学位論文
ニトロピリジンスルフェン酸エステルを基盤とした新規ジスルフィド結合
形成試薬の創製及びそれに基づく環状ペプチド合成法の開発
2021 年 1 月
目 次 略号 3 序論 5 本論 第一章 新規ジスルフィド結合形成試薬の創製 序節 11 第一節 新規ジスルフィド結合形成試薬の着想 15 第二節 3-ニトロ-2-ピリジンスルフェン酸エステル(Npys-OR) 16 誘導体の合成 第三節 Npys-OR 誘導体のジスルフィド形成能の検討 17 第四節 Npys-OR 誘導体 14a-d におけるジスルフィド形成反応の比較 23 第五節 メトキシ誘導体の酸化還元電位測定 26 第六節 固相担持型試薬の合成 27 第七節 固相担持型試薬の機能評価 28 第八節 Npys-OMe の推定反応機構 30 第九節 既知の合成法との比較 32 第十節 小括 34 第二章 メトキシ誘導体を用いた生理活性ジスルフィドペプチドの合成 序節 35
第一節 a-human atrial natriuretic polypeptide の合成 37
総括 71
実験の部 73
引用文献 102
発表文献 107
略号 本論文中に記載した略号を以下に示す。 Ac : acetyl
Acm : acetamidomethyl Ala (A) : alanine
Allocam : allyloxycarbonylaminomethyl Arg (R) : arginine
Asn (N) : asparagine Asp (D) : aspartic acid aq. : aqueous solution Bn (Bzl) : benzyl
Boc : tertiary butyloxycarbonyl
nBu : normal butyl tBu : tertiary butyl
Cys (C) : cysteine 1,2-DCE : 1,2-dichloroethane DCM : dichloromethane DIPCI : diisopropylcarbodiimide DIPEA : N,N-diisopropylethylamine DMF : N,N′-dimethylformamide DMSO : dimethylsulfoxide DTT : dithiothreitol EDT : 1,2-ethanedithiol equiv. : equivalent Fmoc : 9-fluorenylmethoxycarbonyl Gln (E) : glutamic acid
Glu (Q) : glutamine Gly (G) : glycine
HATU : 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
His (H) : histidine
HOAt : 1-hydroxy-7-azabenzotriazole HOBt : 1-hydroxybenzotriazole
Ile (I) : isoleucine IR : infrared spectroscopy Leu (L) : leucine Lys (K) : lysine Me : methyl Met (M) : methionine Mmt : 4-methoxytrityl
NMR : nuclear magnetic resonance Npys : 3-nitro-2-pyridinesulfenyl
Npys-OMe : methyl 3-nitro-2-pyridinesulfenate
Pbf : N-ω-(2,2,4,6,7-pentamethyldihydrobezofuran-5-sulfonyl) PEG : polyethylene glycol
Ph : phenyl
Phe (F) : phenylalanine
iPr : isopropyl
Pro (P) : proline
quant. : quantitative yield rt : room temperature Ser (S) : serine
StBu : tertiary butylthio
STmp : 2,4,6-trimethoxyphenylthio TEA : triethylamine
TFA : trifluoroacetic acid Thr (T) : threonine THF : tetrahydrofuran TIPS : triisopropylsilane Tmob : 2,4,6-trimethoxybenzyl Trp (W) : tryptophan Trt : triphenylmethyl Tyr (Y) : tyrosine Val (V) : valine Xan : xanthenyl
序論 ジスルフィド結合は硫黄原子間で形成される共有結合であり、天然に存在する多く のタンパク質及び一部の生理活性ペプチドでは、当該結合がシステイン(Cys)残基間 で形成される。そして、受容体やリガンド等との分子間相互作用に必須な高次構造を 安定化させ、タンパク質や生理活性ペプチドの生物活性発現に大きく寄与する 1)。さ らに、当該結合により立体構造を剛直化したペプチドは、酵素消化に対して抵抗性を 有する。この様に、ジスルフィド結合は生体分子において重要な構造である。また、 ジスルフィド結合を多数有する天然や生体由来の環状ペプチドは、「Disulfide-rich peptide」と呼称される。具体的には、血糖降下作用を示す「インスリン」2)、抗菌活性 を有する「ディフェンシン」3)、腸管のグアニル酸シクラーゼに作用する「グアニリン」 4) 並びに「耐熱性エンテロトキシン」5)、植物由来の多環状ペプチド「シクロチド」1a, 6) 等を列挙できる。 前述の様に、「Disulfide-rich peptide」は分子内ジスルフィド結合により分子構造が剛 直化されているため、直鎖ペプチドよりも標的分子に対して高い親和性を有している。 そのため、魅力的な創薬シード化合物群であり、これらを基盤とした中分子創薬研究 により医療用医薬品として承認された例もある。その代表例として、「ジコノチド (Prialt ®)」や「リナクロチド(Linzess ®)」は分子内に3 組のジスルフィド結合を持 ち、それぞれ、非オピオイド系重度慢性疼痛治療薬、過敏性腸症候群治療薬として臨 床現場で使用されている(Figure 1)。2000-2016 年の間に承認された Disulfide-rich peptide 由来のペプチド性医薬品を Table 1 に示した1a, 7)。これら以外にも前臨床試験お
よび臨床試験が進行中である1a, 7)。さらに、Disulfide-rich peptide の研究は、医薬品の
リード化合物に留まらず、剛直な立体構造を活かした創薬ツールとしても展開されて いる6a, 8)。
Figure 1. 多環状ジスルフィド構造を持つ承認薬 H CKGKGAKCSRLMYDCCTGSCRSGKC
S
Disulfide-rich peptide の様にジスルフィド結合を含有する環状ペプチドの化学合成で は、所望の高次構造の獲得のために、分子内での正確なジスルフィド結合の構築が重 要となる。ペプチド合成における分子内でのジスルフィド結合の形成手法は「液相法」 と「固相法」の2 つに大別される。 「液相法」はペプチドを含む溶液に酸化剤を加え、ジスルフィド結合を構築する手 法である。一般的には、固相ペプチド合成法(SPPS)により伸長されたペプチドを 固相担体から切り出し、一旦HPLC により精製する。その後、得られた高純度のペ プチドを適切な溶媒に再度溶かし、酸化剤を用いて環化するもので、さらに最終的な 精製過程を経てペプチドを得る手法である(Scheme 1)。本法は SPPS が確立されて 以来、長年研究されており、豊富な実績をもつ手法である。従って、最適な反応条件 を整えれば、非常に有効な手段となる。しかし、潜在的にはいくつかの課題を抱えて いる。1点目は「分子間反応によるoligomer の生成を抑制するために、一般に高希 釈条件下での反応が必要となること」である。2 点目は「難溶性ペプチドへの適用が 厳しいこと」である。即ち、希釈困難な難溶性のペプチドにおいては、液相法でのジ スルフィド結合形成は困難を極める。さらに、3 点目は「高希釈条件下での反応後 に、溶媒の留去が煩雑になりやすいこと」である。これは、ペプチドの回収・精製過 程における収率低下の原因となりやすい。さらに、2 組以上の分子内ジスルフィド結 合を持つペプチドの合成では「多段階の精製工程」が収率低下の原因となる。
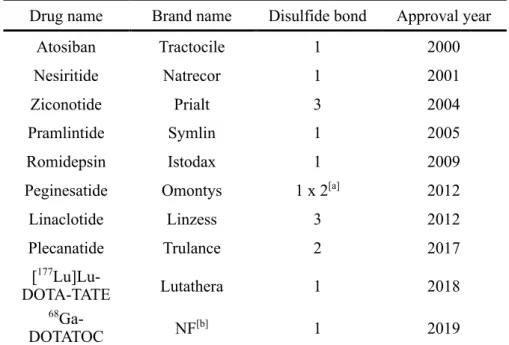
Table 1. Disulfide-rich peptide 由来の承認薬
Drug name Brand name Disulfide bond Approval year Atosiban Tractocile 1 2000 Nesiritide Natrecor 1 2001 Ziconotide Prialt 3 2004 Pramlintide Symlin 1 2005 Romidepsin Istodax 1 2009 Peginesatide Omontys 1 x 2[a] 2012
Linaclotide Linzess 3 2012 Plecanatide Trulance 2 2017 [177 Lu]Lu-DOTA-TATE Lutathera 1 2018 68 Ga-DOTATOC NF[b] 1 2019
にジスルフィド結合を構築できる(Table 2)22)。しかし、Met や Trp 等の酸化反応に敏
感なアミノ酸残基を含むペプチドでは、副反応も観察される22)。
4)スルホキシド-シリル化合物
本法は、TFA 中、シリル化合物(SiCl4, MeSiCl3, トリメチルクロロシラン(TMSCl)
等)とスルホキシド(DMSO, ジフェニルスルホキシド(Ph2SO)等)の共存下におい
て、 Cys 側鎖保護ペプチドを環状の酸化体へ導く23)(Scheme 5)。多様な Cys 側鎖保
護基に対して適用可能であり、反応時間も短いが、Trp 残基側鎖インドール環の塩素 化が同時に進行する23c)。
5)(CF3COO)3Tl
本法は、TFA 中、anisole 共存下において、Cys 側鎖保護ペプチドと (CF3COO)3Tl を
反応させることでジスルフィド結合が形成するものである24)(Scheme 5)。手法 4)と 同様に、多様なCys 側鎖保護基に対して適用可能である有用な酸化剤ではあるが、Trp やMet 残基への副反応も観察された24b)。タリウム及び含有化合物は、経皮または経口 摂取によりタリウム中毒を引き起こすため、使用する際は細心の注意を払わなければ ならない。 Table 2. ヨウ素酸化における反応溶媒及び保護 基と反応速度の関係性22)
solvent Half-time for iodine oxidation
[a]
Trt Acm
MeOH 3-5 sec 1 min AcOH 70-80 sec 40-45 min Dioxane 1 min 1.5-2 h
DMF 25-35 sec 2-3 sec [a] peptide; Boc-Cys(Trt)-Gly-Glu(OtBu)2 or
Boc-Cys(Acm)-Gly-Glu(OtBu)2, conc. of peptide; 5 mM,
前述した酸化剤は「SH 基を保護した Cys 残基間でジスルフィド結合形成を形成で きるか否か」によりグループA(酸化剤 1 及び 2)とグループ B(酸化剤 3-5)に大別 でき、グループA と B の酸化剤を組み合わせて位置選択的なジスルフィド結合の形成 が可能である。 既存のジスルフィド結合形成試薬(酸化剤)は、いずれも素晴らしい特徴を有して おり、それらを活かした手法が研究されてきた。その一方で、いくつかの課題が残存 することも明らかである。これらの課題の克服は、ジスルフィド含有ペプチドの合成 化学のさらなる発展に大きく貢献できると思われる。そこで本論文では、新規ジスル フィド結合形成試薬及びそれを用いた新規環状ペプチド合成法を開発することを目指 した。 本論第一章では、新規試薬の着想から開発達成に至った経緯として、3-ニトロ-2-ピ リジンスルフェニル構造を基盤とした誘導体合成及びジスルフィド形成能の評価を実 施したことについて述べる。第二章では、環状ジスルフィドペプチドに関連した承認 薬や医薬品候補化合物の合成を通じ、第一章で得られた新規試薬の有用性について述 べる。第三章では、更なる効率的合成法の開発を目指し、新規固相環状ジスルフィド ペプチド合成法の開発と自動固相ペプチド合成プロトコールへの適用について述べる。 以下に詳細を論ずる。
Scheme 5. Ph2SO, MeSiCl3又は(CFCOO)3Tl を用いた酸化
Peptide S S Peptide S S (CF3COO)3Tl, TFA, anisole (PG = Acm, Trt, tBu etc.)
PG PG Ph
2SO, MeSiCl3 (PG = Acm, tBu etc.)
本論 第一章 新規ジスルフィド結合形成試薬の創製 序節 序章にて述べた様に、既存の酸化剤を用いたジスルフィド結合形成法には複数の課 題が残っている。そこで、著者は3-ニトロ-2-ピリジンスルフェニル(Npys)基を母核と した新たな酸化剤の開発に着手した。本研究を論ずるにあたり、Npys 基を用いたジス ルフィド結合含有ペプチドの合成に関する研究背景から述べる。 1978 年、Matsueda 及び Aiba により、安定なピリジンスルフェニルハリドとして 3-ニトロ-2-ピリジンスルフェニルクロリド(Npys-Cl, 1)が開発された 25)。その後、
Matsueda らにより、化合物 1 を用いた Boc-Cys(Npys)-OH 及び (Boc-Cys-OH)2の合成
が報告されている(Scheme 6)26)。これは、Npys 保護された SH 基(R 1-S-Npys)と無 保護のSH 基(R2-SH)が共存することで、ジスルフィド交換反応が進行し、所望のジ スルフィド化合物(R1-S-S-R2)が得られることを示唆している(Scheme 7)。即ち、-S-Npys 構造は活性ジスルフィドとして機能し、ジスルフィド結合の構築に利用可能であ ることが明らかとなった。
また、Boc-Cys(Npys)-OH を用いた Lys8-vasopressin(LVP)の合成において、分子内
ジスルフィド交換反応が報告された(Scheme 8)27)。本合成では、ペプチド-樹脂をフ
ッ化水素で処理した際、1 残基目の Cys を保護している Npys 基は残存するが、6 残基 Scheme 6. Npys-Cl を基盤とした Boc-Cys(Npys)-OH 及び (Boc-Cys-OH)2の合成
Scheme 7. 活性ジスルフィドを介したジスルフィド交換反応 Boc Cys OH TEA, DCM rt, 2 h 66% Boc Cys(Npys) OH acetone, 0.1 M phospate buffer (pH 8), rt, 2 h 92% Boc-Cys-OH
目のCys は無保護となり、続く分子内ジスルフィド交換反応を経て、所望の環状ジス ルフィドペプチドが獲得された。さらにChino らは分子間ジスルフィド交換反応を、 a-human atrial natriuretic polypeptide(a-hANP)の逆平行二量体合成へ応用している (Scheme 9)28)。 一方、所属研究室では、Npys-Cl を固相担体に担持させた Npys-Cl 樹脂(2)の開発 に成功し、樹脂2 を基盤とした固相担持型 SH 基選択的ビオチン標識試薬やオリゴア ルギニン導入試薬の開発を達成している(Figure 2)29)。これらの試薬は前述の活性ジ スルフィドに基づく分子間ジスルフィド交換反応を利用したもので、無保護SH 基に 対する選択性を保ちつつ、未反応の試薬や副生成物をろ過のみで除去できるため、高
Scheme 9. Chino らによるa-hANP 逆平行二量体の合成
H SLRRSSCFGGRMDRIGAQSGLGCNSFRY HO YRFSNCGLGSQAGIRDMRGGFCSSRRLS H OH S Acm S Acm SH S Npys
i) disulfide exchange reaction ii) purification iii) I2 oxidation iv) purification H SLRRSSCFGGRMDRIGAQSGLGCNSFRY HO YRFSNCGLGSQAGIRDMRGGFCSSRRLS H OH S S S S
Scheme 8. Matsueda らによる Lys8-vasopressin(LVP)の合成
Figure 3. 固相ジスルフィドライゲーションの概略 A) 活性ジスルフィド形成によるペプチドの固相担持, B) ジスルフィド交換反応 によるペプチドの切り出し, C) 分子内アミド結合形成 H N O Liquid-phase Filtration
A
Peptide A
St-Bu
Wash (H2O X 5)Peptide B
SH
S N O2N N H O S Peptide A Active disulfideC
Peptide A S Peptide B S Disulfide peptide Peptide A S Peptide B SCyclic disulfide peptide
第一節 新規ジスルフィド結合形成試薬の着想 著者は、前節で列挙した手法とそれらの課題を克服すべく、3-ニトロ-2-ピリジンス ルフェニル(Npys)を母格とした新たな酸化剤の開発に着手した。先行研究において用 いられていたNpys-Cl(1)は、塩化物イオン(Cl-)の脱離能が高いため、高い反応性 を持つ。故に、SH 基だけでなく、ヒドロキシ基やアミンを Npys 化する可能性がある 31)。従って、無保護のSH 基間のみのジスルフィド結合形成を目的とした本研究では、 化合物1 は不向きと思われた。そこで、著者は、Matsueda らによって報告された Npys 誘導体である 3-ニトロ-2-ピリジンスルフェン酸エステル(Npys-OR)に着目した (Scheme 10)32)。同氏らは、塩基性条件下において、化合物1 とアルコールを反応さ せ、対応する Npys-OR を獲得していた。しかし、得られた Npys-OR の機能評価は、 化合物6 を用いた H-Pro-Thr-OMe の N 末端 Npys 化及びエステル合成に留まっており、 他の機能に関しては言及していない(Scheme 11)32, 33)。従って、Npys-OR の合成及び 新規な機能評価を通じて、新たな知見が得られると考えた。さらに、Npys-OR を固相 担体に担持させ、固相試薬化することで利便性を高めることが可能であると推測した。
Scheme 10. Matsueda らによる Npys-OR 誘導体合成
第二節 3-ニトロ-2-ピリジンスルフェン酸エステル(Npys-OR)誘導体の合成
まず、本節並びに本章第六節において合成した一連のNpys-OR 及び固相担持型試薬 の共通合成中間体である methyl 6-benzylthio-5-nitronicotinate(11)の合成を実施した。 市販の6-hydroxy-5-nitronicotinic acid(12)を出発原料とし、既存の方法29a)を用い、3
工程で化合物11 を収率 77%にて獲得した。続いて、化合物 11 の Bn 基をアルゴン雰
囲気下、1,2-ジクロロエタン(1,2-DCE)中、pyridine 及び塩化スルフリル(SO2Cl2)に
よりクロロ化することで、methyl 6-chlorothio-5-nitronicotinate(13)を得た29a)。化合物 13 は精製すること無く、次の反応に付した。即ち、化合物 13 を DIPEA 存在下、各種 アルコール又はフェノール(R-OH)と反応させることで、収率 20-81%にて Npys-OR 誘導体(14a-f)を合成した(Scheme 12)。また、誘導体 14a 及び 14f に関しては、結 晶化に成功したため、X 線結晶構造解析からも誘導体の化学構造を確認している。 Scheme 12. 本研究における Npys-OR 誘導体合成 HO O2N N 12 O OH 3 steps, 77% S O2N N 11 (1 eq.) O OMe Bn 1,2-DCE, rt, 1 h SO2Cl2 (2.2 eq.), pyridine (0.5 eq.) S O2N N 13 O OMe Cl or
R-OH, DIPEA (20 eq.) 0 oC to rt, 1.5 h (14b,d,e)
2 steps
第三節 Npys-OR 誘導体のジスルフィド形成能の検討 前節で合成したNpys-OR のうち、最も単純な構造であるメトキシ誘導体 14a を用い て、ジスルフィド形成能を評価した(Table 3)。即ち、モデルペプチドとして、9 残基 のアミノ酸と1 組の分子内ジスルフィド結合からなる下垂体後葉ホルモン oxytocin(15) を選択し、反応条件を検討した。具体的には、還元型 oxytocin(16)溶液に 14a を添 加し、1、3、6 時間後の反応溶液を逆相 HPLC で解析し、ペプチド 15 への酸化反応を 経時的に追跡した。本節にて検討した反応条件は、i)反応溶媒、ii)還元型ペプチド
16 の濃度、iii)誘導体 14a の当量である。また、ペプチド 16 は 20% piperidine/DMF に
よるFmoc 基の脱保護反応と DIPCI、HOBt や HOAt 等の縮合剤によるアミド結合の形 成を繰り返す一般的なFmoc-based SPPS 34) により合成し、逆相HPLC にて分離・精製
後に利用した。
Table 3. 誘導体 14a を用いた oxytocin(15)合成及び反応条件の検討
Entry Solvent (v/v) Conc. of 16 (mM) 14a (eq.) HPLC yield of 15 (%) [a] Oligomer (%) [b] 1 h 3 h 6 h 1 DMF/H2O (1:1) 1 2 55 73 77 16 2 DMF/H2O (1:2) 1 2 59 76 79 16 3 CH3CN/H2O (1:2) 1 2 70 72 86 14 4 CH3CN/H2O (1:3) 1 2 64 71 88 19 5 CH3CN/H2O (1:3) 0.5 2 72 85 87 8 6 CH3CN/H2O (1:3) 0.1 2 63 83 90 2 7 CH3CN/H2O (1:3) 0.1 5 82 92 NT[c] 1[d] 8 CH3CN/H2O (1:3) 0.1 - 0 0 1 0
[a] HPLC yield (%) of oxytocin (15) in the reaction mixture. The yield was calculated by using a calibration curve of 15. [b] Sum of HPLC purities of dimer and trimer after 6 h. [c] Not tested. [d] Sum of HPLC purities of dimer and trimer after 3 h.
i)反応溶媒検討(Table 3, Entry 1-4) DMF または CH3CN と H2O の混合溶媒を用いることで最適な反応溶媒の検討を実施 した。還元型oxytocin(16)を濃度 1 mM になるように溶解した(1 当量, pH 6)。本ペ プチド溶液へ誘導体14a を 2 当量添加し、室温にて反応させた。まず、DMF/H2O = 1:1 混合溶媒中、6 時間後におけるペプチド 15 の HPLC 収率は 77%であった(Entry 1, Figure 4D)。また、DMF/H2O = 1:2 混合溶媒中では HPLC 収率 79%であった(Entry 2, Figure 4H)。一方、有機溶媒を DMF から CH3CN へ変更した CH3CN/H2O = 1:2 混合溶 媒中では、HPLC 収率 86%であり、若干の収率向上が観察された(Entry 3, Figure 5D)。 さらに水の比率を上げたCH3CN/H2O = 1:3 混合溶媒中では、HPLC 収率 88%であった
(Entry 4, Figure 5H)。Entry 1-4 の結果から反応溶媒として、CH3CN と H2O の混合溶
媒の適性が示唆された。しかし、Entry 1-4 において、oxytocin oligomer の副生が確認 された(14-19%)。
ii)還元型ペプチド 16 の濃度検討(Table 3, Entry 5-6)
そこで、oligomer 副生を抑えるべく反応における還元型ペプチド 16 の濃度を検討し た。即ち、ペプチド16 の濃度を 0.5 mM へ低下させたところ、oligomer 副生は 8%と
なり、Entry 4 の結果と比較して半減した(Entry 5, Figure 6D)。さらにペプチド 16 の 濃度 0.1 mM においては、oligomer 副生を 2%まで抑制することに成功した(Entry 6, Figure 6H)。これらの結果から、希釈により還元型ペプチド 16 同士の溶液中での衝突 回数が低減したことで、oligomer 副生が抑制されたと推察される。
iii)メトキシ誘導体 14a の当量検討(Table 3, Entry 7-8)
続いて、Entry 7 に示すように誘導体 14a を 5 当量にて反応を実施したところ、3 時 間で反応が完了し、HPLC 収率が 92%と oxytocin を効率的に得ることに成功した (Figure 7C)。また、oligomer 副生はほとんど検出されなかった(1%)。一方で、誘
導体14a 非存在下におけるペプチド 15 の生成は 1%であることから(Entry 8, Figure
Figure 4. 誘導体 14a を用いた oxytocin(15)合成における条件検討の逆相 HPLC 追 跡(Table 3, Entry 1, 2)
A) before addition of 14a, B) 1 h, C) 3 h, D) 6 h (Table 3, Entry 1), E) before addition of 14a, F) 1 h, G) 3 h, H) 6 h (Table 3, Entry 2); HPLC conditions: a linear gradient starting from 15% to 35% CH3CN in 0.1% aqueous TFA over 20 min at a flow rate of 1.0 mL/min and detection
at 230 nm. *Oxytocin oligomer.
Retention time (min) 10 0 20 A 16 B C D * 16 14a 17
Figure 5. 誘導体 14a を用いた oxytocin(15)合成における条件検討の逆相 HPLC 追 跡(Table 3, Entry 3, 4)
A) before addition of 14a, B) 1 h, C) 3 h, D) 6 h (Table 3, Entry 3), E) before addition of 14a, F) 1 h, G) 3 h, H) 6 h (Table 3, Entry 4); HPLC conditions: a linear gradient starting from 15% to 35% CH3CN in 0.1% aqueous TFA over 20 min at a flow rate of 1.0 mL/min and detection
at 230 nm. *Oxytocin oligomer.
Retention time (min) 10 0 20 A 16 B C D * 16 14a 17
Figure 6. 誘導体 14a を用いた oxytocin(15)合成における条件検討の逆相 HPLC 追 跡(Table 3, Entry 5, 6)
A) before addition of 14a, B) 1 h, C) 3 h, D) 6 h (Table 3, Entry 5), E) before addition of 14a, F) 1 h, G) 3 h, H) 6 h (Table 3, Entry 6); HPLC conditions: a linear gradient starting from 15% to 35% CH3CN in 0.1% aqueous TFA over 20 min at a flow rate of 1.0 mL/min and detection
at 230 nm. *Oxytocin oligomer.
Retention time (min) 10 0 20 A 16 B C D * 16 14a 17
Figure 7. 誘導体 14a を用いた oxytocin(15)合成における条件検討の逆相 HPLC 追 跡(Table 3, Entry 7, 8)
A) before addition of 14a, B) 1 h, C) 3 h (Table 3, Entry 7), D) 0 h, E) 1 h, F) 3 h, G) 6 h (Table 3, Entry 8); HPLC conditions: a linear gradient starting from 15% to 35% CH3CN in 0.1%
aqueous TFA over 20 min at a flow rate of 1.0 mL/min and detection at 230 nm. *Oxytocin oligomer.
Retention time (min) 10 0 20 A 16 B C 16 17
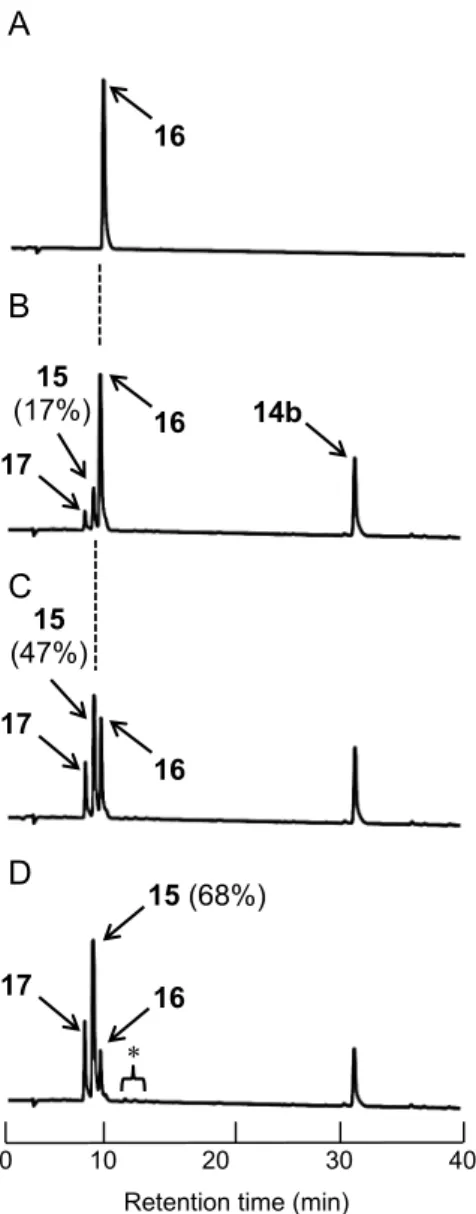
第四節 Npys-OR 誘導体 14a-d におけるジスルフィド形成反応の比較 前節における検討i 及び ii の結果を踏まえ、本章第三節にて合成した 4 種の Npys-OR (14a-d)について、そのジスルフィド形成能を比較した(Table 4)。反応条件とし て、還元型oxytocin(16)を CH3CN/H2O = 1:3(v/v)混合溶媒にて溶解し、0.1 mM ペ プチド溶液(1 当量)を調製した。本ペプチド溶液へ誘導体 14a-d をそれぞれ 2 当量 添加し、室温にて反応を実施した。また、当該反応溶媒への溶解性が乏しいため誘導 体14e-f は比較実験に供することができなかった。反応開始から 6 時間後に逆相 HPLC 分析を実施した。その結果を比較したところ、ペプチド 15 の HPLC 収率はメトキシ
誘導体14a が 90%を示し、最も良好であった(Table 4, Entry 1, Figure
6H)。次いで、n-ブトキシ誘導体14b が 68%(Table 4, Entry 2, Figure 8D)、i-プロポキシ誘導体 14c が
60%であった(Table 4, Entry 3, Figure 9D)。そして、t-ブトキシ誘導体 14d は 29%と反 応性が著しく低下することを確認した(Table 4, Entry 4, Figure 9H)。本結果から、Npys-OR の反応性は、アルコキシ構造の立体障害に主に影響されることが示唆された。一 方で、いずれの誘導体を用いた場合でも、oligomer 及び試薬由来の副生成物 17 以外の 副生成物のピークはHPLC 解析で観察されなかった。さらに、oligomer の副生も 0-2% と低値を示した。本検討より、立体障害の少ないメトキシ誘導体14a がジスルフィド 結合形成において効率的に機能することが明らかとなった。 Table 4. ジスルフィド結合形成能の比較
Entry Compound R HPLC yield of 15 (%)
[a] Oligomer (%) [b] 1 h 3 h 6 h 1 14a Me 63 83 90 2 2 14b n-Bu 17 47 68 1 3 14c i-Pr 15 44 60 1 4 14d t-Bu 4 14 29 0
[a] HPLC yield (%) of oxytocin (15) in the reaction mixture. The yield was calculated by using a calibration curve of 15. [b] Sum of HPLC purities of dimer and trimer after 6 h.
Figure 8. 誘導体 14b を用いた oxytocin(15)合成における条件検討の逆相 HPLC 追跡(Table 4, Entry 2)
A) before addition of 14b, B) 1 h, C) 3 h, D) 6 h (Table 4, Entry 2); HPLC conditions: a linear gradient starting from 15% to 55% CH3CN in 0.1% aqueous TFA over 20 min at a
flow rate of 1.0 mL/min and detection at 230 nm.*Oxytocin oligomer. A
B
C
D
0
Retention time (min)
Figure 9. 誘導体 14c, d を用いた oxytocin(15)合成における条件検討の逆相 HPLC 追跡(Table 4, Entry 3, 4)
A) before addition of 14c, B) 1 h, C) 3 h, D) 6 h (Table 4, Entry 3), E) before addition of
14d, F) 1 h, G) 3 h, H) 6 h (Table 4, Entry 4); HPLC conditions: a linear gradient starting
from 15% to 45% CH3CN in 0.1% aqueous TFA over 20 min at a flow rate of 1.0 mL/min
and detection at 230 nm.
E
F
G
H
Retention time (min)
10 20 0 30 17 16 17 16 15 (4%) 14d 17 16 16 15 (14%) 15 (29%) A B C D
Retention time (min)
第五節 メトキシ誘導体の酸化還元電位測定 メトキシ誘導体 14a の酸化力を検証するため、サイクリックボルタンメトリー 35) による電気化学的な解析を実施した(Table 5)。解析には、0.1 M KCl 含有 20 mM リン 酸緩衝液(pH 6.0):CH3CN(3:1, v/v)を溶媒に用いた。その結果、誘導体 14a にて観 測された還元ピーク電位(Epc)は-0.541 V であり、ジスルフィド結合を有する酸化型 グルタチオン(GSSG)の Epcは-1.550 V であった。これらを比較すると誘導体 14a の Epcが正側であることから、2 組のシステイン残基側鎖 SH 基は誘導体 14a によりジス ルフィドへ酸化され得ることが示唆された。一方、既存の酸化剤として多用されてい るヨウ素(I2)及び Postma, Albericio により 2013 年に報告された N-chlorosuccinimide
(NCS)20) の E pcは、それぞれ0.437 V、-0.175 V であった。これらの値は誘導体 14a の Epcよりもさらに正側である。従って、本節で取り上げた 3 種の酸化剤のなかで、 メトキシ誘導体14a は GSSG に最も近い Epcを示し、温和な酸化剤として機能するこ とが電気化学的手法により示唆された。 Table 5. サイクリックボルタンメトリーを用いた電気化学的解析 Compound Reduction peak potential (Epc)
(V) I2 0.437 NCS -0.175 14a -0.541 GSSG (oxidized glutathione) -1.550
Reduction peak potential (Epc) versus Ag/AgCl was measured by cyclic
voltammetry in CH3CN and 20 mM phosphate buffer (pH 6.0, 1:3, v/v)
第六節 固相担持型試薬の合成 第四節より、最も立体障害が小さいアルコキシ構造を有するメトキシ誘導体14a の 有用性が示唆された。次に、著者は誘導体14a を固相担体上に担持した化合物 18 の合 成に着手した。誘導体14a を固相担体上に担持することで、未反応の誘導体 14a や反 応副生成物であるチオピリドン化合物 17 は固相担体上に保持される。従って、反応 後、それらをろ別することは容易であると推察した。また、ペプチドやタンパク質へ の適用を考慮すると、水系溶媒及び有機溶媒の双方に適応可能な担体を用いる必要が ある。著者は、PEG を基材としたアミノメチル ChemMatrix 樹脂36) を固相担体として 採用した。そして、Npys-Bn 構造を担持させた樹脂 19 を経由し、第二節と同様の反応 経路にて固相担体上で Npys-OMe 構造を構築することにより、固相担持型試薬 18 の 獲得を目指した(Scheme 13)。まず、化合物 11 の加水分解によりカルボン酸 20 を得 た後、HATU、DIPEA による縮合により樹脂 19(Npys-Bn 構造の導入量 = 0.53 mmol/g) を獲得した29b)。本樹脂19 を pyridine 存在下、2% SO
2Cl2/1,2-DCE(v/v)溶液と反応さ
せ、Npys-Cl 樹脂(2)とした後に29b, 30)、室温にて30% DIPEA/メタノール(v/v)溶液
で1.5 時間処理することで、固相担持型試薬 18 を得た(Scheme 15)。固相試薬 18 に おいて、アミノメチルChemMatrix 樹脂に対する Npys-OMe の導入量(= 0.54mmol/g) を元素分析により確認した(Anal. calcd for C161.36H302.72N6O78.68S2: C 53.09, H 8.36, N 2.30;
found : C 52.01, H 8.11, N 2.36)。その結果、固相担体上で Npys-OMe 構造を構築できる ことが明らかになった。 Scheme 13. 固相担持型分子内ジスルフィド形成試薬 18 の合成 MeOH, H2O 0 oC to rt, quant. LiOH・H2O DMF, rt, 3 h Bn S O2N N 20 (4 eq.) O OH
aminomethyl-ChemMatrix® resin (1 eq.), HATU(3.9 eq.), DIPEA (4 eq.)
Figure 11. 固相担持型試薬 18 を用いた oxytocin(15)合成の逆相 HPLC 追跡 A) before addition of resin 18, B) 1 h, C) 6 h, D) 24 h; HPLC conditions: a linear gradient starting from 15% to 35% CH3CN in 0.1% aqueous TFA over 20 min at a flow rate of 1.0
mL/min and detection at 230 nm.*Oxytocin oligomer.
第八節 Npys-OMe の推定反応機構
第九節 既知の合成法との比較
代表的なoxytocin の合成法を Table 6 にまとめ、本手法と比較した。
1) 無保護の Cys 残基間でのジスルフィド結合形成法との比較(Table 6, Entry 2-4) 塩基条件(pH 8)での DMSO 酸化は粗ペプチドの純度が 93%と非常に高いが、反応 完了に24 時間を要する(Entry 2)13c)。一方で、酸性条件でのDMSO 酸化は 2 時間で
反応が完了し、収率は86%である(Entry 3)14b)。また、NCS を用いた当該ペプチドの
合成では、ジスルフィド結合の形成が 15 分で完了し、粗ペプチドの純度は 95%であ った(Entry 4)20)。誘導体14a を用いる本手法(Entry 1)は、Entry3 及び 4 より反応
時間を要するが、3 種の合成法よりも良好な収率で oxytocin 合成を達成している。
第二章 メトキシ誘導体を用いた生理活性ジスルフィドペプチドの合成 序節 前章の oxytocin(15)合成よりメトキシ誘導体 14a が有用なジスルフィド結合形成 試薬である可能性が示唆された。しかし、承認されているペプチド性医薬品はoxytocin (15)と同等若しくは、それ以上に複雑な構造を有する。そこで、本章では、さらに 複雑な環状ジスルフィドペプチドを合成することで、誘導体14a の有用性を示すこと を目標とした。以下に、本章で合成する環状ペプチドを示す。
1)a-human atrial natriuretic polypeptide(a-hANP, 24)38)
a-hANP(24)は 1984 年、Kanagawa と Matsuo により単離・同定されたペプチドホ ルモンであり、ナトリウム利尿ペプチドファミリーに属する。また、第一三共株式会 社より急性心不全治療薬として上市されており、名称は「ハンプ®注射用 1000」であ
る。受容体への結合を介した膜結合型グアニル酸シクラーゼの活性化に基づく細胞内 cGMP の増加により、血管拡張作用および利尿作用等を発現する。28 残基のアミノ酸 からなるペプチド 24 は、分子内に 1 組のジスルフィド結合(Cys7-Cys23)を有する
(Figure 12)。また、Phe8、Met12、Asp13は、受容体との結合を介した活性発現に必須
なアミノ酸残基である 38b)。特に、Met12のスルホキシド体への酸化(Met(O))では、
生理活性の大幅な減弱が知られている38b)。
2)a-conotoxin ImI(a-ImI, 25)
conotoxin 類は、イモガイ属に属する巻貝の分泌液に含まれる神経毒であり、現在ま でに70,000 種類以上が発見されている。また、1 個体から採取される分泌液中には、 50〜200 種の conotxin 類が含有されている39)。a-conotoxin ImI(25)は 1994 年、McIntosh
らによりC. imperialis から単離・同定された conotoxin 類の一種である40)。本ペプチド
はa3b2、a7 及びa9 ニコチン性アセチルコリン受容体と結合し、拮抗剤として機能す る39b)。その活性発現には、Asp5、Pro6、Arg7、Trp10が重要であると報告されている41)。
a-conotoxin ImI の 1 次構造は 12 残基かつ分子内に 2 組のジスルフィド結合(Cys2-Cys8、
Cys3-Cys12)を有する(Figure 13)。
Figure 12. a-human atrial natriuretic polypeptide の一次構造 H
α-human atrial natriuretic polypeptide (α-hANP, 24) SLRRSSCFGGRMDRIGAQSGLGCNSFRY OH
S S
1 7 12
8 13
Figure 13. a-conotoxin ImI の一次構造 H
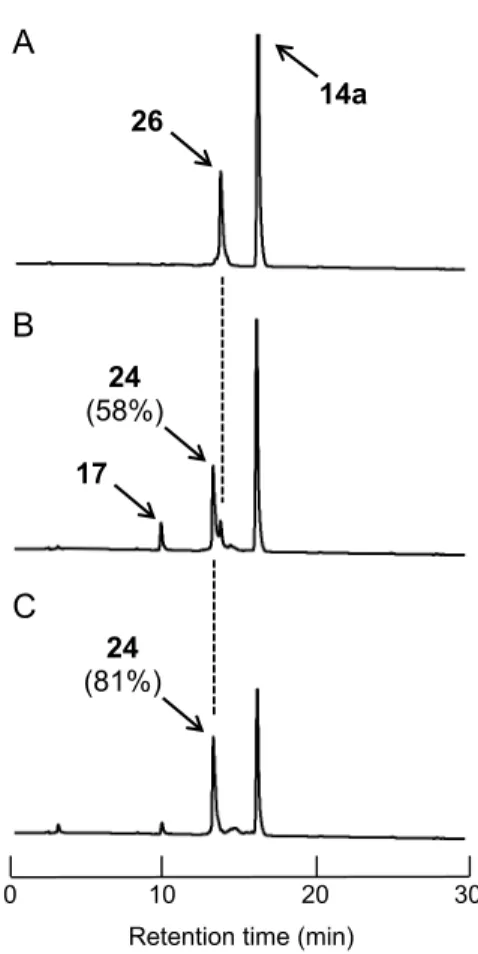
第一節 a-human atrial natriuretic polypeptide の合成 本節では、oxytocin(15)よりも長鎖ペプチドであるa-hANP(24)の合成を実施し た。先ず、第一章第三節で示した一般的なFmoc-based SPPS により還元型a-hANP(26) を単離収率10%で得た。本ペプチドの合成は Fmoc-Tyr(t-Bu)-TrtA-PEG-樹脂(官能基置 換量; 0.44 mmol/g)を用いて、自動固相ペプチド合成機にて実施した(Table 12, 85 頁)。 続いて、前章第三節での反応条件検討結果を参考にし、得られた還元型ペプチド26 を CH3CN/H2O = 1:3(v/v)混合溶媒に溶解し、1 mM のペプチド溶液を調製した。このペ プチド溶液に、5 当量の誘導体 14a を添加することで、7 残基目の Cys(Cys7)と23 残 基目のCys(Cys23)の間で分子内ジスルフィド結合を形成させた(Scheme 16)。本反 応においても、前章と同様に逆相HPLC を用いた解析により、反応を追跡した。その 結果、6 時間後には、ペプチド 26 のピークが 7 割以上減少した(Figure 14B)。さらに 反応を進めたところ、24 時間後にはペプチド 26 の消失が観察された(Figure 14C)。 次に、逆相HPLC による精製を経てペプチド 24 を収率 50%で獲得した。本合成より、 28 残基のアミノ酸からなる比較的高子量ペプチドのジスルフィド結合構築において も、メトキシ誘導体14a は適用可能であることが示された。一方で、既存の合成例は 8 報ある38a, 42-48)。その中でも、2017 年に北条らはヨウ素酸化により Acm 基保護体(27) から収率82%でペプチド 24 を合成している(Scheme 17)48)。収率の面では、Hojo ら の手法には及ばなかったため、今後の検討すべき課題と言える。しかし、本酸化反応 において Met12の酸化は観察されなかった。これは誘導体14a が温和な酸化剤として 機能したためと考えられる。前述したように、ペプチド24 の生理活性には Met 残基 が重要であるため、Met(O)が生じない誘導体 14a によるジスルフィド結合の構築は有 用な手法であると考えられる。
Figure 14. 誘導体 14a を用いたa-hANP(24)合成の逆相 HPLC 追跡
A) 0 h, B) 6 h, C) 24 h; HPLC conditions: a linear gradient starting from 5% to 65% CH3CN in 0.1% aqueous TFA over 30 min at a flow rate of 1.0 mL/min and detection at
230 nm. A B C 17 24 (58%) 14a
Retention time (min)
10 20
0 30
26
24
(81%)
Scheme 17. Hojo らによるペプチド 27 のヨウ素酸化によるa-hANP(24)合成
第二節 a-conotoxin ImI の合成計画 第一章及び前節において、メトキシ誘導体14a を用いて鎖長が異なる 2 種の環状ジ スルフィドペプチドを合成した。それらは分子内に1 組のジスルフィド結合を有して いる。さらに複雑な環状ジスルフィドペプチド合成における誘導体14a の有用性を明 らかにするため、著者は分子内に2 組のジスルフィド結合を有するa-conotoxin ImI(25) の合成を実施した。 分子内にジスルフィド結合を1 組有する前述の oxytocin(15)や a-hANP(24)の 合成では、Cys 残基の側鎖保護基として、酸処理で脱保護可能な Trt 基を採用した。し かし、ペプチド25 配列中の 4 つの Cys を Trt 基で保護した場合、その脱保護で 4 つの 遊離SH 基が生じるため、誘導体 14a による酸化では 3 種のジスルフィド異性体を生 じる可能性を否定できない(Figure 15)。さらに、多くの conotoxin 類において、「globular」 構造をとっている天然物と「ribbon」または「bead」構造をとっている異性体では、生 理活性に大きな差があることが知られているため39, 49)、位置選択的なジスルフィド結 合の形成は非常に重要である。 そこで、著者は2 組のジスルフィド結合のうち、一方を成す Cys 残基側鎖に酸処理 に安定なSH 保護基を、他方に不安定な SH 保護基を導入することで、SH 基の直交型 保護を計画した。即ち、2 残基目の Cys(Cys2)と8 残基目の Cys(Cys8)にTrt 基を、
3 残基目の Cys(Cys3)と12 残基目の Cys(Cys12)に、酸に安定なAcm 基を導入する
ことでメトキシ誘導体14a と既存の酸化剤であるヨウ素を組み合わせた 2 段階の環化
第三節 a-conotoxin ImI における位置選択的ジスルフィド結合形成
まず、前節にて設計した無保護Cys と Acm 保護 Cys が共存する還元型a-conotoxin ImI(28)の合成に着手し、第一章第三節で示した一般的な Fmoc-based SPPS によりペ
プチド28 を単離収率 27%で得た。本ペプチドの合成は Fmoc-NH-SAL 樹脂(官能基置
換量; 0.37 mmol/g)を用いて、自動固相ペプチド合成機にて実施した(Table 12, 85 頁)。 そして、得られたペプチド28 を CH3CN/H2O = 1:3(v/v)混合溶媒に溶解し、1 mM ペ
プチド溶液を調製した。続いて、このペプチド溶液に、2 当量の誘導体 14a を添加し た。即ち、Cys2とCys8間のジスルフィド結合形成反応を誘導体14a による酸化にて実
施した(Scheme 18)。また前章と同様に逆相 HPLC を用いて反応を追跡した(Figure 16)。
その結果、反応開始後4 時間及び 9 時間後の解析から、経時的なペプチド 28 の減 少および所望のペプチド29 の増加が観察された(Figure 16B, C)。その後、27 時間後
では、ペプチド28 が消失した(Figure 16D)。本反応溶液を逆相 HPLC により精製し、
収率61%でペプチド 29 を獲得した(Figure 16E)。本結果から、誘導体 14a は遊離 SH 基間で、位置選択的に分子内ジスルフィド結合を形成することがわかった。尚、ペプ
チド 28 は酸化されやすい Trp 残基を有するが、影響を受けなかった。反応性の高い
Npys-Cl(1)を用いると、一般に側鎖インドール環 2 位で Npys 化を受ける 50)。しか
し、本法ではNpys 化は観察されなかった。これは Npys-Cl(1)とメトキシ誘導体 14a の反応性の違いに起因すると考えられる。
Scheme 18. 誘導体 14a による Cys2 - Cys8間のジスルフィド結合形成 H 28 (1 eq., 1 mM) GCCSDPRCAWRC SH SH S NH2 S CH3CN : H2O = 1 : 3 pH 6, rt, 27 h HPLC purification 61% 14a (2 eq.) H 29 GCCSDPRCAWRC S S S NH2 S MeO S O2N N O OMe
Figure 16. Cys2-Cys8間のジスルフィド結合形成反応の逆相HPLC 追跡
A) 0 h, B) 4 h, C) 9 h, D) 27 h, E) purified peptide 29; HPLC conditions: a linear gradient starting from 5% to 65% CH3CN in 0.1% aqueous TFA over 30 min at a flow rate of 1.0
mL/min and detection at 230 nm. B C D E A 28 17 14a 28 29 (42%) 28 17 29 29 (59%) 29 (77%)
Retention time (min)
10 20
続いて、ヨウ素によりAcm 基で保護されている Cys3とCys12間のジスルフィド結合 形成を行った(Scheme 19)。即ち、合成したペプチド 29 の 50%酢酸水溶液(0.1 mM ペプチド溶液)を調製し、そこへ 0.1 M のヨウ素/メタノール溶液を添加することで、 ヨウ素酸化反応に付した。反応は1 時間で完了したため(Figure 17B)、1 M アスコル ビン酸ナトリウム水溶液の添加により反応を停止させた。得られた反応溶液を逆相 HPLC にて精製し、収率 50%でa-conotoxin ImI(25)の合成を達成した。また、本反応 過程で、既存のジスルフィド結合は特段影響を受けなかった。合成したペプチド25 が
所望のCys 残基間(Cys2-Cys8とCys3-Cys12)でジスルフィド結合を形成されているこ
とを確認するため、標準品(ペプチド研究所より購入)と逆相HPLC における保持時 間を比較した。その結果、Figure 18 に示すように、合成品と標準品の保持時間は一致 した。また、これらの混合溶液を分析した結果、単一ピークのみ検出された。このこ とは、位置選択的なジスルフィド結合が目的通り構築されたことを示唆するものであ る。一方で、当該ペプチドの位置選択的な合成例は3 報あるが、反応条件や単離収率
Scheme 19. ヨウ素酸化による Cys3-Cys12間のジスルフィド結合形成
Figure 17. Cys3-Cys12間のジスルフィド結合形成反応の逆相HPLC 追跡
A) 0 h, B) 1 h; HPLC conditions: a linear gradient starting from 5% to 65% CH3CN in
0.1% aqueous TFA over 30 min at a flow rate of 1.0 mL/min and detection at 230 nm. 50% AcOH aq. rt, 1 h HPLC purification 50% 0.1 M I2/MeOH (10 eq.) H GCCSDPRCAWRC S S S NH2 S α-conotoxin ImI (25) H 29 (1 eq., 0.1 mM) GCCSDPRCAWRC S S S NH2 S Acm Acm A B
Retention time (min)
10 20
0 30
29
等の詳細な記載はされていなかった40b, 41b, 51)。従って、メトキシ誘導体14a と他の酸
化法を組み合わせたジスルフィド結合構築法は新たな環状ペプチド合成法の提供に繋 がるものである。
Figure 18. 本研究で合成したペプチド 25 と標準品の HPLC 解析
A) purified peptide (25), B) commercial peptide 25 (Peptide Institute, Inc.,), C) co-injection; HPLC conditions: a linear gradient starting from 5% to 65% CH3CN in 0.1%
aqueous TFA over 30 min at a flow rate of 1.0 mL/min and detection at 230 nm. A
B
C
Retention time (min)
10 20
0 30
第四節 小括 第二章では、メトキシ誘導体14a の有用性をさらに検討するために、前章よりも複 雑な2 種の環状ジスルフィドペプチドの合成に挑戦し、先ず 28 残基からなり、分子内 に1 組のジスルフィド結合を持つa-hANP(24)の合成を達成した(単離収率; 50%)。 本合成においてMet 残基の酸化は観察されなかった。続いて、分子内に 2 組のジスル フィド結合を持つa-conotoxin ImI(25)の合成も達成した。1 組目のジスルフィド結合 はメトキシ誘導体14a により構築した(単離収率; 61%)。その際、Trp 残基インドール
第一節 既存法とそれらの課題及び新規固相合成法開発の意義 序章でも述べた様に、固相担体上でのジスルフィド結合形成は、固相担体における 擬希釈効果により分子内での環化反応が進行しやすく、さらに試薬や溶媒の除去も簡 便に行える。以下に Fmoc 法に基づく固相ジスルフィドペプチドの既存の合成方法を 列挙する。 1)無保護 Cys 残基間でのジスルフィド結合形成(Scheme 20)54-61) 低濃度の酸による処理で脱保護可能なXan、Mmt、Tmob 基または、還元剤により脱 保護可能なStBu、STmp 基を Cys 側鎖 SH 基の保護基として導入した手法である。こ れらの保護基は、いずれも脱樹脂を伴わず、選択的に SH 基から脱保護される。その 結果、生じた1 組の無保護 Cys 残基を酸化剤によりジスルフィド架橋することで、固 相上で環化構造を構築できる。本反応で用いられる酸化剤は、DMSO 58)、フェリシア ン化カリウム54, 56)、四塩化炭素55)、四臭化炭素57)、NCS 59, 60)やプラチナ(IV)複合体61) である。 2)活性ジスルフィドを介したジスルフィド結合形成(Scheme 21)57,62-64) 本法は、1)にて述べた 2 種の直交型保護基を組み合わせた手法であり、次の 3 工程 を経て、分子内でジスルフィド結合を形成するものである。 i)PG2の還元的脱保護による無保護Cys 残基の獲得 Scheme 20. 無保護 Cys 残基間での固相担体上ジスルフィド結合形成 Peptide S PG1 S PG1 PG2 PG2 Peptide SH SH PG2 PG2 : resin PG : protective group reductive reagent (PG1 = StBu, STmp etc.) or Metal cat. (PG1 = Acm , Allocam etc.)
Peptide
S S
PG2 PG2 1-7% TFA
(PG1 = Xan, Mmt, Tmob etc.) or
第二節 Npys-OMe を用いた固相環状ジスルフィドペプチド合成法の開発
前節でも述べたように、本合成の特徴は固相担体上で直接環状ジスルフィドペプチ ドを構築する点にある。そのため、第一章及び第二章で用いた酸処理(> 95% TFA 溶 液)による SH 保護基に脱保護条件は、樹脂からペプチド鎖を切断してしまうため利 用できない。即ち、Cys 側鎖 SH 基の保護基の変更が必要となる。そこで著者は、 dithiothreitol ( DTT ) や 2-mercaptoethanol ( 2-ME ) 等 の チ オ ー ル 系 還 元 剤 ま た は triphenylphosphine(PPh3)やTris(2-carboxyethyl)phosphine(TCEP)等のホスフィン系還 元剤で選択的に脱保護できるStBu 基を利用することとした(Scheme 23)75)。また、 本合成は固相担体上で行うため、各反応の進行は反応後に得られたペプチド-樹脂の うち、少量(1-3 mg)を脱樹脂し、獲得した粗ペプチドを逆相 HPLC 及び質量分析に よって解析した。 本章ではカルボニル基を有さず、より単純な化学構造であるNpys-OMe(3)をジス ルフィド結合形成試薬として用いることとした。その理由は、i)メトキシ誘導体 14a よりも短い工程で合成可能、ii)固相への担持が目的ではない点である。また、化合物 3 のジスルフィド形成能は oxytocin(15)の液相合成により確認した。具体的な反応条 件は、先ず、還元型oxytocin(16)を CH3CN/H2O = 1:3(v/v)混合溶媒にて溶解し、 0.1 mM ペプチド溶液(1 当量)を調製した。本ペプチド溶液へ化合物 3 を 5 当量添加 し、室温にて反応を実施した(Scheme 24)。3 時間後の反応溶液を逆相 HPLC により 解析した結果、ペプチド15 の HPLC 収率は 95%であった(Figure 19C)。本結果から Npys-OMe(3)はメトキシ誘導体 14a と同等のジスルフィド形成能を有していること Scheme 23. Npys-OMe(3)による固相ジスルフィドペプチド合成の一般式 Peptide S StBu S StBu PG PG Peptide S S PG PG Peptide S S i) deprotection of tBuS groups of Cys residues ii) oxidation on-resin by Npys-OMe (3)
i) final deprotection and cleavage ii) purification
が示唆された。
本節では、第一章と同様の理由から oxytocin(15)をモデルペプチドとし、固相環 状ジスルフィドペプチド合成法の反応条件検討を実施した(Scheme 25)。先ず、ポリ スチレンを基材としたFmoc-NH-SAL 樹脂(官能基置換量; 0.58 mmol/g)を利用し、一 般的なFmoc-based SPPS によって Cys1及びCys6をStBu 基で保護したペプチド-樹脂 31 を合成した(Table 12, 85 頁)。得られた樹脂 31 の一部(1-3 mg)を秤量し、そこに
1 mL の TFA 混合溶液(TFA:TIS:H2O = 95:2.5:2.5, v/v/v)を添加し、1 時間静置するこ
とで、ペプチドの樹脂からの切り出し及びアミノ酸残基側鎖の保護基の脱保護を行っ た(脱樹脂Method A、88 頁)。そして、得られた粗ペプチドを逆相 HPLC によって解 析した。その結果、樹脂31 より切り出されたペプチド 32 の HPLC 純度は 95%であっ
た(Figure 20A)。この樹脂 31 を 0.1 M N-methylmorpholine(NMM)存在下、20% 2-ME/DMF 溶液中にて、8 時間の振とう撹拌することで、Cys1及びCys6のStBu 基を還
元的に脱保護し、2 残基の Cys 側鎖を遊離の SH 基としたペプチド-樹脂 33 を合成し Figure 19. Npys-OMe(3)を用いた oxytocin(15)合成の逆相 HPLC 追跡
A) before addition of 3, B) 1 h, C) 3 h; HPLC conditions: a linear gradient starting from 15% to 35% CH3CN in 0.1% aqueous TFA over 20 min at a flow rate of 1.0 mL/min and
detection at 230 nm.
た。前述と同様に、樹脂33 を少量脱樹脂し、解析した結果、得られた還元型 oxytocin (16)の HPLC 純度は 78%であった(Figure 20B)。続いて、無保護となった Cys1及び Cys6の2 つの SH 基間でジスルフィド結合を形成し、ペプチドを環化させるため、樹 脂33 を DMF 中、2 当量の Npys-OMe(3, 20 mM)存在下で振とう撹拌した。1 時間 後、DMF、メタノール及びジエチルエーテルにより樹脂を洗浄し、化合物 3 を反応系 中から除去することで反応を停止させた。得られたペプチド-樹脂 34 を少量脱樹脂 し、逆相HPLC で解析したところ、目的の環状ジスルフィドペプチドである oxytocin (15)の HPLC 純度は 56%であった(Figure 20C)。本結果から、固相担体上で 1 組の 無保護SH 基は化合物 3 によりジスルフィド結合を形成することが示唆された。しか し、oxytocin の oligomer(36%)も観察された。この結果から、本反応条件では、ジス ルフィド結合形成は分子内のみならず、分子間でも進行することが明らかになった。 そこで、分子間の架橋反応を抑制するために、樹脂、反応溶媒及び化合物3 の濃度 を変化させ、ジスルフィド結合形成反応の反応条件を検討した。先ず、樹脂への官能 基の置換量の効果を検討するため、官能基置換量を0.58 mmol/g から 0.37 mmol/g へ低 下させたFmoc-NH-SAL 樹脂を出発物質とし、前述と同様の手法で樹脂 31 及び 33 を 調製した。少量の樹脂を脱樹脂に付し、逆相 HPLC にて解析した結果、樹脂 31 及び 33 から切り出されたペプチド 32 並びに 16 の HPLC 純度は、それぞれ、97%及び 90% Scheme 25. 固相担体上 oxytocin(15)合成 MeO S O2N N 3 (2 eq., conc.) Fmoc HN
であった(Figure 21A, B)。その後、DMF 中で化合物 3 による環化反応を実施した結 果、ペプチド15 の HPLC 純度は 69%まで向上し、oligomer 形成は 27%まで改善した
(Table 7, Entry 1, Figure 21C)。本結果は分子内反応に有利な反応場を提供する「擬希 釈効果」による影響と考えられる。しかし、同時にoligomer 形成の抑制は、固相担体 に依存する当該効果だけでは不十分であることが示唆された。次に、反応溶媒の効果 を検討した。即ち、DMF から後述の溶媒へ変更し、樹脂 33 と Npys-OMe(3, 2 当量, 20 mM)を反応させた(Table 7, Entry 2-4)。その結果、NMP ではペプチド 15 の HPLC 純度がさらに向上した(HPLC 純度; 86%, Figure 22A)。また、oligomer 形成も 11%ま で減少させることができた(Entry 2)。一方、THF や DCM を反応溶媒として用いた場 合は、ペプチド15 の HPLC 純度が大きく低下し、それぞれ 18%、25%であった(Entry 3, 4)。これらの反応条件では、未反応の還元型ペプチド 16 が確認され、それぞれ 45%、 38%であった。その上、oligomer の副生が増加し、それぞれ 27%、33%であった(Figure Figure 20. 固相担体上 oxytocin(15)合成における逆相 HPLC 解析結果
Each crude peptides were cleavage from A) peptidyl resin 31, B) 33, C) 34 prepared from 0.58 mmol/g Fmoc-NH-SAL resin; HPLC conditions: a linear gradient starting from 15% to 45% CH3CN in 0.1% aqueous TFA over 30 min at a flow rate of 1.0 mL/min and
detection at 230 nm. *Oxytocin oligomer. **Byproduct derived from Fmoc-NH-SAL linker ([oxytocin+106].76)
22B, C)。Entry1-4 で用いた溶媒は、いずれも固相反応で多用されており、固相担体自 体を膨潤させることが知られている77)。しかし、前述したように結果は大きく異なっ
た。従って、固相担体上での分子内ジスルフィド結合形成反応の効率は、反応溶媒に よる固相担体の膨潤性の調節では解決できないことが解った。
Table 7. 反応溶媒条件検討結果 Entry Solvent Conc. of 3
(mM) Time (h) Purity of 15 (%)[a] Oligomers (%)[a] 1 DMF 20 1 69 27 2 NMP 20 1 86 11 3 THF 20 1 18[b] 27[b] 4 DCM 20 1 25[b] 33[b]
[a] Purity of the peptide and oligomers was determined by RP-HPLC analysis of crude products after the treatment of peptidyl resins with TFA:TIS:H2O (95:2.5:2.5, v/v/v) at rt
Figure 21. Npys-OMe(3)を用いた固相担体上 oxytocin(15)合成における溶媒条 件検討の逆相HPLC 解析結果(Table 7, Entry 1)
Each crude peptides were cleavage from A) peptidyl resin 31, B) 33 prepared from 0.37 mmol/g Fmoc-NH-SAL resin, C) 34 prepared under the reaction conditions of Entry 1 of Table 7; HPLC conditions: a linear gradient starting from 15% to 45% CH3CN in 0.1%
aqueous TFA over 30 min at a flow rate of 1.0 mL/min and detection at 230 nm. *Oxytocin oligomer. A B C 32 (97%) H CYIQNCPLG NH2 S S StBu StBu 16 (90%) H CYIQNCPLG NH2 SH SH * 15 (69%)
Retention time (min)
10 20
そこで、著者は「固相担体上に担持されているペプチドに対する溶媒和」78) に着目 した。「ペプチド-樹脂の溶媒和」に関し、Narita 及び Ouchi は、固相担体に担持され たオリゴロイシン((Leu)n, n = 6, 9)が分子間水素結合により集合することを報告して いる78b, 79)。この際、樹脂の膨潤に使われた溶媒は、一般的な固相反応において多用さ れるDCM であった。本知見と Table 7 の結果から、固相担体上においても分子間ジス ルフィド結合形成が進行した原因は、ペプチド鎖部分の溶媒和が不十分なためと考え た。 ペプチド鎖部分の溶媒和を改善する手法として、アクセプター性溶媒とドナー性溶 媒の混合溶媒を用いる場合 80) とドナー性溶媒に塩を添加する場合 81) がある。これら の手法において、アクセプター性溶媒はペプチド鎖のカルボニル基と相互作用し、ド ナー性溶媒はアミド結合のN-H と相互作用する。本研究では、前述の手法から後者を
Figure 22. Npys-OMe(3)を用いた固相担体上 oxytocin(15)合成における溶媒 条件検討の逆相HPLC 解析結果(Table 7, Entry 2-4)
Each crude peptides were cleavage from peptidyl resin 34 prepared under the reaction conditions of Entry 2-4 of Table 7, A) Entry 2, B) Entry 3, C) Entry 4; HPLC conditions: a linear gradient starting from 15% to 45% CH3CN in 0.1% aqueous TFA over 30 min at
a flow rate of 1.0 mL/min and detection at 230 nm. *Oxytocin oligomer. A B C 15 (25%) 15 (86%) * 16 * 15 (18%) * 16
Retention time (min)
10 20
選択し、さらなる反応条件検討を実施した。Table 7 に示す結果を参考に、ドナー性溶 媒としてDMF 及び NMP を選択し、添加する塩を塩化リチウム(LiCl)とした。LiCl を選択した理由は、Li+がカルボニル基と相互作用し、アミド結合のC-N 結合の回転を 抑制することが知られており82)、ペプチド樹脂の膨潤性やペプチド鎖伸長反応が改善 されるとの報告があるためである83)。 まず、LiCl 存在下、DMF 中でペプチド-樹脂 33 を化合物 3(2 当量、20 mM)によ り酸化した結果、ペプチド 15 の HPLC 純度は 88%となり、良好な反応性を反映した
(Table 8, Entry 1, Figure 23A)。特に、本反応における oligomer 形成は 3%と低値であ り、Table 7 の Entry 1 と比較し、劇的に改善された。一方、LiCl 存在下、NMP 中で前 述の酸化反応を実施したところ、oligomer 形成は若干ながら改善されたが、ペプチド
15 の HPLC 純度は 76%と添加前の結果を下回った(Table 8, Entry 2, Figure 23B)。Table
8 における Entry1 及び 2 の結果は Table 7 における Entry1 及び 2 の結果と逆転してい るが、その理由は不明である。しかし、LiCl 存在下では NMP よりも DMF が反応溶媒 として適すると示唆されたため、次に、反応時間を1 時間に、さらに化合物 3 の当量 を2 当量に固定し、LiCl/DMF 中における化合物 3 の濃度を 20 mM から 5 mM へ低下 させた(Table 8, Entry 3, Figure 23C)。しかし、当該反応条件では未反応ペプチド 16 が 観察されたため、反応時間をさらに3 時間へ延長したところ、oligomer 形成を抑制し つつ、ペプチド16 の消失とともに、ペプチド 15 の HPLC 純度が 91%へ向上した(Table
8, Entry 4, Figure 23D)。さらに、化合物 3 非存在下で 0.4 M LiCl/DMF 中、樹脂 32 を 3 時間振とう撹拌した結果、ペプチド15 の HPLC の純度は 7%と低い値を示した(Table
8, Entry 5, Figure 23E)。一方、oligomer 形成は 10%と分子間反応の進行が示唆された。 これらの結果から、LiCl の添加はペプチド鎖の立体構造を変化させ、化合物 3 による 分子内でのジスルフィド結合形成を有利に進行させたと考えられる。
Table 8. LiCl 存在下における反応条件検討の結果 Entry Solvent Conc. of 3
(mM) Time (h) Purity of 15 (%)[a] Oligomers (%)[a] 1 0.4 M LiCl/DMF 20 1 88 3 2 0.4 M LiCl/NMP 20 1 76 8 3 0.4 M LiCl/DMF 5 1 76[b] 10[b] 4 0.4 M LiCl/DMF 5 3 91 2 5 0.4 M LiCl/DMF 0 3 7 10 [a] Purity of the peptide and oligomers was determined by RP-HPLC analysis of crude products after the treatment of peptidyl resins with TFA:TIS:H2O (95:2.5:2.5,
著者は、Npys-OMe(3)を用いた固相環状ジスルフィドペプチド合成法による oxytocin(15)の単離収率を求めるため、Table 8 における Entry 4 の反応条件を用い、 新たにペプチド-樹脂34 を合成した(HPLC 純度; 93%、Figure 33A、91 頁)。そして、 樹脂34 の脱樹脂と逆相 HPLC による単離精製を実施した。その結果、Fmoc-NH-SAL 樹脂から全21 工程で、ペプチド 15 を 65%の単離収率で得ることができた。 既存の固相担体上でのジスルフィド結合形成に基づくoxytocin 合成において、最も 良好な単離収率はSun らにより報告された 51%である(Table 9, Entry 8, Scheme 26)
72)。この例では、固相担体上のジスルフィド結合はAcm 基で保護された Cys 残基間の
直接的なヨウ素酸化反応により成された。Sun らの手法と Npys-OMe(3)を用いた著 者の固相環状ジスルフィドペプチド合成法を比較した結果、本手法の方がoxytocin の 収率において、ヨウ素酸化よりも優れたものであると言える。
Figure 23. Npys-OMe(3)を用いた固相担体上 oxytocin(15)合成における LiCl 共 存溶媒条件検討の逆相HPLC 解析結果(Table 8, Entry 1-5)
Each crude peptides were cleavage from peptidyl resin 34 prepared under the reaction conditions of Entry 1 - 5 of Table 8, A) Entry 1, B) Entry 2, C) Entry 3, D) Entry 4, E) Entry 5; HPLC conditions: a linear gradient starting from 15% to 45% CH3CN in 0.1% aqueous
TFA over 30 min at a flow rate of 1.0 mL/min and detection at 230 nm. *Oxytocin oligomer. **Byproduct derived from Fmoc-NH-SAL linker ([oxytocin+106].76)
A C E B D 15 (88%) ** * * **** 15 (76%) 16 * 15 (7%) 16 * 15 (91%) ** * * 15 (76%)
Retention time (min)
10 20
0 30
Retention time (min)
10 20
Table 9. 固相担体上でのジスルフィド結合形成による oxytocin(15)合成例 Entry Protective group of Cys Oxidative reagent Solvent (v/v/v) Time Crude purity of 15 (%) Isolated yield of 15 (%) Ref. 1 StBu Npys-OMe (3) 0.4 M LiCl/DMF 3 h 93 65 This study 2 STmp NCS DMF 15 min 75 NI[a] 59 3 Mmt Pt(IV) complex[b] DMF/H2O (95:5) 2 h 85 NI[a] 61
4 Acm (CF3COO)3Tl DMF 80 min 72 48 65
5 Tmob (CF3COO)3Tl
TFA/DCM/ anisole (82:15:3)
2 h 60-65 NI[a] 66
6 Xan (CF3COO)3Tl DMF/anisole (19:1) 1 h 80-85 NI[a] 68
7 Acm iodine DCM/MeOH /H2O (6:2.5:0.42) 30 min 73 NI[a] 69 8 Acm iodine DMF 2 h 92 51 72 [a] Not indicated. [b] Pt(IV) complex is [PtCl2(phen)(en)]Cl2 or [PtCl2(bpy)(en)]Cl2.
Scheme 26. Sun らによる固相担体上 oxytocin(15)合成72)
Fmoc HN
Fmoc-Rink amide AM resin (1 eq.) (Fmoc-NH- = 0.55 mmol/g) Fmoc/tBu SPPS Boc CYIQNCPLG 36 tBu Trt Trt S S Boc CYIQNCPLG 35 S tBu Trt Trt S Acm Acm DMF 25 oC, 2.5 h I2 (5 eq.) i) TFA:TIS:H2O (95:2.5:2.5, v/v/v), 30 min x 6 ii) HPLC purification (isolated yield : 51%)
H CYIQNCPLG NH2
S S
第三節 固相担体上での melanin-concentrating hormone の合成 本節では、前節で合成した oxytocin(15)よりも複雑な環状ジスルフィドペプチド の合成を固相担体上で達成するために、Figure 24 に示した melanin-concentrating hormone(MCH, 19 残基, 37)の合成を実施した。MCH は、サケの下垂体から分離され たが、哺乳類の神経細胞からも発見されている84)。特に、摂食行動に関与する脳領域 で大量に観察されており、MCH を動物へ注入すると摂食促進が観察される一方で、 MCH ノックアウトマウスでは食欲減退と体重減少が観察された 85)。また、近年の研 究により、MCH は睡眠覚醒周期や学習または記憶プロセスに関与する可能性が示唆 されているため、興味深いペプチド性ホルモンである86)。 MCH(37)の具体的な合成として、Scheme 27 に示すように、Fmoc-Val-HMPA-PEG 樹脂を用いた。当該樹脂の基材はポリスチレンであるが、その末端に優れた溶媒親和 性を付与するためにPEG リンカーが導入されている。まず、当該樹脂(官能基置換量; 0.19 mmol/g)上に、一般的な Fmoc-based SPPS によって Cys7及びCys16がStBu 基で保
の合成においても、化合物3 を用いる固相環状ジスルフィドペプチド合成が有効に機
能することを確認できた。
Scheme 27. 固相担体上での MCH(37)合成 Fmoc-Val-HMPA PEG resin
Figure 25. 固相担体上での MCH 合成における逆相 HPLC 解析結果 Each crude peptides were cleavage from A) peptidyl resin 38, B) 40, C)
42; HPLC conditions: a linear gradient starting from 5% to 65% CH3CN
in 0.1% aqueous TFA over 30 min at a flow rate of 1.0 mL/min and detection at 230 nm.
Retention time (min)