Studies on a Fish Gene that Relates to
Efficient Growth and Differentiation :
Structure of Myostatin Gene Promoter in
Percoidei Fishes
著者
Eman Mohamed Mamdouh Mohamed Abbas
内容記述
学位授与大学: Osaka Prefecture University(大阪
府立大学), 学位の種類: 博士(理学), 学位記番号:
論理第96号, 学位授与年月日: 2011-09-30, 指導教
員: 加藤幹男.
Studies on a Fish Gene that Relates to Efficient Growth and Differentiation:
Structure of Myostatin Gene Promoter in Percoidei Fishes
(魚類の増殖・分化関連遺伝子に関する研究:スズキ亜目魚類のミオスタチン遺伝子構造)
by
Eman Mohamed Mamdouh Mohamed Abbas
A THESIS
Submitted toGraduate School of Science, Osaka Prefecture University in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy
Under supervision of Dr. Mikio Kato
Department of Biological Science
Graduate School of Science
Osaka Prefecture University, Japan
i
ACKNOWLEDGMENT
Above all, I offer my sincerest gratitude to ALLAH, who made all things possible for me to complete my work, present it in such a satisfactory way and give me the patience to work through
this research.
First and foremost, I would like to express my deepest appreciation and pay my cordial gratitude to my worthy, reverend supervisor Dr. Mikio Kato, Associate Professor of Structural and Evolutionary Genomics, Department of Biological Science, School of Science, Osaka Prefecture University, who has supported me throughout my work with his invaluable help, encouragement, good advice, guidance and patience. His supporting and supervision is not only throughout the duration of this period of study, but also in all my general life matters. He supervised the whole work and gave tremendous constructive comments for the preparation of the thesis. Without him and his efforts this work would not have been completed or written. One simply could not wish for a better or friendlier supervisor.
In particular, thanks are due to the Ministry of High Education in Egypt for granting me the opportunity to come and study in a country like Japan, which facilitated research and thesis with advanced technology. I consider it to be an enormous benefit. It also provided me with the necessary financial support.
It would not have been possible to complete my work without the help and support of the kind people around me.
I am most grateful to Dr. Kouichi Maruyama, National Institute of Radiological Science, Chiba, who particularly assisted me in my work by tutoring me a technique of microinjection into the egg of fish. Special thanks are also to Professor Emeritus Nobuyoshi Shimizu, Advance Research Center for GSP, Keio University and Dr. Atsushi Takayanagi, Department of Molecular Biology, Keio University School of Medicine for their support with the sequencing machine.
I would like to acknowledge and extend my heartfelt gratitude to my best friend Aki, whose kindness, friendship and support have been invaluable on a personal level. She was a good companion and helped me to adapt to living in Japan.
In my daily work I have been blessed with a friendly and cheerful group of fellow students and colleagues whom I would also like to thank, especially Mrs. Erandi Pathirana for assisting in English revision of the written aspect of the thesis. Cordial thanks are due to Osaka Prefecture University, Japan, for providing all facilities and the conductive academic environment to complete the Ph.D. course.
Finally, my absolute and heartiest appreciations go to my dear husband, Khaled, for his personal support and great patience at all times we have lived in Japan. Great thanks to my lovely children, Rana and Mohamed, who are the apple of my eyes and the joy that light up my life.
Last, but by no means least, I extend my sincere thanks and gratitude to my parents: my affectionate father, who always encourages me to acquire more knowledge, especially through studying abroad; and my sweet mother, whose hands always raise to pray for me. In fact, there are no adequate words of thanks to express my respect and appreciation for them.
ii TABLE OF CONTENTS ACKNOWLEDGMENT ... i TABLE OF CONTENTS ... ii LIST OF ABBREVIATIONS ... iv LIST OF TABLES... v LIST OF FIGURES... vi ABSTRACT ... viii
MYOSTATIN – A GENERAL INTRODUCTION ... 1
CHAPTER I ... 5
METHYLATION STATUS AND CHROMATIN STRUCTURE OF THE MYOSTATIN GENE PROMOTER REGION IN THE SEA PERCH Lateolabrax japonicus (PERCIFORMES) ... 5
1.1. INTRODUCTION ... 6
1.2. MATERIALS AND METHODS ...11
1.2.0. Fish Samples ...11
1.2.1. DNA Quantity Measurement ...11
1.2.2. DNA Quality Evaluation ...11
1.2.3. Agarose Gel Electrophoresis ... 12
1.2.4. Primer Design ... 12
1.2.5. Chromatin Analysis ... 13
1.2.6. Methylation Status ... 17
1.3. RESULTS ... 20
1.3.1. Bisulfite Modification of Genomic DNA to Evaluate the Methylation Status ... 20
1.3.2. Sensitivity to MNase in the Myostatin Gene Promoter Region ... 22
1.4. DISCUSSION... 24
CHAPTER II ... 26
STRUCTURE OF MYOSTATIN PROMOTER IN Acanthopagrus latus ... 26
2.1. INTRODUCTION ... 27
2.2. MATERIALS AND METHODS ... 31
2.2.0. Samples Collection ... 31 2.2.1. Isolation and Characterization of Myostatin Promoter Region in Sparidae
iii
Fishes ... 31 2.2.2. Isolation of A. latus Myostatin Promoter and Construction of pAl-MSTN-GFP-1 and Deletion of Constructs ... 45 2.3. RESULTS ... 57 2.3.1 Isolation of Sparidae Myostatin Upstream Sequences (alMSTN, asMSTN Promoter) ... 57 2.3.2. Isolation of alMSTN-1 Upstream Region ... 61 2.3.3. Identification and Localization of alMSTN Promoter Transcription Factor Binding Sites ... 61 2.3.4. Phylogenetic Analysis ... 66 2.3.5. Comparative Characterization of alMSTN Regulatory Motifs with Respect to Other Fish ... 66 2.3.6. Evaluation of Promoter Activity in Transient Expression Assay of GFP in Medaka Embryo ... 72 2.4. DISCUSSION... 76 REFERENCES ... 80
iv
LIST OF ABBREVIATIONS
bp: base pairs
cDNA: complementary DNA kb: kilobase pairs
CpG: Cytosine-Phosphate-Guanine DIG: Digoxigenin
EDTA: ethylenediaminetetraacetic acid EtBr: ethidium bromide
EtOHabs: absolute ethanol F primer: forward primer GFP: green fluorescent protein LB: Luria-Bertani
MNase: Micrococcal nuclease MSTN: myostatin gene
NCBI: National Center for Biotechnology Information
PBS: 1.47 mM KH2PO4, 8.06 mM Na2HPO4, 137 mM NaCl, 2.68 mM KCl PCR: polymerase chain reaction
R primer: reverse primer RNase: ribonuclease
SDS: sodium dodecyl sulfate
STET buffer: 0.1 M NaCl, 10 mM Tris-HCl (pH 8.0), 1 mM EDTA (pH 7.8), 5% Triton X-100
TAE buffer: 40mM Tris-base, 20mM acetic acid, 1mM EDTA. TE buffer: 10 mM Tris HCl, 1 mM EDTA, pH 7.8
TES buffer (DNA isolation buffer): 10 mM Tris-HCl, 140 mM NaCl, 25 mM EDTA, pH 7.8
v
LIST OF TABLES
Table 1. 1. Oligonucleotides used as PCR primers ... 13 Table 1. 2. Summary of bisulfite sequencing: relative percentage of unconverted bases after bisulfite treatment. ... 20 Table 1. 3. Summary of bisulfite sequencing: numbers of unconverted cytosines at each CpG site. ... 21 Table 2. 1. PCR primer sequences with names indicating their specificity ... 41 Table 2. 2. Pairwise comparsion of COI and 18S rRNA sequences ... 44 Table 2. 3. Detection of transcrptinal factor binding site in alMSTN-1 upstream region………...64
vi
LIST OF FIGURES
Figure 1. 1. Conserved sequences in the myostatin gene 5’ flanking region ... 10
Figure 1. 2. Schematic illustration of bisulfite sequencing. ... 19
Figure 1. 3. Sequencing of L. japonicus promoter region. ... 21
Figure 1. 4. Southern blot analysis of TaqI-digested genomic DNA isolated from MNase-treated cell nuclei. ... 23
Figure 2. 1. High molecular weight genomic DNA digested with PstI restriction enzyme. ... 32
Figure 2. 2. Inverse polymerase chain reaction. ... 34
Figure 2. 3. Digestion of pUC19 vector by SmaI restriction endonuclease. ... 34
Figure 2. 4. Genomic PCR of A. latus and A. schlegelii... 40
Figure 2. 5. Schematic illustration demonstrates the inverse polymerase chain reaction process... 43
Figure 2. 6. Second inverse PCR and genomic amplification of Acanthopagrus latus. . 47
Figure 2. 7. Genomic amplification of both A. latus promoter region, GFP coding region and SV40-polyA signal………... 49
Figure 2. 8. Strategy for the construction of plasmid Al-MSTN promoter-GFP. ... 52
Figure 2. 9. Schematic diagram shows all the different constructs. ... 52
Figure 2. 10. Medaka fish Oryzias latipes. ... 53
Figure 2. 11. A method of microinjection of DNA preparation in egg. ... 54
Figure 2. 12. Microinjection into fertilized medaka eggs. ... 56
Figure 2. 13. Genomic PCR for A. latus and A. schlegelii myostatin gene promoter, and part of coding region. ... 57
Figure 2. 14. Sequence alignment of 5’ flanking region of myostatin genes for two alleles of each Acanthopagrus latus and Acanthopagrus schlegelii, Sparus aurat -1 and Lateolabrax japonicus. ... 60
Figure 2. 15. Genomic sequence of the 5’ promoter region of the yellowfin sea bream myostatin... 63
Figure 2. 16. A graphical representation of the transcription binding sites of Acanthopagrus latus myostatin promoter region. ... 65
Figure 2. 17. Phylogenetic tree of teleosts MSTN-1 and -2. ... 68 Figure 2. 18. Sequence comparison of a highly conserved region in the 5’UTR and
vii
proximal promoter of some Percoidei fishes ... 69 Figure 2. 19. Comprtive analysis between analysis between putative binding sites of transcription-regulatory factors of Acanthopagrus latus and Sparus aurata. ... 70 Figure 2. 20. Schematic illustration of comparative analysis of the major regulatory elements of yellowfin sea bream and some fish species. ... 71 Figure 2. 21. Green fluorescent protein expression and activity of A. latus myostatin promoter in eggs of medaka. ... 74 Figure 2. 22. Comparison of the GFP expression of the A. latus myostatin promoter and CMV-GFP promoter in medaka eggs. ... 75
viii
ABSTRACT
Myostatin, also known as growth differentiation factor 8 (GDF-8), is a member of the transforming growth factor-β (TGF-β) family that functions as a negative regulator of skeletal muscle development and growth in mammals. A model for the biological activity of myostatin has been proposed in that myostatin inactivates a specific group of genes, and a negative correlation between myostatin expression and growth/number of muscle fibers has been observed. It is widely known that myostatin-knockout mice exhibit a dramatic increase of skeletal muscle mass that results from a combination of hyperplasia and hypertrophy. Natural mutation of the myostatin gene has been observed in the double muscle breeds of Belgian Blue and Piedmontese cattle, which have significantly more muscle mass than normal breeds do. Myostatin is found almost exclusively in the skeletal muscle in mammals, but appears more ubiquitously in fish, suggesting more diverse functions in their growth and development. A single encoding gene in mammals expresses myostatin, whereas teleost fish possess at least two myostatin genes that are differentially expressed in both muscular and non-muscular tissues. Recently, it was reported that silencing of the myostatin genes resulted in a giant phenotype in zebrafish, and a dominant-negative form of myostatin resulted in the doubling of muscle-fiber number in transgenic medaka.
Myostatin genes have been characterized in several commercially important fishes such as striped bass, white perch, Mozambique tilapia, white bass, Atlantic salmon, rainbow trout, gilthead sea bream, shi drum, catfish species, European sea bass, Croceine croaker, orange spotted grouper, and Japanese sea perch. Worldwide fish consumption has been steadily increasing, and strategies for enhancing skeletal muscle growth of aquaculture species can help to meet the increasing demand for this protein source. It is quite important to determine how myostatin gene expression is regulated and to uncover the protocol that controls the action of myostatin. To gain better understanding of the mechanisms regulating myostatin gene expression, we have analyzed the genomic structures of the myostatin gene in a sea bass, Lateolabrax
japonicus, spanning primary DNA sequences to higher-ordered structures (chromatin
ix
fluorescent protein in the embryos of medaka (Oryzias latipes) driven by a myostatin promoter isolated from a sea bream (Acanthopagrus latus). This trial will assist us in evaluating the environmental effect on myostatin expression in future studies using transgenic medaka fish.
(1) Methylation status of myostatin gene promoter region in Lateolabrax japonicus
DNA methylation at cytosine residues is involved in epigenetic regulation to suppress gene expression through heterochromatinization. However, the methylation status in fish genomes remains unclear. To examine the methylation status in myostatin promoter regions of Lateolabrax japonicus, total genomic DNA was isolated from tissues of the brain, kidney, spleen, liver, heart, eye, muscle, intestine, and gill. The genomic DNA samples were digested with a restriction enzyme and subjected to bisulfite modification. The bisulfite treatment introduces specific changes in the DNA sequences that depend on the methylation status of individual cytosine residues, yielding information on the DNA segments’ methylation status. DNA fragments were treated with sodium bisulfite to convert unpaired cytosine residues to uracil under conditions whereby methylated cytosine remained essentially intact. The modified DNA was used as the PCR template to specifically amplify the sense and antisense strands of bisulfite-treated DNA with their respective PCR primer pairs. The PCR products from each reaction were subjected to a second PCR amplification with nested primers to obtain the DNA fragments that originated from each strand. They were cloned into a plasmid vector and the nucleotide sequences were determined. The efficiency of the bisulfite modification (conversion of C to T) was estimated by counting the numbers of cytosine residues remaining at CpN sites (in the case of N ≠ G), and the conversion frequency was found to be 99% or higher. The number of cytosine residues remaining at CpG sites appeared to be slightly higher than that in the background level in sense strands from the eye and heart, but were identical to that in the background level in complementary strands from the same tissues. The frequency of methylated cytosine was very low in the tissues examined (within the error range of random sampling) regardless of the level of myostatin gene expression. The results may mean that the methylation at CpG is not involved in regulation of the myostatin gene in L. japonicus.
x
(2) Probing the chromatin structures in L. japonicus
Gene expression also reflects the status of the chromatin (euchromatin or heterochromatin) in the region where the gene is situated. The status of the chromatin in particular gene regions is dynamically modulated to control gene expression and other fundamental cellular processes such as proliferation and differentiation. Active chromatin can be distinguished from bulk chromatin by its increased susceptibility to endonuclease. Micrococcal nuclease (MNase) preferentially cleaves chromatin between nucleosomes (naked DNA regions). To examine tissue-specific chromatin structures in myostatin gene regions by MNase as an enzymatic probe, nuclei were isolated from some L. japonicus organs (the liver, eye, kidney, brain, and heart), purified, and treated with MNase. The mixtures were incubated at 37°C for 15, 30, and 60 min, and one-third of the original reaction volumes were collected at each time point. The DNA was isolated from each reaction mixture and subjected to Southern blot hybridization analysis after restriction digestion. A fragment of the L. japonicus myostatin promoter region was used as a probe to detect an approximately 2 kb TaqI restriction fragment. Myostatin gene promoter regions in the brain and eye were highly susceptible to MNase while those in the heart were less susceptible, and those in the kidney and liver were even more resistant to MNase. The results suggest that the myostatin gene is compacted into heterochromatin in tissues where it is not expressed, such as in the liver and kidney, whereas it is susceptible to MNase in the eye and brain, which do express myostatin. According to the results of methylation analysis and MNase probing, DNA methylation may not be involved in regulating heterochromatinization and thus, expression of myostatin, in L. japonicus.
(3) Genomic sequences of myostatin gene promoter regions in Sparidae fishes
The 5′-flanking regions of the myostatin gene were isolated from two sea breams,
Acanthopagrus latus and Acanthopagrus schlegelii, by inverse PCR. Genomic DNA of A. latus and A. schlegelii were digested with restriction enzymes and the fragments were
self-circularized under low DNA concentration. The closed circular DNA molecules were used as the template for PCR with a set of DNA primers for the first-round PCR, after which nested PCR was performed with another set of DNA primers. The PCR products were cloned into a plasmid vector and sequenced by the primer-walking
xi
strategy with internal primers. After the entire sequence was determined, a genomic DNA fragment was amplified with 5′- and 3′-end primers to confirm the genomic sequences. The partial-coding regions of cytochrome oxidase subunit I (COI) and 18S rRNA from the specimens were also amplified and sequenced to confirm identity of species.
Two alleles of the myostatin gene were identified for both A. latus and A. schlegelii. The nucleotide sequences were aligned with the promoter region of the Sparus aurata myostatin gene, showing that the conserved regions spanned approximately 1 kb. The potential cis-acting elements were observed in highly conserved regions between these two Sparidae species. They contain two putative TATA-boxes, one CAAT box, and seven putative E-boxes. Comparative analysis of myostatin gene regulatory regions with those of other Percoidei fishes revealed the occurrence of highly conserved regions approximately 300 bp upstream of the translation start site.
(4) Evaluation of promoter activity in transient expression of GFP in medaka embryos
The genomic fragment was connected to the GFP coding sequence to evaluate the activity of the promoters isolated from A. latus. The recombinant fragments were cloned into a plasmid vector. Six different myostatin fragments in total, truncated at their 5′ ends, were constructed. Some were used for microinjection into the cytoplasm of fertilized medaka fish at the one-cell stage. Embryos were observed under a fluorescence microscope to visualize GFP expression. The expression of the GFP driven by the A. latus myostatin promoter in the medaka embryos was weak but distinctive, with a series of truncated fragments. Based on the current available results, we believe that the genomic fragment obtained from A. latus has minimal promoter activity in medaka embryos. The A. latus promoter might not have worked well in medaka because medaka (Beloniformes) is distantly related to sea bream (Perciformes) evolutionarily, or the A. latus DNA fragment used may have been not enough to express full promoter activity.
xii
Conclusions
It is challenging to clarify the regulatory mechanisms of the myostatin gene in fish. Myostatin expression is suppressed parallel to heterochromatinization but regardless of DNA methylation in L. japonicus. Sequencing of 5′-flanking DNA fragments of the myostatin-coding region in A. latus identified several potential cis-elements and higher similarity to the myostatin gene promoter in other fish species. Weak promoter activity of DNA fragments isolated from A. latus was observed in medaka embryos in a transient expression assay. This assay system may allow us to evaluate the role of promoter elements in vivo. Establishing a transgenic medaka carrying AcGFP driven by an A. latus myostatin promoter will be extremely valuable to efforts to examine expression profiles in response to environmental stimuli under culture conditions in the future.
1
MYOSTATIN – A GENERAL INTRODUCTION
Muscle growth results from the proliferation of myoblasts and their subsequent differentiation into muscle fibers. This process is regulated in vivo through mechanisms that involve cell-to-cell interactions, cell-to-matrix interactions, and extracellular secretory factors including myostatin. Myostatin, also known as growth differentiation factor 8, is a member of the transforming growth factor (TGF-β) superfamily that negatively regulates growth and development of muscle mass in mammals (Lee, 2004). TGF-β superfamily which encompasses a number of peptides, such as inhibins and activins, shares a similar structure a relatively conserved amino acid sequence (De Santis et al., 2008). TGF are involved in important biological functions including cell growth and differentiation (Massagué, 1990). Myostatin gene has an essential role as a negative regulator of myogenesis in several mammalian species (Thomas et al., 2000). Myostatin gene was first characterized in mouse, where it was expressed in developing somites during embryogenesis (McPherron et al., 1997). The lack of evident non-specific defects in null-myostatin mice suggests that mammalian myostatin physiological functions are essentially confined to skeletal muscle. These myostatin "knockout" mice have approximately twice as much muscle as normal mice, and the increase results from combination of hyperplasia and hypertrophy. These mice were subsequently named “mighty mice” (Figure 1B).
The biological activity model proposed for myostatin is experimentally supported by gene inactivation showing a negative correlation between myostatin expression and growth/number of muscle fibers (McPherron et al., 1997). The mutations in the myostatin gene resulted in “double-muscling” phenotype which also happened in two breeds of cattle, Belgian Blue and Piedmontese (Figure 1A), (Grobert et al., 1997; Kambadur et al., 1997; McPherron and Lee, 1997) dogs (Mosher et al., 2007), sheep (Boman et al., 2009) and humans (Schuelke et al., 2004). In cattle, because of its beneficial effects on meat production, several double-muscling breeds have been selectively bred for this trait (Rodgers and Garikipati, 2008). This "double muscle" in Cattle has occurred by a mutation of G–A transition changing a cysteine residue to a tyrosine. This mutation alters one of the residues that are hallmarks of the TGF-β family and are highly conserved during evolution and among members of the gene family
2
(Kambadur et al., 1997). However, mutation at the cystein affectsthe cystine knot structure; in either scenario high concentrationsof Piedmontese myostatin would act as a myostatin mimetic andpossibly disrupt the further downstream signaling of wild-type myostatin. Thus Piedmontese myostatin presents itself as a potential inhibitor of myostatin (Berry et al., 2002). The overexpression of C313Y-MSTN in mice containing the missense mutation exhibits a dominant negative activity and there are two types in the dominant negative form of myostatin, causing either hypertrophy or hyperplasia (Nishi et al., 2002).
The growth of skeletal muscle is due to an increase in muscle-fiber number (hyperplasia) and/or muscle-fiber size (hypertrophy) as seen in myostatin knockout mice and in certain cattle breeds exhibiting the doubled-muscled phenotype, but also human (McPherron et al., 1997; Grobert et al., 1997; Kambadur et al., 1997; Schuelke
et al., 2004). Conversely, muscle wasting was induced in mice either by injection of the
functionally active C-terminal part of myostatin or by releasing this region from the latent propeptide by acid treatment (Zimmers et al., 2002). Interestingly, the process of muscle differentiation and growth is fundamentally different between mammals and fish. In mammals, since the number of muscle fibers is simply dependent on hypertrophy fibers formed during embryonic development. In contrast, numerous species of fish exhibit the unique ability to generate new muscle fibers throughout most of their lives (Rescan, 2008). Myostatin is found almost exclusively in the skeletal muscle in mammals, but seems more ubiquitous in fishes suggesting more diverse functions in fish growth and development (Radaelli et al., 2003; Helterline et al., 2007). In adult mammals, myostatin is primarily expressed in skeletal muscle, which is thought to be the principal target tissue. However, subsequent studies revealed that myostatin is also expressed at lower level in mammary glands (Ji et al., 1998), heart, spleen and brain, and it influences cardiac muscle growth as well as adipogensis (Rodgers and Garikipati, 2008). The myostatin is expressed by a single encoding gene in mammals whereas teleost fishes possess at least two myostatin genes which are differentially expressed in both muscular and non-muscular tissues (Østbye et al., 2007).
The mature myostatin in fish is 109 amino acid long and its sequence is highly conserved compared to other vertebrates (Kerr et al., 2005), differences do exist. In contrast to the well conserved amino acid sequence of mature myostatin, myostatin
3
prodomain varies among fish species in its length and amino acid sequence (Rebhan and Funkenstein, 2008). Conserved nucleotide sequences upstream of the myostatin gene from many different fish species have been identified that share many binding sites for transcription factors (Funkenstein et al., 2009). Significantly, several E-boxes present in the myostatin 5’flanking region are conserved in position and sequence. E-boxes are binding sites for regulatory proteins including the myogenic determination factors MyoD, Myf5, MRF4, and Myogenin, which are activators of the myogenic program and capable of converting many cell types into muscle (Weintraub et al, 1991). In fact, myostatin was already shown to be a downstream target of MyoD (Spiller et al. 2002), and the number of E-boxes upstream of myostatin gene controls the intensity of gene expression, although some E-boxes are more important than others (Salerno et al. 2004).
Myostatin cDNA has been characterized in some commercially important fishes, such as rainbow trout (Rescan et al., 2001), Atlantic salmon (Østbye et al., 2001), Mozambique tilapia (Rodgers et al., 2001), white bass (Rodgers and Weber, 2001), striped bass (Rodgers and Weber, 2001), shi drum (Maccatrozzo et al., 2002), gilthead sea bream (Maccatrozzo et al., 2001a), catfish (Gregory et al., 2004), grouper (Ko et al., 2006), European sea bass (Terova et al., 2006), Croceine croaker ( Xue et al., 2006), Japanese sea perch (Ye et al., 2007). The expression of myostatin mRNA has been detected in a variety of tissues such as muscle, eye, brain, intestine, gill, kidney, heart and spleen, and at different stages of development in some fish species (Maccatrozzo et
al., 2001a, 2001b; Østbye et al., 2001; Rescan et al., 2001; Rodgers and Weber, 2001;
Kocabas et al., 2002; Roberts and Goetz, 2003; Vianello et al., 2003; Xu et al., 2003; Gregory et al., 2004; Biga et al., 2005; Ko et al., 2006; Ye et al., 2007).
To better understand the mechanisms regulating myostatin gene expression in fish, we analyzed the chromatin structure of myostatin gene regions in Lateolabrax japonicus (Sparidae, Perciformes) using micrococcal nuclease (MNase) as an enzymatic probe. Also, we assessed the methylation status of the promoter region of myostatin genes in various tissues from L. japonicus. Controlling myostatin biological activity may be a favorable way to culture high value-added fishes (having large edible protein) and to enhance the skeletal muscle growth of aquaculture species. So, we isolated
4
cis-acting elements and examined the promoter activity by using (GFP) reporter gene
for transgenic in medaka (Oryzias latipes).
Figure 1. "Double-muscling" in myostatin-null animals.
(A) Double-muscling in Belgian Blue cattle breeds (McPherron et al., 1997). Adopted from the website (http://www.healthynewage.com/blog/bull-strong), accessed 13 May, 2011.
(B) Myostatin knock-out mice (McPherron and Lee, 1997). Adopted from the website (http://www.jyi.org/volumes/volume11/issue6/features/iyer.php), accessed 13 May, 2011.
5
CHAPTER I
METHYLATION STATUS AND CHROMATIN STRUCTURE OF
THE MYOSTATIN GENE PROMOTER REGION IN THE
6
1.1. INTRODUCTION
Gene expression is controlled not only by activation of trans-acting factors but also by the status of chromatin (euchromatin or heterochromatin) where the gene exists. Status of chromatin at specific gene regions is dynamically modulated to control gene expression and other fundamental cellular processes, such as proliferation and differentiation (Li, 2002; Felsenfeld and Groudine, 2003; Jaenisch and Bird, 2003). Several biological effects, including chromatin structure modulation, transcriptional gene silencing, and suppression of transposable elements, were shown to be regulated by this epigenetic process (Yang et al., 2007). Epigenetics encompasses heritable alterations in gene expression and chromatin without accompanying changes in the DNA sequence. Several studies showed that these epigenetic modifications have an important impact on transcription in different model systems with respect to the temporal order of events associated with gene silencing. In the late 1990s studies on yeast (Pazin and Kadonaga, 1997), Xenopus (Jones et al., 1998) and mice (Nan et al., 1998) suggested that CpG methylation is the first event that triggers a cascade leading to transcriptional silencing (Meehan et al., 1992). Whether methylation is a cause or an effect of epigenetic silencing, it may provide insight into common mechanisms that eukaryotes use to identify and silence target loci.
The general function of DNA methylation might be conserved between plants and animals, although there are differences in the composition of DNA methylating enzymes and their target sequences between these kingdoms. Studies of epigenetic regulation in plants and mammals are yielding complementary results that reveal the evolutionary conservation of epigenetic mechanisms, including methylation (Habu et al., 2001). DNA methylation is one of the mechanisms for gene regulation, often observed in mammals (5-methyl cytosine occurring at CpG) and plants (5-methyl cytosine occurring at CpNpG). It is involved in epigenetic regulation that is closely related to heterochromatinization that suppresses gene expression and plays essential roles in chromatin structure remodeling (Goll and Bestor, 2005; Klose and Bird, 2006; Reik, 2007; Kuroda et al., 2009; Zhang et al., 2010). DNA methylation at cytosine residues regulates transcription directly by inhibiting the binding of specific transcription factors, and indirectly by recruiting methyl-CpG-binding proteins and their associated
7
repressive chromatin-remodeling activities (Jirtle and Skinner, 2007). These epigenetic alterations are responsible for modulation of developmentally regulated and tissue-specific gene expression (Turek-Plewa and Jagodzin’ski, 2005). A majority of the CpG sites in gene regions are located in the flanking promoter, the first and second exons, and the first introns, which pinpoints the importance of CpG sites in gene regulation (Reamon-Buettner and Borlak, 2007; Tang and Ho, 2007). The importance of genomic DNA methylation is evident by the increasing number of research publications or reviews and grants awarded each year that deal with various aspects of DNA methylation (Boyd et al., 2006).
The most popular technique is bisulfite sequencing, first described by Frommer et
al. (1992), the use of bisulfite treatment of DNA to determine the pattern of methylation.
The bisulfite treatment introduces specific changes in the DNA sequence that depends on the methylation status of individual cytosine, yielding information about the methylation status of a segment of DNA. In this reaction, DNA is first treated with sodium bisulfite to convert cytosine residues to uracil in single-stranded DNA, under conditions whereby methylated cytosine remains essentially non-reactive. The DNA sequence under investigation is then amplified by PCR, with primers specific for methylated and unmethylated DNA, for methylation-specific PCR followed by sequencing can result in revealing the positions of 5-methylcytosine in the gene. The methylation status in the region of satellite DNA flanked by two sub-repeats was analyzed in the liver of Sillago japonica by bisulfite modification and most of the cytosine residues at CpG were methylated (Kato, 1996). Analysis of the global genomic 5-methyl cytosine methylation pattern at CCGG sites of medaka embryo DNA was suggested that the vast majority (>90%) of genomic DNA was methylated at these sites and the extent of methylation at these sites did not change or changed very little during embryogenesis (Walter et al., 2002). On the other hand no evidence of cytosine methylation was found in the CYP1A1 promoter region from hepatic genomic DNA of Killifish Fundulus heteroclitus (Timme-Laragy et al, 2005). In zebrafish Danio rerio, the sex and tissue differences in the level of DNA methylation was studied in a 5’ flanking region of the vitellogenin I gene (Strömqvist et al., 2010). They have shown that the methylation levels of all three CpG sites are higher in adult male liver than in adult female liver, and both females and males have high methylation levels in the CpG
8
positions in the brain which has not expressed vitellogenin.
Eukaryotic genome is packaged within the nucleus in the form of chromatin, which is a complex assembly of DNA, histones, number of non-histone protein and RNA components. This package not only compacts the genome to fit in the nuclear volume, but also has regulatory consequences in the expression of genes (Vasanthi and Mishra, 2008). Several structural as well as functional features of the nucleus have been studied. Regions that are stained bright and correspond to condensed chromatin states are termed as heterochromatin and lightly stained regions representing decondensed chromatin are termed as euchromatin (Passarge, 1979).
Micrococcal Nuclease (S7 Nuclease) is a relatively nonspecific endo-exonuclease that digests single-stranded and double-stranded nucleic acids, but is more active on single-stranded substrates. Cleavage of DNA or RNA occurs preferentially at AT or AU-rich regions yielding mononucleotides and oligonucleotides with terminal 3'-phosphates. The enzyme activity is strictly dependent on Ca2+. A common source is
E.coli cells carrying a cloned nuc gene encoding Staphylococcus aureus extracellular
nuclease (micrococcal nuclease) (Heins et al., 1967). The particular specificity of micrococcal nuclease, which preferentially cuts spacer DNA between nucleosomes (Dingwall et al., 1981), is yielding a regularly spaced ladder of nucleosomal DNA fragments. Active chromatin can be distinguished from bulk chromatin by its increased susceptibility to endonucleases. Micrococcal nuclease prefentially cleaves chromatin between nucleosomes (naked DNA regions). The structure of chromatin in eukaryotes exerts significant influences on many DNA related processes, including transcription, replication, recombination and repair (Li and Arnosti, 2010).
Probing chromatin structure with nucleases is a well-established method for determining the accessibility of DNA to gene regulatory proteins and measuring competency for transcription and a hallmark of many silent genes is the presence of translationally positioned nucleosomes over their promoter regions, which can be inferred by the sensitivity of the underlying DNA to nucleases, particularly micrococcal nuclease (Reese et al., 2008). Nuclease sensitivity and its correlation with gene activity can be viewed as a first step in determining the chromatin structure of a specific gene region and its role in gene expression (Wood and Felsenfeld, 1982). The improved assay had been used to distinguish micrococcal nuclease sensitivity of active chromatin
9
structure for three different genes during rat liver development, an additional step of digesting nuclease-digested DNA with a suitable restriction enzyme prior to Southern blotting hybridization was used (Hamid et al., 1996). Tissue-specific sensitivity of chromatin and vitellogenin gene to micrococcal nuclease after continuous exposure to 17P-estradiol was studied in the liver of Salmo salar fish, showing that the hepatic specificity of vitellogenin synthesis is manifested as structural modulations of the chromatin containing the vitellogenin gene (Waters and von-der Decken, 1992).
The myostatin gene promoter regions in a group of closely related fish (belonging to the suborder Percoidei) are highly conserved and contain several CpG dinucleotides within the conserved regions that could serve as targets for DNA methylation (Figure. 1.1). Sequence comparison between the promoter regions of myostatin-1 (MSTN-1) gene from several teleosts revealed a high conservation of the proximal promoter (~300 bp) region among fish of the order Perciformes and Pleuronectiformes (Funkenstein et
al., 2009).
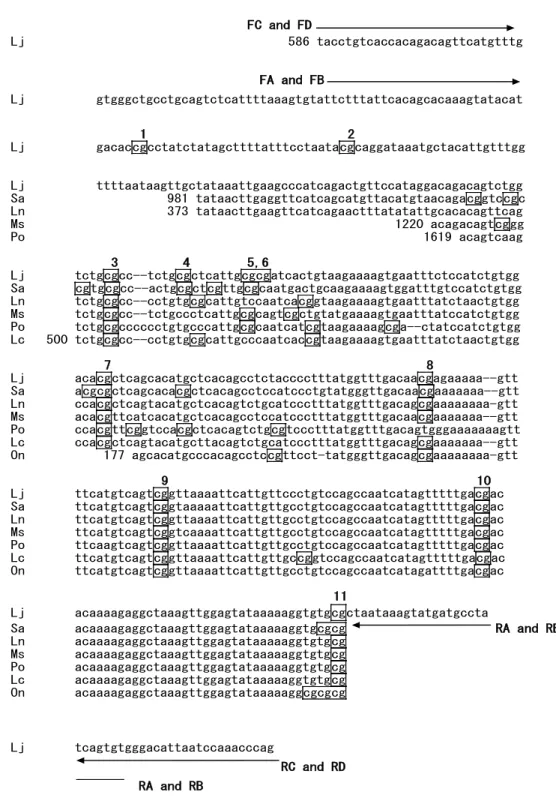
10 FC and FD Lj 586 tacctgtcaccacagacagttcatgtttg FA and FB Lj gtgggctgcctgcagtctcattttaaagtgtattctttattcacagcacaaagtatacat 1 2 Lj gacaccgcctatctatagcttttatttcctaatacgcaggataaatgctacattgtttgg Lj ttttaataagttgctataaattgaagcccatcagactgttccataggacagacagtctgg Sa 981 tataacttgaggttcatcagcatgttacatgtaacagacggtccgc Ln 373 tataacttgaagttcatcagaactttatatattgcacacagttcag Ms 1220 acagacagtcggg Po 1619 acagtcaag 3 4 5,6 Lj tctgcgcc--tctgcgctcattgcgcgatcactgtaagaaaagtgaatttctccatctgtgg Sa cgtgcgcc--actgcgctcgttgcgcaatgactgcaagaaaagtggatttgtccatctgtgg Ln tctgcgcc--cctgtgcgcattgtccaatcacggtaagaaaagtgaatttatctaactgtgg Ms tctgcgcc--tctgccctcattgcgcagtcgctgtatgaaaagtgaatttatccatctgtgg Po tctgcgcccccctgtgcccattgcgcaatcatcgtaagaaaagcga--ctatccatctgtgg Lc 500 tctgcgcc--cctgtgcgcattgcccaatcaccgtaagaaaagtgaatttatctaactgtgg 7 8 Lj acacgctcagcacatgctcacagcctctacccctttatggtttgacaacgagaaaaa--gtt Sa acgcgctcagcacacgctcacagcctccatccctgtatgggttgacaacgaaaaaaa--gtt Ln ccacgctcagtacatgctcacagtctgcatccctttatggtttgacagcgaaaaaaaa-gtt Ms acacgttcatcacatgctcacagcctccatccctttatggtttgacaacgaaaaaaa--gtt Po ccacgttcggtccacgctcacagtctgcgtccctttatggtttgacagtgggaaaaaaagtt Lc ccacgctcagtacatgcttacagtctgcatccctttatggtttgacagcgaaaaaaa--gtt On 177 agcacatgcccacagcctccgttcct-tatgggttgacagcgaaaaaaaa-gtt 9 10 Lj ttcatgtcagtcggttaaaattcattgttccctgtccagccaatcatagtttttgacgac Sa ttcatgtcagtcggtaaaaattcattgttgcctgtccagccaatcatagtttttgacgac Ln ttcatgtcagtcggttaaaattcattgttgcctgtccagccaatcatagtttttgacgac Ms ttcatgtcagtcggtcaaaattcattgttgcctgtccagccaatcatagtttttgacgac Po ttcaagtcagtcggttaaaattcattgttgcctgtccagccaatcatagtttttgacgac Lc ttcatgtcagtcggttaaaattcattgttgccggtccagccaatcatagtttttgacgac On ttcatgtcagtcggttaaaattcattgttgcctgtccagccaatcatagattttgacgac 11 Lj acaaaagaggctaaagttggagtataaaaaggtgtgcgctaataaagtatgatgccta Sa acaaaagaggctaaagttggagtataaaaaggtgcgcg RA and RB Ln acaaaagaggctaaagttggagtataaaaaggtgtgcg Ms acaaaagaggctaaagttggagtataaaaaggtgtgcg Po acaaaagaggctaaagttggagtataaaaaggtgtgcg Lc acaaaagaggctaaagttggagtataaaaaggtgtgcg On acaaaagaggctaaagttggagtataaaaaggcgcgcg Lj tcagtgtgggacattaatccaaacccag RC and RD RA and RB
Figure 1. 1. Conserved sequences in the myostatin gene 5’ flanking region. Homologous
sequences were retrieved from GenBank databases and aligned to identify conserved regions. Sequences are numbered according to the original sequence data. Lj, Lateolabrax japonicus (Genbank accession AY965685); Sa, Sparus aurata (EU881511); Ln, Lates niloticus (EF681885); Ms, Micropterus salmoides (EF071854); Po, Paralichthys olivaceus (DQ997779); Lc, Lates calcarifer (EF672685); On, Oreochromis niloticus (FJ972683). CpG sites are boxed, and those analyzed by bisulfite sequencing are indicated above the lines (from number 1 to 11). Primer sites for methylation analysis are shown by arrows with bold face characters. Primer names (FA, FB, FC, FD, RA, RB, RC, and RD) refer to Table 1.1.
11
1.2. MATERIALS AND METHODS 1.2.0. Fish Samples
Japanese sea bass Lateolabrax japonicus is an important species of the commercial as well as sport fishery and a highly promising species for sea farming in winter (Matsumiya et al., 1982). It naturally inhabits in the costal areas of Japan, China and Korea peninsula (Sun et al., 1994). Fish samples (40 cm to 60 cm SL) were purchased from a fish market at Izumisano Port, Osaka, Japan, and stored at -80C before use.
1.2.1. DNA Quantity Measurement
The amount and concentration of the isolated DNA were determined spectrophotometrically. DNA absorbs UV light so efficiently that optical absorbance can be used as an accurate and rapid measurement of its concentration. The absorbance (A) of the DNA preparations was determined at 260 and 280 nm since a very useful approximation of this absorbance (A) is that double-stranded DNA at 50 µg/ml in aqueous solution has an A260 of 1. Absorbance was also useful as a measure of the purity of DNA. The relevant spectrum was between 230 nm and 320 nm, having the absorbance maximum at 260 nm. Pure DNA has A260/A280 between 1.8 and 2.0. Higher values are often due to RNA contamination and lower values to protein and phenol contamination.
The concentration of the isolated DNA is calculated according to the formula: A (µg/ml) = A260 x 50 x dilution factor
1.2.2. DNA Quality Evaluation
Assessment of the quality and quantity of the isolation DNA is essential to all subsequent manipulations. The integrity of DNA was monitored by agarose gel electrophoresis. Aside from that, a rough estimate of the quantity of DNA band of interest was obtained on an EtBr-stained gel by loading molecular weight marker of known DNA concentration in an adjacent lane. Therefore, the amount of DNA loaded
12
was estimated by visual comparison of the band intensity with that of the standard.
1.2.3. Agarose Gel Electrophoresis
Electrophoresis is used to separate molecules based on their size. DNA has negative charge in solution, so it will migrate to the positive pole in an electric field. In agarose gel electrophoresis, the DNA is forced to move through a sieve of molecular pores made by agarose. Large fragments of DNA move slower than small fragments of DNA. So, the concentration of the gel depends on the fragments lengths beings separated. Agarose gel was prepared by dissolving agarose powder (Wako, Japan) to a desired molecular volume in 1x TAE buffer [40mM Tris base (Wako), 20mM acetic acid (Wako), 1mM EDTA (Bio-Rad)], and the mixture was heated in a microwave oven until the agarose totally dissolved. Then the agarose solution was cooled down until it reached 50°C and the solution was casted into a gel mold to solidify. A suitable comb was used to make wells. The gel was transferred to the electrophoresis chamber containing running buffer (1x TAE). Samples were mixed with 1x loading buffer [0.02% Bromophenol blue, 0.02% Xylene cyanol FF, 50% Glycerol and 1% SDS-Wako, Japan], and loaded together with molecular weight marker onto the wells for electrophoresis at a voltage of 100 V for 30 min until nucleic acids were fully separated. Then the gel stained by ethidium bromide (1 µg/ml EtBr, stock 5 mg/ml) and visualized by FMBIO-II Ultra-Veiw (Takara-Bio).
1.2.4. Primer Design
Generally, primers for polymerase chain replication (PCR) experiments ranged from 18-30 nucleotides in length, have guanine/cytosine content greater than 50% and a melting temperature (Tm) from 50-70°C. The following are things to avoid when designing primers. Mismatches between primers and target, especially at the 3' end of the primer, can occur if there are multiple T's at the 3' end or if there are runs of identical nucleotides, especially 3 or more G's or C's. To reduce hairpins and primer-dimer, avoid complementarity especially at the 3' end.
The oligonucleotides used as PCR primers in the present study are listed in (Table 1.1). Primers were designed to amplify bisulfite-treated genomic DNA, or were used to
13 amplify the probe for Southern blot hybridization.
Primer name Sequence ( 5'- 3' ) Position in AY965685
FA TTCTTTATTCACAACACAAAATATACAT 647-674 FB TTTTTTATTTATAGTATAAAGTATATAT 647-674 RA ATATTGATAGGTATTATATTTTATTAG 1023-1049 RB ACACTAATAAACATCATACTTTATTAA 1023-1049 FC TACCTATCACCACAAACAATTCATATTT 586-613 FD TATTTGTTATTATAGATAGTTTATGTTT 586-613 RC TTGGGTTTGGATTAATGTTTTATATTGA 1043-1060 RD CTAAATTTAAATTAATATCCCACACTAA 1043-1060 LjaMSTNP2F CCTATCTATAGCTTTTATTTCCTAATA 682-708 LjaMSTNR1 CACACCTTTTTATACTCCAACTTTA 986-1010
Table 1. 1: Oligonucleotides used as PCR primers
Names, sequences and targeted genomic regions are shown.
1.2.5. Chromatin Analysis
1.2.5.1. Nuclei preparation
Individual fish were dissected after thawing under running tap water, and selected tissues (liver, eye, kidney, brain and heart) were isolated. Each tissue was homogenized using a Teflon homogenizer in TES buffer (10 mM Tris-HCl, 140 mM NaCl, 25 mM EDTA, pH 7.8), after which cells were harvested by centrifugation at 3000 g for 10 min. The cell pellets were then resuspended in the same buffer and centrifuged again. The resultant washed cells were suspended in 10 volumes (relative to the packed-cell-volume) of hypotonic buffer (10 mM Tris-HCl, 15 mM KCl, 0.5 mM MgCl2, pH 7.8) and left for 10 min on ice. Once swollen, the cells were centrifuged at 1000 g, and the cell pellet was resuspended in 5 volumes of hypotonic buffer. The cells were then homogenized using a Dounce glass homogenizer (B-pestle), and the nuclei were harvested by centrifugation and resuspended in the hypotonic buffer.
14
1.2.5.2. Micrococcal nuclease (MNase) treatment
Micrococcal nuclease (MNase, Sigma-Aldrich) was added to the nuclei suspensions (40 µg/ml), after which the mixtures were incubated at 37C for 15, 30 or 60 min, and one-third of the original reaction volumes was collected at each time point. The nuclease reactions were terminated by adding aliquots (1/20 of the reaction volume) of 0.5 M EDTA, after which the DNA was isolated from each reaction mixture.
1.2.5.3. Isolation of genomic DNA from MNase-treated cell nuclei
An equal volume of TES buffer containing 1% SDS and 0.5 mg/ml Proteinase K (Invitrogen Life Technologies) was added to the MNase-treated nuclei mentioned above, after which the reaction mixtures were incubated for 30 min at 50C. The DNA was then extracted with phenol, precipitated with ethanol and then dissolved in TE buffer (10 mM Tris HCl, 1 mM EDTA, pH 7.8). Total DNA isolated from the MNase-treated nuclei and the intact genomic DNA (as a control) was digested with the restriction endonuclease TaqI (Takara-Bio). The reaction mixture was 100 µl volume containing 10-15 µg of DNA from control or treated DNAs, 1x High buffer (Nippongene) and 10 units of TaqI enzyme. The samples were incubated in water-bath for one hour at 65C. DNA fragments were extracted by adding equal volume from phenol and precipitated with ethanol. After the centrifugation, the DNA pellet was rinsed with 80% ethanol, and dried by a vacuum drier (GLD-101 HITACHI) for 5 min, and then dissolved in TE buffer.
1.2.5.4. Southern blotting hybridization
Southern blotting hybridization allows visualizing a specific DNA fragment against the background of a complex genome. Generally, genomic DNA is digested with one or more restriction enzymes, and the fragments are transferred from the gel to a membrane and then exposed to a solution containing a DNA probe. This technique relies on the hydrogen bonds that hold together complimentary adenine-thymine and guanine-cytosine base pairs in double-stranded DNA. At temperatures above 90C, or a pH greater than 10.5, these hydrogen bonds are disrupted, causing the complementary strands to denature into single strands. Under proper conditions of salt content,
15
temperature, and pH, complementary single-stranded molecules can renature to restore the original duplex DNA molecule. This process is known as hybridization. A single stranded DNA probe, derived from either a restriction fragment, a plasmid containing a cloned sequence of interest, or a synthetic oligonucleotide, is employed to recognize and hybridize to its complementary sequence in a sample of genomic DNA. A radioactive, colorimetric, or chemiluminescent group attached to the probe allows areas of probe hybridization to be visualized (Southern, 1975).
- Reagents
Denaturation buffer: 0.5 M NaOH, 1.5M NaCl.
Neutralization buffer: 0.5 M Tris, 1.5 M NaCl (pH 7.5) adjusted by HCl. 20x SSC buffer: 3 M NaCl, 0.3 M Na3 Citrate (pH 7.0) adjusted by HCl.
Prehybridization buffer: 5x SSC, 0.1% (w/v) N-lauroylsarcosine, 0.2% (w/v) SDS,
1% (w/v) blocking reagent.
Blocking buffer: 1% (w/v) blocking reagent, 10% Neutralization buffer. Wash buffer 1: 2x SSC, 0.1% SDS.
Wash buffer 2: 0.1x SSC, 0.1% SDS.
Wash buffer 3: 0.1 M Tris-HCl, 0.15 M NaCl.
Detection buffer: 0.2 M Tris-HCl pH (9.5), 0.1 M NaCl, 0.1 M MgCl2.
- Southern blotting procedure
1. Intact and nuclease-digested DNA (10-15 µg per sample) fractionated by 1.2% TAE agarose gel electrophoresis (20V. overnight).
2. The gel was stained with EtBr and visualized by FMBIO- II Ultra-Veiw (Takara-Bio). 3. Determination of the marker’s size under an ultraviolet transilluminator.
4. Denaturation the DNA by gently shaking the gel in 2 volumes of denaturation buffer for 30 min.
16 was repeated.
6. The fractionated DNA on the agarose gel was transferred to nylon membranes (Hybond™-N+, Amersham Biosciences) using the capillary blot procedure and kept overnight (Southern, 1975).
7. The place of the marker was labeled by making small pores on the membrane related to the markers’ fragments.
8. For binding the nucleic acids to the membrane, CL-1000 Ultraviolet cross-linker was used for 30 seconds, and the membrane was dried inside the oven at 70C for 10 min. 9. For prehybridization, 30 µg from junk bacterial DNA was added to 20 ml hybridization buffer, heat-shocked for 3 min., transferred to ice and then added to the membrane for 4 hours incubation at 65C inside (DNA oven HI-80R, Kurabo).
10. The probe was then labeled using a PCR DIG Probe Synthesis Kit (Roche) with a pair of DNA primers, LjaMSTNP2F and LjaMSTNR1. The 50 µL reaction mixture contained 0.5 ng of DNA template, 1x PCR buffer with MgCl2, 200 µM dNTP (PCR DIG labeling mix), 0.4 μM of each primer and 2.6 U enzyme mixture. The PCR was run as followed: initial incubation at 95C for 5 min, followed by 35 cycles (30 sec at 95C, 30 sec at 55C, and 2 min at 72 C); and a final extension of 7 min at 72 C.
11. The hybridization step was carried by the diluted probe with 15 ml hybridization buffer after heat-chock for 3 min., shielded by ice and incubated overnight at 65C in the same previous oven.
- Washing the membrane
1. The probe was removed and the membrane was washed by wash buffer 1 for 10 min. twice at 60C.
2. Secondly, the membrane was washed twice by wash buffer 2 for 10 min. at same previous temperature.
3. For 30 min., the membrane was blocked with shacking at room-temperature.
4. Anti-Digoxigenin-AP, Fab fragments (2.6 units) was added to the new volume (10 µl) of blocking buffer for detection and gently shacked by hand for 30 min.
17
5. The membrane was washed with wash buffer 3 for 15 min. by shacking.
6. Finally, it was rinsed with detection buffer 2 min., packed between plastic sheets with NBT/BCIP detection buffer, and incubated 2 hours at 37C until bands became clearly visible. The membranes which have clear signals were scanned by Epson GT-X700 scanner.
1.2.6. Methylation Status
1.2.6.1. Bisulfite treatment of genomic DNA
Fish was dissected, and tissue samples from brain, kidney, spleen, liver, heart, eye, muscle, intestine and gills were homogenized in TES buffer, after which the total genomic DNA was isolated as described previously (Sambrook and Russell, 2001). High molecular weight of DNA was dissolved in TE buffer. Genomic DNA samples (2µg) were then digested with EcoRI (New England Biolabs) in the reaction mixture (100 µl), extracted with phenol, precipitated with ethanol and then dissolved in TE buffer. The quantitiy of DNA fragments was estimated spectrophotometrically, and the resultant DNA fragments (500 ng) were adjusted to 20 µl volume from each, and denatured by boiling for 3 min. After that, they were subjected to bisulfite modification using EZ DNA Methylation-GoldTM Kit (Zymo Research) according to the manufacture’s instruction. Bisulfite treatment converts cytosines to uracils, but 5-methylcytocines remains intact. Thus, the combination of bisulfate treatment and following PCR enable us to detect methylated cytosines occurring in genomic DNA (Clark et al., 1994; Kato, 1996). A set of primers (FC and RC) was used for PCR to amplify the antisense strand of the bisulfite treated DNA, while another set of primers (FD and RD) was used for PCR to amplify the sense strand of bisulfite treated DNA. Each PCR product was then subjected to a second PCR with a set of nested primers: FA and RA for antisense strand amplification and FB and RB for sense strand amplification. The PCR was performed with Ampli Taq Gold 1X PCR Master Mix (Applied Biosystems), with 0.4 μM of each primer and the mixture (25µl) in a GeneAmp PCR System 9700 (Applied Biosystems). The amplification protocol entailed initial
18
denaturation at 95C for 5 min followed by 35 cycles of 95C for 30 sec, 50C for 30 sec and 72C for 2 min, and a final elongation at 72C for 7 min (Figure 1.2). The resultant PCR products (related to each tissue) were then precipitated with ethanol (2.5 volumes of 100% ethanol) in 0.3 M sodium acetate, rinsed with 80% ethanol, and dissolved in sterile water. The PCR products were blunt-ended in parallel with phosphorylated at 5’-end by Blunting Kination Ligation Reagent Kit (Takara-Bio) according to the manufacturer’s instruction, and cloned into the HincII site of pUC118 vector to transform HIT™ JM-109 competent cells, (RBC Science) (Figure 1.2).
1.2.6.2. Colony-direct PCR
This technique was used to quickly screen for plasmid inserts directly from E.
coli colonies. White colonies (10-20 from each plate) were picked to 25 µl PCR reaction
mixture and amplified with M13 primers. After preincubating the reaction mixture for 5 min at 94 °C, a cycling protocol was applied entailing 30 cycles of 94 °C for 30 sec, 55 °C for 30 sec and 72 °C for 1 min, followed by incubation at 72 °C for 7 min.
1.2.6.3. Sequencing reactions
The plasmid DNAs which have prospective inserts were firstly purified by Hi Yield PCR DNA Fragments Extraction Kit (RBC-Bioscience), and then dideoxy-mediated termination method was performed for DNA sequencing analysis using Big Dye Terminator ver. 3.1 Cycle Sequencing Kit (Applied Biosystems) and ABI3730 Sequencer (Applied Biosystems) with M13 primers. The sequencing PCR reaction performed at 96°C for 2 min, followed by 25 cycles of 10 sec at 96°C, 5 sec at 50°C, and 4 min at 60°C, followed by a final elongation step at 60°C for 1 sec. The results from sequencing were applied to Chromas Lite (Technelysium Pty, Ltd.) for editing. The homology search and multiple sequence alignment were done by GENETYX-MAC ver. 13 (Genetyx Co, Ltd., Tokyo).
19
Figure 1. 2. Schematic illustration of bisulfite sequencing. PCR amplification for sense and
antisense strands, 2% agarose gel electrophoresis of the PCR products from the variety of tissues (spleen, kidney, brain, liver, heart, eyes, intestine, muscle and gill). The amplicons were inserted into HincII site for pUC118 vector.
sense strand antisense strand target region (393 bp) Amplification • Blunting • Kination • Ligation • Transformation • Colony PCR • Sequencing FB FD RB RD RA RC FA FC 1500 1000 500 18 17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 M
•Fragments from 1-9 for antisense strand • Fragments from 10-18 for sense strands
bp 393 pUC118 3162bp HincII 2896 lac Z Plac
20
1.3. RESULTS
1.3.1. Bisulfite Modification of Genomic DNA to Evaluate the Methylation Status
We initially determined the nucleotide sequences of the PCR products obtained with bisulfite-treated DNA (5-18 clones each from the respective samples) (Tables 1.2 and 1.3). The efficiency of the bisulfite modification (conversion of C to T) was estimated by counting the numbers of cytosines remaining at CpNs (N≠G), and the conversion frequency was found to be 99% or higher (Figure 1.3).
Tissues Sense strand Antisense strand
C at CpG C at CpN (N≠G) G at CpG G at NpG (N≠C) intestine 0.00 0.32 4.55 0.68 brain 1.52 0.66 1.52 0.61 spleen 2.10 0.49 1.82 0.36 liver 1.52 0.53 1.52 0.91 eyes 6.36 0.48 0.00 1.12 kidney 0.76 0.66 1.01 1.01 gill 3.50 0.37 1.52 0.30 muscle 1.52 0.32 1.07 0.75 heart 5.68 0.40 0.91 0.55
Table 1. 2: Summary of bisulfite sequencing: relative percentage of unconverted bases after
bisulfite treatment.
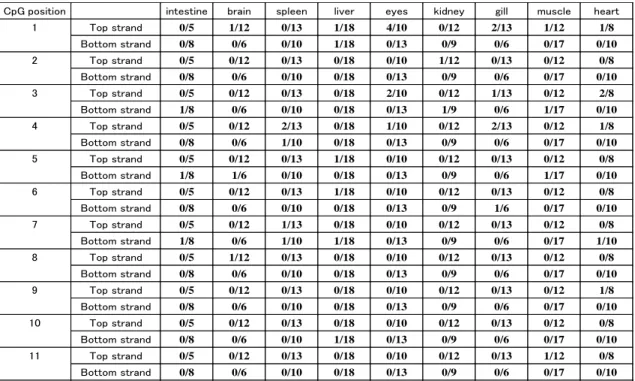
The number of cytosine residues remaining at CpGs appeared to be above the background level in the sense strands from eye and heart (methylation frequency: 6.36% and 5.68%, respectively), but were at the background level in the complementary strands from these tissues. This may indicate that the methylation frequency was within the error range of the random sampling. In any case, the frequency of methylated cytosine was very low in the tissues examined, regardless of the level of myostatin gene expression. This runs counter to the suggestion drawn from the results of Ye et al., 2007, and may mean that CpG methylation is not involved in regulating myostatin gene expression in L. japonicus.
21
Figure 1. 3. Sequencing of L. japonicus promoter region. Bisulfite-modified genomic DNA,
was subjected to the PCR with the primers were shown in Table 1.1. The PCR amplicons were inserted in pUC118 vector and sequenced. The converted cytosine nucleotides to thymine nucleotides are indicated by black arrows.
CpG position intestine brain spleen liver eyes kidney gill muscle heart
1 Top strand 0/5 1/12 0/13 1/18 4/10 0/12 2/13 1/12 1/8 Bottom strand 0/8 0/6 0/10 1/18 0/13 0/9 0/6 0/17 0/10 2 Top strand 0/5 0/12 0/13 0/18 0/10 1/12 0/13 0/12 0/8 Bottom strand 0/8 0/6 0/10 0/18 0/13 0/9 0/6 0/17 0/10 3 Top strand 0/5 0/12 0/13 0/18 2/10 0/12 1/13 0/12 2/8 Bottom strand 1/8 0/6 0/10 0/18 0/13 1/9 0/6 1/17 0/10 4 Top strand 0/5 0/12 2/13 0/18 1/10 0/12 2/13 0/12 1/8 Bottom strand 0/8 0/6 1/10 0/18 0/13 0/9 0/6 0/17 0/10 5 Top strand 0/5 0/12 0/13 1/18 0/10 0/12 0/13 0/12 0/8 Bottom strand 1/8 1/6 0/10 0/18 0/13 0/9 0/6 1/17 0/10 6 Top strand 0/5 0/12 0/13 1/18 0/10 0/12 0/13 0/12 0/8 Bottom strand 0/8 0/6 0/10 0/18 0/13 0/9 1/6 0/17 0/10 7 Top strand 0/5 0/12 1/13 0/18 0/10 0/12 0/13 0/12 0/8 Bottom strand 1/8 0/6 1/10 1/18 0/13 0/9 0/6 0/17 1/10 8 Top strand 0/5 1/12 0/13 0/18 0/10 0/12 0/13 0/12 0/8 Bottom strand 0/8 0/6 0/10 0/18 0/13 0/9 0/6 0/17 0/10 9 Top strand 0/5 0/12 0/13 0/18 0/10 0/12 0/13 0/12 1/8 Bottom strand 0/8 0/6 0/10 0/18 0/13 0/9 0/6 0/17 0/10 10 Top strand 0/5 0/12 0/13 0/18 0/10 0/12 0/13 0/12 0/8 Bottom strand 0/8 0/6 0/10 1/18 0/13 0/9 0/6 0/17 0/10 11 Top strand 0/5 0/12 0/13 0/18 0/10 0/12 0/13 1/12 0/8 Bottom strand 0/8 0/6 0/10 0/18 0/13 0/9 0/6 0/17 0/10
Table 1. 3: Summary of bisulfite sequencing: numbers of unconverted cytosines at each CpG
site.
The ratios (number of methylated cytosines at CpG) / (number of sequences examined) are shown.
The CpG positions are defined in Figure. 1.1.
22
1.3.2. Sensitivity to MNase in the myostatin gene promoter region
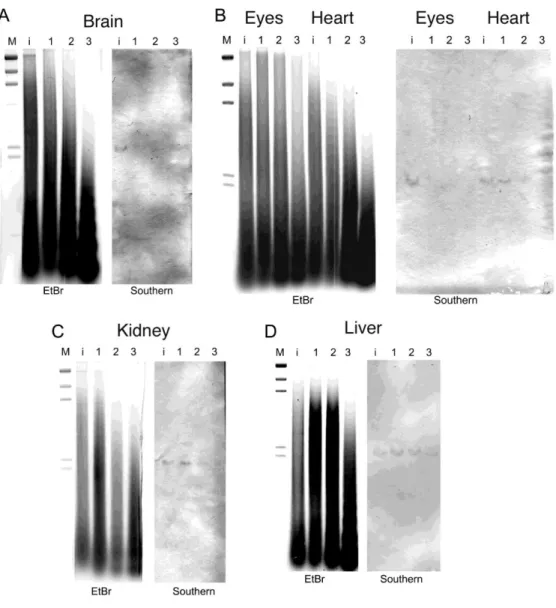
Southern blot hybridization analysis was carried out to evaluate the structure of the chromatin in the myostatin gene promoter region. DNA in the heterochromatin would be more resistant to MNase, which dominantly degrades naked DNA. Myostatin gene regions in brain and eye, which express myostatin (Ye el al., 2007), indicated at the bottom of Table 1.3), were highly susceptible to MNase (Fig. 1.4A, B). By contrast, the myostatin promoter region was less susceptible in heart, and those in kidney and liver are even more resistant to MNase (Fig. 1.4B~D), which is consistent with the lower level of myostatin gene expression in these organs. We have examined several fish individuals for chromatin analyses, and the profiles of MNase digestion were reproducibly observed.
As known, the level of myostatin gene expression was highest in muscle. We have also tried to examine the chromatin structure of myostatin gene in muscle, gills and intestine. Isolation of intact nuclei, however, was very difficult for these tissues. Most of muscle consists of fibrous protein (actin and myosin fibers), and during the homogenization of the muscle, nuclei are destroyed by mechanical shearing and/or the other factors. As for gills and intestine, which also express the myostatin gene, the preparation of nuclei contained a high activity of endogenous nuclease, so that the “nucleosome ladders” were not observed after MNase treatment. It was not possible to avoid the effects of nonspecific digestion (shearing, endogenous nucleases, etc.), and these tissues were not submitted to Southern blot analysis.
23
Figure 1. 4. Southern blot analysis of TaqI-digested genomic DNA isolated from MNase-treated
cell nuclei. Southern blots were probed with digoxigenin-labeled DNA fragments covering position 682 to 1010 of AY965685. Lane M, lambda DNA-HindIII digest size marker; lane i, genomic DNA isolated from intact cell nuclei; lane 1, DNA fragments isolated from cell nuclei after 15 min of MNase treatment; lane 2, DNA fragments isolated from cell nuclei after 30 min of MNase treatment; lane 3, DNA fragments isolated from cell nuclei after 60 min of MNase treatment. Panel A, cell nuclei isolated from brain; panel B, eye and heart; panel C, kidney; panel D, liver.
24
1.4. DISCUSSION
Although it is known that many active genes promoters for transcription in the human genome show little DNA methylation (Weber et al., 2007). DNA methylation at cytosines has not been well studied as an epigenetic trait in fish. There is reportedly the absence of genome-wide changes in DNA methylation during early embryogenesis in zebrafish (Macleod et al., 1999), though DNA methylation in the promoter region appears to correlate with vitellogenin I gene expression in the same fish (Strömqvist et
al., 2010). In the medaka fish Oryzias latipes, by contrast, the vast majority of CCGG
sites are methylated during early embryonic development, and the extent of the methylation at these sites does not change, or changes very little, during the remaining stages of embryogenesis (Walter et al., 2002).
In the present study, we analyzed the methylation status of the 5’ flanking region of the myostatin gene in various tissues from L. japonicus. Bisulfite treatment and subsequent PCR revealed nearly complete conversion of cytosines to thymines at CpNs (N≠G), so that very few cytosines remained at CpGs (Table 1.2 and 1.3). This may mean that the methylation system is not utilized for tissue-specific regulation of myostatin gene. Sequence comparison among closely related species has shown that the 5’ flanking regions of myostatin genes are highly conserved (Figure 1.1). The majority of the variation within these conserved regions is caused by indels at contiguous nucleotides (oligo-C or oligo-A) and transitional substitutions. While several CpG sites are situated within the conserved regions, transitional substitutions (CpG to TpG or CpG to CpA) are observed in the unconserved CpG sites. Methylated cytosines tend to change to thymines through spontaneous deamination (Ehrlich et al., 1986), so that 5-methyl-CpG may be substituted by TpG or CpA on an evolutionary time scale. Thus DNA methylation may not be involved in myostatin gene regulation, but may instead reflect evolutionary processes. These results are consistent with the results that the promoter region of the CYP1A gene in killifish were not methylated, suggesting that CpG methylation is not the mechanism responsible for the lack of CYP1A induction in this fish and some other epigenetic mechanism may exist (Timme-Laragy et al. 2005).
Our MNase assays suggest myostatin gene is compacted into heterochromatin in tissues where it is not expressed, such as liver and kidney, whereas it is susceptible to