大規模ゲノム復元のための
de novo
アセンブリアルゴリズムに関する
研究
2015
年
3
月
遠藤友基
宇都宮大学大学院工学研究科
システム創成工学専攻
内容梗概
大規模ゲノムの解読は,生命システムの解明だけでなく,医療科 学,薬学,農学などの多様な応用が考えられ,様々な種を対象にゲノ ム解読の研究が行われている.ゲノム解読はシーケンサから得られた データを元に行われるが,近年,次世代シーケンサの登場によりシー ケンシングのコストパフォーマンスは飛躍的に向上し,短時間で大量 のデータを生成できるようになった.次世代シーケンサは対象の塩基 配列の短い断片(リード)を大量に出力するため,それを正しく並べ 替えて元の塩基配列の決定するためのアルゴリズムが必要になる.そ のようなアルゴリズムはde novo アセンブリアルゴリズムと呼ばれ, 様々な手法が提案されている.特にVelvetは消費メモリや計算時間な どについてパフォーマンスに優れており,コンティグの精度も比較的 高いため,最も普及しているアセンブリ手法の一つとなっている.し かし,リードの総量が数十Gbp(bp : 塩基対)を越える大規模なデータ のアセンブリを行う場合,Velvetを用いても実行時に要求されるメモ リが非常に膨大になってしまいメモリ不足となってしまう.そこで本 研究では,次世代シーケンサから得られた大量のデータに対して,大 規模なゲノムのアセンブリが可能となる,消費メモリの少ないde novo アセンブリアルゴリズムを提案する.実験では,E.coli K-12 strain MG1655及びヒトの14番染色体から得られたリードに対して,本手法 とVelvet及びSOAPdenovoとでアセンブリを行った.その結果,本 手法はE. coliに対しては他手法の約20%,ヒト14番染色体に対して は他手法の約60%の消費メモリ量でde novoアセンブリが可能である ことを確認した.De novo Assembly Algorithm for Huge Genomes with
Massively Short Read Sequencing
Yuki Endo
Abstract
Sequencing the whole genome of various species has many ap-plications, not only in understanding biological systems, but also in medicine, pharmacy, and agriculture. In recent years, the emergence of high-throughput next generation sequencing technologies has dra-matically reduced the time and costs for whole genome sequencing. These new technologies provide ultrahigh throughput with a lower per-unit data cost. However, the data are generated from very short fragments of DNA. Thus, it is very important to develop algorithms for merging these fragments. One method of merging these fragments without using a reference dataset is called de novo assembly. Many algorithms for de novo assembly have been proposed in recent years. Velvet and SOAPdenovo2 are well-known assembly algorithms, which have good performance in terms of memory and time consumption. However, memory consumption increases dramatically when the size of input fragments is larger. Therefore, it is necessary to develop an alternative algorithm with low memory usage. In this paper, we propose an algorithm for de novo assembly with lower memory. In our experiments using the E.coli K-12 strain MG 1655, the av-erage maximum memory consumption of the proposed method was approximately 20% of that of the popular assemblers. Moreover, in the experiments using human chromosome 14, the average amount of memory of our method was approximately 60% of that of the popular assemblers.
発表論文
• 学・協会誌発表論文
1. Yuki Endo,Fubito Toyama,Chikafumi Chiba,Hiroshi Mori and Kenji Shoji, “A Memory Efficient Short Read De Novo Assem-bly Algorithm,” IPSJ transaction on Bioinformatics, Vol.8, pp.2-8,2015.
• 国際会議等発表論文
1. Yuki Endo, Fubito Toyama, Chikafumi Chiba, Hiroshi Mori and Kenji Shoji, “De Novo Short Read Assembly Algorithm with Low Memory Usage,” BIOINFORMATICS 2014 - Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms, pp.215-220, 2014. • 学会・研究会等講演論文・ポスター発表 1. 遠藤友基,外山史,東海林健二,宮道壽一,“大規模ゲノム復元の ための de novo アセンブリアルゴリズムの開発,” FIT2011,第 2分冊,pp.569-570,2011. 2. 遠藤友基,“大規模ゲノム復元のための de novo アセンブリアル ゴリズム,” NGS 現場の会第二回研究会,2012 年 5 月 24 25 日, ホテル阪急エキスポパーク. 3. 遠藤友基,“大規模ゲノム解読のための de novo アセンブリアル ゴリズムの開発,” 生命情報科学若手の会第四回研究会,2013 年 3月 1 3 日,自然科学研究機構岡崎コンファレンスセンター.
目 次
内容梗概 i Abstract ii 目 次 v 1 まえがき 1 2 DNAとゲノム 7 2.1 DNAと塩基配列 . . . 7 2.2 ゲノム . . . 9 3 塩基配列の決定方法 13 3.1 シーケンシング . . . 13 3.2 de novoアセンブリ . . . 19 3.3 リードのフォーマット . . . 22 4 de Bruijnグラフを用いた de novo アセンブリアルゴリズム 25 4.1 de Bruijnグラフを用いたアセンブリの概要 . . . 25 4.2 k-mer整数 . . . 26 4.3 ハッシング . . . 28 4.4 de Bruijnグラフの構築 . . . 30 4.5 エッジの除去とコンティグの生成 . . . 34 4.6 de Bruijnグラフを用いた de novo アセンブラ . . . . 365 提案手法 39 5.1 k-mer整数の登録とグラフの構築 . . . 41 5.2 分岐と閉路の除去 . . . 45 5.3 部分グラフの連結とコンティグの生成 . . . 48 6 実験 55 6.1 E.coli の de novo アセンブリ . . . . 56 6.2 ヒトの 14 番染色体の de novo アセンブリ . . . . 58 6.3 考察 . . . 61 7 むすび 63 7.1 本研究のまとめ . . . 63 7.2 今後の課題・展望 . . . 64 謝 辞 67 参考文献 69 vi
第
1
章 まえがき
近年多くの生物を対象に実施されているゲノムプロジェクト,そしてそ れに伴うに解析技術の発展によって,遺伝子やタンパク質の構造といった 生命の持つ ”情報 ”を大量に得ることが可能となってきた.一方,それら の大量の情報から生物学的な意味を抽出することが困難となる場面も増加 してきた.このような課題に取り組むために,バイオインフォマティクス (bioinformatics)と呼ばれる分野の重要性が注目されてきている.バイオイ ンフォマティクスとは,生物学 (biology) と情報学 (information science) が 融合した新たな学問・技術であり,実験等で得られる膨大な生命が持つデー タからコンピュータを用いて生物学的な発見を行う分野である.バイオイ ンフォマティクスの研究対象分野は様々であるが,その中でも重要な分野 の一つとして生物の持つゲノムの解読がある.ここでゲノム(genome)と は,”gene(遺伝子)”と集合をあらわす”-ome”を組み合わせた言葉であり, 一般に遺伝情報全体のことを指す.これを解読することは遺伝子の役割を 解明するための重要なステップとなる.ゲノムを解読するためには DNA の 全塩基配列を解読することが必要であるが,この塩基配列は非常に長く,そ の並び方を決定するために必要なデータも膨大な量となるため,現在のコ ンピュータの処理能力をはるかに超えてしまう非常に困難な問題となる.そ のような問題を解決するためのアルゴリズムの開発がバイオインフォマティ クスの主要な研究分野であり、その成果は塩基配列の決定に大きく貢献し てきた. 特にヒトゲノムのような大規模なゲノムの解読は,生命システ ムの解明だけでなく,医療科学,薬学,農学などの多様な応用が考えられ るため,生命に関する多くの分野において重要な要素となっている. ゲノムを解読するために塩基配列を決定することはシーケンシングと呼第 1 章 まえがき ばれる.シーケンシングの最も基本的な手法として,マクサム-ギルバート 法 (化学分解法)[1],サンガー法 (酵素法)[2]がある.マクサム-ギルバート法 は,特定の塩基の位置で DNA 分子を切断する化学的な処理を行う方法であ るが,この方法は充分な量の DNA と,取り扱いに注意を要するヒドラジン などを必要とするので,現在,全塩基配列の決定にはサンガー法を用いるの が一般的になってきている.サンガー法では,一本鎖 DNA から酵素 (DNA ポリメラーゼ) を用いて相補的なポリヌクレオチド鎖を形成し,特定の塩基 の位置で酵素反応を停止させることによって,短い塩基配列の断片 (リード) を得る.このリードを繋ぎ合わせることで,元の全体の塩基配列を再現す る. このサンガー法は様々な改良が加えられ,2000 年代前半には全自動で比 較的多くのリードを生成できる装置が登場した.シーケンシングを行う装 置はシーケンサと呼ばれ,リードを生成する処理は並列化によってより高 速化されていき,読み取ることのできるリードの長さは 1000bp(base pair: 塩基対) 程度まで増加し読み取り精度も向上した.それに伴い,それまで手 作業で行われていたリードの重複部分からの元の全塩基配列の決定をコン ピュータで行う技術が必要となってきた.この技術はアセンブリアルゴリズ ムと呼ばれ,そのうちとくに未知である塩基配列を決定するものは de novo アセンブリアルゴリズムと呼ばれる.de novo アセンブリアルゴリズムはバ イオインフォマティクスの分野の中でも重要なテーマの一つとなっている. de novoアセンブリでは,シーケンサで生成されるリードの長さが長ければ 長い程,数が多ければ多いほど再現率は高くなるが,その分要求されるコ ンピュータ資源は大きくなる.また,シーケンサの読み取り誤差への対応 や,反復配列と呼ばれる同じ配列が反復して (特に数回以上) 出現する領域 への対応が必要である.特により短いリードでアセンブリを行おうとした 場合,リードの比較・探索が高速にできる反面,アセンブリの位置を決め る段階で反復配列が含まれていると,正しい並び方を得ることが困難にな る.シーケンサが出力するリードの長さ・正確性を重視した改良が進むに つれてそのコスト・解析速度が大幅に増大し,大規模な DNA 鎖の塩基配列 2
第 1 章 まえがき 決定のためにはより低コストにシーケンシングを行う必要が出てきた.そ のため近年では,読み取るリードの長さを犠牲にし,より低コストで短時 間で大量のリードを得る方法が主流となってきた.生成されるリードが短 いほど,リードを読み取る処理の並列数を大幅に増やすことができ,且つ 読み取りに必要な試薬を削減できる.すなわち,短いリードの生成であれ ば,シーケンシングのスループットを飛躍的に高めることできる.この短 いリードを短時間に大量に生成することを目的とした装置は次世代シーケ ンサと呼ばれ,2005 年以降,様々な次世代シーケンサが登場した.次世代 シーケンサの出力するリードの長さは数十∼数百 bp であり,従来のシーケ ンサのリードの長さと比較すると遥かに短く,その読み取り精度も比較的 低い.しかし,短時間で極めて多くのリードを出力することができる.そ のため,次世代シーケンサによって得られる大量の,しかし精度の低いデ ジタルデータを用いた塩基配列の決定を行うためのアセンブリアルゴリズ ムが必要となってきた. 次世代シーケンサに対応した de novo アセンブリアルゴリズムとして, Edena[3],SSAKE[4],VCAKE[5],Velvet[6],ABySS[7],SOAPdenovo[8]など が挙げられる.これらのアセンブリアルゴリズムはそのアルゴリズムの特徴 によって大きく 2 種類に分類される.一つは Overlap-Layout-Consensus(OLC) 法を用いたものであり,Edena,SSAKE,VCAKE がこれに分類される. OLC法は比較的早期に提案された手法で,各リードをノードとして表現 し,重複する塩基配列があるノード同士を辺で結んだグラフを作成し,全 てのノードを通るパスを探索する.そしてパスを辿りながらノードの文字 列を連結していくことで長い塩基配列 (コンティグ) を求める.ここで,コ ンティグとはリードに対してアセンブリを行うことで得られるある程度長 い塩基配列であり,最も理想的な場合では 1 本のコンティグが元の DNA の 塩基配列となる.この手法はノード間の重複する塩基配列の長さがある程 度以上必要となるため,比較的長いリードのアセンブリに用いられる.も う一つのグループとして de Bruijn グラフ[9] を用いたものがあり,Velvet,
第 1 章 まえがき
ABySS,SOAPdenovo がこれに分類されている.de Bruijn グラフとは,任 意のノードが k 文字の文字列に対応し,辺で連結された両ノードの各文字 列は k− 1 文字だけ重複するという特徴を持つグラフである.これらの手法 では,まず全てのリードから k 文字の塩基配列を抽出してノードとし,次 に k− 1 文字の重複があるものを辺で結ぶことで de Bruijn グラフを構築す る.そして,得られた de Bruijn グラフ内のパスからコンティグを求めてい る.de Bruijn グラフを用いた手法は短いリードのアセンブリに向いており, 次世代シーケンサの出力するリードにより適している.
de Bruijn グラフを用いたアルゴリズムのうち,Velvet や SOAPdenovo が 比較的少ない消費メモリ量で高精度のコンティグが得られるので,現在で は広く普及している.しかし,数 Gbp を越える大規模な全塩基配列を決定 する場合,これらのアルゴリズムを用いても,実行時に要求される消費メ モリ量が非常に膨大でメモリ不足となりやすい[10] .空きメモリ量が不足し た場合,プログラムが強制終了されてしまう可能性もあるが,ほとんど場 合はスワップと呼ばれる OS の機能が利用される.スワップとは,スワップ 領域と呼ばれる専用の保存領域をメインメモリの代替領域として使用する 機能である.一般的なコンピュータの OS は,このスワップを行うことでプ ログラムの続行を試みる.しかし,スワップ領域はハードディスク等の補 助記憶の一部であり,メインメモリと比較してアクセス速度は極めて遅い. その結果,コンピュータの動作速度が著しく低下し,特に大規模な de novo アセンブリでは実行時間が大幅に増加してしまう.そのため,de novo アセ ンブリではその消費メモリ量が重要な課題となっている.
本研究では,de Bruijn グラフを用いた de novo アセンブリアルゴリズム の特徴と課題を明らかにし,次世代シーケンサから得られた大量のリード を用いて,大規模なゲノムに対しても de novo アセンブリが可能となるよ うに,消費メモリ量の少ない de novo アセンブリアルゴリズムを提案する. 本論文で提案するアルゴリズムは,Velvet と同様に de Bruijn グラフを用い てアセンブリを行うが,実際に構築するグラフでは,どのノードに辺があ 4
第 1 章 まえがき るかという情報は保持せずに,あるノードに対してどのノードとの間に辺 が存在するかを必要な時に 4.2 節で述べる k-mer 整数を用いて効率的に調べ る.これにより,大量の辺情報を保持する必要が無くなるため,大幅なメモ リ消費量の削減が実現できる.又,入力データの文字列をバイナリデータ として表現することで更なるメモリ削減を行っている.実験では,E.coli K-12 strain MG1655及びヒトの 14 番染色体から得られたリードに対して, 本手法,Velvet 及び SOAPdenovo でアセンブリを行い,消費メモリ量を比 較した.その結果,本手法は E. coli に対しては他手法の約 20%,ヒト 14 番 染色体に対しては他手法の約 60%の消費メモリ量で de novo アセンブリが 可能であることを確認した. 以下,第 2 章では DNA と塩基配列,ゲノムの役割とそれらの関係につい て,第 3 章では一般的な塩基配列の決定方法について述べる.第 4 章では de Bruijnグラフを用いた de novo アセンブリアルゴリズムについて説明す る.第 5 章で提案手法のアルゴリズムについて説明し,第 6 章では実験とそ れに対する考察を述べる.最後に第 7 章で本研究についてまとめる.
第
2
章
DNA
とゲノム
ゲノムとは膨大な量の遺伝情報であり,それらの情報には遺伝子や遺伝 子の発現の制御に関わるものなど,その生物に必要な全ての情報が全て含 まれている.遺伝子の発現とは,遺伝子の持つ情報がタンパク質の合成を通 じて細胞の構造や機能へと変換される過程のことを指す.ゲノムを解読す るためには,DNA と呼ばれる物質を構成する分子の並び方 (全塩基配列) の 決定が必要となる.本章では,まず DNA の構造と塩基配列について述べ, ゲノムの役割と塩基配列との関係について論ずる.2.1
DNA
と塩基配列
DNA(Deoxyribo Nucleic Acid) とは,デオキシリボ核酸と呼ばれる核酸 の一種であり,高分子生体物質である.真正細菌・古細菌においては細胞質 に環状 DNA として存在する.人間のような真核生物においては通常は細胞 核内に存在し,細胞分裂の際にヒストンと呼ばれるタンパク質に巻きつき, 更に幾重にも折りたたまれ,染色体として現れる.DNA は,ヌクレオチド (Nucleotide)と呼ばれる物質によって構成されている.ヌクレオチドが鎖の ように連なったものはポリヌクレオチド (polynucleotide) と呼ばれ,DNA はこのポリヌクレオチドが二本平行に結合しており,二重らせん構造と呼 ばれるらせん状の構造をとっている.ヌクレオチドには塩基と呼ばれる物 質が含まれており,その種類によって図 1 に示すようなアデニン (adenine), シトシン (aytosine),グアニン (guanine),チミン (thymine) の 4 種類の塩基 があり,各塩基はそれぞれ A,C,G,T と略される.各ポリヌクレオチド の塩基が水素結合で結ばれることによって,DNA の二重らせん構造を形成
第 2 章 DNA とゲノム している.塩基は一方の鎖の塩基が決まればもう一方の鎖に対応する塩基 も決まるという性質 (塩基の相補性) を持っており,A と T,C と G という 組み合わせで結ばれている.図 2 に相補的に結合された二本鎖の模式図を示 す.ヌクレオチドは含まれている塩基によってその種類が決まるため,DNA の (ポリヌクレオチド) の長さは bp(base pair:塩基対) という単位で表記され る.生物の種によってその長さは大きく異なり,短いものでは数 kbp から数 Mbp,特に真核生物などの DNA は数 Gbp という膨大な長さである.例え ば,ヒトの持つ DNA の長さは約 3Gbp である.また細胞分裂時には,DNA はヒストンと呼ばれるタンパク質に巻き複雑に折りたたまれ,複数に分割さ れる.この構造は染色体と呼ばれる.ヒトの場合は,二本鎖の DNA が 23 組 の染色体へと分割される.DNA のヌクレオチドの結合順を塩基の種類に注 目して記述したものを塩基配列と呼び,DNA の塩基配列は”AACCCGT...” のように,一文字の略称で記述される.詳細は後述となるが,生物の持つ 遺伝情報はこの塩基配列の形で保持されており,上記のような塩基配列の 並び方を求めることが遺伝情報の解析において非常に基本的かつ重要な作 業となる. 図 1. 4 種類の塩基 8
第 2 章 DNA とゲノム 図 2. DNA の相補的二本鎖構造
2.2
ゲノム
2.1 節で述べたように,塩基配列の並び方には,生物の持つ全ての遺伝情 報が含まれている.遺伝情報とは,遺伝現象によって親から子に伝わる情 報であり,生物を構築する細胞の形質発現や複製に関わる情報のことを指 す.遺伝情報の最小単位は遺伝子と呼ばれ,ヒトは約 2 万 3 千個の遺伝子を 持つと言われている.そして,生物の持つ全ての遺伝情報 (遺伝子) のこと をまとめてゲノム (genome) と呼ぶ.つまり,ゲノムを解読することによっ て,様々な遺伝子が生物の肉体の構築にどのように関与しているのかを明 らかにすることができる.遺伝情報は様々な種類があり,代表的なものと して生物の肉体を構成する各種のタンパク質の構造や,その生成の制御に 関与するものがある.タンパク質は約 20 種類あるアミノ酸が鎖上に結合し たものであり,生物の神経細胞や骨、臓器といったものはすべてタンパク 質から構成されている.塩基配列には,発現するタンパク質の種類を決定 するアミノ酸配列や,発現するタイミング等の全ての情報が含まれている. 例えば,塩基配列のコーディング領域と呼ばれる領域では,トリプレット (triplet)と呼ばれる 3 つの塩基が 1 種類のアミノ酸を意味しており,この並第 2 章 DNA とゲノム び方によってアミノ酸の結合順序が決まる.アミノ酸に対応するトリプレッ トはコドン (codon) と呼ばれ,表 1 に示すように 20 種のアミノ酸とコドン が対応している.また,表 2 は表 1 のアミノ酸名と表記方法と対応を示して いる.このコドンの並び方 (アミノ酸配列) から生物に必要なタンパク質が 合成される.2.1 節では DNA の塩基は A,C,G,T で表現されると述べた が,実際は DNA から RNA(RiboNucleic Asid) と呼ばれる生体物質 (DNA と同様の核酸の一種) へ塩基配列がコピー (転写) が行われ、RNA からタン パク質が合成される.RNA では塩基 T の代わりに U(uracil) が用いられる ため,表 1 では T の代わりに U が対応している.RNA は DNA と同様核酸 の一種ではあるが,DNA は遺伝情報の全てが保存されたものであるのに対 し,RNA はその情報の一時的な処理・運搬・翻訳等を担う物質である.ま た,RNA からタンパク質を作ることは翻訳と呼ばれ,DNA から RNA へ の転写,RNA からタンパク質への翻訳という一連の流れはセントラルドグ マ (central dogma) と呼ばれる.この他にも塩基配列には様々な遺伝情報が コードされており,タンパク質の合成に関与するものの他にも様々な情報が 含まれている.例えば,いつ,どのような条件になったら転写を開始するか といった,転写の活性を調節する特殊な塩基配列などがある.このように, 塩基配列は様々な情報を持っており,その並び方を調べることでゲノムを解 析することができ,その生物が持つ様々な性質を知ることができる.また, 塩基配列の並び方は一生変わることがないため,それによって決定される 各個体の性質は一生変わることがはない.塩基配列の全てがタンパク質合 成に関わるものではなく,そもそも未だ解明されていない領域も多いため, 塩基配列の並び方を決定しただけではゲノムを完全解読することはできな い.しかし,全塩基配列の決定は,生物の持つ遺伝情報全体,生物一個体 を形成するのに必要な情報を明らかにするの非常に重要なステップである. 10
第 2 章 DNA とゲノム 表 1. コドンとアミノ酸の対応 2塩基目 1塩基目 T C A G 3塩基目 U UUU:Phe UUC:Phe UUA:Leu UUG:Leu UCU:Ser UCC:Ser UCA:Ser UCG:Ser UAU:Tyr UAC:Tyr UAA:終止 UAG:終止 UGU:Cys UGC:Cys UGA:終止 UGG:Trp U C A G C CUU:Leu CUC:Leu CUA:Leu CUG:Leu CCU:Pro CCC:Pro CCA:Pro CCG:Pro CAU:His CAC:His CAA:Gln CAG:Gln CGU:Arg CGC:Arg CGA:Arg CGG:Arg U C A G A GUU:Ile GUC:Ile GUA:Ile GUG:Met GCU:Thr GCC:Thr GCA:Thr GCG:Thr GAU:Asn GAC:Asn GAA:Lys GAG:Lys GGU:Ser GGC:Ser GGA:Arg GGG:Arg U C A G G AUU:Val AUC:Val AUA:Val AUG:Val ACU:Ala ACC:Ala ACA:Ala ACG:Ala AAU:Asp AAC:Asp AAA:Glu AAG:Glu AGU:Gly AGC:Gly AGA:Gly AGG:Gly U C A G 表 2. アミノ酸の表記方法 アミノ酸名 3文字表記 アラニン (Alanine) Ala アルギニン(Arginine) Arg アスパラギン (Asparagine) Asn
アスパラギン酸 (Asparastic acid) Asp
システイン(Cysteine) Cys
グルタミン(Glutamine) Gln
グルタミン酸 (Glutamic acid) Glu
グリシン (Glycine) Gly ヒスチジン(Histidine) His イソロイシン (Isoleucine) Ile ロイシン (Leucine) Leu リジン (Lysine) Lys メチオニン(Methionine) Met フェニルアラニン (Phenylalanine) Phe プロリン (Proline) Pro セリン (Serine) Ser スレオニン(Threonine) Thr トリプトファン (Tryptophan) Trp チロシン (Tyrosine) Tyr バリン (Valine) Val
第
3
章 塩基配列の決定方法
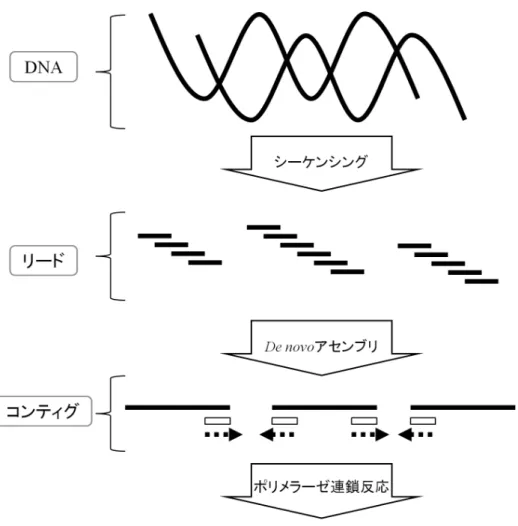
ゲノムを解読するためには,まず全塩基配列を決定することが必要とな る.一般的に,塩基配列を決定するには幾つかの手順が必要となる.DNA の一般的な塩基配列決定の流れを図 3 に示す.まず未知の DNA から次世代 シーケンサを用いてシーケンシングを行うことで,断片的な塩基配列の情 報 (リード) を得る.次世代シーケンサでは非常に大量のリードを出力する ことができるが,その長さは DNA に対して非常に短いため,そのまま遺伝 子解析等に利用することはできない.そこで,リード間の重複関係を利用 することでリード同士をつなぎ合わせていき,より長い塩基配列 (コンティ グ) を生成する.理想的には 1 本のコンティグが元の DNA の塩基配列とな ることであるが,ほとんどの場合は複数本のコンティグとなる.複数のコ ンティグ間には隙間 (ギャップ) があるため,必要に応じて PCR 法と呼ばれ る手法によって DNA の増幅を行うこことでギャップを埋める.このように, リードから複数のコンティグを生成することで元の DNA の塩基配列を再構 成する.本章では,一般的な塩基配列の決定の各手順について述べる.3.1
シーケンシング
シーケンシングとは,本来は塩基配列の並び方を決定すること自体を指 すが,次世代シーケンサを利用した塩基配列の決定では,リードを生成す る (断片的な塩基配列の決定) のことを意味することが多い.そのため,本 論文でも断片的な塩基配列の決定をシーケンシングと呼ぶ.シーケンシン グの基本的な原理としてはマクサム-ギルバート法とサンガー法が一般的で あるが,ここではより一般的なサンガー法について説明し,近年主流となっ第 3 章 塩基配列の決定方法
図 3. 一般的な塩基配列決定の流れ
第 3 章 塩基配列の決定方法
てきている次世代シーケンサについて解説する.
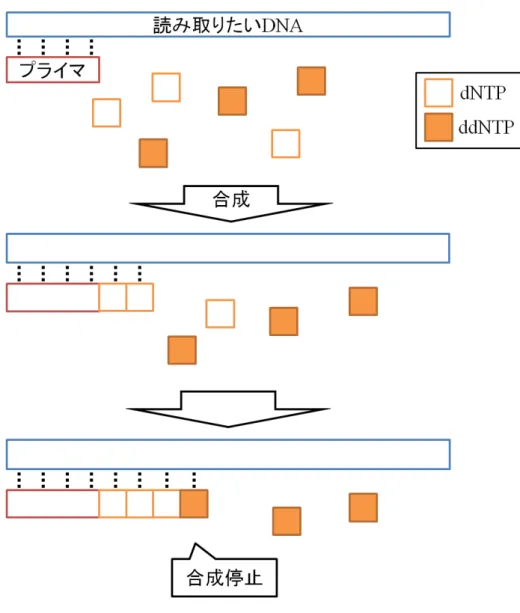
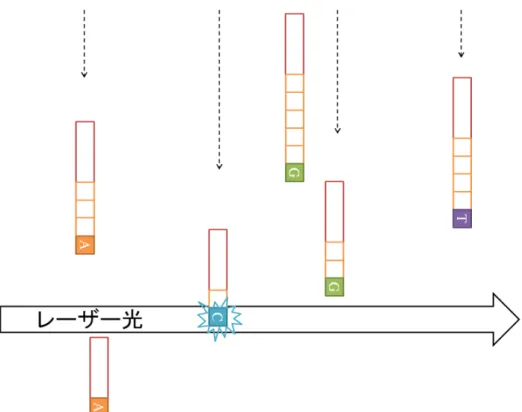
まず,図 4 に示すように,解読したい DNA(鋳型 DNA) とその一部と相補 的な短い DNA(プライマ DNA) を,DNA 合成することができる酵素 (DNA ポリメラーゼ) と DNA を構成するデオキシヌクレオチド (dNTP) の共存さ せることで酵素反応を起こさせる.この酵素反応により,鋳型 DNA に相補 的なデオキシヌクレオチドがプライマ DNA に次々に結合し,二本鎖 DNA の合成が行われる.しかし,デオキシヌクレオチドの特定の原子を本来とは 異なる原子で置換したデオキシヌクレオチド (ジデオキシヌクレオチドと呼 ぶ) が結合されると,酵素反応が止まる.ここで,特定の原子とはヌクレオ チドの連結に必要な原子である.例えばアデニンではヒドロキシル基が連 結に本来必要であり,これを水素原子で置換している.各デオキシヌクレオ チドとジデオキシヌクレオチドの共存下で上記の合成反応をさせると,デ オキシヌクレオチドが結合されれば合成は進行し,ジデオキシヌクレオチ ドが結合されれば合成が停止する.両者はランダムで結合されるため,様々 な長さの塩基配列の集団が得られることになる.この性質を利用し,ジデオ キシヌクレオチド (ddNTP) をターミネータ (鎖停止ヌクレオチド) として一 種類のみ加えることで,相補的な配列のポリヌクレオチド鎖を合成し,特 定の塩基の位置でその合成反応を停止させることができる.このとき,デ オキシヌクレオチド自体,またはターミネータを蛍光標識しておく.その 後,電気泳動によって二本鎖を分離する.分離の際に短い塩基配列ほど速 く移動するため,図 5 に示すように塩基配列の長さの短い順に流れてくる. これらにレーザー光を当ててターミネータの蛍光標識の発色を次々に読み 取っていき,塩基配列の並びを得る. 第 1 章で述べたように,サンガー法は多数の研究により改良が重ねられ, 生成できる断片の長さは 1000bp まで増加したが,そのコスト・必要な時間 も増大し,大規模な DNA の塩基配列の決定に用いるのは困難となった.そ のため,読み取るリードの長さを数十∼数百 bp と短くすることで,読み取 りの並列数を大幅に増加させたシーケンサが主流となっていった.このよう
第 3 章 塩基配列の決定方法
なシーケンサは次世代シーケンサと呼ばれ,従来型と比較して高速・低コス トでリードを読み取ることが可能である.従って,短時間で大量のリードを 低コストで生成することができるという特徴を持つ.代表的な次世代シーケ ンサにはイルミナ社の Genome Analyzer(GA) がある.GA は従来型のシー ケンサと比較して飛躍的にスループットが向上している.例えば,GA を用 いてヒトゲノムからリードを生成するとき,従来型のシーケンサと比較して 必要な時間・コストはそれぞれ 1/160,1/52000 に削減でき,従来型のシー ケンサでは 1 ランあたり 300kbp の塩基を読み取るのに対し,次世代シーケ ンサでは 27Gbp もの塩基を読み取ることができる.近年では,このような 次世代シーケンサで短い大量のリードを生成することが主流となってきて おり,それに対応するアセンブリアルゴリズムが必要となってきている.更 に,低コストでより多くのリードを生成することを重視した結果,ほとん どの次世代シーケンサでは読み取り精度が従来型と比較して低下し,生成 するリードにはシーケンスエラーが多く含まれている.ここでシーケンス エラーとは,シーケンサから出力されたリードに含まれる読み取りミスで ある.例えば,”AACCGGTTAACCGGTT”という塩基配列をシーケンサ によって処理した時,”AACCGCTT”や”CCGGN TAA”(’N ’ は読み取りに 失敗している) のようなリードが出力されてしまう.シーケンサ自身の対策 として,読み取り精度の向上を目指した改良や,出力できるリード長を伸 ばすことで上記二つの問題に対応したシーケンサもある.また,単純に同 じ領域に対して読み取り・複製を何度も行うことで,シーケンスエラーをあ る程度減少させることも可能である.しかし,最新のシーケンサでもその 読み取り精度は 100%ではなく,完全に対策されているとは言えない.以上 のことから,アセンブリアルゴリズムは大量のリードを効率的に扱うこと に加えて,これらの問題に対する処理をどのように行うかが重要になる. 16
第 3 章 塩基配列の決定方法
第 3 章 塩基配列の決定方法
図 5. 発色による塩基配列読み取りの模式図
第 3 章 塩基配列の決定方法
3.2
de novo
アセンブリ
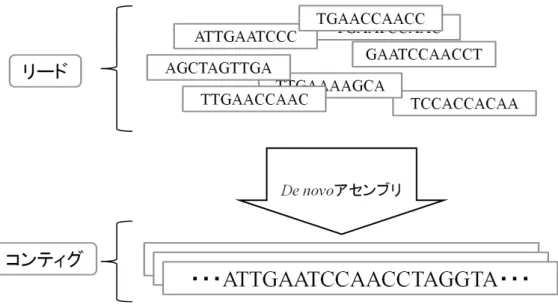
現在では次世代シーケンサと呼ばれる装置の登場によって大量のリード を短時間で取得できるようになった.しかし,ここで得られるリードは元の 全塩基配列の” どこか” の領域であり,得られたリードが実際の元の配列の どの領域を表しているのかは知ることができない.そのため,複数のリー ド間で配列の重複関係を調べ,リードを連結していくことでより長い塩基 配列 (コンティグ) を生成する.図 6 に示すように,このリードからコンティ グを得る技術をアセンブリと呼び,とくにそれまで未知であった DNA の塩 基配列の決定を行う技術を de novo アセンブリと呼ぶ.現在,次世代シーケ ンサの登場と改良に伴い様々なアルゴリズムが提案されており,それを利 用したプログラム・ソフトウェアは de novo アセンブラと呼ばれる.一方, 既に全塩基配列が決定されている DNA の塩基配列に対して,シーケンサに よって得られたリードがこの既知の DNA 鎖のどの一部分になるかを決定す る,マッピングアセンブリと呼ばれる手法がある.この手法は,類似する種 族の生物や,同種の生物の個体差を解析するのに有効とされる.本研究で は未知の全塩基配列の決定が目的であるため,本稿では前者の de novo ア センブリアルゴリズムについての検討を行う. アセンブリする上で反復配列への対応やシーケンスエラーの発生という 問題は避けられず,使用するリード長によっては完全に元の DNA を再現す ることはできない[11].ここで,反復配列とは,塩基配列上において同じ配 列が反復して出現することを言い,主に真核生物など特に進化した動植物に 多く見られる.例えば,”ACGT”という塩基配列が”ACGTACGTACGT...” のように何度も連続して出現したり、”AAA...”のような一文字の繰り返し や,更に数十 bp の配列が反復して現れることもある.例えば,反復配列が リード長より長い場合,繰り返されている配列が何回出現しているのかを リードから判断することはできない.ただし,そのような場合でもある程 度長いコンティグを決定することはできる.コンティグに対しアセンブリ で決定できなかった領域,すなわち元の全塩基配列の内のコンティグ以外第 3 章 塩基配列の決定方法
図 6. リードからアセンブリを行う流れ
の部分はギャップと呼ばれる.
ギャップ部分の配列が決定できない限り,全塩基配列の完全な再現は不可 能である.しかし,PCR 法 (Polymerase Chain Reaction,ポリメラーゼ連 鎖反応)[12]と呼ばれる手法によって,数十 bp の短いギャップならその部分 の配列を決定することができる.PCR 法は,酵素法に似た DNA 増幅の手 法であり,DNA 上で特定の領域だけを選択的に増幅させることができる. DNAを構成するポリヌクレオチドは非常に不安定な結合であるため,温度 変化によって簡単に切断・再結合が可能である.PCR 法はそれを利用し,3 段階の温度変化を何度も繰り返すことで DNA の一部分だけを選択的に増 幅させる.PCR 法に必要な試薬等は微量であり,増幅に要する時間も短く, サーマルサイクラーと呼ばれる全自動の卓上用装置を用いることで比較的 容易に増幅することができる.そのため,de novo アセンブリ等によってあ る程度の長さのコンティグを生成することができれば,この技術を利用す ることで多少のギャップは埋めることができ,最終的に元の塩基配列のより 多くの領域を再現できる.また,コンティグのようにギャップが残っていた 20
第 3 章 塩基配列の決定方法 り並べ方が不確定である配列はドラフト配列 (ドラフトゲノム) とも呼ばれ る.ある程度の長さのコンティグを持ったドラフト配列であれば,その情 報からおおよそのゲノムを明らかにすることができる.実際にヒトのドラ フト配列が決定されたことにより,ヒトの持つ遺伝子の数や構造などのヒ トゲノムの輪郭が明らかにされている[13] [14].更に,ドラフト配列とその生 物の近縁種の遺伝子情報用いることで,新たな遺伝子の構造や遺伝子機能 の予測にも利用することができる.ドラフト配列の情報は様々なゲノム解 析に対して有効に利用することができる.以上のことから,de novo アセン ブリの結果が複数のコンティグであっても,その情報はゲノム解析に対し て様々な面から貢献できる.
第 3 章 塩基配列の決定方法
3.3
リードのフォーマット
リードの種類として,シングルエンドリード (single-end read)・ペアエン ドリード (paired-end read) がある.シングルエンドリードは DNA 断片の 片側をシーケンシングする手法である.これに対して、ペアエンドリード は断片の両側からシーケンシングする方法である.具体的には,ペアエン ドリードは一つ目の配列情報,二つ目の配列情報,これらの配列に挟まれ た領域 (インサート) のサイズの三つの情報から成り立っている.ペアエン ドリードのインサートのサイズは両端の配列の距離であり,塩基配列の情 報だけでは知りえなかった,より大きな構造を把握するための手掛かりと なる.de novo アセンブリでは,コンティグ間の関係を調べるために利用す ることが多い.インサートの情報だけでギャップを直接埋めることはできな いが,生成したコンティグ間の距離や位置関係を決定したり,繰り返し配 列の解決に利用することができる. また,リードにはシーケンシングのクオリティスコアと呼ばれる情報を 別途利用する場合がある.クオリティスコアは各塩基ごと設定されており, 一般的には対応する塩基のシーケンシングエラーの確率を表している.塩 基配列のシーケンスデータ (ACGT からなる文字列) のみを保存する場合, FASTAフォーマットと呼ばれるテキストファイルの記述方式が用いられて いる.FASTA フォーマットでは,1 つのシーケンスのデータは “>” で始まる 1行のヘッダと、2 行目以降の実際のシーケンス文字列で構成される。ヘッ ダでは、“>” の次にシーケンスデータを識別するための文字列を記述し、続 けてそのシーケンスデータを説明する文字列を記述する.“>” で始まる別 の行が出現すると、そこでシーケンスデータが区切られ、別のシーケンス データが始まる。一方,塩基配列のシーケンスデータとクオリティスコアを 併せて保存する場合は,FASTQ フォーマット[15]と呼ばれる記述形式が用い られている.FASTQ フォーマットでは,塩基配列とクオリティスコアは各 1文字の ASCII 文字で表さている.FASTA フォーマットと FASTQ フォー マットは,次世代シーケンサー等から出力された塩基配列のデータを保存
第 3 章 塩基配列の決定方法
する際のフォーマットとして標準的な記述形式となっており,公開されてい るリードのほとんどがこれらの形式で表されている.
第
4
章
de Bruijn
グラフを用いた
de novo
アセンブリアルゴリズム
一般的な de novo アセンブラでは de Bruijn グラフを用いたものが多く, 提案手法においても de Bruijn グラフを用いた代表的なアセンブラである Velvetをベースに,消費メモリ量を削減するようにアルゴリズムを改良し ている.本章では,Velvet や SOAPdenovo 等の de novo アセンブラの多く に用いられている,de Bruijn グラフを用いた de novo アセンブリアルゴリ ズムの原理と課題について述べ,提案手法の実現方針について論じる.
4.1
de Bruijn
グラフを用いたアセンブリの概要
de Bruijn グラフを用いたアセンブリでは,図 6 で示したようにリードか らコンティグを求めるものであるが,アセンブリの問題を de Bruijn グラフ の構築とグラフの余分な枝を取り除く問題に帰着させ,それを解くアルゴ リズムを実装している.de Bruijn グラフを用いたアセンブリではまず,全 てのリードから k-mer(k は整数) と呼ばれる kbp の塩基配列を取り出し,そ れらをノードとする de Bruijn グラフを構築する.このように構築した de Bruijnグラフにはシーケンスエラーや反復配列等の原因によって生じた分 岐や閉路が多く含まれるため,不要なエッジを削除することでそれを解決 し,グラフ内のノードを辿ることでコンティグを生成する.前章で述べた ように,リードから未知の元の塩基配列をすべて決定することは困難であ り,とくに大規模なものに対して正しい全塩基配列を再現するのは極めて 困難である.そのため Velvet や SOAPdenovo といった de Bruijn グラフを 用いた de novo アセンブラではコンティグを複数本出力する.第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
4.2
k-mer
整数
de Bruijn グラフを用いたアセンブリでは,まず入力したすべてのリード に対し k-mer のパターンを抽出する.多くの de novo アセンブラでは,そ のまま文字列として扱うのではなく k-mer 整数と呼ばれる形式に変換する ことで,k-mer を整数として扱う.k-mer とは k 文字の塩基配列のパターン のことを指し,k-mer 整数とは各 k-mer に一対一で対応づけた整数である. 具体的には,各塩基 A,C,G,T をそれぞれ 0,1,2,3 の数値に対応させ て塩基配列を 4 進数として表現し,この 4 進数で表された塩基配列を 10 進 数で表した値が k-mer 整数となる.すなわち,各 k-mer は 0 から 4k− 1 ま での整数に対応している.例えば,”ACGTA”の 5-mer 塩基配列に対応するk-mer整数は 108 となる.表 3,表 4 では例として 3-mer と 5-mer の整数と の対応を示す.
k-mer 整数を用いてリードを扱うことはメモリ使用量の面で有利であると 言える.例えば,k-mer をそのまま文字列として表した場合,一般に kbyte のメモリを消費するのに対し,k-mer 整数では同じ kbyte で 4k-mer 分の塩 基列を表すことができ,メモリ使用量は 1/4 に節約できる.一般に計算機で は文字の操作よりもバイナリデータの操作の方が高速で行える.そのため, 配列同士の比較やある配列の探索を行う場合,k 文字の文字列よりも k-mer 整数で扱う方がより高速に処理できる.
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
表 3. k -mer と k -mer 整数の対応表 (3-mer の場合)
k -mer Quaternary Binary
k -mer integer (decimal) AAA 0 0 0 AAC 1 1 1 AAG 2 10 2 AAT 3 11 3 ACA 10 100 4 ACC 11 101 5 ACG 12 110 6 ACT 13 111 7 AGA 20 1000 8 : : : :
表 4. k -mer と k -mer 整数の対応表 (5-mer の場合)
k -mer Quaternary Binary
k -mer integer (decimal) AAAAA 0 0 0 AAAAC 1 1 1 AAAAG 2 10 2 AAAAT 3 11 3 AAACA 10 100 4 AAACC 11 101 5 AAACG 12 110 6 AAACT 13 111 7 AAAGA 21 1000 8 : : : : ACGGT 1223 01101011 107 ACGTA 1230 01101100 108 ACGTC 1231 01101101 109 : : : :
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
4.3
ハッシング
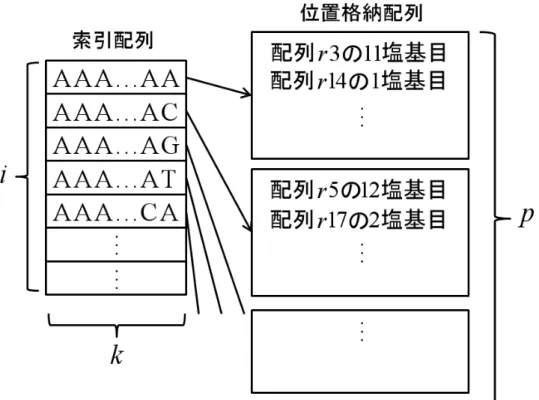
de Bruijn グラフを用いたアセンブラの多くは,k-mer のパターンそのも のだけでなく,リードの中に含まれる各 k-mer が出現する回数・位置などを ハッシュテーブルと呼ばれるデータ構造を用いてメモリ上に格納し,素早く 呼び出せるようにしている.このようなデータ構造を用いることによって, k-merの位置情報の参照を比較的高速で行うことができ,消費メモリ量も抑 制される.k-mer の出現回数や位置情報は,後の余分なエッジの削除で重要 な役割を持つ (エッジの削除については次節以降で述べる).ハッシュテー ブルの単純な実装例を図 7 に示す.図 7 の例では索引配列と位置格納配列と 呼ばれるデータ構造を用いている.索引配列とは k-mer 整数の配列であり, k-merのパターンの数だけ必要となる.図 7 の例では文字列として表現され ているが実際は整数であり,実装方法によっては配列の添え字として表現 される.また,位置格納配列は対応する k-mer のリード上の出現位置や出 現回数を表しており,各要素が索引配列の各要素と一対一に対応している. 例えば,図 7 の例では,AAA...AA という k-mer は,3 番目のリードの 11 塩 基目から出現,14 番目のリードの 1 塩基目から出現... ということを意味し ている.このようなハッシュテーブルを用いることで,k-mer そのものと各 k-merの出現位置や出現回数を表現することができる.しかし,索引配列の 大きさ i と位置格納配列 p の分だけの消費メモリ量, i = 4k (1) p = (l− k)n (2) を必要とする.ここで,l はリードの長さ,n はリードの総数であり,k は 索引配列の要素の長さ,すなわち k-mer における k である.この関係式か ら,要求されるメモリ資源はリードの長さ l,リードの総数 n に依存し,k にも依存することがわかる.例えば,k-mer の k が 1 増加すると索引配列は 4倍必要となり,それだけ消費メモリ量が増加する.そのため多くのアセン ブラでは,膨大な k-mer の全てのパターンを保存するために,上記のよう 28第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
なハッシュテーブルに様々な改良や工夫を施すことで,消費メモリ量の削 減に取り組んでいる.
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
4.4
de Bruijn
グラフの構築
ハッシュテーブルに格納された各 k-mer の情報を元に,図 8 に示すような de Bruijnグラフと呼ばれるグラフを構築する.de Bruijn グラフとは,任意 のノードが k 文字の文字列に対応し,エッジで連結された両ノードの各文字 列は k− 1 文字だけ重複するという特徴を持つ有向グラフである.まず,4.2 で全てのリードから得た k-mer をノードに対応する文字列とし,次に k−1 文 字の重複がある二つのノードを有向エッジで結ぶことで de Bruijn グラフを 構築する.例えば,”AACCGCTT”というノード v1に対し,”ACCGCTTG” というノード v2が存在するならば,v1から v2へのエッジを生成する.ある ノードから k− 1 文字の重複 (有向エッジ) を持つノードは,図 9 に示すよう に最大で 4 種類存在する. 図 8. de Bruijn グラフの例 (3-mer の場合) 30
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム de Bruijn グラフを利用した de novo アセンブリにおいて最も理想的な グラフは,元の全塩基配列の両端の配列に対応する二つの端点 (次数が 1 の ノード) と,次数が 2(入次数と出次数がそれぞれ 1) のノードのみで構成さ れた連結グラフである.このようなグラフであれば,ある端点からもう一 方の端点までノードを辿っていき,辿ったノードに対応する文字列 (k-mer) を連結していくことで,全塩基配列を生成することができる.このように, グラフ上で隣接しているノード同士を辿った際に,両端の 2 ノードの次数が 1,それ以外のすべてのノードの次数が 2 の経路 (系列) を単純パスと呼ぶ. 実際には,反復配列やシーケンスエラーが含まれるため,単純パスになる ことはほとんどない.例えば,入次数 (或いは出次数) が 2 以上あるのノー ドが存在する場合,ノードを辿る際に分岐が生じてしまう.図 9 に示した ように入次数 (或いは出次数) は高々4 であるが,実際に k-mer から構築さ れた de Bruijn グラフにはこのような分岐が大量に発生する.また,パスの 始点と終点が同じノードとなるパス (閉路) となる場合も非常に多い.元の 塩基配列に同じ配列が何度も反復している領域 (反復配列) が存在する場合 にこのような閉路になりやすく,リードの配列情報のみでは反復配列を正 確に求めることは困難となる.実際に k-mer から生成された de Bruijn グラ フは,これらの分岐や閉路が大量に,かつ複雑に連結された状態となって おり,始点と終点までのパスが一意となる単純パスはほとんど存在しない. これらの問題となるグラフについては 5 章でより詳細に説明する. 多くの de Bruijn グラフを用いたアセンブラでは,単純パスを得るために 様々な工夫を行いグラフを表現している.例えば,代表的なアセンブラで ある Velvet では,グラフ全体をそのまま用いるのではなく,分岐や閉路を 含まない単純グラフを部分グラフとし,グラフの簡略化を行っている.検 出された部分グラフは単純グラフであるため,部分グラフ内のノードを全 て一度ずつ通る単純パスを一つ持つ.得られた部分グラフを一つのノード と見做すことで,グラフ全体の簡略化を行う.その後,反復配列やシーケ ンスエラーが原因と考えられる余分なエッジを除去し,単純パスを辿るこ
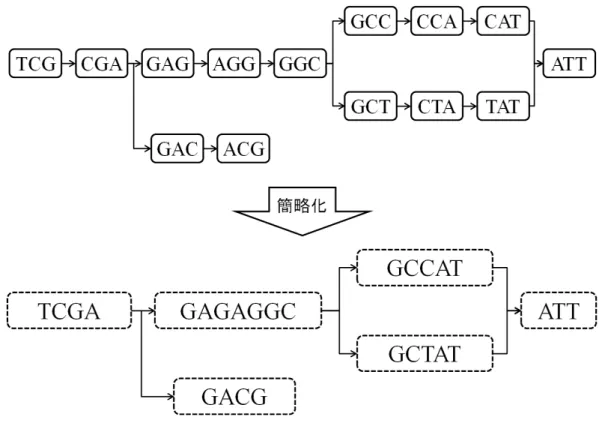
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
とでコンティグを生成する.Velvet における k=3 のときの de Bruijn グラ フとそれを簡略化したグラフの例を図 10 に示す.
図 9. 有向エッジで接続されている 4 種のノードの例 (3-mer の場合)
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
4.5
エッジの除去とコンティグの生成
既に述べたように,k-mer から構築した de Bruijn グラフは分岐や閉路を 多く含んでおり,正しいコンティグを得ることは困難となる.そのため多く の de Bruijn グラフを用いた de novo アセンブラでは,誤っていると思われ るエッジをグラフから除去する.例えば,Velvet では分岐が発生している ノードにおいて,分岐先の各ノードに対応する k-mer の数やその出現位置, そしてどちらのノードを辿ればより長いコンティグを得られるか等を調べ る.そして,より正しいと思われるノードへのエッジを選択し,そうでな い方のエッジは除去する.この処理を繰り返し行うことで,より正確でよ り長いコンティグの生成を行っている.ただし,例外として分岐している パスが同じ閉路に含まれている場合はエッジ除去の対象としない.例えば, 反復配列または同一の配列に挟まれた領域などが存在する場合,図 11 のよ うなグラフとなる.図 11 では,4.4 節で述べた簡略化を行った結果,閉路が 形成されてしまった例である.このような場合,”ACCTCCGAGCT”とい う配列は少なくとも二回以上繰り返されている反復配列である可能性が高 く,必ずしも間違ったパスではないと考えられるため,エッジの除去は行わ ない.多くの de Bruijn グラフを用いた de novo アセンブラでは,効率よく de Bruijnグラフから単純パスを求めるため,このような余分なエッジの除 去 (選択) やグラフの簡略化などの様々な工夫を行っている.de Bruijn グラATTGGACC ACCTCCGAG
GAGCTACC
GAGTTTAA
図 11. 閉路を含むグラフの例
フを用いた de novo アセンブリでは,上記のような方法で de Bruijn グラフ
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム を構築した後,図 12 に示すようにグラフの単純パスの始点から終点までの ノードを辿ることで,それに対応する k-mer の塩基配列を連結しコンティ グを出力する.図 12 では,破線で囲まれたノードによる単純パスから,各 ノードを辿ることでコンティグを求めている.図 12 では破線で囲まれてい ないノードがいくつか残っている.これらのノード群から短いコンティグを 生成することも可能であるが,このようなコンティグを生成するかはアセ ンブラの仕様や設定によって異なる. de Bruijn グラフを用いた de novo アセンブラでは,全てのノードを含んだ一つのグラフを構築し,元の全塩 基配列を復元するようなコンティグに対応する単純パスを得ることが理想 的である.しかし,分岐や閉路,或いはギャップやシーケンスエラーによっ て,複数のグラフ・単純パスとなる場合がほとんどである.そのため,実際 の de novo アセンブリでは,よほど小規模ゲノムでない限り,出力は複数の コンティグとなる. 図 12. de Bruijn グラフからコンティグを生成する例
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム
4.6
de Bruijn
グラフを用いた
de novo
アセンブ
ラ
de Bruijn グラフを用いた de novo アセンブラとして,Velvet や SOAP-denovoが現在では広く普及している.実際にこれらのアセンブラは数々の ゲノムプロジェクトに利用されており,様々な生物種のゲノム解読に貢献 している[17] [16].しかし,数 Gbp を越える大規模な全塩基配列を決定する場 合,これらのアルゴリズムを用いても,実行時に要求される消費メモリ量 が非常に膨大でメモリ不足になりやすい.これに対し,de Bruijn グラフを コンパクトなデータ構造で表現することを目指した研究がある[18] [19] [20] [21]. 文献[18] [19]
では,簡潔データ構造 (succinct data structure) と呼ばれるデー タ構造を用いて de Bruijn グラフをコンパクトなデータ構造で表現してい る.しかし,これらの手法では,いかにしてグラフをコンパクトに表現する かという点に焦点が当てられており,リードあるいは k-mer を読みこむ処 理や,グラフを構築する前処理などを含めた,アセンブリ全体の効率につ いては詳細に検討されていない.簡潔データ構造を利用した de Bruijn グラ フは非常にコンパクトであり,一度構築してしまえば展開等の操作を行う ことなく高速にデータの参照を行うことができるが,その構築には時間的・ 空間的なコストが必要となる.また,それによって表現された de Bruijn グ ラフの変更に複雑な処理が必要であったり,変更の内容によっては新たに グラフを再構築する必要がある. 一方,文献[20] [21]においても de Bruijn グラフをコンパクトなデータ構造を 実現しており,消費メモリ量を減少させることに成功している.しかし,実 際には k -mer を読みこむ処理を行う際に比較的大容量の外部記憶装置 (HDD など) を利用することで実現している.本章で述べてきたように,k -mer の 読み込み処理は de novo アセンブリの各ステップの中でも重要な処理ある が,そのコストも de novo アセンブリの中で最も大きな処理の一つでもあ る.実際に,文献[20] で行われた実験においても,この操作はアセンブリの ステップの中で最も時間がかかっている.主記憶 (メモリ) と HDD 等の外部 36
第 4 章 de Bruijn グラフを用いた de novo アセンブリアルゴリズム 記憶のアクセス速度は非常に大きな差があり,実装方法の工夫によっては全 てメインメモリで処理することも可能ではあるが,文献[20] [21]では詳細には 言及されておらず,de novo アセンブリの結果であるコンティグの精度等に 関しても評価がなされていない.このことから,これらの研究においても いかにしてグラフをコンパクトに表現するかという点に焦点が当てられて おり,de novo アセンブリ全体の効率については詳細に検討されていない. 一方,本研究の目的は消費メモリ量の削減であるが,その方針は上記の研 究の方針とは異なる.本研究では de Bruijn グラフのデータ構造のサイズの みに注目するのではなく,k -mer の読み込み処理やグラフ構築に要するコス ト等も含めた,アセンブリ全体における最大の消費メモリ量を抑え,効率 よく de novo アセンブリを行うことができる手法の提案を目的とした.ま た,de novo アセンブリの結果であるコンティグについても検討している.
第
5
章 提案手法
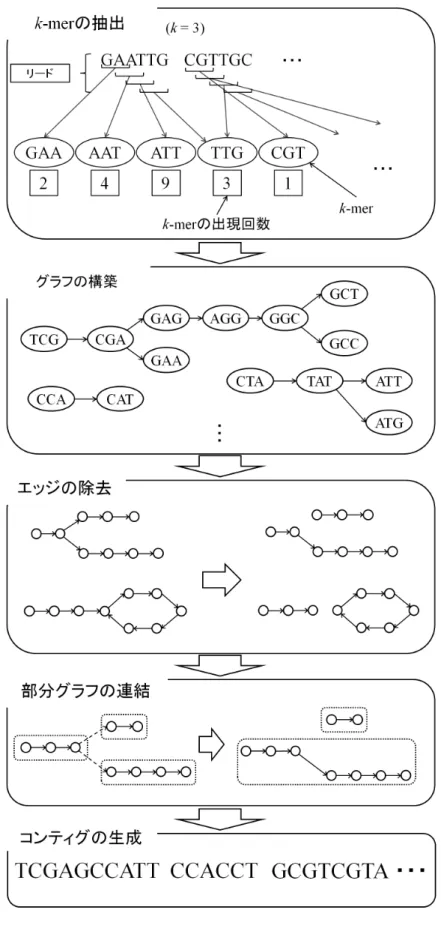
本章では,消費メモリ量を大幅に削減した de novo アセンブリアルゴリズ ムである提案手法について説明する.提案手法では 4 章で述べた,de novo アセンブリの問題を de Bruijn グラフを用いてコンティグを求めるアルゴリ ズムとして実装する.提案手法の全体の流れを図 13 に示す.まず,入力と なるリードから k-mer の全てのパターンを登録する.この時,各 k-mer の パターンがリード中に出現する回数も登録しておく.次に,登録した k-mer から de Bruijn グラフを構築する.そして、構築したグラフを複数の部分グ ラフに分割する。このとき、各部分グラフは分岐による曖昧さや閉路を持 たず、単純な経路 (単純パス) を必ず一つ持つように分割される。分割され た各部分グラフを、より長い単純パスを持つように連結する。最後に、連 結された各部分グラフ内のノードを辿り、コンティグを生成する.ただし, 本手法ではグラフを構成する要素の表現や,メモリ上に保持する情報の厳 選などの工夫によって,消費メモリ量の削減を行っている.本章の各節で はその詳細について述べる.第 5 章 提案手法
図 13. 全体の処理の流れ 40
第 5 章 提案手法
5.1
k-mer
整数の登録とグラフの構築
一般的な de Bruijn グラフを用いたアセンブラと同様,本手法でもすべて のリードからすべての k-mer のパターンを調べ,それらを k-mer 整数とし てハッシュテーブルに登録する.図 14 に本手法の k-mer のパターンの抽出 の流れを示す.本手法では,まずリードを一本ずつ読み込み k-mer を抽出 する.得られた k-mer を k-mer 整数に変換し,単純なハッシュテーブルに 登録していく.この処理を全てのリードに対して行う. 図 14. 本手法における k-mer の抽出処理 この時,第 4 章で述べたように,Velvet などのようなアセンブラの多くで は,その k-mer の出現回数やリード内における位置などの情報も併せて登 録している.これらの情報を利用することで,コンティグの長さや精度を 高めることができる.しかし,大規模なゲノムのアセンブリでは,必要な リードの数は大幅に増加してしまい,それに伴い k-mer のパターン数も膨第 5 章 提案手法 大なものとなる.それに伴い k-mer に付随する情報も大幅に増加し,それ を保持するのに費やす消費メモリ量も飛躍的に増加するという問題が生じ る.それに対し,本手法では k-mer 整数を登録する際にその k-mer の出現 回数のみを登録することで,消費メモリ量の削減を図っている.後述とな るが,de Bruijn グラフ上に発生する分岐に対してはこの出現回数のみを利 用することで解決する.また本手法では,図 15 に示すような非常に単純な ハッシュテーブルを用いて k -mer のパターン及び出現回数をメモリ上に格 納する.本手法でのハッシュテーブルは,k -mer のパターンを基に生成され た数値 (ハッシュ値) を添え字とした配列となっている.ハッシュテーブル の各要素には,k -mer 整数と出現回数が格納されているレコードへの参照情 報が格納されている.ある k -mer を参照する場合は,まずハッシュ値を計 算し,ハッシュテーブル上のハッシュ値に対応する要素からレコードを参照 することで,目的の k -mer を参照することができる.これにより,図 7(29 項) で示したような索引配列は必要なく,k が変化してもテーブルの大きさ は一定であるため,消費メモリ量を削減することができる.尚,本手法で は相補鎖を考慮し,互いに相補的な k -mer は同一の k -mer として扱う. 次に,登録した k-mer を用いて de Bruijn グラフを構築する.グラフ理論 におけるグラフは本来ノードとエッジの集合で表される.そのため,Velvet などの de Bruijn グラフを用いた手法では,de Bruijn グラフを構成するノー ド,ノード間のエッジ情報を用いることでグラフを表現する.しかし,大 規模なゲノムのアセンブリではリードの数が大幅に増加し,それに伴って
k-merの数も膨大なものとなる.そのような膨大な k -mer から de Bruijn グ ラフを構築しようとすれば、膨大な数のノード・エッジをメモリ上に保持 する必要があるため,消費メモリ量が大幅に増加するという問題が生じる. そこで本手法では,de Bruijn グラフのエッジで連結された両ノードの各 文字列は k− 1 文字だけ重複するという特徴を持つ有向グラフという特徴に 着目した.de Bruijn グラフにおいてあるノードがエッジを持っているかを 考える時,そのノードに対応する k -mer と k− 1 文字重複する k-mer を持つ 42
第 5 章 提案手法
第 5 章 提案手法 ノードが存在すれば,それらノード間にはエッジが存在するとみなすこと ができる.すなわち,全てのノードの存在を調べることができれば,全て のエッジの存在を調べることができる.本手法では,このような de Bruijn グラフの特徴を利用し,ノードのみで de Bruijn グラフを表現する.具体的 には,メモリ上にはノードに関わる情報のみを保存し,エッジに関わる情 報を一切保存しない.ノードに関わる情報とは,k -mer 整数,k -mer の出現 回数,分岐・閉路の検出に必要なラベルである.ラベルの詳細については 5.2節で述べる.あるノードがエッジを持つか否かは,パスを探索する際に ノード v の k-mer 整数を左シフトしたものに 0∼ 3 を足すことでその k-mer と k− 1 文字重複している k-mer を調べ,もし重複している k-mer が存在す ればそのノード v′に対して v は有向エッジがあるものとする.このように, de Bruijnグラフを仮想的に表現することで,大幅に消費メモリ量を削減し ている.ここで,k-mer”ACGTA”に対応するノードのエッジの有無を調べ たいときの例を図 16 に示す.まず”ACGTA”の k-mer 整数である 108 の 2 進 表記 “0001101100” を左に 2 ビットシフトすることで”CGTAA”の k-mer 整 数である 660(2 進表記では “0110110000”) を得る.そしてこの値に 0∼ 3 を 足した 660∼ 663 をハッシュテーブルから検索し,存在すればエッジがある と見做す.ただし,出現回数の低い k-mer はシーケンスエラーである確率 が非常に高いため,そのような k-mer は無効とし,ハッシュテーブルから 存在を確認しても無いものとしてみなす.実験 (第 6 章) では,出現回数が 1回以下の k-mer は無効とした.本手法ではエッジをこのように仮想的に表 現しており,有向エッジをあらかじめ調べて情報として保存する必要が無い ため,大幅に消費メモリ量の削減を行うことができる.本手法では有向エッ ジの登録は行われないため,全ての k-mer の登録の完了と共に de Bruijn グ ラフの構築は完了となる. 44
第 5 章 提案手法 図 16. k = 5 の時のエッジの有無の計算の例