複素環キノン系化合物の新規合成と医薬素材の探索
西山 卓志
Novel synthesis of heterocyclic quinone compounds and search for pharmaceutical materials
Takashi Nishiyama
ABSTRACT
We are developing the synthesis of biologically interesting carbazole-1,4-quinone compounds, including natural products by tandem cyclic reactions. In this report, we describe the new synthesis of carbazole-1,4-quinones as follows; 1) the synthesis of carbazole-
1,4-quinones using a tandem RCM-dehydrogenation reaction, 2) a novel one-pot synthesis of carbazole-1,4-quinone by consecutive Pd
catalyzed cyclocarbonylation, desilylation, and oxidation reactions.
These new reactions were applied to the synthesis of carbazole-1,4- quinone alkaloids and ellipticine quinones, and evaluation of these antiproliferative activity against HCT-116 and HL-60 cells.
1. はじめに
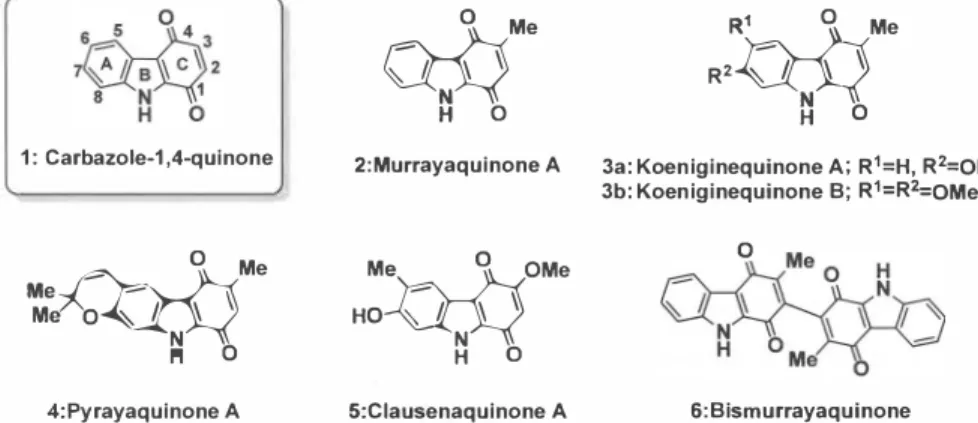
キノン系化合物は、 高等植物、 菌類、 細菌類および動物界を通じて自然界に広く分布して おり、 生体内では細胞呼吸における電子伝達系や酸化還元の補酵素として重要な役割を担っ ている。 また、 抗がん剤である mitomycin C 、 daunomycin や mitoxantrone は、 天然由来のキノ ン構造を含む医薬品であり、 現在も臨床で使用されている。 一方、 近年では、 様々な種を起 源としてキノン構造を含む新しい縮合複素環系化合物が単離・構造決定されてきており、 さ らに抗腫瘍活性のみならず、 抗原虫活性、 抗菌活性、 抗マラリア活性などの生物活性を有す ることが併せて報告されている。 !)このようにキノンを含む縮合複素環化合物は
、新しい医 薬品やそのリ
ード化合物となる可能性を多分に秘めていると考えることができる。
Carbazolequinone は、 三環性含窒素縮合複素環キノン類の中では、 最も単純な構造を有する
ものの
一つであり、 生物活性としてこれまでに強心作用、 抗腫瘍作用、 抗結核作用、 神経保 護作用などが報告
2)されている (Figure 1) 。 中には、 民間薬として利用されているものもあり、
これらの構造的特徴とその生物活性への興味から多くの研究グループにより全合成研究
2)が
盛んに行われている。著者は、この点に着目し、新しい医薬素材の候補化合物として、キノ ン系縮合複素環化合物を設定し、最適な医薬品あるいはリ
ード化合物の候補化合物の合成に 挑戦している。本総説では、金属触媒を用いた新規carbazole-1,4-quinone構造の合成法の開発 を基盤とし,標的とした天然物群の基本骨格の構築と全合成研究、および抗腫瘍活性化合物 の探索研究を行った経緯について述べる。
`
Me
R
2R
` : Me
1: Carbazol e -1,4-quinon e 2: M urrayaquinon e A 3a: Ko 3b: Ko e e nigin nigin e e quinon quinon e e A; R B; R
11=H, R =R2=0
2=0 Me Me
ご口 Me HO M : `O Me
4:Pyrayaquinon e A 5:Claus e naquinon e A
゜
6:Bismurrayaquinon e
Figure 1: Carbazole-1,4-quinone alkaloids.
2.三環性Carbazole-1,4-quinone骨格の新規合成法の開発
2-1. Ring Closing Metathesis (RCM)反応を利用したcarbazole-1,4-quinone骨格構築法の確 立とその応用
近年目覚しい発展を遂げた遷移金属触媒を用いた炭素
ー炭素結合形成反応の中で、オレフィ ンメタセシス反応
3)は、幅広くかつ驚異的に有機合成化学の発展に寄与してきた。本反応は、
二種類の異なる オレフィン間で結合の組換えが起こる触媒反応であり、反応形式により①交 差メタセシス(CM: Cross Metathesis)、②エンインメタセシス(Enyne Metathesis)、③アルキン
Olefin metathesis reaction B /
A +DF
C
catalyst
c /
A +D
B
①Cross Metathesis (CM)
A [Ru] A
t
+B 』—→ -
H2C=CH
2B J
②Enyne Metathesis ③A/kyne Metathesis
こ
H2 竺パしニ二 ol
④Ring-Opening Metathesis (ROM)
〇+『 R 竺し
C
CH
2 翠R
⑤Ring-Closing Metathesis (RCM)
ご 庄 ニ
CH 2 - H
2C=
CH
2。
Scheme 1: Types of olefin metathesis.
-24-
メタセシス (Alleyne Metathesis) 、 ④開環メタセシス (ROM: Ring Opening Metathesis) と⑤閉環 メタセシス (RCM: Ring Closing Metathesis) に分類される (Scheme 1) 。
中でも RCM 反応は、 有機合成化学の分野では最も頻繁に用いられており
、近年、 医薬品 や生理活性天然物の環状構造形成に、 RCM 反応が活用される例が多く報告
4)されている。 し かしながら、 窒素や酸素などのヘテロ原子を含む複素環化合物の合成に RCM 反応を利用し た例はあまりなく、 数例が報告されているのみである。 そこで、 著者は、 複素環化合物であ る carbazole-1,4-quinone 骨格合成に、 RCM 反応を鍵反応とする新たな含窒素複素環キノン類 の合成法の確立を目指した。
Murryaquinone A (2) は、 古川ら
”によって Murraya euchrestifolia Hayata から単離・構造決定 された carbazolequinone アルカロイドである。 Murrayaquinone A (2) は、 生物活性として、 強 心作用
”と P-388 細胞に対する細胞増殖抑制作用
6)が報告されており、 これまで多くの研究 グル
ープによって、 形式合成を含む全合成が報告
7)されている。 そこで、 著者は、 新たな含 窒素複素環キノン類の合成法の確立を目指し、 murrayaquinone A (2) を標的化合物とした全合 成を計画した。 その基本的な考えは、 2,3 —ビスアクリロイルインド
ール(10)を鍵化合物とす る RCM 反応による炭素
ー炭素結合形成によって carbazole-1,4-quinone (11) の 2,3 位結合部を 構築するものである。 3-iodoindole-2-carbaldehyde (7)を出発原料とし、 一酸化炭素挿入クロ スカップリング反応、 次いで Grignard 反応に付し
、得られた 2 級水酸基を Mn かにより酸化 することで鍵化合物である 2,3 —ビスアクリロイルインド
ール(10)を合成した。 これに対し、
Grubbs 触媒存在下 RCM 反応を行ったところ、 目的とする carbazole-1,4-quinone (11) を合成す ることができた (Scheme 2) 。
J---snBu R
3O:j_CHO
CO (1 atm), BHT PdCl
2(dppf) M
llOM 70 °C, 24 h DMF
琴叫 a
a, 1 u
. x M
, n
40� 3 ,
h 疇 - 闊' O O M R ゜
/10a: 48%
10b:-
疇。 M
lOM
Sa: 70%
8b: 51%
Grubbs
2nd toluene 70 °c, 1 h 72%
<7'MgB
『心 MO O M R OH
THF 0 °C, 4 h
9a: 66%
9b: 26%
`如M Me
11a
Scheme 2: Synthesis of N-MOM-carbazole-1,4-quinone (lla)
しかしながら、 2 級水酸基を酸化し、 鍵化合物である 2,3-bis(acryloyl)indole 10 を合成する
ステップが低収率か基質によっては合成できず、 問題を残した。 そこで
、合成ルートを改善
する目的で、 その前駆体であるアリルアルコ
ール9に対して、 RCM 反応を行ったところ閉環
反応と脱水素反応が連続して進行し、 carbazole-1,4-quinone (11) が 一 挙に生成することを見出 した。 このように、 RCM 反応とともに脱水素反応がタンデム型に進行する例は数例報告
4)さ れており、 著者らの基質にも同様の現象が起こっていると考えられた。 この結果から本現象 を利用した carbazole-1, 4-quinone 骨格合成の効率的な新規合成法になると考え、 種々検討した 結果、 酸素気流中、 toluene 溶媒、 70 ℃で加熱する条件で、 反応収率 90-93%まで反応条件を 改良することができた。 最後に保護基である MOM 基を 6M 塩酸で処理することで、 標的化 合物である murrayaquinone A (2) の全合成を達成することができた (Scheme 3)。 以上の結果か ら、 タンデム RCM
ー脱水素反応を鍵反応とした carbazole-1,4-quinone 骨格構築に対する効率 的な合成法を確立することができた。 そして 、 本法において 3-iodoindole-2-carbaldehyde 7 よ り 4 工程、 総収率 43%で murrayaquinone A (2) の全合成を達成することができた。
`戸ら , 竺切、 12 □ 11a:93% (5min) 麟〗: 99% Me
2: M urrayaquinon e A
ヽ�↑ ~
へや 11b: 90% (10 mi n)
Scheme 3: Synthesis of murrayaquinone A (2) by RCM and dehydrogenation reaction.
2-2. 分子内 CO 挿入シクロカルボニレ
ーション反応を用いた carbazole-1,4
—Q.Jinone 骨格構築法 の確立とその応用
前述したタンデム RCM
ー脱水素反応により carbazole-1,4-quinone 構造の短工程での合成 法を確立できた。 しかし、 Grignard 反応によって得られる本反応の鍵中間体であるアリルア ルコ
ール 9 が基質によっては収率が低く問題を残した。 この原因として、 3 位のアクリロイ
ル基が Grignard 試薬によって求核攻撃を受けやすいことが考えられた。 そこで、 更なる
一般性を向上させるため、 合成ルート改善を検討した。 3-iodoindole-2-carbaldehyde 7 に対し、
vinylmagnesium bromide による Grignard 反応を行い、 高収率で 2- アリルインド
ール 12 を合成 した。 12 の二級水酸基を TBS 基で保護した後、 再度、 一酸化炭素気流中 PdCh(dppf) 存在下、
O も OM C
HO :ご二
BrMg へ ゜ \ー 臣 1 -
TBSCI imidazole
DMF 50
°C, 12 h 1)否SnBu
393%
PdCl
2(dppf)
□ \ ; co(1 atm),
BHTO ゜
\
:3 M ゜T - BS 口ご: : (:互三)
Scheme 4: New synthesis of carbazole-1,4-quinone (llb) by one pot reaction.
-26-
tributy I(viny I)tin と反応を行った。 その結果、 予想していた 3—アクリロイルインド
ール 14 は 得られず、 最終目的物である carbazole-1,4-quinone llb が低収率ながら得られた (Scheme 4)。
そこで、 本反応の現象を検証すると共に
、carbazole-1,4-quinone の新規合成法となりうるか 最適条件の検討を行った。 この現象について詳細に検討した結果、 一酸化炭素存在下、 DMF 中 PdCh(dppt) 触媒
、有機スズ試薬 (tributyl(vinyl)tin)、 BHT 存在下で cyclocarbonylation 反応を 行い、 反応終了時、 酸素気流中、 TBAF 処理することで目的とする carbazole-1,4-quinone を反 応収率 60%で得ることができ、 本反応条件を one pot 反応を用いた carbazole-1,4-quinone の新 規合成法の最適条件として確立することができた。
以上の結果をもとに
、one-pot 反応に関する反応メカニズムを考察した (Scheme 5)。 まず、
3—ヨ
ードインド
ール7と0価の Pd 触媒が酸化的付加することによりアリ
ールパラジウム錯体 14 を形成する。 次いで、 パラジウムに
一酸化炭素が配位後、 挿入がおこりアシル錯体 15 を 形成する。 次に有機スズ化合物とのトランスメタル化が進行し、 従来であれば
、還元的脱離 が進行し、 3 ーアクリロイルインド
ール 14 が生成するはずである。 しかし、 化合物 16 は構造 中に含まれるアルケンに対して cyclocarbonylation 反応が進行し
、続いて P —脱離が起こってカ ルバゾ
ール 18 が生成後
、TBAF による脱シリル化、 酸化反応が連続しておこり、 目的とする carbazole-1,4-quinone llb が生成したものと考えている。 以上の結果から、 これまで報告例の 少ない孤立したアルケンに対する cyclocarbonylation、 続いて脱シリル化
、酸化反応が連続し て起こり、 キノン部が
一挙に形成される one-pot 反応が進行する新たな carbazole-1,4-quinone 骨格合成法を確立することができた。
HO � O
')\...__
PdCl2(dppf)
:〗ー疇
IOTB S B HT↓
0人: B S
MOM MOM
Pd(O)MOM
11b 7
20 \
L`
/ 。
XIこ
L:L`〉 元 add ition - P 閃 ")
い。 M ゜ T B S \f3-H
el i m ina ti on 口 B S
`TBene /:t
” 。 co
:
r
ed uctw
eIns
ertIO n
L へ L Bu3 S nl : / 屯 ゜TBS
\ I \ - B S
< e
lImmat
IOn \\ `s ごこ S
e: a B 9::ヽO n
MOM 14 17
Scheme 5: Our plausible mechenism for this reaction.
3. タンデム環化反応を利用した carbazole-1,4
—q_,1inone 誘導体の合成と生物活性試験
Carbazole-1,4-quinone 誘導体を使った抗腫瘍活性評価試験は、 2000 年糸魚川ら
2b)によって
、2ーメチルおよび 3ーメチルカルバゾ
ールキノン誘導体を用いてヒトロ腔類表皮がん細胞株 KB、
ヒト皮膚がん細胞株 SK-MEL-5、 結腸細胞株 Colo-205、 ヒト大腸がん由来細胞株 HCT-8 に対 する in vitro 試験を実施し、 特に 3-methyl-6-methoxycarbazole-1,4-quinone がそれぞれの細胞に 対し、 高い活性を示すことが報告されている。 しかしながら、 carbazole-1,4-quinone の A 環上 に置換基を有する誘導体の合成および活性評価試験に関する報告例は、 少なく限られていた。
そこで
、carbazole-1,4-quinone 骨格のキノン部の置換基と A 環であるベンゼン環部への様々な
置換基を導入した誘導体合成を行い、 得られた carbazole-1,4-quinone 誘導体を使い
、構造
ー活 性相関研究を行うことで、 新しい抗がん剤となりうる医薬品素材の探索研究を実施した。
3-1. One pot 反応を利用した carbazole-1,4
—CJ.Jinone alkaloid, koenigineCJ.Jinone 類の全合成 Koeniginequinone A (3a) および B (3b) は、 1998 年 Chowdhury ら
8)により Murraya koenigii
Speng から単離・構造決定された carbazole-1,4-quinone アルカロイドである。 これまでに
全合成については数例報告
9)されているが、 生物活性に関する報告は少ない。 そこで、
cyclocarbonylation、 脱シリル化および酸化反応が連続して進行する one-pot 反応を鍵反応とし た koeniginequinone A (3a) および B (3b) の合成を計画した。 出発原料である N-MOM インド
ール 21 を propenylmagnesium bromide を用いた Grignard 反応に付し、 次いで、 imiadazole 存在下
TBSCI 処理することで 2 級水酸基をシリル基で保護した。 シリルエ
ーテル 23 を
一酸化炭素存
在下 PdC'2(dppf)、 BHT と共に tributyl(vinyl)tin を DMF 中 70 ℃で反応させると one-pot 反応が 進行し
、目的の carbazole-1,4-quinone 24 が低収率ながら得られた。 続いて、 6 M 塩酸存在下、
MOM 基の脱保護を行ったが、構造不明物を与えるのみで koeniginequinone A (3a) および B (3b) を得ることができなかった (Scheme 6)。
TBSCI imidazol D
e..
M
F
汽M CH0 戸戸`:
e50
°C, 12 h R
2R 二八
MO
M§ OTBS
Me21a ( a: R1=H, R
2=0
Me21b[b:R1=R
2=O
Me1)公SnBu
3PdCl
2(dppf) CO (1 atm), BHT
D
MF 70
°C, 20 h 2) TBAF und
er 0
2R
2R `
MeM
O
M゜
24a: 30%
24b:41%
22a 22b
6
MHCI IJ”,.
Me
OH 60
°c, 1 h
23a: 97% from 21a 23b: 87% from 21 b
R
2R `
Me3a: Ko
enigin
equinon
eA 3b: Koenigin
equinon
eB Scheme 6: New synthesis of carbazole-1,4-quinone alkaloid, koeniginequinones (3)
using one pot cyclocarbonylation reaction.
- 28 -
そこで、
koeniginequinoneA
(3a)および
B (3b)の全合成を達成すべく、 インドール窒素原 子の保護基として
BOM基と
SEM基を選択し、 再度合成を検討した。
N-BOMインドール
26と
N-SEMインド
ール
27に対し、 先程と同様、
2工程で
2ーアリルインドール
30, 31へと誘 導した。 合成した
N—保護体
30, 31に対し、
one-pot cyclocarbonylation反応を行い、
carbazole-1,4-quinone
をそれぞれ中程度の反応収率で合成できた。 最後に、 窒素位の脱保護を検討し
た結果、
N-BOM carbazole-1,4-quinone 32a, 32bを液体
NHJ中金属
Naによる
Birch還元に付 し、
BOM基の除去を行ったところ
koeniginequinoneA
(3a)および
B (3b)を良好な収率で得 ることができた
(Scheme 7)。 以上の結果から、
koeniginequinone類の合成に
3種の保護基を 検討し、
MOM基および
SEM基に比べて
BOM基が保護基として最適であること見出し、
one-pot cyclocarbonylation
反応を利用した
koeniginequinoneA
(3a)および
B (3b)の全合成を
3-iodoindole-2-carbaldehyde 25
から
5工程で達成することができた。
R’R
り'
H
,
cooBOMCI, K2C03
R2 R
-v:/- 1
---- I cHO
Me �MgBr R'•
叫
' 闊
H DMF, rt, 12 h •
or THF
SEMCI, NaH R' 3 0 °C, 20 min 25a
25b
DMF, rt, 12 h rー・...._1 I ....? 1
^.. 、
TBSCI imidazole
夏
26a: R3=BOM (92%) 26b: R3=BOM (91%) 27a: R3=SEM (94%) 27b: R3=SEM (98%)
1) <?'SnBu3
PdCl2(dppf) CO (1 atm), BHT DMF, 70 °C, 20 h DMF R
2
R
` T
M
B
e
s 2) TBAF, 02 o『ai『
50 °C, 12 h
30a: R3=BOM (80% from 26a ) 30b: R3=BOM (92% from 26b) 31a: R3=SEM (85% from 27b) 31b: R3=SEM (61% from 27b)
28a: R3=BOM 28b: R3=BOM 29a: R3=SEM 29b: R3=SEM
R2R
亨 e
32a: R3=BOM (69%) 32b: R3=BOM (39%) 33a: R吐SEM(59%) 33b:R吐SEM(37%)
R2
R
:言:三三:し`
Me R2Rし心
Me予ご>
R2R1\-IN:,MeSEM 32a 3a: Koeniginequinone A : 33a
: 0 50°c,1h 3a: Ko
□
eniginequinone A32b ;R=H, R1 2=OMe (78%)
:
33b ;R=H, R2=O1 Me(48%)3b:Koeniginequinone B : 3b: Koeniginequinone B
;R=R2=O1 Me(72%) : ;R=R2=O1 Me(52%)
Scheme 7: New synthesis of carbazole-1,4-quinone alkaloid, koeniginequinones (3) using one pot cyclocarbonylation reaction.
3-2. タンデム RCM- 脱水素反応を応用した carbazole-1,4-quinone 誘導体の合成と生物活性評価 試験
上述したように cyclocarbonylation を含む one-pot 反応を鍵反応とした koeniginequinone 類の 全合成を 3-iodoindole-2-carbaldehyde 25 から 5 工程で達成し、 同時に carbazole-1,4-quinone の 窒素原子の保護基として BOM基が最適であることを述べた。 しかし、 koeniginequinone A (3a)
に比べ、 koeniginequinone B (3b) は
、鍵反応である one-pot 反応によるキノン部構築のステッ プが低収率と carbazole-1,4-quinone 誘導体の合成を行う上では、 問題を残した。 そこで、 もう 1 つの方法であるタンデム RCM
ー脱水素反応を鍵反応とした koeniginequinone 類の全合成を 検討することとした。 3-Iodoindole-2-carbaldehyde 25 よりインド
ール窒素原子に保護基を導入 することなく、 一酸化炭素挿入クロスカップリング後、 Grignard 反応を行い、 2 工程で合成し
た 2ーアリルアルコ
ールに対して酸素気流中、 第 2 世代 Grubbs 触媒存在下、 タンデム RCM
脱水素反応を行ったところ、 koeniginequinone A (3a) および B (3b) を 3 工程で合成する条件を 見出し、 両化合物共に工程数の短縮と総収率の改善に成功した。
そこで、 本)レ
ートを多置換 carbazole-1,4-quinone 誘導体の合成へと応用することとした。 ベ ンゼン環上に種々の置換基を有する 3-iodoindole-2-carbaldehyde 34 より 2 工程で合成した 2- アリルアルコ
ール 36 に対し、 タンデム RCM
ー脱水素反応を行うことで目的とする A 環上に 種々の置換基をもつ carbazole-1,4-quinone 誘導体 37 を 24 種類合成できた (Scheme 8)。
I
; 2 co(1 atm)
,BHT
R
3illr.1-1n �Cl2 (dppf)
\ / \ N CHO
H 340 R 2
R
3心
36
DMF
Grubbs 2 nd und
e『0
2tolu
en
eO R ぅ ノ R1
R
3Q ぐ/ニ
N CHO THF
H 35
o R 2 Carbazol
e-1,4-quinon
es R
3□ R1 ,
R1= ;ぷ°ら: I F -
eb□': ; e Cl,
N
; R 2 =H or M
eH 0
37
; R
3=H or M
eヽr ヽヘ^� ‘^ ~で ` ^,ふ
Scheme 8: Synthesis of carbazole-1,4-quinone derivatives (37).
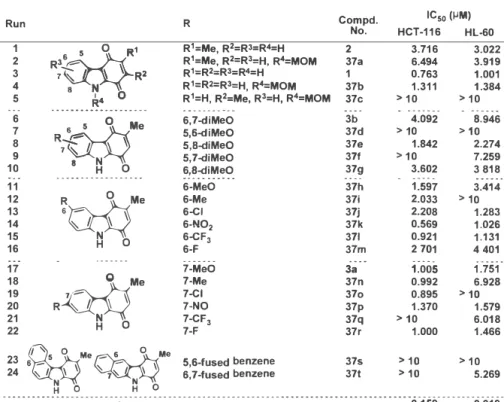
そして、 これら誘導体を用いたヒト大腸癌由来細胞株 HCT-116 細胞およびヒト白血病細胞 HL-60 に対する細胞増殖抑制活性評価試験を実施した。 その結果、 carabzole-1,4-quinone の 6 位に比べ、 7位へ電子供与基や電子吸引基の区別なく置換基(メチル基、 クロロ基、 ニトロ 基、 トリフルオロメチル基、 フルオロ基)を導入すると、 活性が強くなる傾向であった。 ま た、 一連の関係性はみられないが強い活性を示す 3-methyl-6-nitrocarbazole-1,4-quinone (37h) を見いだすことができた。 さらに、 インタ
ーカレ
ーションの作用を期待して構造中に平面性 をもたせる目的でさらにベンゼン環が縮環した 2 種の四環性 benzocarbazole-1,4-quinone 37s, 37t を合成し活性評価を行ったが、 全く活性を示さなかった (Table 1)。 次のステップとして、
-30-
Table 1: Evaluation of cell growth inhibitory activity by MTT assay against HCT-116 and HL-60 cell lines.
Run R Compd.
No. HCT- 11
IC6
50 (µM)HL-60
-946274碑818 巫四悶 401迅 73 -301114i ;8 0 2 ー ー
>->-2
2 -2738911
- 5
函邸邸涵g⑳邸泣
70両
-4
0103-122002-1-11
-
>
> -
m-�bm78"79-mn刀九717-a
- 3
- 33333る333333
60000: -eeeee- iiMiMiMiMiM� 0 02巳 io
ば且知知翌
�9キ�le i65556
-666666
-
sl`:HOO 心
R 予 一 R6 - 〗
:67890i123456--
1 -1111 11 -
12345
R
1;Me,R
2=Rl=R
4=H 2 R
1=Me, R
2=R
3=H, R
4=MOM 37a R
1=R
2:R
3:R
4=H 1 R
1=R2=R3=H, R
4=MOM 37b R
1=H, R
2=Me, R
3=H, R
4=MOM 37c
3.716 6.494 0.763 1 .3 1 1
3.022 3.919 1.00 1 1.384
> 1 0 > 1 0
789012111222
7-N 7-Me 7-CI 7-NO 7-CF
37-F
3a 37n 370 37p 37q 37r
1 .005 0.992 0.895 1.370
> 10 1 .000
1 .75 1 6.928
> 10 1.579 6.018 1.466
::麟
H OMe q 芯
H 0Me g: :::字:悶 □ : 盟 凜 > 1 �.269
· · • · •...............---- -- -- -- --- -- -- --- -- --- -- -- - - · • - - - --0 - - - -
Camptothecin 0.159 0.0 1 9
carbazole-1,4-quinone 誘導体を使った詳細な作用メカニズムの解明を実施する予定である。
4. 四環性 ellipticine quinone 誘導体の合成と細胞増殖抑制試験
Ellipticine (38) は、 1959 年 Ochrosia elliptica から Goodwin ら
)0)により単離・構造決定され
た四環性 pyrido[4,3-b ]carbazole アルカロイドであり、 生物活性として、 高い抗腫瘍活性と抗
マラリア活性を有することで注目され、 多くの研究グル
ープにより全合成および構造
ー活性 相関研究が実施されてきた。
II)特に、 2 位にアルキル基を導入した四級塩
、6 位へのアルキル 基の導入、 9 位にメトキシ基あるいは水酸基を導入することで生物活性が増強することがこ れまでに報告されている。 12> Ellipticine 誘導体である celiptiniurn (39) が乳がんの骨髄転移治療 薬として臨床応用されている (Figure 2)。
13
)この ellipticine (37) の全合成研究の中、 1984年Gribble ら
14)により ellipticine (37) の合成
中間体として ellipticine qui none (45) が初めて報告された。 2004 年 Bernardo ら
6)は、 in vitro
試験においてヒト子宮頚癌由来細胞株 Hela 細胞に対する細胞毒性試験を実施し、 ellipticine
qumone (45) が高い抗腫瘍活性を示すことを報告している。 しかし
、未だ ellipticine quinone 誘
導体を使用した生物活性評価試験に関する報告は少なく限られている。 この点に着目し、 著
ellipticinium salts OHorOMe
\ Me 1 。 rigin:
90d:P
2
B'. 乞::ば 、 aese:Iliptica
6 N Antitumor activities
I H Me •DNA inte
『Calation
N-6 alkylation • Topoisomerase II inhibitor Antimararial activity 38: Ellipticine
HO
`
H Me
f39: Celiptinium Figure 2: Structure of ellipticine and analogue
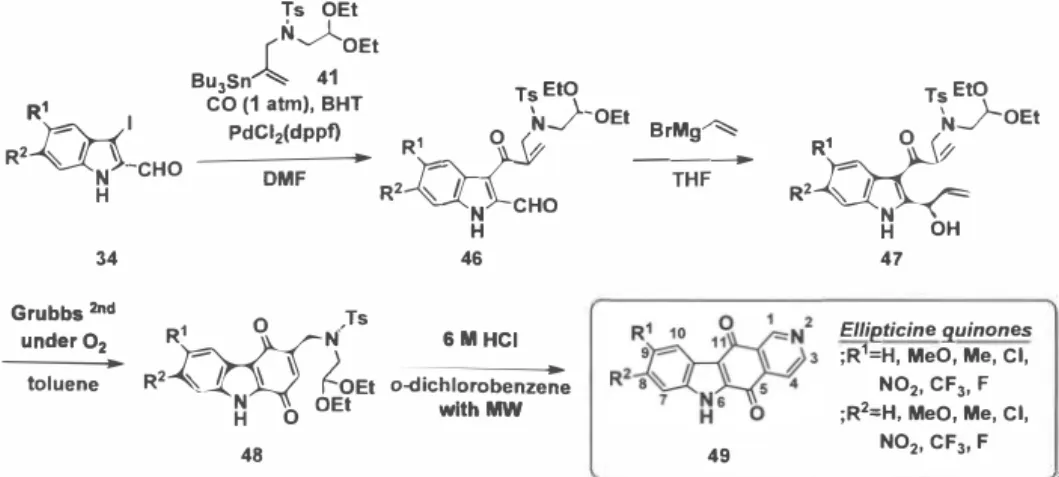
者は、新たな抗腫瘍活性化合物の探索研究を実施するキノン系縮合複素環化合物の基質とし てellipticine quinone (4 5)を設定した。今回開発した2つの合成法のうちタンデムRCM
ー脱水
素化反応を鍵反応としたellipticine quinine 合成法の開発を検討した。
3-lodoindole-2-carbaldehyde (40)を有機スズ試薬41と共に 一 酸化炭素気流中、Pd触媒存在下 反応させ、3—アクリロイルインド ー ル42を収率64%で得た。次に、42をGrignard 反応に付 すことで鍵前駆体のアリルアルコ ー ル4 3を収率79%で誘導できた。アリルアルコ ー ル 43を 、 酸素気流中、第2世代Hoveyda-Grubbs触媒存在下toluene中70℃でタンデムRCM ー 脱水素反 応を行ったところ望むcarbazole-1,4-quinone (44)を反応時間5 分 、 収率58%で合成した。最 後に、ピリジン環形成の最適条件を検討した結果、carbazole-1,4-quinone 44を6M塩酸存在下 o-dichlorobenzene中、マイクロ波照射下 、 70 ℃で加熱したところ 、 Pomeranz-Fritsch反応に続 いて芳香化が進行 し、 反応収率81%でellipticine quinine (45)を合成することができた。以上 のように 、 ellipticine quinine (45)のピリジン環形成のために必要なdiethoxyethylamino- methyl 基を3位にもつcarbazole-1,4-quinone 4 4合成を行うための分子設計を行い 、 合成ル ー トを検 討した結果 、 3-iodoindole-2-carbaldehyde (40)から4工程 、 総収率15%と、効率的なellipticine quinine (45)の新規合成ル ー トを確立することができた(Scheme 9)。
Ts OEt
|N ツ 人OEt B u
3Sn 上 41
l PdCl
2(dppf), BHT,
o co(1 atm)
N CHO
H 80 °C, 17 h DMF
40 64%
TsEtO
J TsE
t O
k ... /-OEt
`こ胃〗 42 79% 43 �_}-OEt
Hoveyda 2nd under 0 2
toluene
70 °C, 5 min 又 H 灯 0 O 心 O N E 、 t T • 0-dichlorobenzene 6MHCI 責 H ゜ °
with MW
58% 44 150 °C, 30 min 81% 45: Ellipticine quinone Scheme 9: Synthesis of ellipticine quinone (45) by tandem RCM-dehydrogenation.
-32-
次に、 本合成法を活用し、 本研究の目的の 一 つである未だ報告例の少ない A 環部へ置換基導
入した ellipticine quinone 誘導体合成を行い、 細胞増殖抑制活性評価試験を実施した。 出発原料
であるベンゼン環上に置換基を持つ 3-iodoindole-2-carbaldehyde 34 から先ほどと同様の方法で、
2 工程で誘導したアリルアルコ ー ル 47 に対し、 タンデム RCM
ー脱水素反応、 続く Pomeranz
Fritsch 反応に付すことで、 4 工程で ellipticine quinone 誘導体 49 を合成した (Scheme 10) 。 合成した 14 種類の ellipticine quinone 誘導体を用いて、 ヒト大腸癌由来細胞株 HCT-116 細 胞およびヒト白血病細胞 HL-60 に対する細胞増殖抑制活性評価試験を実施した (Table 2) 。
TS
OEt
ょ Nふ。Et
Bu
3Sn� 41
CO
(1 atm), B
HT Ts
Etq
R
1R
2-0:( ご R
10 / h_)-0
EtBrMg ヘ
廿
CHODMF 祀こ〕: ↓ 一面
→N CH
O
HtE ゜ ご
H 2町 R
34 46 47
Grubbs
2nd
,.,.
Ts
under 0
2R
'. ON 6M
HCI
� R
2� ド。
Et�
廿o
゜Etwith MW
48 49
Ellipticinequinones
; R
1=H, Me
O, Me,
Cl, N0
2,
CF
3, F
; R
2=
H, Me
O, Me,
Cl, N0
2,
CF
3, F
3