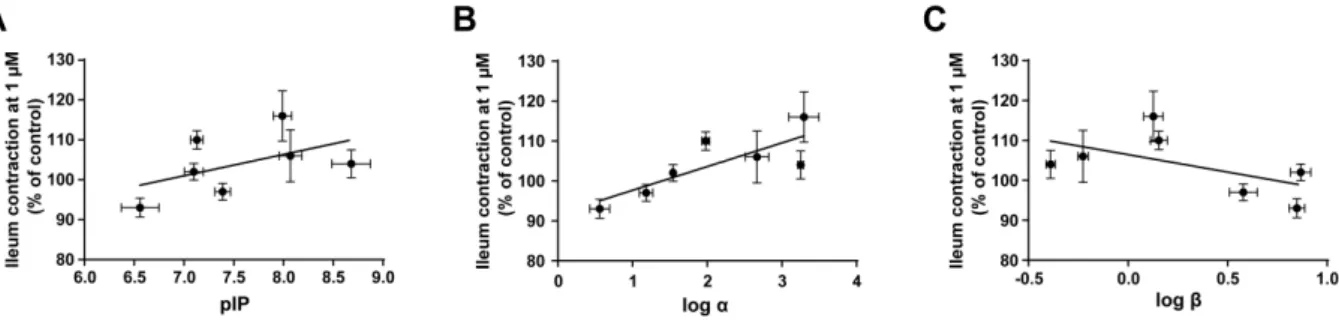
impairment in AD. However, a selective M1 PAM, benzyl quinolone carboxylic acid (BQCA), induced diarrhea in mice via M1R activation. Moreover, BQCA augmented electric field stimulation-induced ileum contraction in an in vitro Magnus assay. Thus, I established a drug screening strategy to discover novel M1 PAMs producing potent cognitive improvement with minimized GI side effects. I assessed several PAM parameters of various M1 PAMs by using in vitro binding and functional analyses, and evaluated these M1 PAMs in the Magnus assay. The combined results revealed a significant correlation between augmentation of ileum contraction and their α-value, a PAM parameter associated with the binding cooperativity between acetylcholine and M1R. Indeed, in evaluations of in vivo profiles of two M1 PAMs with high and low α-value, the M1 PAM with low α-value showed wider safety margin between cognitive efficacy and diarrhea induction than the high α-value M1 PAM. These results suggest that fine adjustment of the α-value could be a key for discovering M1 PAMs yeilding potent cognitive improvement with a lower risk of GI side effects.
3. Exploration of translational biomarker for a novel M1 PAM TAK-071 in clinical development for the treatment of AD.
Based on the novel screening strategy for M1 PAMs, TAK-071 with low α-value was identified as a clinical candidate. In this study, I explored changes in quantitative electroencephalogram (qEEG) power bands, with scopolamine challenge, as a non-invasive translational biomarker for the effect of TAK-071 in cynomolgus monkeys. In line with clinical observations, scopolamine increased theta and delta power bands in a dose-dependent manner. Next, the effects of TAK-071 on scopolamine-induced qEEG spectral changes were examined. TAK-071 suppressed the scopolamine-induced increases in alpha, theta, and delta power bands. These results suggest that changes in theta and delta power bands in the context of scopolamine challenge could be used as translational biomarkers for the evaluation of TAK-071 in clinical studies.
In the current study, I demonstrated that RAR agonist Am80 showed prominent neuroprotective effects against dopaminergic neuronal death via recruitment of BDNF signaling in rat midbrain slice cultures. The neuroprotection by Am80 was also confirmed in a dopaminergic neurodegeneration model in mice in vivo. Thus, RAR agonists including Am80 could be novel and fundamental therapeutics for PD. Moreover, in the present study, I revealed a novel screening strategy for M1 PAMs with minimized GI dysfunction, leading to identification of TAK-071 as a clinical candidate. In addition, I determined the utility of qEEG analysis under the scopolamine challenge paradigm as a translational biomarker for TAK-071 efficacy in non-human primate. Overall, our findings suggest a safer M1 PAM is a valuable approach to treat AD, and would support the clinical development of M1
PAMs as effective drugs for symptomatic relief of AD without severe adverse effects.
目 次
緒 言
... 5第一章 新規パーキンソン病治療薬としての
RARアゴニスト
Am80の可能性
... 9実験方法
... 9実験結果
... 13第一節 炎症性応答によるドパミンニューロン死に対するレチノイドの作用
... 13第二節 炎症性応答によるドパミンニューロン死に対する
Am80の保護作用機序
... 18考 察
... 23第二章 新規アルツハイマー病治療薬としての
M1 PAM M1 PAMの副作用低減を目的としたスクリーニング戦略の構築
... 26実験方法
... 27実験結果
... 31第一節
M1 PAMの消化管症状への影響と副作用低減に繋がる
PAMパラメータの同定
... 31第二節
M1 PAMの結合協調性と消化管副作用の低減の検証
... 35考 察
... 38第三章 新規アルツハイマー病治療薬としての
M1 PAM TAK-071トランスレーショナルバイオマーカーとしての定量的脳波解析の可能性
... 40実験方法
... 41実験結果
... 43考 察
... 52総括および結論
... 54謝 辞
... 56引用文献
... 57緒 言
神経変性疾患は慢性進行性の神経機能不全を呈する疾患であり、特定の脳領域または脊髄にお ける神経細胞の消失を特徴とする。神経変性疾患の例としては、アルツハイマー病やパーキンソ ン病、筋萎縮性側索硬化症、ハンチントン病および多発性硬化症が挙げられる。これらのうち、
アルツハイマー病とパーキンソン病は最も有病率の高い、代表的な加齢性の神経変性疾患であり、
世界的に見て数百万人の高齢者が罹患している
1,2。世界的な高齢者人口の増加にともなって、パ ーキンソン病およびアルツハイマー病は、先進国と新興国のいずれにおいても、医療経済的負担 が大きく、公衆衛生上の重大な課題となっている。これら疾患に関して、遺伝学的素因や環境因 子など、病因の解明は進められているものの、未だ不明な点は多い。また、現在のパーキンソン 病およびアルツハイマー病の治療は対症療法に依存している一方で、既存の治療薬では重篤な副 作用や病態進行に伴う有効性の低下が問題視されており、神経変性を遅らせる根本的治療薬はな い。将来的には、これら神経変性疾患の原因となる分子メカニズムの解明が、有効な治療薬の開 発に繋がると考えられ、神経保護作用を有する治療薬や副作用の少ない症状改善薬の開発が、基 礎的研究および臨床開発の両面で非常に注目されている
3。
パーキンソン病は安静時振戦、無動、筋固縮、姿勢反射障害を主症状とする
2。パーキンソン 病の有病率は
65歳以上で
1%、
85歳以上になると
5%までに上昇することから、病態の進行にお いて加齢の影響が大きいとされている
4。中脳黒質緻密部ドパミンニューロンの選択的な変性・脱 落により、その投射先である線条体におけるドパミン遊離量が減少し、運動機能障害を生じるこ とが明らかとなっている
5。このドパミンニューロン死を引き起こす正確な病理学的メカニズムは 解明されていないが、炎症性応答やミトコンドリア機能不全、興奮毒性、異常タンパク質の凝集 が、パーキンソン病の進行に関わる重要な因子と考えられている。近年、他の脳部位に比べて中 脳黒質緻密部にミクログリアが高密度で存在すること、また、パーキンソン病患者の剖検脳にお いて活性化ミクログリアの蓄積が確認されていることから、中脳黒質緻密部に特異的なドパミン ニューロン死とミクログリアの活性化による炎症性応答との関連性が示唆されている
6,7。現在、
パーキンソン病に対する第一選択はドパミン補充療法で、ドパミンの前駆物質であるレボドパが 用いられる。また、ドパミン作動薬であるプラミペキソールやロピニロール、ブロモクリプチン も線条体でのドパミン受容体を活性化する目的で使用される
8。このように、パーキンソン病治療 の選択肢が対症療法に限られているため、神経変性を阻止して病態の進行を抑制する神経保護治 療薬への期待が高まっている。
レチノイドとは、ビタミン
Aの活性代謝物である
all-trans-retinoic acid(
ATRA)や同様の活性 をもつ合成化合物の総称であり、核内受容体であるレチノイン酸受容体(
RAR)またはレチノイ ド
X受容体(
RXR)に結合する。
RARおよび
RXRには、それぞれ
α、
β、
γの
3種類のサブタイ プが存在し、標的遺伝子の転写制御を介して細胞の分化・増殖および形態形成に関与することが 知られている
9。
RARは発生期のみならず、成体の脳内にも発現していることが確認されており、
近年、中枢神経系における記憶学習といった高次脳機能制御にも深く関与することが示唆されて
いる
10。さらに、
ATRAが、培養ミクログリアの活性化に伴って生じる誘導型一酸化窒素合成酵 素や複数の炎症性サイトカインの発現を抑制することが報告され
11,12、ミクログリアの機能制御 に
RARを介したシグナル伝達が寄与すると考えられる。このような背景から、炎症性応答に伴う 中脳ドパミンニューロン死に対するレチノイドの作用を検証し、その作用機序を解明することは、
パーキンソン病に対する根本治療をもたらす新規治療薬開発に繋がると考えられる。
アルツハイマー病は認知症の主要因とされ、中核症状である進行性の認知記憶障害と、幻覚や 妄想、抑うつ、攻撃的行動、徘徊といった行動・心理症状によって特徴づけられる
1。
65歳以上 の高齢者の罹患率は
11%であり、
85歳以上の高齢者では
33%にまで罹患率が上昇することは
13、 加齢が本疾患の主なリスク因子であることを浮き彫りにしている。
1970年代後半からの神経伝達 物質の研究により、大脳皮質におけるアセチルコリン(
ACh)量の低下がアルツハイマー病の主 要因であるとするコリン仮説が最も有力な仮説として提唱されている
14。これまでにアメリカ食 品医薬品局が承認した
5種の治療薬のうち
4種が
AChの加水分解を抑制するアセチルコリンエス テラーゼ阻害薬(
AChE-I)である
15。
AChE-Iによって軽度の症状改善が認められる一方で、吐気 や嘔吐、下痢といった副作用を引き起こす。そのため有効性と安全性の高い新規治療薬が求めら れている。
複数の報告から、
ACh受容体サブタイプのうちムスカリン
M1受容体(
M1R)の活性化がアル ツハイマー病における認知機能障害の改善に有効であることが示唆されている
16,17。一方、臨床 および非臨床試験において
M1Rの
ACh結合部位に直接作用するオルソステリックアゴニストが 顕著な薬効を発揮したが、
M1Rに対する選択性の低さが原因で消化管症状といった重篤な副作用 が出現し、これらの薬物はいずれも臨床開発段階で開発中止に至った
16,17,18。また、
ACh結合部 位の構造は、ムスカリン受容体の全サブタイプにおいて相同性が高く、
M1R選択的なオルソステ リックアゴニストを創出することは困難である。そこで近年、
M1Rの
ACh結合部位とは異なるア ロステリック部位に結合することで
M1Rへの選択性を示す活性化薬、すなわちアロステリックア ゴニストおよびポジティブアロステリックモジュレーター(
PAM)が注目されている。なかでも、
M1R
選択的な
PAM(
M1 PAM)はそのメカニズムとして、内因性リガンドである
AChが存在する 条件下でのみ
M1Rシグナル伝達を増強することから、受容体脱感作の懸念が低いという点でも特 に注目されている。このような背景から、選択的な
M1 PAMの創出が、副作用の少ない新規アル ツハイマー病治療薬に繋がると考えられる。
中枢神経系の治療薬開発の前期臨床試験において、開発化合物が脳内の目的とする標的に結合
して期待通りの作用を示すか否かを確認することは、開発の成功確率を上げる上で非常に重要と
なる。その際、非臨床と臨床を繋ぐ非侵襲性の定量的バイオマーカーが必須となる。中枢神経作
用薬については一般的にポジトロン断層法(
PET)が使用され、脳内におけるリガンドと標的分
子との結合や分布を評価することが可能である。しかしながら
PETによる評価は、標的に対する
最適な放射性リガンドを入手できるか否かで制限され、また化合物と標的の相互作用を検出でき
る一方で、その機能的側面は評価できない。副作用を低減した新規
M1 PAMを臨床開発に進める
上で非臨床と臨床を繋ぐバイオマーカーが必要となるが、
M1Rのアロステリック部位に結合する
最適な放射性リガンドはなく、
PETに代わるトランスレーショナルバイオマーカーを探索する必
要がある。
そこで著者は、これらの背景をもとに、根本治療を目指す新規パーキンソン病治療薬としての
RARアゴニストの可能性、および、
M1 PAMについては、臨床開発を見据えた上での副作用を低 減した新規アルツハイマー病治療薬としての可能性を明らかにするために研究を進めた結果、以 下の新知見を得た。
(1)
インターフェロン
-γ(
IFN-γ)
/リポ多糖(
LPS)処置により、中脳培養切片内のグリア細胞が 活性化され、一酸化窒素(
NO)産生量の上昇を伴うドパミンニューロン死が惹起された。
RARα/β
アゴニストであるタミバロテン(
Am80)は
IFN-γ/LPSによるドパミンニューロン死
に対して、ミクログリアの活性化抑制ではなく、脳由来神経栄養因子(
BDNF)の発現誘導お よびその下流シグナルの活性化を介して神経保護効果を示した。
(2) M1R
への選択性が高い
M1 PAMであっても、マウスにおいて
M1Rを介して下痢を誘発するこ とが判明した。複数の
M1 PAMを用いた検証により、マウス単離回腸の収縮作用と
PAMパラ メータの一つ (
AChと
M1Rとの結合協同性を示す
α値) との間に強い正の相関性を見出した。
副作用症状を回避した安全域の広い新規
M1 PAMを見出すためのスクリーニング戦略を構築 した。
(3)
副作用を低減した
M1 PAMの臨床開発化合物として同定した
TAK-07119に対するトランスレ ーショナルバイオマーカーとして、定量的脳波(
qEEG)解析に着目した。サルにおいて非選 択的ムスカリン受容体アンタゴニストであるスコポラミン存在下での
qEEGパワースペクト ル変化に対する
AChE-I(ドネペジル) 、
M1/M4Rアゴニスト(
xanomeline)および
TAK-071の 作用を検証したところ、
θと
δ波帯域に対する作用がトランスレーショナルバイオマーカー として有望であることを明らかにした。
これらの研究成果について、以下に三章に分けて論述する。
なお、本文中および図中で使用した略号は以下の通りである。
ACh; acetylcholine
AChE-I; acetylcholinesterase inhibitor ATRA; all-trans-retinoic acid
Am80; tamibarotene
BDNF; brain-derived neurotrophic factor BQCA; benzyl quinolone carboxylic acid BSA; bovine serum albumin
CHO; Chinese hamster ovary Compound A;
3-fluoro-2-((2-(4-(1-methyl-1H-pyrazol-4-yl)benzyl)-3-oxo-2,3-dihydro-1H-isoindol-4-yl)oxy)benzonitrile Compound B;
7-(((1S,2S)-2-hydroxycyclohexyl)oxy)-2-(4-(1-methyl-1H-pyrazol-3-yl)benzyl)isoindolin-1-one Compound C;
3-((1S,2S)-2-hydroxycyclohexyl)-6-((6-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)methyl)benzo[h]quinazoli n-4(3H)-one
Compound D;
2-(2-fluorophenyl)-5-(4-(1H-pyrazol-1-yl)benzyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one Compound E; 2-(4-(1-methyl-1H-pyrazol-4-yl)benzyl)-7-(2-(piperidin-1-yl)ethoxy)isoindolin-1-one Compound F; 2-(4-(1-methyl-1H-pyrazol-4-yl)benzyl)-7-(1H-pyrazol-5-yl)isoindolin-1-one
CREB; cAMP-response element binding protein EFS; electric field stimulation
ERK; extracellular signal-regulated kinase GDNF; glial cell line-derived neurotrophic factor GFAP; glial fibrillary acidic protein
IFN-γ; interferon-γ
iNOS; inducible nitric oxide synthase KO; knockout
LPS; lipopolysaccharide
MAP-2; microtubule-associated protein 2 MAPK; mitogen-activated protein kinase M1R; muscarinic M1 receptor
M4R; muscarinic M4 receptor NO; nitric oxide
PAM; positive allosteric modulator PBS; phosphate buffered saline PET; positron emission tomography PI3-kinase; phosphatidylinositol 3-kinase qEEG; quantitative electroencephalogram RAR; retinoic acid receptor
RXR; retinoid X receptor
RT-PCR; reverse transcription-polymerase chain reaction TAK-071;
4-fluoro-2-[(3S,4S)-4-hydroxytetrahydro-2H-pyran-3-yl]-5-methyl-6-[4-(1H-pyrazol-1-yl)benzyl]-2,3-dihy dro-1H-isoindol-1-one
TH; tyrosine hydroxylase WT; wild-type
第一章
新規パーキンソン病治療薬としての RAR アゴニスト Am80 の可能性 RAR 活性化を介した中脳ドパミンニューロンの保護作用
レチノイン酸受容体(
RARα、
βおよび
γ)は細胞核内に存在し、
all-trans-retinoic acid(
ATRA) や他のレチノイドと結合して活性化する転写因子である。
RARはレチノイド
X受容体(
RXRα、
βおよび
γ)と呼ばれる別の核内受容体とヘテロ二量体を形成する。中脳黒質ドパミンニューロン にはビタミン
Aを
ATRAへ変換する酵素が高発現していることから、
ATRAが黒質線条体経路の 神経発生と維持に重要な役割を果たすと考えられる
9,20。また、
RARαおよび
βの中脳ドパミンニ ューロンでの発現も確認されており、
RARを介したシグナル伝達がドパミンニューロンにおける 遺伝子発現制御に関与することが示唆される
21。加えて、パーキンソン病患者の死後脳解析の結 果、中脳黒質内ドパミンニューロンにおいて
ATRA合成必須酵素であるレチンアルデヒドデヒド ロゲナーゼの
mRNA発現量が顕著に減少していたことからも
22、
RARがパーキンソン病におけ るドパミンニューロン死の制御に深く関与する可能性が示唆される。
パーキンソン病の発症過程において、ミクログリアの活性化を伴う炎症性応答の関与が広く知 られており、中脳黒質における活性化ミクログリアの蓄積に伴うドパミンニューロン変性がパー キンソン病患者および動物モデルにおいて確認されている
7,23,24。また、中脳でのミクログリアの 活性化によってドパミンニューロン特異的に細胞死が引き起こされることが、
in vitroおよび
in vivo試験系の両方で認められている
25-27。さらに、活性化ミクログリアに対する
ATRAの作用も 報告されており、
ATRAによる
RARβの活性化はミクログリアの活性化状態に依存し、リポ多糖
(
LPS)による刺激条件下で抗炎症作用を示した
11。
本章では
RARアゴニストによるレチノイドシグナル活性化の炎症性ドパミンニューロン死に 対する作用を検証し、新規パーキンソン病治療薬としての可能性を検討する。なお、本研究では
RARアゴニストとして
Am80(タミバロテン)を使用した。
Am80は、再発または難治性の急性 前骨髄球性白血病の治療薬として日本でも上市されており、
RARαおよび
βに選択性を有する合 成レチノイドである。
実験方法
試薬
本章で用いた試薬は以下の通りである。
ラット組換え型インターフェロン
-γ(
IFN-γ) 、ヒト組換え型脳由来神経栄養因子(
BDNF)およ
びラット組換え型グリア細胞由来神経栄養因子(
GDNF)は全て
Pepro Tech(
Rocky Hill, NJ)より
購入した。
LPS(
Escherichia coli, serotype 0111; B4)は
Sigma(
St. Louis, MO)より、
K252aは富士
フィルム和光純薬株式会社(
Osaka, Japan)よりそれぞれ購入した。
PD98059および
LY294002は
Merck Millipore(
Darmstadt, Germany)より購入した。抗ヒト
BDNFポリクローナル抗体と、その
対照となる
IgYは
Promega Corp.(
Madison, MI)より購入した。
Am80、
TAC-101および
HX630は
既報
28-30の通り合成されたものを、乙卯研究所 首藤紘一博士より供与いただいた。
培養中脳切片の調製および維持
生後
2-3日齢の
Wistar/STラット(日本
SLC株式会社
, Shizuoka, Japan)に氷中で低温麻酔をか けた。ハサミで断頭した後、頭蓋内より全脳を取り出し、氷冷した
Hanks液に浸した。
Hanks液 は、
NaCl(
137 mM) 、
KCl(
5.4 mM) 、
Na2HPO4(
3.4 mM) 、
K2HPO4(
0.5 mM) 、
D-グルコース(
5.6 mM)および
HEPES(
2.4 mM)からなる溶液を、
95% O2/5% CO2に
30分以上曝気し、
1N NaOHにて
pHを
7.2に調整後、孔径
0.22 μmのフィルターで濾過滅菌したものである。約
1分間氷冷し た後、全脳を半球に切断し、
tissue slice chopper(
NARISHIGE、
Tokyo, Japan)により厚さ
350 μmの中脳冠状切片を作製した。培地にはウマ血清含有培地を使用し、その組成は、
50% minimum essential medium/HEPES(
Thermo Fisher Scientific Japan、
Tokyo, Japan) 、
25% Hanks balanced salt solution(
Thermo Fisher Scientific Japan)および
25%非働化ウマ血清(
Thermo Fisher Scientific Japan) からなり、本混合物に
6.5 mg/mL D-グルコース、
2 mM L-グルタミン、
10 U/mL penicillin-G/10 μg/mL streptomycin(
Thermo Fisher Scientific Japan)を添加したものである。実体顕微鏡下にて中脳黒質 および腹側被蓋野を含む切片を分離し、直径
30 mmの
Millicell-CM insert membrane(孔径
0.4 μm、
polytetrafluoroethylene製
; Milipore、
Bedford, MA)上に
6枚の切片を置き、
34°C、
5% CO2環境下で 静置界面培養を行なった。培地交換は切片作製の翌日から隔日で行なった。
薬物処置
薬物処置は培養後
16-18日に、薬物を含む
0.7 mLの無血清培地で満たされた
6穴プレートに、
6
枚の切片を含む
insert membrane自体を移し変える方法で実施した。無血清培地は、
75% minimum essential medium/HEPES、
25% Hanks balanced salt solutionからなり、本混合物に
6.5 mg/mL D-グル コース、
2 mM L-グルタミン、
10 U/mL penicillin-G/10 μg/mL streptomycinを添加したものである。
免疫組織化学および組織学的評価
薬物処置終了後の培養切片を
4%パラホルムアルデヒドを用いて固定した。
0.02%過酸化水素 含有メタノールで
30分間処理することにより、内因性ペルオキシダーゼを失活させた。
0.2%Triton-X
で
30分間、膜の可溶化を行ない、
10%ウシ胎仔血清(
FCS; JRH Bioscience、
Lenexa, KS) 含有
PBSで
30分間ブロッキングを行なった後、
3%ウシ血清アルブミン(
BSA)溶液でウサギ由 来抗チロシンヒドロキシラーゼ(
TH)抗体(
Merck-Millipore、
Darmstadt, Germany)を
500倍に希 釈した一次抗体反応液中で
4°Cで一晩反応させた。ビオチン標識抗ウサギ抗体(
Vector Laboratories、
Byrlingame, CA
)を
1% BSA溶液で
200倍希釈した二次抗体反応液と室温で
1時間反応させた後、
アビジンビオチン複合体 (
Vectastain Elite ABC kit; Vector Laboratories) を室温で
1時間反応させた。
その後、
0.07% 3,3 -diaminobenzidine tetrahydrochlorideおよび
0.005%過酸化水素により抗原を可視
化させた。抗
TH抗体で染色される細胞をドパミンニューロンと同定した。明瞭な輪郭と突起を
有する神経細胞を生存細胞として、顕微鏡観察により単位面積(
520× 670 μm2)当たりの
TH陽
性細胞を計数することにより薬物作用を評価した。
ミクログリアのマーカーである
CD11bに関する免疫組織化学では、マウス由来抗
CD11b(
clone OX-42; Serotec Ltd.、
Oxford, UK)抗体を
300倍に希釈した一次抗体反応液と、ビオチン標識抗マ ウス抗体(
Vector Laboratories)を
1% BSA溶液で
200倍希釈した二次抗体反応液を用いた。
RARα
と
RARβの発現確認には特異的な細胞マーカーとの二重蛍光染色法を用いた。ウサギ由 来抗
RARα抗体(
500倍希釈、
sc-551; Santa Cruz Biotechnology、
Santa Cruz, CA) 、ウサギ由来抗
RARβ抗体(
500倍希釈、 )
sc-552; Santa Cruz Biotechnology) 、マウス由来抗
TH抗体(
1000倍希釈、
Sigma
) 、マウス由来抗
microtubule-associated protein抗体(
MAP-2; 500倍希釈、
Sigma) 、マウス由 来抗
CD11b抗体(
500倍希釈)およびマウス由来抗
glial fibrillary acidic protein抗体(
GFAP; 1000倍希釈、
Sigma)を一次抗体として使用した。
Alexa Fluor 488標識抗マウス
IgG (H+L)抗体(
1000倍希釈、
Molecular Probes、
Eugene, OR)を細胞マーカータンパクの検出に使用した。
Alexa Fluor 594標識抗ウサギ
IgG (H+L)抗体(
1000倍希釈、
Molecular Probes)を
RARαおよび
βの検出に使用し た。
TH
とリン酸化
TrkBおよび、
THとリン酸化
extracellular signal regulated kinase(
ERK)の共染 色にも二重蛍光染色法を用いた。マウス由来抗
TH抗体(
1000倍希釈、
Sigma) 、ウサギ由来抗
TrkB抗体(
phospho-Y515、
500倍希釈、
Abcam, Cambridge, MA)および、ウサギ由来抗リン酸化
ERK1/2抗体
[phospho-p44/42 MAP kinase (Thr202/Tyr204)、
1000倍希釈、
Cell Signaling Technology, Danvers, MA]を一次抗体として使用した。
Alexa Fluor 488標識抗マウス
IgG (H+L)抗体(
1000倍希釈、
Molecular Probes
) 、および
Alexa Fluor 594標識抗ウサギ
IgG (H+L)抗体(
1000倍希釈、
MolecularProbes
)を二次抗体として使用した。
一酸化窒素(
NO)産生量測定
IFN-γ/LPS
の処置により培養組織から遊離された
NO量を、
NOの代謝物である亜硝酸を指標と
してグリース反応により定量化した。薬物処置終了後、培地上清
100 μLをグリース試薬(
1%sulfanilamide
、
0.1% N-(1-naphthyl)ethylenediamine dihydrochlorideおよび
2.5% phosphoric acidの水溶
液)
100 μLと
22-25°Cで
10分間反応させ、ジアゾカップリングにより生成したアゾ化合物の吸光
度を波長
540 nmで測定した。検量線の作製には亜硝酸ナトリウムを用いた。
半定量的逆転写ポリメラーゼ連鎖反応(
RT-PCR)解析
薬物処置後の中脳切片を
400 μLの
ISOGEN試薬(株式会社ニッポンジーン、
Tokyo, Japan)に 採 取し、 氷冷 下で組 織破砕 して 全
RNAを 抽出し た。
RNA量 は
glyceraldehyde-3-phosphatedehydrogenase
(
GAPDH)の
mRNAを用いて補正した。使用したプライマーを以下に示す。
Bdnf
(
D10938)
Forward primer, 5 -GTGACAGTATTAGCGAGTGGG-3産物
: 213bp Reverse primer, 5 -GGGTAGTTCGGCATTGC-3 TrkB(
M55291)
Forward primer, 5 -CACCAACCATCACATTTCTC-3産物
: 265bp Reverse primer, 5 -ATCTGTCTCTCGTCCTTCCC-3Gapdh
(
M17701)
Forward primer, 5 -GCCAAGGTCATCCATGACAAC-3産物
: 351bp Reverse primer, 5 -AGTGTAGCCCAGGATGCCCTT-3RT-PCR
には
PrimeScript RT-PCR Kit(
RR014A; TaKaRa、
Shiga, Japan)を用いた。
42°C 30分、
95°C 5分および
4°C 5分の条件で逆転写反応を行なった。また
94°C 30秒、
54°C 30秒および
72°C 1.5分を
30サイクル実施して、
Bdnfと
TrkBの
PCR反応を行なった。
Gapdhの
PCR反応には
94°C 30秒、
60°C 30秒および
72°C 1.5分を
20サイクル実施した。
PCR産物は
1.5%アガロースゲル電気泳 動に供して検出した。
In vivo
実験
これまでに報告されている手順にしたがって
31、
8-10週齢の
C57BL/6マウスの片側の中脳黒質 に
LPSを微量注入した。ペントバルビタール(
45 mg/kg)の腹腔内投与によってマウスに麻酔を かけ、リン酸緩衝食塩水(
PBS)に溶解させた
1 μg/μL LPSを片側脳の定位座標に注入した。注入 部位の座標は、
bregmaから
2.8 mm後方、正中線から
1.3 mm外側、硬膜表面から
4.5 mm腹側の 位置とした。
LPS注入側の反対側には同量の
PBSを注入した。注入部位の同定には
1%phthalocyanine blue
溶液(
Sigma)を用いた。
30ゲージの針を用いて溶液
1 μLを
1分以上かけて注 入し、注入後
3分間、針を維持した後、引き抜いた。
Am80は
0.5%カルボキシメチルセルロース 溶液に容量
10 mg/mLで懸濁し、連続した
5日間(
1日一回、午後
4時) 、マウスに
10 mg/kgで経 口投与した。対照群となる動物には溶媒を投与した。動物への
LPS注入は
Am80の投与開始
2日 目の午前
9時から
12時の間で実施した。
LPS
投与後
7日目に、ペントバルビタールでマウスに再度麻酔をかけ、
PBSに続き
4%パラホ ルムアルデヒド溶液で経心灌流固定を行なった。脳を単離し、さらに
2日間
4%パラホルムアル デヒド溶液で固定した後、
4°Cで一晩
20%スクロース溶液に浸漬した。凍結後、中脳冠状切片を
厚さ
30 μmで作製し、
120 μmごとに注入部位付近の
5枚を選び、スライドに封入した。培養中脳
切片で使用した
TH抗体を用いて免疫組織化学を行なった。一次抗体と二次抗体の希釈倍率は、
それぞれ
500倍と
300倍であった。免疫反応性は
Vectastatin Elite ABC Kitにより可視化した。中 脳黒質内の外側部分における
TH陽性細胞数を各切片で計数し、各脳半球について
5枚の切片の 平均値をデータ化した。
統計解析
実験結果は平均値±標準誤差(
SEM)で表記した。統計学的有意性は
one-way ANOVAでの検
定を行なった後、
Student-Newman-Keuls testによって評価した。有意確率が
5%未満の場合を統計
学的に有意であるとした。
実験結果
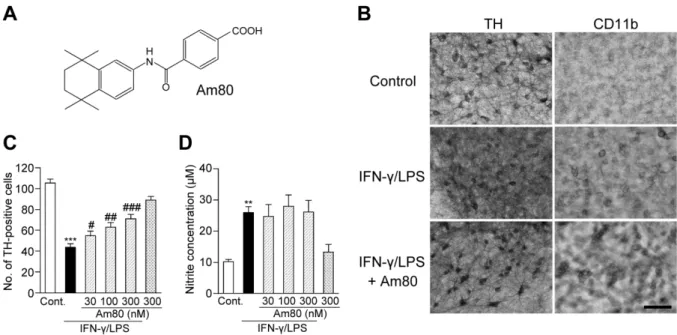
第一節 炎症性応答によるドパミンニューロン死に対するレチノイドの作用
培養中脳切片における炎症性ドパミンニューロン死に対する
RARアゴニスト
Am80の作用 これまでの報告と一致して
32、培養中脳切片への
IFN-γ(
50 ng/mL)の
24時間処置と、それに 続く
LPS(
10 μg/mL)の
72時間処置により、
TH陽性ドパミンニューロン数の有意な減少が観察 された(図
1-1Bおよび
C) 。残存した
TH陽性細胞は神経突起が萎縮しており、対照群に比べて 状態が悪化していた(図
1-1B左) 。
CD11bに対する免疫組織化学の結果から、
IFN-γ/LPS処置に より、ミクログリアの形態も円〜楕円状の活性化型へと変化している様子が観察された(図
1-1B右) 。
RARアゴニストの
Am80(
30-300 nM)を
LPSと
72時間共処置すると、
IFN-γ/LPS誘発性ド パミンニューロン死は
Am80の濃度依存的に抑制された(図
1-1C) 。なお本研究の予備的検討に おいて、
Am80単独の
72時間処置の場合
10 μMまで濃度を上げても
TH陽性細胞数に影響がない ことを確認している。
Am80によるドパミンニューロンの保護作用は、
TH陽性生存細胞数が増加 することに加えて、
TH陽性の樹状突起の形状が良好に維持されていることからも示された(図
1-1Bおよび
1C) 。以後の試験では、ドパミンニューロンに対して明らかな保護作用を示した濃度
である
300 nMを
Am80の処置濃度として使用した。
これまでに、
IFN-γ/LPSによるドパミンニューロン死の誘導は、活性化ミクログリアに発現す る誘導型一酸化窒素合成酵素による
NOの産生に依存することが報告されている
26,32。本研究の 実験条件においても、
IFN-γ/LPS処置によって培地上清中の亜硝酸塩濃度が上昇したことから、
NO
産生量が増加していることが示唆された(図
1-1D) 。しかし、
Am80は
IFN-γ/LPSの誘発する
NO産生量の上昇には何ら影響を及ぼさず(図
1-1D) 、またミクログリアの活性化型への形態変化
に対しても著明な効果を及ぼさなかった(図
1-1B) 。したがって、
Am80のドパミンニューロン保
護作用はミクログリアからの
NO産生の抑制によるものではないことが示唆された。
図
1-1ラット培養中脳切片における
IFN-γ/LPS誘発性ドパミンニューロン死に対する
Am80の作用
(A)Am80 の化学構造式。 (B)培養中脳切片における
TH(左)およびCD11b(右)に対する免疫組織化学の典型画像。対照群の切片(最上段) 、
IFN-γ(
50 ng/mL)の
24時間処置に続き、
72時間
LPS(
10 μg/mL)処置した切 片(中段)および
IFN-γの
24時間処置後に、72 時間
LPSと同時に
Am80(300 nM)処置した切片(下段)。画像 中のスケールバーは
100 μm。 (
Cおよび
D)
IFN-γ/LPS処置によるドパミンニューロン生存細胞数(
C)および培 地中の亜硝酸濃度(
D)の変化に対する
Am80の作用。
Am80は、
IFN-γ(
50 ng/mL)の
24時間処置後の切片に、
72
時間
LPS(
10 μg/mL)と共処置した。各群で検討された切片数は
16-20枚。亜硝酸濃度には
4-5サンプル分の値 を使用し、独立した
4回の試験を実施した。**P < 0.01、***P < 0.001(vs. 対照群) 。#P < 0.05、##P < 0.01、###P
< 0.001
(
vs. IFN-γ/LPS処置群) 。
培養中脳切片における炎症性ドパミンニューロン死に対する
RARアゴニスト
TAC-101および
RXRアゴニスト
HX630の作用
Am80
と同様に選択的
RARアゴニスト活性を有する
TAC-101(
Am555S) の作用を検討した
29,33。
TAC-101は
Am80とは異なる置換基を有する化学構造を特徴とする (図
1-2A) 。
TAC-101(
0.3-3 μM) の処置は、
IFN-γ/LPSによる生存ドパミンニューロンの減少を有意に抑制した(図
1-2B) 。一方で、
TAC-101
は
IFN-γ/LPSによる培地中の亜硝酸濃度の上昇には影響しなかった(図
1-2C) 。
核内受容体である
RARは、一般に
RXRとヘテロ二量体を形成して標的遺伝子の発現を調節す ることから
9、
RXRアゴニストも
RARアゴニストと同様のドパミンニューロン保護効果を示す可 能性が考えられた。そこで、
RXRアゴニストとして
HX630を使用し
34、同様の検討を行なった
(図
1-2D) 。
HX630 (1-10 μM)は、
IFN-γ/LPSにより誘発される生存ドパミンニューロン数の減少 および亜硝酸濃度の上昇のいずれに対しても顕著な作用を示さなかった(図
1-2Eおよび
2F) 。
図
1-2ラット培養中脳切片における
IFN-γ/LPS誘発性ドパミンニューロン死に対する
RARアゴニスト (
TAC-101) および
RXRアゴニスト(
HX630)の作用
(
A)
TAC-101の化学構造式。 (
Bおよび
C)
IFN-γ/LPS処置によるドパミンニューロン生存細胞数(
B)および培 地中の亜硝酸濃度(C)の変化に対する
TAC-101の作用。TAC-101 は、IFN-γ(50 ng/mL)の
24時間処置後の切片 に、
72時間
LPS(
10 μg/mL)と共処置した。各群で検討された切片数は
15-38枚。亜硝酸濃度には
5-9サンプル分 の値を使用し、独立した
3回の試験を実施した。 (
D)
HX630の化学構造式。 (
Eおよび
F)
IFN-γ/LPS処置による ドパミンニューロン生存細胞数(
E)および培地中の亜硝酸濃度(
F)の変化に対する
HX630の作用。
HX630は、
IFN-γ(50 ng/mL)の24
時間処置後の切片に、72 時間
LPS(10 μg/mL)と共処置した。各群で検討された切片数は
14-19枚。亜硝酸濃度には
3-4サンプル分の値を使用し、独立した
3回の試験を実施した。
*P < 0.05、
**P < 0.01、
***P < 0.001(vs.
対照群) 。##P < 0.01、###P < 0.001(vs. IFN-γ/LPS 処置群) 。
培養中脳切片における炎症性ドパミンニューロン死に対する
RARアゴニスト(
Am80)と
RXRア ゴニスト(
HX630)の共処置の効果
先述の通り、
RARの作用に関する古典的メカニズムでは、
RARは
RXRとヘテロ二量体を形成 することにより標的遺伝子のレチノイン酸応答配列上で転写調節因子として機能すると考えられ ている
9。そこで
RARと
RXRの協調性の観点から、
HX630が
Am80のドパミンニューロン保護 作用に対して相乗効果を発揮するか検討した。
Am80(
30 nM)を
IFN-γ処置時から併用し、
HX630(
3および
10 μM)を
Am80および
LPSと
72時間共処置した条件下では、
Am80によるドパミン
ニューロン保護作用が有意に阻害され、予想とは逆の結果が得られた(図
1-3A) 。また、これま での結果と同様に、培地中の亜硝酸濃度には変化が認められなかった(図
1-3B) 。
図
1-3ラット培養中脳切片における
IFN-γ/LPS誘発性ドパミンニューロン死に対する
RARアゴニスト (
Am80) と
RXRアゴニスト(
HX630)の共処置による作用
(
Aおよび
B)
IFN-γ/LPS処置によるドパミンニューロン生存細胞数(
B)および培地中の亜硝酸濃度(
C)の変化
に対する
Am80と
HX630共処置時の作用。Am80(30 nM)は、IFN-γ(50 ng/mL)の
24時間処置時から
72時間の
LPS(
10 μg/mL)処置時も含めて計
96時間処置した。
HX630(
1-10 μM)は
IFN-γと
Am80での
24時間処置後の
切片に、
72時間の
LPSおよび
Am80と共に処置した。各群で検討された切片数は
17-35枚。亜硝酸濃度には
5-8サンプル分の値を使用し、独立した
3回の試験を実施した。
***P < 0.001(
vs.対照群) 。
###P < 0.001(
vs. IFN-γ/LPS処置群) 。+P < 0.05。
In vivo
でのドパミンニューロン変性に対する
Am80の作用
雄性
C57BL/6マウスの片側中脳黒質内に
LPS(
1 μg)を注入し、ドパミンニューロン変性を惹
起した。
LPS注入後
7日目において注入側における
TH陽性細胞数は反対側に比べて有意に減少 した(図
1-4Aおよび
4B) 。
LPS注入の前日から
Am80(
10 mg/kg)を連続
5日間経口投与したと ころ、ドパミンニューロン数の減少は有意に抑制された(図
1-4Aおよび
4B) 。
Am80(
10 mg/kg) の経口投与による全身性の副作用は確認されなかった。
図
1-4 Am80の
in vivoでのドパミンニューロン保護作用
(A)中脳上方に
LPS(PBS 1 μL中に
1 μg)を注入した7日後におけるマウス中脳切片(矢頭は黒質を示す)の
典型画像(左) 。反対側には同量の
PBSを注入した(右) 。マウスに溶媒または
Am80(
10 mg/kg)を
LPS注入前
日から連続して
5日間経口投与した。画像中のスケールバーは
500 μm。(B)LPS 誘発中脳黒質ドパミンニューロ
ン数の減少に対する
Am80の作用を
TH陽性細胞数で評価した。 溶媒投与群は
10例、
Am80投与群は
8例。
*P < 0.05。
第二節 炎症性応答によるドパミンニューロン死に対する Am80 の保護作用機序
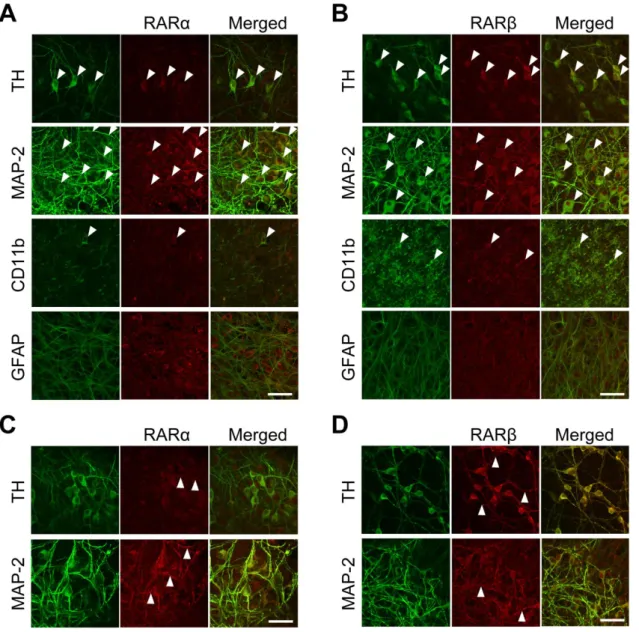
培養中脳切片における
RARαおよび
RARβタンパク質発現
Am80
が標的とする細胞種を同定するため、培養中脳切片において
RARαおよび
RARβと細胞 種特異的マーカーとの免疫組織化学を実施した。
RARαの免疫反応は
TH陽性細胞の大多数で検出 され、全般的なニューロンのマーカーである
MAP-2陽性細胞とも共局在した(図
1-5A) 。一方で、
CD11b
で同定されたミクログリアの一部において
RARαの発現が検出されたものの、大部分のミ
クログリアでは
RARαの発現は認められなかった。また、
GFAP陽性細胞であるアストロサイト にも
RARαの発現は観察されなかった。
RARβについても同様の発現が確認され、
TH陽性ドパミ ンニューロン、
MAP-2陽性ニューロン、および
CD11b陽性ミクログリアの一部に検出されたが、
GFAP
陽性アストロサイトでの発現は観察されなかった(図
1-5B) 。加えて、各
RARアイソフォ
ームの細胞内局在を検討したところ、
RARαが細胞質に顕著に発現していたのに対し、
RARβは細
胞質および核内の両方に発現が認められた。発現細胞種に関する同様の結果は、
Am80(
300 nM)
を
72時間処置した後の切片についても得られた。ただし
Am80処置後の組織では、ドパミンニュ
ーロンを含む神経細胞の神経突起において
RARαおよび
RARβの発現が著明に増加していた(図
1-5Cおよび
5D) 。
図
1-5ラット培養中脳切片における
RAR発現細胞種の同定
RARα
および
RARβに対する免疫反応は細胞種特異的なマーカーに対する免疫反応と共に検出した。ドパミンニ ューロンマーカーとして
TH、全般的なニューロンのマーカーとして
MAP-2、ミクログリアのマーカーとして
CD11b、およびアストロサイトのマーカーとしてGFAP
を使用した。 (A)二重免疫蛍光組織化学における共焦点
画像では、未処置の培養中脳切片における各細胞種マーカー(左) 、
RARα(中央) 、マージ画像を示す。 (
B)二重
免疫蛍光組織化学における共焦点画像では、未処置の培養中脳切片における各細胞種マーカー(左) 、
RARβ(中
央) 、マージ画像を示す。矢頭はいずれの抗体にも陽性であった細胞の典型例を示す。 (
Cおよび
D)
300 nMの
Am80を
72時間処置した際の培養中脳切片における
RARα(C)およびRARβ(D)の典型画像。矢頭はRAR陽性
の神経突起を示す。画像中のスケールバーは
50 μm。
Am80
の神経保護作用への神経栄養因子の関与
レチノイドの中枢神経系に対する作用の一部に、神経栄養因子やそれらの受容体をコードする 複数の遺伝子の発現制御が関わることが報告されている
35。
Am80のドパミンニューロン保護作 用における神経栄養因子シグナルの関与を検討するため、
IFN-γ/LPS誘発性ドパミンニューロン死 に対する代表的な神経栄養因子の作用を検証した。その結果、ニューロトロフィンファミリーの 一つである
BDNF(
3-30 ng/mL)がドパミンニューロンに対して濃度依存的かつ有意な保護作用を 示すことを見出した(図
1-6A) 。次に、生理的条件下および病態時にドパミンニューロンの生存 に主要な役割を果たすとされる神経栄養因子である
GDNFの作用を検討した
36。
GDNF(
3-30ng/mL
) は
IFN-γ/LPS誘発性ドパミンニューロン死に対して有意な効果を示さなかった (図
1-6B) 。
Am80
の神経保護作用における
BDNF関連シグナルの関与の可能性を検証するため、
BDNFお よびその高親和性受容体である
TrkBの
mRNA発現量への影響を検討した。半定量的
RT-PCR解
析から、
Am80(
300 nM)の処置により培養中脳切片における
BDNF mRNA発現量が時間依存的
に増加することが示された(図
1-6C) 。それに対し、同濃度の
Am80は最長
48時間の処置を行な
っても
TrkB mRNA発現量には何ら影響を及ぼさなかった(図
1-6D) 。
図
1-6ラット培養中脳切片における
IFN-γ/LPS誘発性ドパミンニューロン死に対する神経栄養因子の作用
(A および
B)IFN-γ/LPS処置によるドパミンニューロン生存細胞数の減少に対する
BDNF(A)および
GDNF(B)
の作用。
BDNFおよび
GDNFは、
IFN-γ(
50 ng/mL)の
24時間処置後の切片に、
LPS(
10 μg/mL)と共に
72時間 処置した。各群で検討された切片数は
11-21枚。***P < 0.001(vs. 対照群) 。###P < 0.001(vs. IFN-γ/LPS 処置群) 。
(
Cおよび
D)半定量的
RT-PCR解析を用いた、
BDNF(
C)および
TrkB(
D)の
mRNA発現量に対する
Am80の
作用。各
mRNA量は
GAPDH mRNA量によって補正した。培養中脳切片に
Am80(
300 nM)を提示した時間で処
置し、各群には
6枚のスライスを一つのサンプルとして
RT-PCRに供した。グラフ上に対応する
mRNAの典型例
を示した。BDNF および
TrkBの検討にはそれぞれ各群
5サンプル用いた。**P < 0.01(vs. 対照群) 。
Am80
の神経保護作用に対する
BDNFシグナル阻害の影響
Am80
の神経保護作用における
BDNFシグナルの寄与について検討する目的で、
TrkBの特異的 阻害薬である
K252aを用いた。
K252a(
0.3-3 μM)の
Am80との同時添加により、
Am80のドパミ ンニューロン保護作用は濃度依存的に抑制された(図
1-7A) 。次に
TrkBの下流シグナルにある
phosphatidylinositol 3-kinase(
PI3-kinase)の関与を、その阻害薬である
LY294002を用いて検討し た。 これまでの報告で
PI3-kinaseは
BDNFの神経栄養活性に関わる分子とされている
37。
LY294002の処置により、
Am80によるドパミンニューロン生存細胞数の増加が抑制され、
30 μMで統計学的 に有意な抑制作用が認められた(図
1-7B)。次に
ERKシグナルの関与を検討するため、
mitogen-activated protein kinase
(
MAPK)
/ERK kinase阻害薬として
PD98059を使用した。
PD98059(
10-100 μM)の
Am80との共処置によっても、
Am80のドパミンニューロン保護作用が減弱した
(図
1-7C) 。さらに、抗
BDNF中和抗体の作用を検証した。ドパミンニューロンに対する
Am80の保護作用は、抗
BDNF中和抗体の共処置により阻害されたが、対照として使用した
IgYでは明 確な作用が認められなかった(図
1-7D) 。
図
1-7ラット培養中脳切片における
Am80のドパミンニューロン保護作用に対する
BDNFシグナルの影響
Am80によるドパミンニューロン保護作用に対する
TrkB阻害薬
K252a(A)、PI3-kinase 阻害薬
LY294002(B)、
MAPK/ERK阻害薬
PD98059の影響。
K252a、
LY294002および
PD98059と
Am80は、
IFN-γ(
50 ng/mL)での
24時間処置後の切片に、72 時間
LPS(10 μg/mL)と共に処置した。各群で検討された切片数は8-20枚。***P < 0.001
(
vs.対照群) 。
###P < 0.001(
vs. IFN-γ/LPS処置群) 。
+P < 0.05、
++P < 0.01、
+++P < 0.001。 (
D)
Am80によるド
パミンニューロン保護作用に対する抗
BDNF抗体の影響。抗
BDNF中和抗体または
IgY(各
5 μg/mL)と
Am80は、
IFN-γ(
50 ng/mL)での
24時間処置後の切片に、
LPS(
10 μg/mL)と共に
72時間処置した。各群で検討され
た切片数は
13-21枚。***P < 0.001(vs. 対照群) 。###P < 0.001(vs. IFN-γ/LPS 処置群) 。++P < 0.01。n.s.は有意差
なしを示す。
Am80
の作用における
TrkBシグナルの関与をさらに明らかにするため、 培養中脳切片のドパミ ンニューロンにおける
TrkBのリン酸化レベルの変化について検証した。培養中脳切片に
Am80(
300 nM)を
72時間処置したところ、
TH陽性細胞におけるリン酸化
TrkBの免疫反応性が増大
した(図
1-8A) 。また、この
Am80の作用は抗
BDNF中和抗体を同時に処置することによって遮 断された(図
1-8A) 。
ERK
は
TrkBの下流の分子として機能するだけでなく、レチノイドの非ゲノミック作用にも関 与するため
38、次に
Am80による
ERKの活性化について免疫組織化学で検証した。
Am80処置開 始後
24および
72時間の時点でドパミンニューロンにおける
ERK1/2のリン酸化が増大した(図
1-8B) 。また、抗
BDNF中和抗体を
Am80と同時に処置した場合、処置後
24時間ではリン酸化
ERK1/2
の増加が見られたのに対し、処置後
72時間の
ERK1/2リン酸化は抑制された(図
1-8B)。
図
1-8ラット培養中脳切片のドパミンニューロンにおける
TrkBおよび
ERK1/2のリン酸化に対する
Am80の影 響
(
A)抗
BDNF中和抗体(
5 μg/mL)の非存在下または存在下において培養中脳切片に
Am80(
300 nM)を
72時間 処置し、TH(赤)およびリン酸化型
TrkB(緑)陽性細胞を免疫組織化学で検出した共焦点顕微鏡画像。矢頭は両抗体に対する陽性細胞を示す。画像中のスケールバーは
50 μm。 (
B)抗
BDNF中和抗体(
5 μg/mL)の非存在下ま たは存在下において培養中脳切片に
Am80(
300 nM)を
24または
72時間処置し、
TH(赤)およびリン酸化
ERK1/2(緑) 陽性細胞を免疫組織化学で検出した共焦点顕微鏡画像。 矢頭は活性化
ERK1/2を有する
TH陽性細胞を示す。
画像中のスケールバーは
50 μm。考 察
成体の脳内における
RARの役割は、発達期に比較するとほとんど明らかにされていないが、
本研究において
RARが病態時の神経細胞死を制御する可能性が見出された。本章における検討で は、 (
1)
RARの活性化がミクログリアによる炎症性障害から中脳黒質ドパミンニューロンを保護 する一方で、ミクログリアの活性化自体には影響しない、 (
2)臨床適用可能な
RARアゴニストの 経口投与によって
in vivoでの中脳黒質ドパミンニューロン変性が抑制される、および(
3)
RAR刺激は、
BDNFシグナルの活性化を介してドパミンニューロン保護作用を発揮する、という
3つ の新たな知見を得た(図
1-9) 。
図
1-9活性化ミクログリアによるドパミンニューロン死に対する
Am80の保護作用機序
初代培養細胞等を用いた複数の報告において、レチノイドがミクログリアからの
NOや炎症性 サイトカインの産生を阻害することが示されている
11,12。また、パーキンソン病患者の中脳黒質 の病理学的特徴の一つとして活性化ミクログリアの蓄積が知られており、
NOがドパミンニュー ロン変性に関与する可能性も示唆されている
6,7,24。本研究においては、炎症性応答に伴うドパミ ンニューロン変性を再現するため、培養中脳切片への
IFN-γ/LPS処置モデルを使用した
32。本モ デルについては、ドパミンニューロン死の誘導において
NOが主要な役割を果たすことが明らか にされている
26。当該モデルを用いて
RARアゴニストである
Am80の作用を検討したところ、
Am80
は
IFN-γ/LPSの誘導するドパミンニューロン死に対し顕著な抑制作用を示した(図
1-1C) 。
その一方で、
Am80は
NO産生に影響しなかったことから(図
1-1D) 、これまでの報告とは異なり、
Am80