6Li同位体交換によるリチウムイオン電池正極材中
のLi拡散機構の研究
著者
長谷川 源
学位授与機関
Tohoku University
博士学位論文
6
Li 同位体交換によるリチウムイオン電池正極材中の
Li 拡散機構の研究
Study on Li diffusion in cathode materials of
lithium-ion batteries by
6Li isotope exchange
長谷川 源
目次
第
1 章
序論
1
1.1
リチウムイオン電池の歴史とこれから
1
1.2
リチウムイオン電池の起電力
6
1.3
リチウムイオン電池内での様々な電荷の移動過程
8
1.4
リチウムイオン電池における動力学
10
1.4.1
活物質
―電解質界面におけるイオン交換速度
10
1.4.2
活物質内部におけるイオン・電子の輸送
13
第
2 章
リチウムイオン電池と
Li 拡散
15
2.1
拡散係数と電池の大きさ
15
2.2
拡散係数の定義
17
2.3
拡散方程式
18
2.3.1
拡散方程式の解
18
2.3.2
リチウムイオン電池における境界条件
20
2.4
正極における拡散係数測定法とその現状
22
2.4.1
電気化学的手法
22
2.4.2
NMR による手法
24
2.4.3
μ
+SR による手法
26
2.4.4
中性子線による手法
27
2.4.5
SIMS による手法
29
2.5
本研究の目的
30
第
3 章
Li
xMn
2O
4多結晶配向薄膜の拡散現象
32
3.1
緒言
32
3.2
実験方法
35
3.2.1
Li
xMn
2O
4薄膜の作製
35
3.2.2
ステップ同位体交換法
36
3.2.3
PITT 測定
37
3.3
実験結果と考察
38
3.3.1
LMO 薄膜の特性評価
38
3.3.2
Li
xMn
2O
4薄膜における
Li トレーサー拡散係数
40
3.3.3
Li
xMn
2O
4薄膜における
Li 化学拡散係数
50
3.3.4
Li
xMn
2O
4薄膜における
Li トレーサー拡散係数の温度依存性
54
3.4
まとめ
56
第
4 章
Li
xCoO
2多結晶配向薄膜の拡散現象
58
4.1
緒言
58
4.2
実験方法
60
4.2.1
Li
xCoO
2薄膜の作製
60
4.2.2
ステップ同位体交換法
61
4.2.3
DFT 計算
61
4.3
実験結果と考察
62
4.3.1
LCO 薄膜の特性評価
62
4.3.2
Li
xCoO
2薄膜における
Li トレーサー拡散係数
65
4.3.3
Li
xCoO
2薄膜における
c 軸方向拡散モデル
70
4.3.4
トレーサー拡散係数と化学拡散係数の比較
76
4.4
まとめ
78
第
5 章
単結晶
Li
xCoO
2における拡散係数測定
80
5.1
緒言
80
5.2
実験方法
82
5.2.1
電気化学測定と電気化学的
Li 脱離
82
5.2.2
同位体トレーサー測定
82
5.3
実験結果と考察
84
5.3.1
LCO 単結晶の電気化学特性
84
5.3.2
Li
xCoO
2単結晶における
ab 面内のトレーサー拡散係数
88
5.3.3
Li
xCoO
2単結晶における
PITT 測定
92
5.4
まとめ
95
参考文献
97
第1章 序論
1.1
リチウムイオン電池の歴史とこれから
図1.1 化学電池の模式図 電池,特に化学電池は大きく分けると3 つの部材で構成されている.正極,負極,電解質の 3 つである.化学電池は正極―負極間における酸化還元反応を制御することで化学エネルギ ーを電気エネルギーへ変換する.イオンのみが透過できるフィルターとして電解質を使用する ことで電子の流れを外部回路へ取り出すことを可能にしている(図 1.1).その化学電池の中で 近年注目を浴びているのがリチウムイオン電池である. リチウムイオン電池は電気化学反応に Li を用いる二次電池である.この電池はモバイル端 末の急速な発展を支えてきたデバイスであり,また近年,脱炭素社会の実現に向けて重要な 役割を担うと期待されている.その期待度の大きさは 2019 年のノーベル化学賞受賞[1]からも わかる. そもそも Li は電池の構成元素として高いポテンシャルを持っている.一つは非常に低い標 準電極電位(Li/Li+で−3.05 V)を有する点(表 1.1 参照),もう一つは単体において 0.53 g cm−3 と非常に密度が軽い元素という点である.高エネルギー密度な電池を作るアプローチは 1. 反応して得られる電子 1 つ当たりのエネルギーを大きくする. 2. 反応にかかわる原子 1 つあたりの平均質量を小さくする. の2 通りであるため,いかに Li がおあつらえ向きな特性をしているかわかるであろう.しかし, 単体のLi 金属は反応性が高く,水分が存在する環境や大気中では扱えない.したがってこの e− e− e− M+ e−正極
負極
電解質
表1.1 Li と地殻中の主要な元素における標準電極電位[3] 電極反応 25°C における標準電極電位(V) Li++ e−→ Li −3.05 Al3++ 3e−→ Al −1.68 Fe2++ 2e−→ Fe −0.44 Ca2++ 2e−→ Ca −2.84 Mg2++ 2e−→ Mg −2.36 K++ e−→ K −2.93 Ti2++ 2e−→ Ti −1.63 一般的な化学電池の電解質は水溶液である.Li を用いた電池を開発するためにはまず, 適切な非水系電解液を開発しなければならなかった. 今日のリチウムイオン電池につながる非水系電解液の報告は 1958 年の William S. Harris の炭酸プロピレン(Propylene carbonate : PC)にはじまる[2].環状エステル溶媒中の金属メッキ に関する彼の研究の中で PC はアルカリ金属イオンに対して非常に良い電気化学特性を示し た.この発見は徐々に社会へ浸透していき,炭酸エステル系の電解液が今現在もリチウムイオ ン電池の主要な電解液となっている. 実用に耐えうる電解液を獲得したことにより,Li を利用した二次電池の開発は新たな段階へ とうつる.当時,負極材はLi 金属が想定されていたためそれに適合した正極材の探索が重点 的に行われた.M. Stanley Whittingham は TiS2の化学的インターカレーションが全Li 組成(0 < x < 1)によって可能であることに着目し 1976 年に LixTiS2を用いた二次電池の実証を行った [4].電池の起電力は 2.5 V,初期電流密度は 10 mA cm−2 を記録し,𝑥Li + TiS
2⇄ Li𝑥TiS2と
いう単相反応が可逆的に行われることが示された.LixTiS2 を用いた二次電池の開発は Whittingham の共同研究先である Exxon Research and Engineering Company によって 45 Wh の大型セルを作成するところまでいたったが[5],サイクル劣化によって生じる Li デンドライトを 抑えることができずにいた.このデンドライトは正極に到達するほど大きく成長し,短絡や火災 などの危険性があったため,商業的な開発は一度頓挫してしまう. Li デンドライトの問題を回避するため,負極に Li 金属を用いないロッキングチェア型セルの 開発が盛んになった.このセルのコンセプトは酸化還元反応に使用する稼働イオンを正極・負 極の両方でイオンのまま貯蔵するというものである.Li を金属化させないため必然的に動作電 位は金属Li 負極に劣るが,安全性の極めて高い電池が作れると期待された. 負極にインターカレーション材料を使うデメリットを補うほどの高電位の正極材が 1980 年に John B. Goodenough のもとで研究していた水島公一によって報告された.LiCoO2である[6]. LixTiS2と類似した層状構造をもっており,Li/Li+で 4–5 V と非常に高い電位で動作する.この 正極材は動作電位の余裕をもたらしたうえに,合成段階でLi が挿入されていたため負極材探 索の幅を大きく広げた.
様々な負極材の検討が行われる中,1985 年に吉野彰によって当初困難だと思われたグラ ファイト負極を使用したリチウムイオン電池を完成させる[7].もともと吉野はポリアセチレンを対 象に実験を行っていたが,すぐに他の炭素材料に着手し,石油コークスを熱処理して得られる 結晶性の低い人工黒鉛によって溶媒分子の分解を抑制することに成功した.なお,現在は結 晶質のグラファイト負極に対しても還元しづらい溶媒組成が知られている[8].グラファイト負極 はLi/Li+において~ 0.5 V で Li の挿入脱離反応が起きるため,結果として LiCoO 2の非常に高 い動作電位を最大限に生かすことができた.その後,1991 年に Sony がリチウムイオン電池を 初めて商品化するにいたる. リチウムイオン電池が市場に登場した 1990 年代,IT バブルのもと開発・普及が進んだ携帯 電話やノートパソコンなどの小型電子機器によって小型で高容量な蓄電池需要が増大した. それに伴い国内における鉛蓄電池以外の蓄電池の生産も増加した(図 1.2).リチウムイオン 電池は登場当初,コストと安全性において優位であったニッケル水素電池に遅れをとっていた が,日本でのIT バブル崩壊後,一気にシェアが逆転する.その後 2010 年代あたりで生産個 数はおおよそ頭打ちになっている. 0 200 400 600 800 1000 1200 1400 19890 1994 1999 2004 2009 2014 2019 1000 2000 3000 4000 5000
生産量
/
百万個
ニッケルカドニウム ニッケル水素 リチウムイオン ITバブル崩壊 リーマンショック 東日本大震災生産量
/
M
Ah
暦年
ニッケルカドニウム ニッケル水素 リチウムイオン図1.3 車載用電池がリチウムイオン電池の生産に占める量[14, 15] 一方,容量ベースで見てみるとリチウムイオン電池の生産される総容量は増加傾向のままで ある.つまり,単セル当たりの容量が大きくなっていることが言える.この傾向はIT バブル崩壊 を機に生産個数が頭打ちになったニッケル水素電池においても同じである.単セル当たりの 容量増加の需要は,普及時に貢献した小型電子機器によるものではなく,ハイブリット車や電 気自動車などの大型原動機によって生じたものである.実際,リチウムイオン電池において, 車載用途の生産数は年々増加しており,2019 年には 6 割を占めるようになっている(図 1.3). 電力貯蔵技術は電気自動車の他に再生可能エネルギーを用いた発電システムにも必要と されている.再生可能エネルギーの多くは発電出力の制御ができない.それゆえに再生可能 エネルギーは電力需要に合わせて供給することが困難である.特に普及の進んでいる太陽光 発電においては,日の当たる日中しか発電できないため,図 1.4 のように他の発電方式で補う 需要(見かけの需要)の時間変化が急激になるダックカーブ現象が問題になりつつある[16]. 再生可能エネルギーの出力平滑化のためには電力貯蔵の仕組みが重要となる.その一つと して大型のリチウムイオン電池を用いた貯蔵施設が利用されている. リチウムイオン電池はこれからさらに大きな出力・容量を求められるようになる(図 1.5).従来 の小型電子機器の用途においては「いかに小さい空間に大きなエネルギーを詰め込むことが できるか」を重点的に考えられてきた.この考え方は大出力用途においても重要であり続ける であろう.しかし大出力用途の需要に応えていくためには「いかに大きく作れるか」を考えること
も重要になってくる.この問いは「ある程度の時間でどの程度の大きさまでなら電池反応が進 行するのか」という問いに言い換えることができる.この問いに答えるためにはLi+イオンの輸送 特性を知らなければならない.純粋な材料単体における Li の輸送特性はもちろん,「どの程 度の大きさの粒子ならよいのか」や「その粒子をどのように並べたらよいのか」といった広い意 味でのLi 輸送特性の知見が求められている. 図1.4 2020 年 11 月 4 日における九州電力管轄での電力需要と見かけの電力需要[17] 図1.5 大型リチウムイオン電池の活用例.左上:電動タンカー[18], 03時 06時 09時 12時 15時 18時 21時 0 20 40 60 80 100 120
電力需要
/ GW
時刻
太陽光発電
火力,水力,原子力等
見かけの需要
1.2
リチウムイオン電池の起電力
前節でふれたとおり,リチウムイオン電池をはじめとする化学電池は酸化還元反応から取り 出せるエネルギーを電気エネルギーへ変換するデバイスである.もう少し科学的な表現を用い るならば,反応前後のGibbs 自由エネルギー差Δ𝐺から起電力𝐸をとりだしている.すなわち 𝐸 = −Δ𝐺 𝑛𝑒 (1.1) となる.ここで𝑒は電気素量であり,𝑛は反応に寄与した電子数である. ためしに平衡条件を考慮して負極を Li 金属にしたリチウムイオン電池の起電力に関して求 めてみる.ここで電気化学ポテンシャル𝜇̃を次のように定義する. 𝜇̃ = 𝜇 + 𝑧𝑒𝜙 (1.2) 𝜇は化学ポテンシャル,𝑧は符号を含めた電価数,𝜙は外場電位である.外場を含めた熱力 学状態を議論する場合,電気化学ポテンシャルで記述した方が見通しがよい. Li 組成 x におけるリチウムイオン電池のホスト・ゲスト系正極 LixH-電解質と Li 金属負極- 電解質における電極反応は以下のように表せる. 正極: Li𝑥H + d𝑥Li++ d𝑥e−⇄ Li𝑥+d𝑥H (1.3) 負極: Li ⇄ Li++ e− (1.4) 一般にホスト・ゲスト系化合物 LixH の自由エネルギーは Li 組成に依存する.したがって満 たすべき平衡条件は正極: 𝜇̃Lielectrolyte+ + 𝜇̃ecathode− (𝑥) = 𝜇Licathode(𝑥) (1.5)
負極: 𝜇̃Li+ electrolyte + 𝜇̃eLi metal− = 𝜇LiLi metal (1.6) となる.電極間の電位差は配線など同種金属間で測定される.図 1.8 では集電体を同種金属 M,M’であるとしている.同種金属であれば電場のないときの化学ポテンシャル𝜇eM−,𝜇eM−′は等 しい.すなわち,観測される電位差𝐸は式(1.2)より次のようになる[21]. 𝐸 = −𝜇̃e− M − 𝜇̃ e− M′ 𝑒 (1.7) 図1.6 電池内の電気化学ポテンシャルと平衡条件 負極:Li 電解質 正極:LixH 集電体 (M’) 集電体 (M) − − + − −′
平衡状態において正極―集電体間ならびに負極―集電体間における電子の電気化学ポ テンシャルは等しい.よって式(1.5)–(1.7)より観測される起電力𝐸は 𝐸 = −𝜇̃e− cathode− 𝜇̃ eLi metal− 𝑒 = − 𝜇Licathode(𝑥) − 𝜇LiLi metal 𝑒 (1.8) となる.各電極間での平衡条件を考慮した結果,式(1.1)を得ることができた. Li 金属における化学ポテンシャル𝜇LiLi metalは Li xH 正極の Li 組成には左右されない.その ため𝜇LiLi metalをエネルギー原点と考えれば Li 金属負極におけるリチウムイオン電池の起電力 は以下のようになる. 𝐸(𝑥)(vs. Li/Li+) = −𝜇Li cathode(𝑥) 𝑒 (1.9) すなわち,Li 金属負極を用いた電池の起電力は正極の化学ポテンシャルの大きさを観測し ていると考えてよい.仮にホスト・ゲスト系化合物 LixH における自由エネルギーの組成依存性 がLi サイト内の配置エントロピーのみで決まるのであれば 𝐸 = 𝐸0− 𝑘B𝑇 𝑒 ln 𝑥 1 − 𝑥 (1.10) となる[23].ただし𝑘Bはボルツマン定数である.この式は Li サイトにおける Li を還元体,空孔 を酸化体とした Nernst の式と見ることができる.式(1.10)を室温に関してプロットしたものを図 1.7 左に示す.参考のために LixTiS2の放電曲線を図 1.7 右に示した.放電レートが試料に対 して十分に遅ければ平衡状態の電位―組成曲線(OCV 曲線)とほぼ一致する.実際に LixTiS2 の放電曲線は式(1.10)のふるまいと定性的には一致している.さらに精度よく再現する ためにはLi 同士の相互作用や正極内での Li 拡散などを加味しなければならない[22]. 図1.7 室温(25°C)における Nernst の式(左)と LixTiS2の放電曲線(右)[4] 0.0 0.2 0.4 0.6 0.8 1.0 -0.2 -0.1 0.0 0.1 0.2 E - E0 (V) x in LixH
1.3
リチウムイオン電池内での様々な電荷の移動過程
図1.8 リチウムイオン電池内部における電子と Li+イオンの移動過程 リチウムイオン電池内の輸送過程を議論するためにはその内訳をどのようにモデル化する かが重要である.リチウムイオン電池の模式的な内部構造を図 1.8 に示す.リチウムイオン電 池が動作するためには Li+イオンのみが動けばいいというわけではない.電子も動かなければ 電池反応は進行することができない.もっとも単純なモデルにおいても電子とLi+イオンの移動 過程は9 つある. 1. 正極-集電体における接触抵抗 2. 正極内部の電子伝導 3. 負極内部の電子伝導 4. 負極-集電体における接触抵抗 5. 負極内部の Li+イオン拡散 6. 負極-電解質界面の電荷移動抵抗 7. 電解質内部の Li+イオン伝導 8. 正極-電解質界面の電荷移動抵抗 9. 正極内部の Li+イオン拡散 実際のリチウムイオン電池ではさらに多くの要素が存在する.図 1.9 では正極を拡大したと きの模式図を示している.一般的な正極は様々な材料が混ざった合材電極となっている.電 池反応に寄与する正極活物質の他に電子伝導性を担保する導電助剤や粒子同士の接合や 集電体に固定するために混ぜるバインダーなどが存在する.全固体電池であれば電解質を 負極 電解質 正極 界面 界面 集電体 集電体 e -e- e -Li+⑤
④
③
②
①
⑥
⑦
⑧
⑨
Li+バインダーに混ぜ込むこともある.したがって正極内部においても,多数の異種界面や同種界 面が存在し,Li+イオンと電子はそこを超えて伝導しなければ電池反応を起こすことができない. 充放電におけるレート特性や仕様に対するセルのサイズ限界はこれらの移動過程がお互い にかかわりあって決まる.基本的にはその中で最も遅い過程がその電池の特性を制限してい る.幾重にも折り重なった移動過程の中から,電池特性のみで律速過程を判断することは非 常に困難である.それは水の滴る様子から目詰まりを起こしているフィルターを的確に言い当 てることに等しい(図 1.10).出来上がった電池の特性が何によって決まっているかを理解する ためには,なるべく単純なモデル電極を用いることで考慮すべき移動過程を減らし,個々の輸 送過程に注目した測定を行うことが必要である. 図1.9 正極内部での様々な電荷の移動過程 図1.10 律速過程を電池特性のみから読み解く困難さ 負極 電解質 正極 導電助剤等 活物質 Li+ e
-?
律 速 過 程1.4
リチウムイオン電池における動力学
リチウムイオン電池には多くの輸送過程が存在するが,本節ではリチウムイオン電池におけ るLi や電子の移動過程の基本である反応速度論による「界面でのイオン交換」の取り扱いと, 現象論的な「バルクでの電荷輸送」の取り扱いに関して述べる.1.4.1
活物質―電解質界面におけるイオン交換速度
活物質―電解質でのLi+の交換反応を考える.交換反応は次のように定義できる.Li+(electrolyte) + VLi(active material) + e−⇄ Li(active material) (1.11)
電解質のLi+濃度を𝐶 el,内のLi サイト濃度を𝑛Li,活物質内のLi サイト占有率を𝑥としたとき, 反応速度𝑗は以下のようになる[23]. 𝑗c= 𝑘c𝑛Li(1 − 𝑥)𝐶el (1.12) 𝑗a= 𝑘a𝑛Li𝑥 (1.13) ただし𝑘c,𝑘aはそれぞれ,カソード電流(還元電流)とアノード電流(酸化電流)の反応速度 定数とする.反応速度定数𝑘はアレニウス則にしたがうとし,電位差Δ𝜙 = 𝜙act− 𝜙elにおける 活性化エンタルピーを𝐻c(Δ𝜙),𝐻a(Δ𝜙)とすれば 𝑘c(Δ𝜙) = 𝐴cexp [− 𝐻c(Δ𝜙) 𝑘B𝑇 ] (1.14) 𝑘c(Δ𝜙) = 𝐴aexp [− 𝐻a(Δ𝜙) 𝑘B𝑇 ] (1.15) 電極反応が平衡のときの電位差Δ𝜙eqとしたとき,正逆反応速度はともに平衡反応速度𝑗eqと 一致する.このときの𝑗eqは 𝑗eq = 𝐴cexp [− 𝐻c(Δ𝜙eq) 𝑘B𝑇 ] 𝑛Li(1 − 𝑥)𝐶el = 𝐴aexp [− 𝐻a(Δ𝜙eq) 𝑘B𝑇 ] 𝑛Li𝑥 (1.16) となる.したがって exp [𝐻c(Δ𝜙eq) − 𝐻a(Δ𝜙eq) 𝑘B𝑇 ] =𝐴c 𝐴a 1 − 𝑥 𝑥 𝐶el (1.17) となる. 図1.11 活物質―電解質界面におけるイオン交換反応
電解質
活物質
𝑗
c
𝑗
a
Li
+𝜙
el
𝜙
act
ここで交換反応における反応経路を考えてみる.電位差Δ𝜙の変化は小さいものとし,以下 の仮定を設けて平衡電位差の変化と活性化エンタルピーの変化を考える. 1. エネルギー曲線の形の変化は小さい. 2. 電位差Δ𝜙の変化は片側のエネルギー曲線の平行移動として説明される. 3. 活性化障壁を決める反応座標𝜉近傍においてエネルギー曲線は 1 次関数に近似できる. 電位差Δ𝜙 = 0の活性化エンタルピーを𝐻0とするとき𝐻c(Δ𝜙),𝐻a(Δ𝜙)はそれぞれ 𝐻c(Δ𝜙) = 𝐻0+ 𝛼𝑒Δ𝜙 (1.18) 𝐻a(Δ𝜙) = 𝐻0− (1 − 𝛼)𝑒Δ𝜙 (1.19) となる.𝛼は対称因子または透過係数と呼ばれ,通常,1/2 に近い値をとる. 式(1.18),(1.19)を式(1.17)に代入すると以下の関係を得る. exp (𝑒Δ𝜙eq 𝑘B𝑇 ) =𝐴c 𝐴a 1 − 𝑥 𝑥 𝐶el (1.20) また式(1.16)に式(1.18)–(1.20)を代入すれば平衡反応速度𝑗eqは 𝑗eq = (𝑘c0)1−𝛼(𝑘a0)𝛼𝑛Li(1 − 𝑥)1−𝛼𝐶el1−𝛼𝑥𝛼 (1.21) となる.ただし,𝑘c0= 𝐴cexp[−𝐻0⁄𝑘B𝑇],𝑘a0 = 𝐴aexp[−𝐻0⁄𝑘B𝑇]とする. 図1.12 電荷移動過程におけるエネルギー曲線 エ ネ ル ギ ー 反応座標 ξ 活物質 (Φact) Li (Active material) 電解質 (Φel) Li+(Electrolyte) + e -eΔΦ = e (Φact - Φel) αeΔΦ (1 - α)eΔΦ H0 dξ
ここでさらに過電圧𝜂 = Δ𝜙 − Δ𝜙eqに対する反応電流について考える.式(1.12),(1.13)に 式(1.20),(1.21)を代入すればカソード電流密度𝑖cとアノード電流密度𝑖aは以下のように求まる. |𝑖c| = 𝑒𝑗c= 𝑒𝑗eqexp [− 𝛼𝑒𝜂 𝑘B𝑇 ] (1.22) |𝑖a| = 𝑒𝑗a= 𝑒𝑗eqexp [ (1 − 𝛼)𝑒𝜂 𝑘B𝑇 ] (1.23) となる.したがって,正味の電流密度𝑖は 𝑖 = |𝑖a| − |𝑖c| = 𝑖ex{− exp [− 𝛼𝑒𝜂 𝑘B𝑇 ] + exp [(1 − 𝛼)𝑒𝜂 𝑘B𝑇 ]} (1.24) となる.ただし,交換電流密度𝑖ex= 𝑒𝑗eqとしている.式(1.24)を Butler-Volmer の式という. 過電圧𝜂に対する電流密度𝑖の変化を図 1.13 に示す.過電圧が小さい領域では一次関数 的だが,大きくなるほど指数関数的に大きくなる.また,アノード方向とカソード方向における反 応の非対称性も過電圧が大きいほど顕著になることもわかる.過電圧が温度因子に対して十 分小さいとき(𝑒𝜂 𝑘⁄ 𝐵𝑇≪ 1),電流密度は次のように近似できる. 𝑖 ≅ 𝜂𝑑𝑖 𝑑𝜂|𝜂=0 = 𝑒𝑖ex 𝑘B𝑇 𝜂 (1.25) このときの比例係数はオームの法則のアナロジーから電荷移動抵抗𝑟ctと呼ばれている.し たがって,電荷移動抵抗𝑟ctは以下のように定義される. 𝑟ct= 𝜂 𝑖 = 𝑘B𝑇 𝑒𝑖ex (1.26) 図1.13 過電圧と電流密度 -4 -2 0 2 4 -10 -5 5 10
i / i
eqeη / kT
α = 0.3 α = 0.4 α = 0.51.4.2
活物質内部におけるイオン・電子の輸送
荷電粒子は電場と濃度勾配(化学ポテンシャル勾配)を駆動力にして輸送される.荷電粒 子 i において,クロスタームを無視すれば電気化学ポテンシャル勾配∂𝜇̃i/ ∂𝑥を用いて流束𝑗i は次のように表せる[24]. 𝑗i= − 𝜎i 𝑧i2𝑒2 ∂𝜇̃i ∂𝑥 (1.27) ただし,𝜎iは伝導度である.活物質は一般に Li+と電子の両方が動く混合伝導体である.し たがって,活物質内の電流密度𝑖は任意の場所で Li+の流束𝑗 Li+,電子の流束𝑗eを用いれば以 下のように表せる. 𝑖 = 𝑒𝑗Li+− 𝑒𝑗e− (1.28) ここで活物質内部では局所平衡が成り立つとすれば,以下の条件を満たす. 𝜇Li= 𝜇̃e−+ 𝜇̃Li+ (1.29) したがって式(1.27)–(1.29)より,Li+の流速と電子の流速はそれぞれ以下のようになる. 𝑗Li+ = − 1 𝑒2 𝜎e−𝜎Li+ 𝜎e−+ 𝜎Li+ ∂𝜇Li ∂𝑥 + 𝜎Li+ 𝜎e− + 𝜎Li+ 𝑖 𝑒 (1.30) 𝑗e− = − 1 𝑒2 𝜎e−𝜎Li+ 𝜎e−+ 𝜎Li+ ∂𝜇Li ∂𝑥 − 𝜎e− 𝜎e−+ 𝜎Li+ 𝑖 𝑒 (1.31) ここで,第一項は中性のLi による拡散流速𝑗Liを表している. 𝑗Li= − 1 𝑒2 𝜎e−𝜎Li+ 𝜎e−+ 𝜎Li+ ∂𝜇Li ∂𝑥 = −𝐷̃Li ∂𝐶Li ∂𝑥 (1.32) ただし,𝐷̃Liは正味のLi 化学拡散係数,𝐶Liは活物質中のLi 濃度とする.このとき𝐷̃Liは以下 のように表せる. 𝐷̃Li= 1 𝑒2 𝜎e−𝜎Li+ 𝜎e−+ 𝜎Li+ ∂𝜇Li ∂𝐶Li (1.33) 一般に電池に使用される正極材料等では𝜎e− ≫ 𝜎Li+であるため 𝑗Li+ = − 𝜎Li+ 𝑒2 ∂𝜇Li ∂𝑥 = 𝑗Li (1.34) 𝑗e− = − 𝜎Li+ 𝑒2 ∂𝜇Li ∂𝑥 − 𝑖 𝑒 = 𝑗Li− 𝑖 𝑒 (1.35) となる.一般的な正極材料等においてLi+は電子とともに中性Li として拡散している.ここで図1.14 のようなセルの電位差を𝐸0= −𝜇Li0/𝑒から𝐸f= −𝜇Lif /𝑒に変化させたときを考え る.活物質―電解質界面の位置座標を𝑥 = 𝐿,活物質―集電体界面の位置座標を𝑥 = 0とし たとき,Li 金属を参照極にした電位の式(1.9)は次のように一般化される[25]. 𝐸 = −𝜇Li(0) − 𝜇̃Li+(0) + 𝜇̃Li+(𝐿) 𝑒 (1.36) この式は各界面で平衡条件を与えることで得られる.Δ𝐸により過渡電流が流れるとき電気化 学ポテンシャル差𝜇̃Li+(𝐿) − 𝜇̃Li+(0)は式(1.27),(1.30)を用いて次のようになる[26, 27]. 𝜇̃Li+(𝐿) − 𝜇̃Li+(0) = ∫ −𝑒2 𝜎Li+𝑗Li +d𝑥 𝐿 0 = −𝑒𝐿 𝜎e−+ 𝜎Li+𝑖(𝑡) + 𝜎e− 𝜎e−+ 𝜎Li+[𝜇Li(0) − 𝜇Li(𝐿)] (1.37) ただし𝑖(𝑡)は活物質内において電荷保存則が満たされるとし場所によらない.式(1.34), (1.35)と同様に𝜎e− ≫ 𝜎Li+とすれば式(1.36)は式(1.37)より 𝐸f= − 𝐿 𝜎e−𝑖(𝑡) − 𝜇Li(𝐿) 𝑒 (1.38) となる.したがって過渡電流𝑖(𝑡)は 𝑖(𝑡) = −𝜎e− 𝑒𝐿 [𝜇Li(𝐿) − 𝜇Li f ] (1.39) と表せる.また式(1.34),(1.35)より次の関係を得られる. 𝑖(𝑡) 𝑒 = − 𝜎Li+ 𝑒2 ∂𝜇Li ∂𝑥 − 𝑗e−(𝑥, 𝑡) (1.40) 電解質界面では電子は流れないため,式(1.39),(1.40)より次の境界条件を得る. 𝑖(𝑡) 𝑒 = − 𝜎e− 𝑒2𝐿[𝜇Li(𝐿) − 𝜇Lif ] = − 𝜎Li+ 𝑒2 ∂𝜇Li ∂𝑥 |𝑥=𝐿 (1.41) 以上より電子伝導が速い系ではLi の化学ポテンシャル𝜇Liに関する拡散方程式のみで活物 質内の物質輸送を議論できることがわかる. 図1.14 活物質中におけるイオンと電子の輸送 Li 金 属 電解質 活物質 集 電 体
𝑗
Li+𝑗
e− Li+𝑖
e -x = L x = 0第2章 リチウムイオン電池と
Li 拡散
2.1
拡散係数と電池の大きさ
1.4.2 項では活物質内における物質輸送は専ら Li の拡散現象のみで説明ができることを示 した.つまり充電時間に対してどの程度の活物質の大きさが適切か拡散係数から見積もれるこ とがわかる.拡散係数𝐷に対して活物質の大きさ𝐿,拡散時間(充電時間)𝑡diffとしたとき,次元 解析から以下の関係が成り立つ. 𝐷 ≅ 𝐿 2 𝑡diff (2.1) 式(2.1)より拡散時間の等高線図が作れる(図 2.1).製品開発において充電時間の目安とさ れている時間は1 時間である.電池の容量に対して 1 時間で充電できる電流を 1 C ということ がある.拡散係数と活物質サイズの関係において拡散時間が 1 時間になる等高線を太線で, その他代表的な時刻の等高線を破線で示した.製品化を見据えるためには,この太線の等高 線より下の範囲で模索する必要がある.例えば10−10 cm2 s−1の場合では活物質の大きさは6 × 10−4 cm(6 μm)未満でなければならない. 図2.1 拡散係数と活物質の大きさから見積もられる拡散時間の等高線図 10-14 10-12 10-10 10-8 10-6 10-5 10-4 10-3 10-2 10-1L
(
c
m)
D (cm
2s
-1)
10-4 10-2 100 102 104 106 108 1010 1012t
diff(s)
10 m in 1 h 1 da y 1 ye ar 1 m in薄膜電池において活物質の大きさが充放電のレート特性にどのように影響与えるかは, Dudney[28]や松田[29]によって検討されている.薄膜電池は通常のバルク型電池と比べて, 正極・電解質・負極の大きさや形がわかりやすいため,定量的評価を行いやすい.松田らはLi 金属|Li3PO4|LiCoO2で構成された薄膜電池の LiCoO2正極の膜厚を変えてレート特性の違い を評価した.図 2.2 に示すように膜厚が厚くなることで放電電流の大きさに対する使用可能な 容量の減少が顕著になる.これは拡散係数に対して放電電流が大きいとき,電解質界面近傍 において放電が先に進行してしまい,正極内部の Li を利用できていないためにおこる.松田 は電荷の移動過程が正極内の拡散によって律速されていることを仮定し,定電流下における 放電容量の膜厚依存性(図2.3)から LiCoO2正極のおおよそのLi 拡散係数を~10−12 cm2 s−1 と見積もっている. 図2.2 正極の膜厚が 0.32 μm(左)と 6.7 μm(右)の薄膜電池における放電曲線[29] 図2.3 電流密度 5 μA cm−2における放電容量の膜厚依存性[29]
2.2
拡散係数の定義
拡散係数という物理量は現象論に強く根差している.したがって,拡散種によっていくつか 拡散係数を定義し使い分ける必要がある. ⚫ 自己拡散係数𝑫𝐬 𝐟 ある原子が自身の格子中を化学ポテンシャル勾配がない環境で移動していく現象を自己 拡散という.ランダムウォークによる原子の移動は空孔などの欠陥を利用して生じる.自己拡散 係数は平均二乗変位⟨𝑅2(𝑡)⟩から見積もることが可能であり,等方的な媒体中では Einstein-Smoluchowski の関係式より以下のように表せる[30, 31]. 𝐷self= lim𝑡→∞ ⟨𝑅2(𝑡)⟩ 6𝑡 = 𝑑2 6𝜏̅ (2.2) ただし𝑑はジャンプ距離,𝜏̅は原子の平均滞在時間である.この拡散係数は𝜏̅を観測すること で核磁気共鳴(NMR)や準弾性中性子散乱(QENS),ミューオンスピン緩和法(μ+SR)などで 測定が可能である.また,PFG-NMR を用いることで直接的に観測することも可能である. ⚫ 伝導度拡散係数𝑫𝝈 直流伝導度𝜎は𝐶をキャリア濃度,𝑧をキャリアの価数としたとき Nernst-Einstein の関係式より 以下のように表せる[23, 30]. 𝐷𝜎 = 𝑘B𝑇 𝐶𝑧2𝑒2𝜎 (2.3) この関係式を用いて伝導度から定義される拡散係数を伝導度拡散係数𝐷𝜎という.キャリア 同士の相互作用が無視できる場合は𝐷selfと一致する. ⚫ 化学拡散係数𝑫̃ 組成傾斜などによって化学ポテンシャル勾配が生じる.これを駆動力に移動する現象を化 学拡散という.化学拡散流束𝑗̃は絶対易動度𝐵を用いて次のように表せる[23, 31]. 𝑗̃ = −𝐶𝐵𝜕𝜇 𝜕𝑥= −𝐶𝐵 𝜕𝜇 𝜕𝐶 𝜕𝐶 𝜕𝑥 = −𝐷̃ 𝜕𝐶 𝜕𝑥 (2.4) 絶対易動度𝐵と伝導度𝜎には𝜎 = 𝐶𝑧2𝑒2𝐵の関係がある.したがって式(2.3)より 𝐷̃ = 𝐶 𝑘B𝑇 𝜕𝜇 𝜕𝐶𝐷𝜎= Θ𝐷𝜎 (2.5) となる.このときのΘを熱力学因子という.基本的にリチウムイオン電池においては化学拡散 によって反応が進行する. ⚫ トレーサー拡散係数𝑫∗ 化学的に均一な媒体中に同位体元素を置換し拡散させた系から得られる拡散係数をトレー サー拡散係数𝐷∗という.厳密には同位体効果が存在するがその効果は 一般に𝐷 2⁄𝐷1= √𝑚1⁄𝑚2程度であるため多くの場合は無視できる[30].そのような場合,トレーサー拡散係数 ∗2.3
拡散方程式
本節ではLi 拡散の解析に使用する拡散方程式の解とその境界条件に関して述べる.特に 境界条件に関する記述は界面での Li 交換速度と内部の Li 拡散のどちらが律速過程である かを判別するうえで非常に重要なものとなっている.2.3.1
拡散方程式の解
濃度𝐶(𝑥, 𝑡),拡散係数𝐷における拡散方程式は以下のように与えられる. 𝜕𝐶 𝜕𝑡 = − 𝜕𝑗 𝜕𝑥= 𝐷 𝜕2𝐶 𝜕𝑥2 (2.6) ただし,𝐷は濃度によらないとする.電池の拡散解析において有用な平板解を紹介する.ま ずは表面濃度𝐶(𝐿, 𝑡)が𝑡 ≠ 0のとき一定,𝑥 = 0が壁になっている場合,すなわち 𝐶(𝑥, 0) = 𝐶0 (2.7) 𝐶(𝐿, 𝑡) = 𝐶f= const. (𝑡 ≠ 0) (2.8) −𝐷𝜕𝐶 𝜕𝑥|𝑥=0 = 0 (2.9) のとき,解は以下のように与えられる[32]. 𝐶(𝑥, 𝑡) − 𝐶0 𝐶f− 𝐶0 = 1 −4 𝜋∑ (−1)𝑛 2𝑛 + 1cos [ (2𝑛 + 1)𝜋𝑥 2𝐿 ] exp [− (2𝑛 + 1)2𝜋2𝐷𝑡 4𝐿2 ] ∞ 𝑛=0 (2.10) この解は理想的な活物質―電解質界面において電位ステップを与えた場合や,同位体置 換を行った場合などに適用できる.しかし,実際の系では理想的な界面交換が行われず,内 部の拡散と競合することが多々ある.そのとき比例定数𝑎を用いて次のように境界条件をおく. 図2.4 表面濃度が一定のときの拡散プロファイル𝐶(𝑥, 0) = 𝐶0 (2.11) −𝐷𝜕𝐶 𝜕𝑥|𝑥=𝐿 = −𝑎[𝐶f− 𝐶(𝐿, 𝑡)] (2.12) −𝐷𝜕𝐶 𝜕𝑥|𝑥=0 = 0 (2.13) 具体的に比例定数𝑎がどのような物理量と関係するかは 2.3.2 項でふれる.以上の境界条件 から次の解を得ることができる[32, 33]. 𝐶(𝑥, 𝑡) − 𝐶0 𝐶f− 𝐶0 = 1 − ∑ 2Λ cos (𝑏𝑛𝐿 ) exp (−𝑥 𝑏𝑛 2𝐷𝑡 𝐿2 ) (Λ2+ Λ + 𝑏 𝑛2) cos 𝑏𝑛 ∞ 𝑛=0 (2.14) ただし,Λと𝑏𝑛は以下の関係を満たす. Λ =𝑎𝐿 𝐷 (2.15) 𝑏 tan 𝑏 = Λ (2.16) 図 2.5 に規格化時刻𝐷𝑡 𝐿⁄ 2= 0.1における式(2.14)の振る舞いを示す.破線は表面濃度が 一定の時の解である式(2.10)をプロットしたものである.Λが大きくなるほど交換速度が速くなり, 拡散が律速になっていく.解析を行う際,Λが交換律速か拡散律速を見分ける重要なパラメー タであることがわかる. 2
0.0
0.2
0.4
0.6
0.8
1.0
0.0
0.2
0.4
0.6
0.8
1.0
(C
C
0)
/
(C
fC
0)
x / L
Λ = 50
Λ = 5.0
Λ = 1.0
Λ → ꝏ
Dt / L
2= 0.1
2.3.2
リチウムイオン電池における境界条件
2.3.1 項で紹介した一定濃度のソースに接触したときの拡散方程式の解は定電圧における 過渡電流の挙動解析や,同位体置換において有用である.以下,同位体置換時のトレーサ ー拡散における境界条件と,電気化学測定時の化学拡散における境界条件につて考える. ⚫ 同位体置換における境界条件 一般に同位体トレーサー実験では化学組成の変化はない.したがって,注目すべきは平衡 反応速度𝑗eqとなる.電解質中の 6Li と7Li 同位体濃度をそれぞれ𝐶el6Li,𝐶el7Liとし,正極中の同 位体濃度を𝐶ca6Li,𝐶 ca7Liとすれば,アノード方向(電解質→正極)における 6Li の流束𝑗a6Liとカソ ード方向(正極→電解質)における6Li の流束𝑗 c6Liはそれぞれ |𝑗a6Li| = 𝑗eq 𝐶el6Li 𝐶el6Li+ 𝐶el7Li= 𝑗eq𝑁el 6Li (2.17) |𝑗c6Li| = 𝑗eq 𝐶ca6Li(𝐿) 𝐶ca6Li(𝐿) + 𝐶ca7Li(𝐿) = 𝑗eq𝑁ca6Li(𝐿) (2.18) となる.ただし𝑁el6Liは電解質中における 6Li 同位体相対濃度,𝑁 ca6Liは正極中の 6Li 同位体相 対濃度とする.したがって,正味の6Li 同位体の流束𝑗 6Liは以下のようになる.𝑗6Li= −|𝑗a6Li| + |𝑗c6Li| = −𝑗eq[𝑁el6Li− 𝑁ca6Li(𝐿)] (2.19)
式(2.19)より,次の境界条件が成り立つ. 𝑗eq[𝑁el6Li− 𝑁ca6Li(𝐿)] = −𝐷∗ ∂𝐶ca6Li 𝜕𝑥 | 𝑥=𝐿 (2.20) ただし𝐷∗はトレーサー拡散係数である.同位体トレーサー実験において基本的に観測でき る物理量は相対濃度である.したがって右辺の濃度も正極中のLi 濃度𝐶ca6Li+ 𝐶 ca7Li= 𝐶caLiで規 格化する必要がある.同位体相対濃度の拡散方程式において式(2.12)は次のようになる. −𝑗eq 𝐶caLi [𝑁el6Li− 𝑁ca6Li(𝐿)] = −𝐷∗ 𝜕𝑁ca6Li 𝜕𝑥 | 𝑥=𝐿 (2.21) このときの式(2.12)における比例定数𝑎は以下のように表せる[34]. 𝑎 = 𝑗eq 𝐶caLi (2.22) 図2.6 同位体置換における境界条件
電解質
正極
𝑗
c
𝑗
a
6Li+𝑁
el
6Li
𝑁
ac
6Li
7Li+x
=
L
x
= 0
⚫ 電気化学測定における境界条件 1.4.2 項と同様にセルの電位差を𝐸0= −𝜇Li0/𝑒から𝐸f= −𝜇Lif /𝑒に変化させたときを考える. 界面との交換速度を考慮すると過渡電流𝑖(𝑡)が流れる際,過電圧が生じる.したがって式 (1.38)は電荷移動抵抗𝑟ctを用いて次のように表せる. 𝐸f= − ( 𝐿 𝜎e−+ 𝑟ct) 𝑖(𝑡) − 𝜇Li(𝐿) 𝑒 (2.23) 化学ポテンシャルにおける境界条件の式(1.41)は 𝑖(𝑡) 𝑒 = − 1 𝑒2 1 𝐿 𝜎e−+ 𝑟ct [𝜇Li(𝐿) − 𝜇Lif ] = − 𝜎Li+ 𝑒2 ∂𝜇Li ∂𝑥 |𝑥=𝐿 (2.24) となる[33].ここで𝐿 𝜎⁄ e− ≪ 𝑟ctであれば式(1.26)より − 𝑖ex 𝑒𝑘B𝑇 [𝜇Li(𝐿) − 𝜇Lif ] = − 𝜎Li+ 𝑒2 ∂𝜇Li ∂𝑥 |𝑥=𝐿 (2.25) と表せる.正極中のLi 組成濃度𝐶caLiに関する化学拡散の表式にすると − 𝑖ex 𝑒𝑘B𝑇 𝜕𝜇Li 𝜕𝐶caLi [𝐶caLi(𝐿) − 𝐶Lif ] = −𝐷̃ ∂𝐶caLi ∂𝑥 |𝑥=𝐿 (2.26) と記述できる.ただし,𝜇Li0 − 𝜇 Li f は組成依存性を一次関数で近似できる程度に微小であるとす る.したがって式(2.12)における比例定数𝑎は以下のように表せる. 𝑎 = 𝑖ex 𝑒𝑘B𝑇 𝜕𝜇Li 𝜕𝐶caLi (2.27) 交換電流密度𝑖exは平衡反応速度𝑗eqを用いて𝑖ex = 𝑒𝑗eqと表せた.よって,比例定数𝑎は熱 力学因子Θを用いれば次のようになる. 𝑎 =𝑗eq 𝐶caLi 𝐶caLi 𝑘B𝑇 𝜕𝜇Li 𝜕𝐶caLi = Θ𝑗eq 𝐶caLi (2.28) これは式(2.22)で示したトレーサー拡散のときの比例定数𝑎を熱力学因子で補正したもので あり,化学拡散係数𝐷̃と伝導度拡散係数𝐷𝜎(キャリア同士の相互作用を無視したときトレーサ ー拡散係数𝐷∗と等価)の関係式(2.5)と同様の関係となっている.
2.4
正極における拡散係数測定法とその現状
2.4.1
電気化学的手法
電気化学的手法はその簡便さから最もリチウムイオン電池の正極材に対して行われている 手法である.直流伝導度測定に関してはLi+イオン伝導と電子伝導との切り分けが困難なため 報告はない.その代わり PITT(Potentiostasic Intermitted Titration Technique)[35]や GITT (Galvanostatic Intermitted Titration Technique)[36]といった手法が盛んに行われている.PITT は電位ステップをあたえて過渡電流を観察する手法であり,GITT は一定時間電流を流した後 の電位の緩和を観察する手法となっている.その解析式は拡散方程式をもとにしており,例え ばPITT であれば式(1.41)より式(2.14)を電解質界面で微分することで過渡電流𝐼(𝑡)を求めるこ とができる[33]. 𝐼(𝑡) = −𝐹𝑆𝐷̃∂𝐶ca Li ∂𝑥 |𝑥=𝐿 = 2Δ𝑄𝐷̃ 𝐿2∑ Λ2 Λ2+ Λ + 𝑏 𝑛2 exp (−𝑏𝑛 2𝐷̃𝑡 𝐿2 ) ∞ 𝑛=0 (2.29) Λが十分に大きければ Li+イオンの移動過程は正極内の拡散が律速となる.そのとき式 (2.29)は次のように表せる. 𝐼(𝑡) = 2Δ𝑄𝐷̃ 𝐿2∑ exp [− (2𝑛 + 1)2𝜋2𝐷̃𝑡 𝐿2 ] ∞ 𝑛=0 (2.30) 式(2.23)は短時間近似と長時間近似でそれぞれ特有の振る舞いを示す.短時間近似の場 合(𝐷̃𝑡 𝐿⁄ 2≪ 1)は Cottrell の式として知られており 𝐼(𝑡) ≅Δ𝑄 𝐿 √ 𝐷̃ 𝜋𝑡 (2.31) と表せる.一方で長時間近似の場合(𝐷̃𝑡/𝐿2≫ 1)は次のように表せる. 𝐼(𝑡) ≅ 2Δ𝑄𝐷̃ 𝐿2exp (− 𝜋2𝐷̃𝑡 𝐿2 ) (2.32) 式(2.31),(2.32)ともに適切な横軸を用いれば直線となる.リチウムイオン電池の正極材にお ける多くの研究では簡便である近似式を用いた解析を行っている.しかし近似式を用いた解 析には注意が必要である.界面における反応速度が律速の場合(Λ ≪ 1),任意の時間で 𝐼(𝑡) = Δ𝑄𝑎 𝐿exp (− 𝑎𝑡 𝐿) (2.33) の関係が成立する.したがって式(2.33)と同様に指数関数型の減衰を示す.式(2.32)と式 (2.33)の大きな違いは適用できる時間領域である.近似式を用いる場合は測定された時間領 域と得られ𝐷̃が,適用できる時間領域(𝐷̃𝑡/𝐿2 ≫ 1)と矛盾しないか精査する必要がある.
代表的な正極材である LiCoO2と LiMn2O4での報告例を表2.1,表 2.2 に示す.大雑把な 傾向としてバルク電池では値が少し大きく,薄膜系では少し小さめの𝐷̃が測定されている.
表2.1 電気化学的手法による LiCoO2の化学拡散係数
表2.2 電気化学的手法による LiMn2O4の化学拡散係数
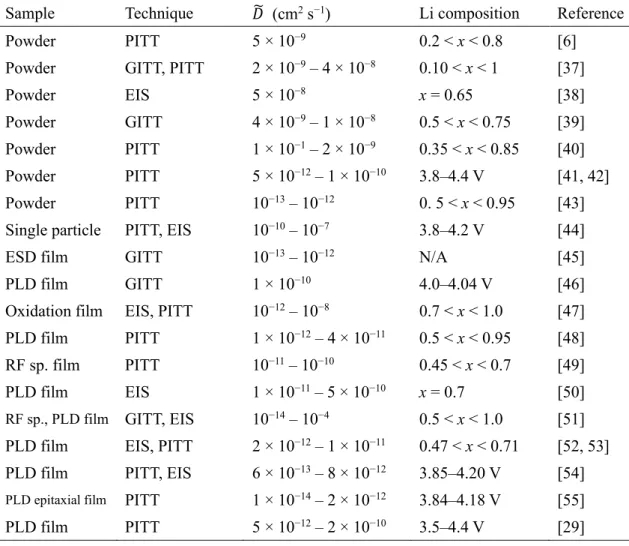
Sample Technique 𝐷̃ (cm2 s−1) Li composition Reference Powder PITT 5 × 10−9 0.2 < x < 0.8 [6] Powder GITT, PITT 2 × 10−9 – 4 × 10−8 0.10 < x < 1 [37] Powder EIS 5 × 10−8 x = 0.65 [38] Powder GITT 4 × 10−9 – 1 × 10−8 0.5 < x < 0.75 [39] Powder PITT 1 × 10−1 – 2 × 10−9 0.35 < x < 0.85 [40] Powder PITT 5 × 10−12 – 1 × 10−10 3.8–4.4 V [41, 42] Powder PITT 10−13 – 10−12 0. 5 < x < 0.95 [43] Single particle PITT, EIS 10−10 – 10−7 3.8–4.2 V [44] ESD film GITT 10−13 – 10−12 N/A [45] PLD film GITT 1 × 10−10 4.0–4.04 V [46] Oxidation film EIS, PITT 10−12 – 10−8 0.7 < x < 1.0 [47] PLD film PITT 1 × 10−12 – 4 × 10−11 0.5 < x < 0.95 [48] RF sp. film PITT 10−11 – 10−10 0.45 < x < 0.7 [49] PLD film EIS 1 × 10−11 – 5 × 10−10 x = 0.7 [50]
RF sp., PLD film GITT, EIS 10−14 – 10−4 0.5 < x < 1.0 [51]
PLD film EIS, PITT 2 × 10−12 – 1 × 10−11 0.47 < x < 0.71 [52, 53] PLD film PITT, EIS 6 × 10−13 – 8 × 10−12 3.85–4.20 V [54] PLD epitaxial film PITT 1 × 10−14 – 2 × 10−12 3.84–4.18 V [55] PLD film PITT 5 × 10−12 – 2 × 10−10 3.5–4.4 V [29]
Sample Technique 𝐷̃ (cm2 s−1) Li composition Reference Powder GITT 7 × 10−9 – 3 × 10−8 0.08 < x < 0.96 [56] Powder PITT 6 × 10−11 – 5 × 10−10 4.05–4.2 V [57] ESD film PITT 6 × 10−11 – 5 × 10−10 4.05–4.2 V [57] PLD film GITT 2.5 × 10−11 4.0–4.04 V [46] ESD film PITT 7 × 10−13 – 5 × 10−11 3.9–4.3 V [58] PLD film GITT 8 × 10−12 – 9 × 10−11 0.5 < x < 1.0 [59] ESD film PITT 2 × 10−12 – 6 × 10−11 3.9–4.3 V [60] PLD film PITT 2 × 10−12 – 8 × 10−11 3.85–4.45 V [61]
2.4.2
NMR による手法
核磁気共鳴(NMR)は元素選択性に優れている.したがって,電気化学的手法で起こりうる キャリアの不確かさがない.拡散測定においてはパルス磁場勾配 NMR(PFG-NMR)[62]が直 接的な手法として知られている.しかし正極材においては電子スピンの大きい遷移金属などの 影響によって緩和時間が短くなることから,拡散時間を長くできない.したがって正極材にお いてPFG-NMR の適用は困難である. NMR は緩和時間やスペクトルの半値幅の温度依存性からジャンプ時間を決めることも可能 である.𝑇2緩和または半値幅は核スピンにおける歳差運動のコヒーレンスを反映している.低 温ではイオンの滞在時間が長くなり,局所磁場の揺らぎによって歳差運動の位相がずれてくる. それが短い緩和時間となりスペクトルは幅広になる.高温になりイオンの滞在時間が短くなると 局所磁場の揺らぎがイオンの移動によって平均化される.その結果コヒーレンスは維持されス ペクトルは先鋭化する.これを「運動による先鋭化(Motional narrowing)」と呼ぶ.半値幅Δ𝑊と ジャンプ滞在時間𝜏̅には次の関係がある. 1 𝜏̅= Δ𝑊 tan [𝜋2 (Δ𝑊Δ𝑊 RL) 2 ] (2.34) ただしΔ𝑊RLはイオンが全く動かないときの半値幅である.中村はこの手法で LixCoO2にお けるLi の滞在時間𝜏̅を見積もっている[63].半値幅の温度依存性は急峻なため先鋭化後の半 値幅とΔ𝑊Rlの中央値が滞在時間𝜏̅を算出するに適している温度点であることが経験的に知ら れている[64].x = 1.0 では 400 K において 10−12 cm2 s−1相当の滞在時間が観測されているが, x = 0.6 まで Li を脱離すると 180 K の低温で 10-12 cm2 s−1相当の滞在時間が観測されている. この結果はLi 脱離によって Li 自己拡散係数が速くなることを示唆している. 図2.8 LixCoO2における半値幅Δ𝑊の温度依存性[63]NMR は周辺環境の異なる同種イオンを分離できる場合がある.化学シフトの違いを固体中 のサイトの違いに帰属できる場合,スペクトル間における核スピンの交換時間をイオン同士の 交換に対応すると解釈できれば,そのサイト間でのジャンプ頻度を見積もることが可能である. Verhoeven[65]は LiMn2O4においてMAS-NMR を行い,それぞれを Li が存在するサイトの違 いによるピークだと帰属した(図 2.9).2D MAS-NMR ではスピン同士の交換時間依存性から 平均滞在時間𝜏̅を決定した.Li サイト(8a)から格子間サイト(16c)へのジャンプと逆向きのジャ ンプではその頻度が異なっており,これは8a サイトと 16c サイトで Li に対するポテンシャルの 深さが異なるためとしている(図2.10).得られた滞在時間𝜏̅から算出される Li 自己拡散係数の 値は345 K で 3 × 10−16 cm2 s−1となっている.
図2.9 LiMn2O4における7Li-MAS-NMR スペクトル(左)と 2D MAS-NMR スペクトル(左). 右図a は 285 K で,右図 b は 380 K で測定した結果[65].
2.4.3
μ
+SR による手法
ミューオンスピン緩和法(μ+SR)とは μ+を試料に打ち込み,トラップされた μ+のスピン緩和を 測定するものである.μ+スピンは崩壊時に放出される陽電子の異方性から測定される.共鳴周 波数が異なるものの緩和時間と拡散に関する考え方はNMR とほぼ同じである.ただし μ+自身 が質量数の小さい水素原子核のように振る舞うため,通常は μ+(疑似的な H+)の拡散現象の 情報が得られる.リチウムイオン電池の正極材などのようにトラップされたμ+よりも十分に速く動 く核スピンが存在する場合は目的のイオンに関する拡散現象をとらえることが期待できる. LiCoO2において,杉山[66, 67]は LixCoO2中のμ+はO2−と安定的な結合状態にあるとし[66, 68, 69],150–300 K の温度範囲において.μ+スピン緩和に寄与するのはLi+の拡散によるもの だとした.観測されたジャンプ頻度𝜈 = 1 𝜏̅⁄ から300 K の Li 自己拡散係数は x = 0.75 で 1 × 10−10cm2 s−1であると報告している[67].杉山の研究グループは Li xMn2O4においてもμ+SR に よるLi 拡散係数測定を試みており,250 K において x = 0.92 での Li 自己拡散係数は 1 × 10−11 cm2 s−1と見積もっている[70]. 図2.11 LixCoO2におけるジャンプ頻度𝜈 = 1/𝜏̅の温度依存性[66] 図2.12 LixCoO2における300 K でのジャンプ頻度𝜈から計算される Li 自己拡散係数[67]. 実線はVan der Ven によるモンテカルロシミュレーションの結果[71].2.4.4
中性子線による手法
内殻電子の数が少ない Li のような軽元素は X 線などでは測定しづらいが,中性子線だと それなりの散乱断面積を持つ.したがって軽元素に着目した構造解析などでは中性子線はし ばしば用いられる. Li に関しては 6Li と 7Li の間には吸収断面積の違いが 104倍程度と非常に大きいという特 徴がある[72].この特性に着目し,高井は中性子線ラジオグラフィー(NR)を用いた同位体トレ ーサー実験をLiMn2O4で行った[73].7LiMn2O4(6Li / 7Li = 0.1 / 99.9)に6LiNO3水溶液(6Li / 7Li = 95 / 5.0)を薄く塗布し 300–800°C 範囲でアニールを行い拡散プロファイルの測定を行っ た(図2.13).600°C 近傍で活性化エネルギーが変化しており,その値は高温側で 1.1 eV,低 温側では0.77 eV であった.600°C より低温での結果を参考に室温へ外挿すると 1.3 × 10−14 cm2 s−1というLi トレーサー拡散係数の値を得ている. 図2.13 天然存在比(6Li / 7Li = 7.5 / 92.5)で規格化した温度 800°C,拡散時間 3 h におけ る6Li 同位体濃度の拡散プロファイル(左)と LiMn 2O4におけるLi トレーサー拡散係数の温 度依存性(右)[73]中性子線には X 線等には見られないユニークな特徴として非干渉性散乱断面積が大きい 原子核が存在するという点があげられる.1H に関しては特に非干渉性が強く,準弾性散乱 (QENS)を用いたダイナミクスの測定が行われている.QENS における拡散係数測定では得ら れたスペクトルを Bragg ピークと原子核の拡散による準弾性散乱ピークに分けられる.すなわ ち,散乱関数𝑆(𝑄, 𝜔)は以下のようになる. 𝑆(𝑄0, 𝜔) = 𝐴(𝑄0)𝛿(𝜔) + 1 𝜋 Γ𝑄0 Γ𝑄20+ ℏ2(𝜔 − 𝜔 0)2 (2.35) 𝛿(𝜔)は Bragg ピークに対応するデルタ関数だが,実際は装置分解能程度の幅を持つ.準 弾性成分はLorentz 関数で表される.半値幅Γ𝑄0は散乱ベクトルの大きさ𝑄0が小さい領域にお いては次のように表せる. Γ𝑄0= 𝑄0 2𝐷 self (2.36) Li に関しても非干渉性散乱断面積を有しており,蒲沢によって LiMn2O4のLi 拡散係数測 定が試みられている[74].400 K において LiMn2O4のLi 自己拡散係数は 1.8 × 10−8 cm2 s−1と 見積もっている. 図2.14 中性子線散乱におけるエネルギースペクトル(b)と準弾性成分の半値幅Γ𝑄0より見積 もられたLi 自己拡散係数(c)[74]
2.4.5
SIMS による手法
同位体トレーサー実験を行ううえで放射光施設や放射線同位体に頼らない測定法の一つ に二次イオン質量分析法(SIMS)があげられる.SIMS は試料表面を一次イオンビームによっ て二次イオンを生成しそれを回収後,質量分析を行う表面分析法である.質量分析法は電 場・磁場を用いて分離する方法と飛行時間によるものがある. リチウムイオン電池の正極に対して SIMS による同位体トレーサー実験行った例として桑田 のLiMn2O4薄膜の実験があげられる[75].PLD で作製した6LiMn2O4薄膜(7Li : 6Li = 5 : 95) を天然存在比のnatLi 電解液(7Li : 6Li = 92.5 : 7.5)に浸漬してイオン交換を行い,拡散対を用 意した.温度範囲は 250–550°C で拡散実験を行い,トレーサー拡散係数の温度依存性を測 定した.得られたLi トレーサー拡散係数は NR で測定された高井の結果[73]と一致した. 図2.15 LiMn2O4薄膜における各温度での同位体拡散プロファイル[75]2.5
本研究の目的
第1 章ではリチウムイオン電池の開発方針が大型化・高出力化へ変わりつつあることを示し, そこにはLi+イオンや電子の移動過程の理解が重要であることを説明した.第2 章ではその中 でも活物質の Li 拡散係数は電池の大きさを決定づけるパラメータであることを説明し,リチウ ムイオン電池の正極材料において拡散係数測定がどのように行われてきたかを代表的な正極 材である LiCoO2 と LiMn2O4 を中心に説明した.2.4 節における拡散係数の測定結果を LiMn2O4に関しては図2.17,LiCoO2に関しては図2.18 にまとめた. 今までの拡散係数測定には大きく 2 つの問題点がある.1 つ目はリチウムイオン電池とうい 系そのものが複雑であるという点である.ここを解決するためにはまずなるべく単純なモデル電 極で測定を行う必要がある.また測定手法そのものも次の条件を満たすことが望ましい.(1)電 子イオン混合導体への適用,(2)界面交換速度およびバルク拡散係数の決定,(3)粒界拡散へ の適用本研究ではこの点を満たす測定法として SIMS による同位体トレーサー実験に着目し た.SIMS は以上の 3 点に関して実際に運用されてきた実績がある[34, 76–79].2 つ目はリチ ウムイオン電池が実際に動作する環境での拡散係数の情報が不足している点である.具体的 には Li 組成依存性に着目した測定が少ない点,室温における測定が少ない点が挙げられる. リチウムイオン電池は基本的には室温で動作するように設計されている.また,充放電の際に は Li+イオンを挿入脱離することで電池反応が進行させている.つまり充放電過程の際に正極 の Li 組成は大きく変化しているのである.それにも関わらす Li 組成依存性に関しては, LiMn2O4では電気化学的手法以外の測定法では化学量論組成でしか議論がなされておらず, LiCoO2においてもせいぜい2 点程度の測定しかなされていない.また,室温での測定が電気 化学的にしか評価されていないのは従来の手法での測定可能な拡散係数の大きさが,室温 における正極のLi 拡散係数の大きさに対応していないためだといえる. 以上の観点から本研究ではLi 組成依存性をはじめとするリチウムイオン電池の動作環境に おける正極材のLi 拡散係数の測定法を確立し,リチウムイオン電池正極材料における拡散現 象の理解を深めることを目的とする. 第 3 章,第 4 章では単相の正極を得ることを目的とし薄膜系における正極材の Li 拡散係 数測定を行った.ここでは新たにステップ同位体交換法を開発することで室温での拡散係数 測定を実現し,代表的な正極材料であるLixMn2O4薄膜とLixCoO2薄膜それぞれに対して Li 組成依存性とその拡散現象の仕組みに関して議論を行った. 第 5 章においては LiCoO2単結晶を用いてその電気化学特性と LiCoO2の結晶構造に基 づくLi 拡散係数に関して議論した.
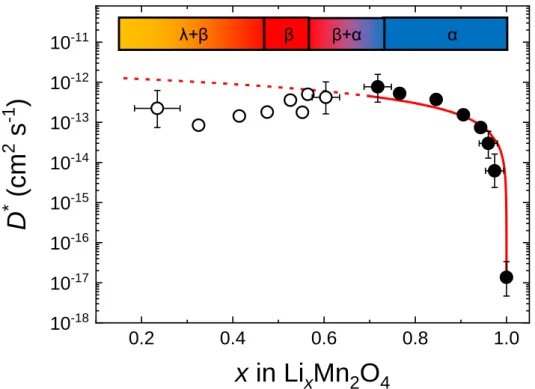
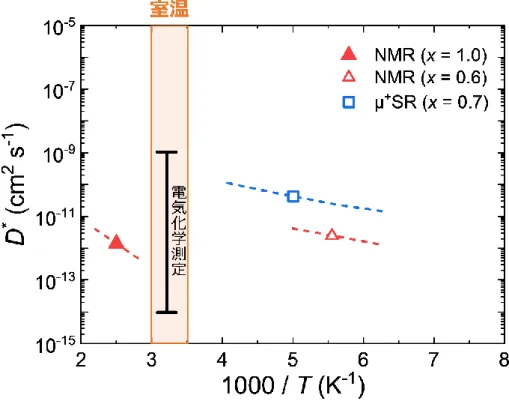
図2.17 LiMn2O4における拡散係数測定の先行研究のまとめ[75]
第3章
Li
xMn
2O
4多結晶配向薄膜の拡散現象
3.1
緒言
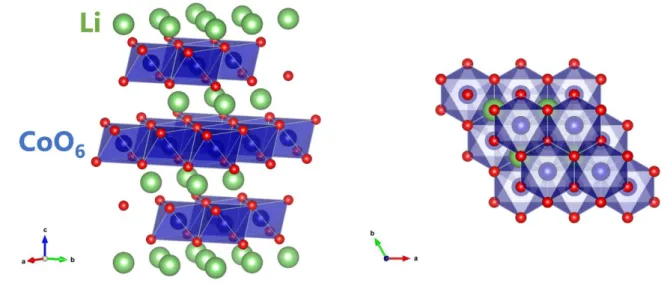
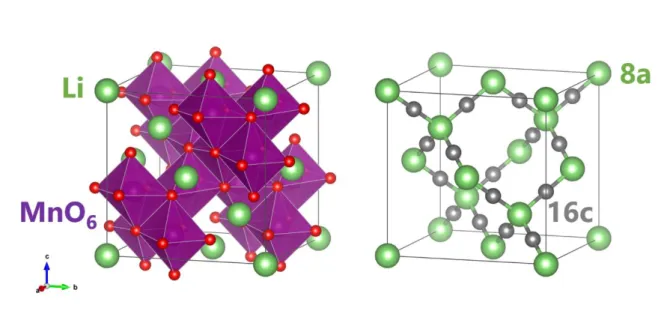
LiMn2O4(LMO)は主要なリチウムイオン電池の正極材料の一つである.構造は立方晶系の スピネル型構造をしており空間群𝐹𝑑3̅𝑚に属する.この材料も 1983 年に Goodenough の研究 グループによって発見されており[80],スピネル型正極の基となる材料である.この材料系は 5V 級正極である LiNi0.5Mn0.75O4やLiCoMnO4などが存在し,リチウムイオン電池の高出力化 への貢献を期待されている材料系の一つである. LMO の Li サイトの並びはいわゆるダイヤモンド構造(8a サイト)をしており,その中間に 16c 格子間サイトが空位で存在している(図3.1).LMO 内で Li+イオンは8a サイトから 16c 格子間 サイトを経由し,隣接した8a-Li 空孔サイトへ移動すると考えられている[81, 82]. Li の挿入脱離にともない LMO は構造相転移を起こすことが知られている[83–85]. LixMn2O4において,格子長の変化からα 相(0.74 < x < 1.0),β 相 (0.44 < x < 0.62),λ 相(x < 0.04)の 3 つに分類されている[84, 85].それぞれの相の間には二相共存領域が存在する.室 温では2 段階のプラトーが観測され(図 3.2),1 段目(~4.0 V)は α 相の Li 固溶領域と α 相― β 相の二相共存領域に対応し,2 段目(~4.15 V)は β 相―λ 相の二相共存領域に対応してい る.λ 相は同じ立方晶である λ-MnO2のLi 固溶体となっている. LMO における Li 拡散係数測定は 2.5 節でふれたように様々な方法で行われている.おも な報告を図3.3 に示す.特に化学量論組成に関しては様々な検討が行われてきたが,実際に 電池が動作する環境はLi 脱離している領域である.Li 拡散係数における詳細な Li 組成依存 性の報告はない.特に室温近傍では化学拡散係数と他の測定法の室温への外挿値は一見 矛盾しているようにも思える.Li+イオンが8a サイトから 16c 格子間サイトを経由して空の 8a サ イトへホッピングするのであれば Li 自己拡散係数は Li 空孔濃度の変化に強く依存するはず である.本研究では特に化学量論組成からLi が脱離される近傍での振る舞いに直目し,従来 の電気化学測定(実際に電池が動作す環境での Li 化学拡散)と化学量論組成における他の 自己拡散がどのように対応するか議論を行う. この目的のためには,Li 脱離した LixMn2O4薄膜において室温でのトレーサー拡散を測定 する必要があるが,室温では Li 拡散が速すぎて検出可能な濃度プロファイルを形成すること ができない.そこで本研究では,室温の薄膜試料でもトレーサー拡散測定を可能にする「ステ ップ同位体交換法」を開発した.この測定法はこれまでの空間プロファイル測定の代わりに, LixMn2O4薄膜中の 6Li 同位体濃度の時間依存性を測定する手法である.この手法を用いて 異なるLi 組成の LixMn2O4薄膜からLi トレーサー拡散係数𝐷∗のLi 組成依存性を求め,PITT で測定した化学拡散係数𝐷̃と比較し議論を行った.図3.1 LiMn2O4の結晶構造(左)と8a Li サイトと 16c 格子間サイト(右)
3.2
実験方法
3.2.1
Li
xMn2O4
薄膜の作製
⚫ PLD による LiMn2O4薄膜の製膜
パルスレーザー堆積法(PLD)により LMO 薄膜を作製した.ターゲットには LiMnO2とMnO が質量比1:1 の混合された LiMn2Oxペレット(豊島製作所)を用いた.基板はPt / Cr コートし
た SiO2ガラス基板(仙台石英硝子製作所)を使用した.ターゲットと基板を真空チャンバー内 に設置後10−4 Pa の圧力まで真空排気した.その後,酸素分圧 20 Pa,基板温度 500°C で製 膜を行った.レーザーは波長193 nm の ArF エキシマレーザーCOMPexPro 205(Coherent 社) を使用し,ターゲット上の照射エネルギー密度を 1 J cm−2,繰り返し周波数を 15 Hz に設定し た.製膜した薄膜は,X 線回折(XRD),Cyclic Voltammetry 測定(CV 測定)及び誘導結合プ ラズマ原子発光分光法(ICP-AES)を用いて基礎的な特性評価を行った.XRD パターンは, CuKα 線を用いた X 線回折装置(リガク,RINT-2100V)によって測定し,スキャン速度 2.0° min−1,測定範囲 10–90°で 2θ スキャンを行った.ICP-AES は Optima 3300XL(Perkin Elmer 社)を用いた. ⚫ 電気化学的手法によるLi 脱離 作用極としてLMO 薄膜を用い,対電極および参照極として Li 金属を用いた三極式ビーカ ーセルを作製しLixMn2O4薄膜中のLi 組成を電気化学的に制御した.電解液は 1 mol L−1の LiClO4 / PC(富山薬品工業)を使用した.ポテンショ/ガルバノスタット(Bio-Logic 社:SP-150 または VMP3)を用いて定電流充電を行うことでLiを脱離した.電位が目標値に到達後,電流 がバックグラウンドレベル(< 200 nA)まで減少するまで電位を維持した.LixMn2O4,LiClO4,Li 金属の同位体比は,天然存在比(natLi:7Li 92.4%,6Li 7.6%)であった. ⚫ 6LiClO 4 / PC 電解液の作製 同位体イオン交換を行うための6LI 電解液の合成を行った.6LiClO 4粉末は文献[86]を参考 に6Li
2CO3(Cambridge Isotope Laboratories 社:6Li 95%,7Li 5%)と HClO4(和光純薬)を遊離 反応させ合成した.6Li 2CO3にHClO4の水溶液をビーカー中で白色固体が溶解するまで滴下 添加し,pH = 7 に達した時点で反応が完了したと判断した.得られた6LiClO 4粉末を200℃で 真空乾燥し,PC(キシダ化学)に 1 mol L−1になるよう溶解した.得られた6LiClO 4電解液の含 水率はKarl Fischer 電量滴定を AQ-300(平沼産業)を用いて測定した.その結果,含水率は 100 ppm 以下であった.また,電気化学的な安定性を評価するため作製した 6LiClO
4電解液 を用いてLMO 薄膜の充放電試験を行い,可逆的な Li 挿入抽出を確認した.
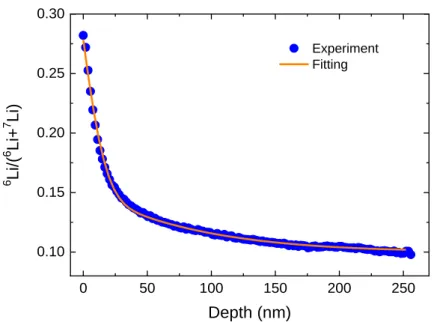
![図 3.3 LMO における様々な拡散係数測定結果[75]](https://thumb-ap.123doks.com/thumbv2/123deta/5888907.1047716/39.892.196.714.138.582/図33LMOにおける様々な拡散係数測定結果75.webp)