昆虫寄生糸状菌由来化合物 metarhizin 類の構造に
関する研究
著者
曲 翠玉
学位授与機関
Tohoku University
平成 23 年度修士論文
昆虫寄生糸状菌由来化合物 metarhizin 類の構造に
関する研究
東北大学大学院薬学研究科
分子薬科学専攻
医薬資源化学分野
学籍番号 B0YM1010 曲 翠玉
目次
序論 1 本論 第一章 Metarhizin E および F のフラノン環に相当するモデル化合物のデザインおよび 合成研究 第一節 モデル化合物 17 のデザインと逆合成 8 第二節 モデル化合物 rac-17 の合成検討 13 第三節 ラセミ体でのモデル化合物 rac-18 の合成 15 第四節 考察と今後の展望 17 結語 19 実験の部 20 第一章の実験 21 謝辞 27 参考文献 28本論文中において以下の略記を用いた. Ac : acetyl Bz : benzoyl calcd. : calculated CD : circular dichroism DBU : 1,8-diazabicyclo[5.4.0]undec-7-ene DIBAL : diisobutylaluminium hydride DMAP : 4-(dimethylamino)pyridine DMF : N,N-dimethylformamide DMI : 1,3-dimethyl-2-imidazolidinone DMP : Dess-Martin periodinane
EIMS : electron impact mass spectroscopy Et : ethyl
HPLC : high performance liquid chromatography HR : high resolution
LHMDS : lithium bis(trimethylsilyl)amide Me : methyl
MS : mass spectrometry
NMR : nuclear magnetic resonance Ph : phenyl
PTSA : p-toluenesulfonic acid rt : room temperature
TBAF : tetrabutylammonium fluoride TBDPS : tert-butyldiphenylsilyl
TLC : thin layer chromatography UV : ultraviolet
序論
昆虫寄生糸状菌は,広く土壌中に分布しており,その多くは特定の昆虫に特異的に寄生 して増殖し,栄養分の奪取,組織の破壊,毒素の産生などにより昆虫を死に至らしめる. カイコやミツバチといった有用昆虫も侵すために,これまで寄生菌に対する研究は主に昆 虫病理学的観点から進められてきた.その結果,黒彊病菌 Metarhizium anisopliae から 5 個 のアミノ酸と 1 個のオキシ酸からなる環状デプシぺプチド destruxin A (1) が硬化病の原因 物質として単離された (Figure 1).1 Destruxin 類の発見を契機に,Aspergillus ochraceus からaspochracin (2),2 白彊病菌 Beauveria bassiana から beauvericin (3)3 や bassianolide (4)4 などの殺
虫性物質が次々と単離された (Figure 1).
Destruxin A (1)
(from Metarhizium anisopliae) (from Aspergillus ochraceus) Aspochracin (2)
Beauvericin (3) Bassianolide (4) (from Beauveria bassiana)
一方,昆虫寄生糸状菌からは新規医薬品のリード化合物となるようないくつかの有望な 化合物も単離されている.このような化合物の例として,冬虫夏草 Paecilomyces cicadae から単離された免疫抑制物質である myriocin (5)5 や Cordyceps militaris から単離された抗生
物質である cordycepin (6)6 などが挙げられる (Figure 2). このようにいくつかの寄生菌からは,有用な二次代謝産物も単離されている.しかし, 感染例が少ないために入手困難な種や,人工培養が困難な種も多く,他の微生物に比べる と昆虫寄生糸状菌の成分研究は十分に行われているとは言い難い.したがって昆虫寄生糸 状菌は未利用菌類として,新規生物活性物質の探索源になり得ると考えられる. 代表的な昆虫寄生糸状菌は,分類学的には主に鞭毛菌類,接合菌類,子嚢菌類,および 不完全菌類に含まれている.それらの中でも比較的容易に人工培養ができる不完全菌類の 昆虫寄生糸状菌に着目し,これまでに当研究室では Beauveria 属や Hirsutella 属, Metarhizium 属などから数種の菌の培養条件検討および成分研究を行ってきた.
Metarhizium 属は不完全菌類の不完全糸状菌綱に分類され,M. album や M. anisopliae, M.
anisopliae majus など 8 種の菌を含む.最も普通種である M. anisopliae は,冒頭でも述べた
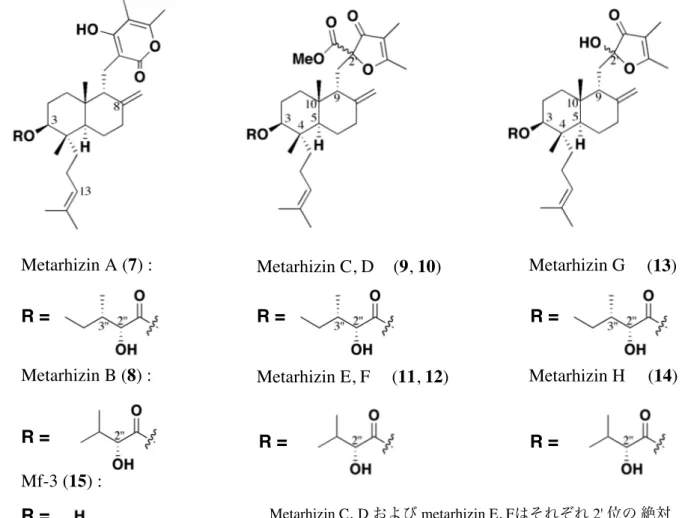
ように黒彊病菌として知られ,病原力が強く寄主範囲も広いため,二次代謝物の研究が広 く行われている7 .一方で同属の他の 7 種に関しては感染例が少なく,成分研究の報告例 も少ない8 .当研究室の喜多山は,その中の一種である M. flavoviride の成分研究におい て,ショウジョウバエ胚由来 S2 細胞9 に対する毒性を指標として新規化合物である metarhizin A~H (7~14) および既知化合物 Mf-3 (15) を単離した (Figure 3).10,.14 Mf-3 (15) は 既に Penicillifer superimpositus より単離され,骨再吸収を阻害することにより骨代謝異常 に対する効果が期待される BR-050 と同一の化合物であった.11
(from Paecilomyces cicadae)
Myriocin (5) Cordycepin (6) (from Cordyceps militaris) Figure 2. Myriocin (5) と codycepin (6) の構造
Metarhizin A (7) および metarhizin B (8) は種々のがん細胞に対して選択的かつ強力な増殖 抑制作用を示した.その構造活性相関研究により,活性の発現に 3 位の側鎖の長さや形は 非常に重要であることが示されている.12
ところで,metarhizin A~H (7~14) のうち,これまでに当研究室の喜多山により metarhizin A (7) の絶対配置が決定され,星により metarhizin B (8) の絶対配置が決定された.
Metarhizin A (7) については,viridoxin A (16)13 へ誘導することにより 3S, 4S, 5R, 9R, 10R,
2"R, 3"S であることが示されている (Figure 4).Viridoxin A (16) はコロラドイモムシ (Leptinotarsa decemlineata) の個体に対する殺虫性を指標に M. flavoviride から初めて見つけ られた二次代謝産物である.
Figure 3. 昆虫寄生糸状菌 Metarhizium flavoviride より単離された化合物
Metarhizin C, D および metarhizin E, Fはそれぞれ 2' 位の 絶対 配置のみが異なるジアステレオマー Metarhizin A (7) : Metarhizin B (8) : Mf-3 (15) : Metarhizin C, D Metarhizin E, F Metarhizin G Metarhizin H (9, 10) (11, 12) (13) (14) R = R = R = R = R = R = R =
Metarhizin B (8) の 3, 4, 5, 9, 10 位の絶対配置については,metarhizin A (7) の NMR および CD スペクトルと metarhizin B (8) のそれらを比較することにより両化合物で同一であるこ とが示されている.また,2" 位の絶対配置は,2 位の絶対配置が既知の (R)-2-hydroxy-3-methylbutanoic acid と Mf-3 (15) を縮合させて metarhizin B (8) へと誘導することにより決定 された.誘導した metarhizin B (8) の NMR スペクトルデータおよび比旋光度が天然物のも のと一致したことにより 2"S であることが示されている (Figure 5). Figure 4. Viridoxin A (16) の構造 Figure 5. Mf-3 (15) と立体既知化合物を用いた metarhizin B (8) への誘導 + Metarhizin B (8) Mf-3 (15) (R)-2-Hydroxy-3-methylbutanoic acid (S) 2''
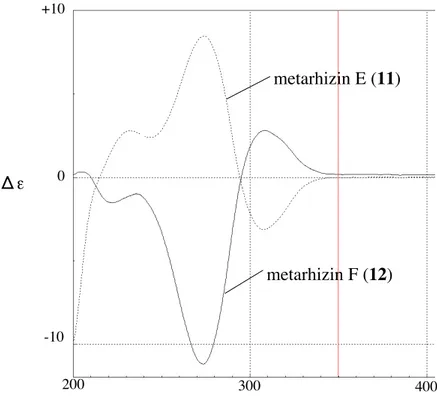
一方,metarhizin C~H (9~14) の相対および絶対配置については単離された量が微量で あったために十分な検討が行われておらず, metarhizin C, D (9~10)についてNOESY スペク トルの相関から 3, 4, 5, 9, 10 位の相対配置が決定されているのみである. デカリン環部分および 3 位の側鎖部分の絶対配置については,その生合成を考慮すると metarhizin A (7) および metarhizin B (8) と同一の絶対配置をとることが予想される.しかし 構造的に大きく異なる metarhizin C~F (9~12) (Figure 6) のフラノン環部分の 2' 位について は,周囲にプロトンが少ないため NMR スペクトルから得られる情報が少なく,推測も出 来ず未決定のままであった10. ところで,CD スペクトルによる絶対構造の決定は,天然物のように微量の化合物で あっても行うことができる非常に有用な手法である.天然より得られた Metarhizin E (11) および metarhizin F (12) の CD スペクトルの結果より,11 ではそれぞれ 275 nm 付近に負の Cotton 効果,310 nm 付近に正の Cotton 効果が観察されたのに対し,12 ではその逆の現象 が観察されている (Figure 7).Metarhizin E (11) および metarhizin F (12) は 2' 位の絶対配置 のみが異なる立体異性体であることが分かっており,それに近接する α, β- 不飽和カルボ ニルが発色団であると考えられるので,ここで観察された Cotton 効果は 2' 位の絶対配置 の影響によるものであると推測できる. Figure 6. Metarhizin C~F (9~12) の構造 Metarhizin E, F (11, 12) Metarhizin C, D (19, 10)
そこで本研究では metarhizin C~F (9~12) の 2' 位について,フラノン環部位に相当するモ デル化合物と天然物との CD スペクトルの比較により絶対配置を決定することを目的とし た.モデル化合物として metarhizin C~F (9~12) のデカリン環部分をシクロヘキシル基に置 換した化合物 (R)-17 および (S)-17 をデザインした (Figure 8).その CD スペクトルを天然 物のものと比較することで,発色団である α, β- 不飽和カルボニルに近接する metarhizin E (11) および metarhizin F (12) の 2' 位の不斉炭素の絶対構造を検討することができるはずで ある.
Figure 7. Metarhizin E (11) および metarhizin F (12) のCD スペクトル
0 +10 400 300 200 metarhizin E (11) Δε Wavelength [nm] metarhizin F (12) -10 Figure 8. モデル化合物 (R), (S)-17 (R)-17 (S)-17 2 2
モデル化合物 17 の合成においては 2 位の四級炭素の立体の制御が鍵となる.筆者は 3-オキソ吉草酸メチルを出発原料に用いて,カルベノイドを経由する転位反応15を利用する ことで, 2 位の不斉中心を構築する合成経路と考察した.化合物 17 に関しては,シクロ ヘキシル基の立体障害のため合成には至らなかったものの,シクロヘキシル基の代わりに ブチル基を導入した化合物 18 を新たなモデル化合物としたところ,そのラセミ体の合成 を行うことができた (Figure 9). これらの詳細について第一章で述べる. Figure 9. モデル化合物 (R)-18 および (S)-18 (R)-18 (S)-18 2 2
本論
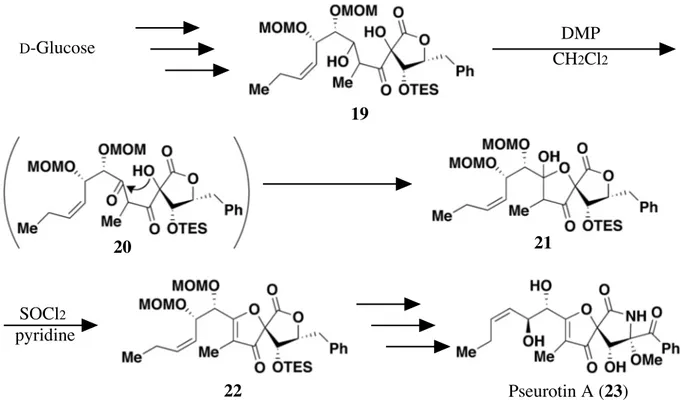
第一章 Metarhizin E および F のフラノン環に相当するモデル化合物のデザインおよび 合成研究 第一節 モデル化合物 17 のデザインと逆合成 Metarhizin E (11) および metarhizin F (12) は,分子量が同一で 1H および 13C NMR スペク トルにおけるほとんどのシグナルも一致している化合物である.そして,NMR スペクト ルにおいてシグナルに差がみられた部分を比較することにより,2' 位の絶対配置のみが異 なる立体異性体であると推測されている (Figure 10).10 このフラノン環を含む 2' 位について は,周囲にプロトンが少ないため NMR スペクトルより得られる情報が少なく,絶対配置 を決定するには構造変換などのアプローチが必要である.しかしこれらの化合物は単離さ れた量が微量であり,化学変換による検討は困難であることが予想された. 序論でも述べたように,天然より得られた metarhizin E (11) および metarhizin F (12) の CD スペクトルの結果より,metarhizin E (11) ではそれぞれ 275 nm 付近に負の Cotton 効 果,310 nm 付近に正の Cotton 効果が観察されたのに対し,metarhizin F (12) ではその逆の 現象が観察された (Figure 7).この違いは 2' 位の絶対配置を含むフラノン環部位による影 Metarhizin E, F (11, 12) Figure 10. Metarhizin E, F (11, 12) の構造 2'響と推測できる.したがって,2' 位が制御されたフラノン環部位を含む部分構造を合成 し,天然物と CD スペクトルを比較することで metarhizin E (11) および metarhizin F (12) の 絶対配置を決定できると考えられる.そこで,モデル化合物として metarhizin E, F (11, 12) のデカリン環部分をシクロヘキシル基に置換した化合物 (R)-17, (S)-17 をデザインした (Figure 11). モデル化合物 (R)-17, (S)-17 の合成において困難なところは,フラノン環の構築と 2 位 不斉中心の制御である.Aoki らは,pseurotin A (23) の全合成を行った際に,metarhizin E, F (11, 12) のフラノン環部位に類似した構造を構築した.まず,1,4-ジオール体 19 に Dess-Martin periodinane を用いて二級水酸基を酸化し,得られたケトンに対して γ 位の三級 水酸基が攻撃することで,γ-ラクトール 21 を合成した.その後,pyridine 存在下 thionyl chloride を作用させて脱水反応を行いフラノン環を有する化合物 22 を合成したと報告して いる(Figure 12).16 (R)-17 (S)-17 Figure 11. モデル化合物 (R)-17, (S)-17 2 2
この手法を参考に,化合物 (S)-17 のフラノン環は,化合物 30 のような 1,4 ジオールの 二級水酸基の酸化によって得られたケトンに,三級水酸基が攻撃することで分子内環化し て,更に脱水反応をおこなうことで構築することができると考えられる (Figure 13).な お,これらの反応の過程では,2 位の不斉中心は保持されることから, 2 位が S 配置 の化合物 30 が得られればよいことになる.
Figure 12. Aoki らによる pseurotin A (23) の全合成16
D-Glucose DMP CH2Cl2 SOCl2 pyridine Pseurotin A (23) 19 22 21 Figure 13. (S)-17 の逆合成解析 (1) 20 脱水 (S)-17 30 2 環化 酸化 2 2 2 2
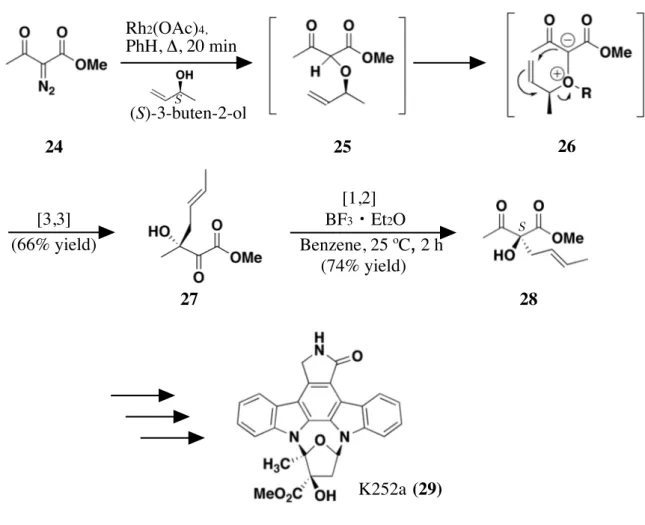
続いて,化合物 30 の 2 位の立体の制御である.Wood らは,K252a (29) の全合成を 行った際に,30 に類似した光学活性化合物を合成中間体として得ている15.まず,ロジウ ム (Ⅱ) アセテート存在下,ジアゾ化合物 24 に光学活性な (S)-3-buten-2-ol を作用させるこ とで,水素-酸素結合への挿入反応が起こり中間体 25,更に環化および転位反応を経て, α-ケトエステル 27 とした.続いて,ルイス酸を作用させることで [1,2] 転位が起こり, β-ケトエステル 28 を合成した (Figure 14).この合成法では,(S)-3-buten-2-ol の不斉中心は 最終的に化合物 28 の 2 位に転写されており,光学活性な 3 級アルコールを得ることが 可能となる. Rh2(OAc)4, PhH, Δ, 20 min (S)-3-buten-2-ol BF3・Et2O Benzene, 25 ºC, 2 h (74% yield) (66% yield) [3,3] [1,2] 24 25 26 28 27 K252a (29)
Figure 14. Wood らよる K252a (29) の全合成15
S S
この合成経路を基づいて,モデル化合物 (S)-17 の逆合成解析を行った.前述のように, 化合物 30 を酸化して,環化および脱水反応を行うことで, (S)-17 を得られる.化合物 30 は化合物 31 に対して接触還元とアルドール反応を行うことにより得ることとした.ま た,化合物 31 は前述のようにジアゾ化物 32 と (S)-2-methylenecyclohexanol との反応によっ て,2 位にメチルシクロヘキセニル基が結合した化合物 31 を得ることができる.最後 に,化合物 32 は 3-オキソ吉草酸メチルをジアゾ化することにより得られる (Figure 15). Figure 15. 化合物 (S)-17 の逆合成解析 (2) 環化 脱水 転位 アルドール反応 (S)-17 30 31 32 (S)-2-methylenecyclohexanol 還元 3-オキソ吉草酸メチル ジアゾ化 2 2 2
第二節 モデル化合物 rac-17 の合成検討
前節で述べた逆合成解析に基づいて,モデル化合物のラセミ体 rac-17 の合成を行っ た.
まず,中間体 2-メチレンシクロヘキサノール (33) の合成を行った.Helen らは,エポ キシド類縁体を p-toluenesulfonic acid と 1,3-dimethylimidazolidin-2-one に作用させて,アリ ルアルコールを得られると報告した18.この手法を参考に,1,2-epoxy-1-methylcyclohexane を原料として用いて,2-メチレンシクロヘキサノール (33) の合成を行った (Scheme 1). 続いて,出発原料である 3-オキソ吉草酸メチルを 4-アセトアミドベンゼンスルホンア ミドでジアゾ化させ,化合物 32 が得られた.次に,ジアゾ化物 32 と 2-メチレンシクロ Scheme 2. 中間体 31 の合成検討 4-AcNHC6H4SO2N3, DBU CH3CN, rt, 92% 1) 33, Rh2(OAc)4, 110 ºC 2) BF3・Et2O, Benzene, rt 3-オキソ吉草酸メチル 32 31 30 rac-17 PTSA, DMI CH2Cl2, rt, 46% Scheme 1. 2-メチレンシクロヘキサノール (33) の合成 33 1,2-epoxy-1-methylcyclohexane
ホウ素ジエチルエーテル錯体存在下で転位反応を行った.しかし,目的中間体 31 を合成 することができなかった.これはメチルシクロヘキセニル基の嵩高さが転位にとって障害 になったことが原因であると考えられる (Scheme 2).
第三節 ラセミ体でのモデル化合物 rac-18 の合成 前節の結果より,シクロヘキセニル基は立体障害のために,導入および転位反応を引き 起こしにくいと考えられる.したがって,より立体障害の小さい置換基を用いることにし た.そこで,metarhizin E (11), F (12) のデカリン環部位を少しでも再現するものとして,ブ チル基に変換したモデル化合物 18 (Figure 15) をデザインした. ジアゾ化物 32 にロジウム (Ⅱ) アセテート存在下で 3-buten-2-ol を作用させ,ブテニル 基を導入し,更に三フッ化ホウ素ジエチルエーテル錯体により転位反応を行ったところ, 化合物 34 を合成することができた.続いて,化合物 34 の三級水酸基を TES 基で保護し たのち,ブテニル基の接触還元することにより,36 を得た.次に,トリエチルアミン存 在下,ジシクロヘキシルボロントリフラートを作用させて,化合物 36 のケトンをエノ レートにとし,更にアセトアルデヒドとアルドール反応を行うことで,化合物 37 を合成 することが出来た17.化合物 37 に Dess-Martin periodinane を作用させたところ,生じたケ トンと γ 位の三級水酸基との間でヘミアセタールを形成し,更に,自動的に脱水反応 を起こり, rac-18 を合成することができた(Scheme 3). Figure 15. モデル化合物 , (R)-18 および (S)-18 (R)-18 (S)-18 2 2
1) 3-buten-2-ol, Rh2(OAc)4, 110 ºC 2) BF3・Et2O, Benzene, rt, 40% TESOTf pyridine, 50 ºC, 60% 1) c-(Hex)2BOTf, Et3N, 0 ºC, 2) acetaldehyde, CH2Cl2, rt 10% (2 steps) Pd(OH)2/C EtOH, rt, 70% CH2Cl2, rt 30% DMP 32 34 36 35 37 38 rac-18 Scheme 3. モデル化合物 rac-18 の合成 2 2
第四節 考察と今後の展望 本章では,当研究室において昆虫寄生菌 Metarhizium flavoviride より単離された新規化合 物のうち,これまでに未決定であった metarhizin E (11) および F (12) の 2' 位の絶対構造を 明らかにすることを目的として研究を行った.その方針としては,天然物の CD スペクト ルをそれに対応する構造を有したモデル化合物の CD スペクトルと比較することにし た.デカリン環部分をシクロヘキシル基で置換したフラノン化合物 (R)- および (S)-17 を デザインし,その合成検討を行った.その結果,モデル化合物 17 はシクロヘキシル基の 嵩高さのために合成することができなかったものの,デカリン環部位をブチル基で置き換 えたモデル化合物 rac-18 の合成を得ることができた. 今後,(S)- および (R)-3-buten-2-ol の合成を行うことで,モデル化合物 (S)-18 と (R)-18 を 合成し,それら の CD スペクトルmetarhizin E (11) および F (12) の CD スペクトルと比較 して,絶対配置の決定を行う予定である. なお,(S)-(+)-3-buten-2-ol の合成法に関しては,Hirama らによって報告された手法19を参 考に合成する予定である.具体的には,出発原料であるL-(-)-乳酸エチルの水酸基を TBDPS 基で保護して,エステル部分を DIBAL でアルデヒドへ還元し,メチルトリフェニ ルホスホニウムブロミドと Wittig 反応により,中間体 40 を得た.更に, 化合物 40 を水酸 基の脱保護,保護および加水分解反応を行うことで,(S)-(+)-3-buten-2-ol へ導く予定であ る (Scheme 4).また,出発原料の L-(-)-乳酸エチルのエナンチオマーである D-(+)-乳酸エ チルを用いて同様の反応を行うことで (R)-18 を合成することができると考えられる.
39 40 42 41 TBDPSCl, Im DMF, rt, 80% 1) DIBAL, CH2Cl2, -80 ºC TBAF 2) Ph3PCH3Br, LHMDS THF, -20 ºC(5% 2steps) (PhCO)2O, Et3N, DMAP, CH2Cl2 THF LiOH THF L-(-)-乳酸エチル (S)-(+)-3-buten-2-ol Scheme 4. (S)-3-buten-2-ol の合成方針 (S)-18 2
結語
昆虫寄生糸状菌は,代謝産物の研究がまだ充分に行われていない未利用天然資源の一つ である.また一般に宿主である昆虫の体内に侵入して増殖し,栄養分の奪取,組織の破 壊,毒素の産生などにより昆虫を死に至らしめるという特徴から,細胞増殖抑制作用や免 疫抑制作用等の働きをもつ生物活性物質を産生している可能性がある.そのような観点か ら,昆虫寄生菌は新規生物活性物質の探索源として有用な天然資源であると考えられる. 昆虫寄生菌の一種である Metarhizium flavoviride の成分研究において,当研究室では新規 化合物 metarhizin A~H (7~14) および既知化合物 Mf-3 (15) を単離した.これらの化合物 は,ジテルペノイドにピロン環あるいはフラノン環が結合した特徴的な骨格を有してい る.特にフラノン環構造を有する metarhizin C~H (9~14) は天然由来化合物としては非常に 珍しい構造であるといえる. 本研究では,まだ未決定であった metarhizin E (11), F (12) の 2' 位の絶対構造の決定を目 的に研究を行った.Metarhizin E (11), F (12) の CD スペクトルは互いに逆のコットン効果を 示したことから,フラノン環部位を有したモデル化合物との CD スペクトルの比較により 絶対配置を決定することができると考えた.本研究ではそのようなモデル化合物 (R)-17, (S)-17, (R)-18, (S)-18 をデザインし,合成検討を行った.その結果,化合物 rac-18 の合成を 達成した.今後,モデル化合物 (R)-18, (S)-18 の合成を行い,これらの CD スペクトル研 究よりmetarhizin E (11), F (12) の 2' 位の絶対配置を決定していく予定である. 未利用天然資源である昆虫寄生菌の成分研究がさらに進み,新規構造を有する化合物や 新規生物活性物質が様々な研究の対象として数多く供されることを期待する.実験の部
マススペクトルは日本電子 JEOL JMS-DX 303 型質量分析計,JMS-700 型質量分析計お よび JMS-T 100 GC 型質量分析計を使用した.NMR スペクトルの測定は JEOL GX-500 型 核磁気共鳴装置および JEOL AL-400 型核磁気共鳴装置を使用し,内部標準物質として TMS を用いた.化学シフト値は ppm で表し,結合様式は,一重線 : s,二重線 : d,三重 線 : t,四重線 : q,二分裂した二重線 : dd,三分裂した二重線 : td,多重線 : m,幅広いシ グナル : br. で表した.カラムクロマトグラフィーの担体には Silica gel 60 (70-230 mesh ASTM, Merck) を用いた.カラムクロマトグラフィーの移動相には担体 1 g に対して 2 mL の溶媒を 1 fraction として用い,試料を順次溶出させた.HPLC は LC-908W (Japan Analytical Industry Co., Ltd.) を用い,GPC 用分離カラムとして JAIGEL-GS310 (φ 21.5 mm x 500 mm) (Japan Analytical Industry Co., Ltd.) を用いた.TLC は TLC aluminium sheets Silica gel 60F254 (0.25 mm, Merck) を用い,検出は UV (254, 365 nm) 照射下における蛍光およびアニスアルデヒド硫酸溶液噴霧後の加熱発色およびリンモリブデン酸溶液噴霧による加熱発色 により行った.試薬は,市販のものを精製せずにそのまま用いた.
第一章の実験
Methyl 2-diazo-3-oxopentanoate (32) の合成
Methy 3-oxovalerate (200.0 mg, 1.5 mmol) を CH3CN (5.0 mL)
に溶かし,4-acetamido-benzenesulfonamide (432.4 mg, 1.8 mmol), DBU (268.l µL, 1.8 mmol) を順次加え,室温で 2 時間攪拌した.反応液に 1M NaOH を加え EtOAc で 3 回抽出し,得られた有機層を 水・飽和食塩水で順次洗浄し,無水硫酸ナトリウムで乾燥後,減圧下溶媒を留去した. 化合物 32 を得た.32: colorless oil. 1 H NMR (400 MHz, CDCl3) δ 1.13 (3H, t, J = 7.6 Hz), 2.86 (2H, q, J = 7.6 Hz), 3.84 (3H, s); 13 C NMR (100 MHz, CDCl3) δ 8.2, 33.7, 52.0, 161.1, 193.3;EIMS: m/z 156 [M]+ , 125, 113, 84, 69, 57 (base); HREIMS: m/z 156.0530 [M]+ (156.0535 calcd. for C6H8N2O3). 2-Methylenecyclohexanol (33) の合成 1,2-Epoxy-1-methylcyclohexane (150 mg, 1.33 mmol) を CH2Cl2 (5.0 mL) に溶かし,
1,3-dimethylimidazolidin-2-one (288.4 µL, 2.66 mmol), p-toluenesulfonic acid (114.5 mg, 0.67 mmol) を加え 2 時間攪拌した.反応液を水を加え EtOAc で 3 回抽出し,得られた有 機層を飽和食塩水で洗浄し,無水硫酸ナトリウムで乾燥後,減圧下慎重に溶媒を留去し た.残渣をシリカゲルクロマトグラフィーに付し,n-hexane-EtOAc (9:1) 溶出画分より 33 (68.4 mg, 46%) を得た.33: colorless oil. 1 H NMR (400 MHz, CDCl3) δ 1.40~1.49 (3H, m), 1.61~1.65 (1H, m), 1.77~1.81 (2H, m), 1.95~2.00 (2H, m), 2.41 (1H, br), 4.11 (1H, t, J = 7.2 Hz), 13
151.6; EIMS: m/z 112 [M]+
, 97, 83, 41 (base); HREIMS: m/z 112.0982 [M]+
(112.0888 calcd. for C7H12O).
Methyl (E)-2-hydroxy-2-propanoylhex-4-enoate (34) の合成
化合物 32 (192.8 mg,1.24 mmol) を benzene (8.0 mL) に溶かし, 3-buten-2-ol (118.7 µL, 1.36 mmol), rhodium (II) acetate (4.4 mg, 0.01 mmol) を順次加え,反応液を 110 ºC で予熱 したオイルバスに入れて,20 分還流したのち,室温で冷却した.反応液に boron trifluoride diethyl ether complex (194.7 µL, 1.55 mmol) を加え,室温で 2 時間攪拌した.減圧下溶 媒を留去した.残渣をシリカゲルクロマトグラフィーに付し,n-hexane-EtOAc (9:1) 溶 出画分を集め減圧下溶媒留去した.残渣を HPLC (column: JAIGEL-GS310, elutant: EtOAc, wave length: 250 nm, flow rate: 5 mL/min) により分離・精製し,34 (108.9 mg, 44%) を得 た.34: colorless oil. 1 H NMR (400 MHz, CDCl3) δ 1.10 (3H, t, J = 7.2 Hz), 1.60 (3H, d, J = 6.4 Hz), 2.55~2.66 (2H, m), 2.67~2.74 (2H, m), 3.78 (3H, s), 4.13 (1H, s), 5.30 (1H, m), 5.59 (1H, m); 13 C NMR (100 MHz, CDCl3) δ 7.5, 18.0, 30.4, 38.8, 53.2, 83.3, 123.1, 130.6, 171.2, 207.2; EIMS: m/z 200 [M]+ , 144 (base), 102; HREIMS: m/z 200.1067 [M]+ (200.1205 calcd. for C10H16O4). (E)-Methyl 2-propanoyl-2-(triethylsilyloxy)hex-4-enoate (35)
化合物 34 (120.5 mg, 0.60 mmol) を pyridine (4.0 mL) に溶かし,trimethylsilyltrifluoro-methanesulfonate (474.2 µL, 4.10 mmol) を加え,50 ºC で 5 時間攪拌した.反応液に水 を加え EtOAc で 3 回抽出した.得られた有機層を水・飽和食塩水で洗浄し,無水硫
酸ナトリウムで乾燥後,減圧下溶媒を留去した.残渣をシリカゲルクロマトグラフィー に付し,n-hexane-EtOAc (99:1) 溶出画分より 35 (144.2 mg, 76%) を得た.35 colorless oil.
1 H NMR (400 MHz, CDCl3) δ 0.65 (6H, t, J = 8.0 Hz), 0.96 (9H, q, J = 8.0 Hz ), 1.02 (3H, t, J = 7.6 Hz), 1.62 (3H, d, J = 6.8 Hz), 2.58 (1H, m), 2.61 (2H, q, J = 7.2 Hz) 2.72 (1H, dd, J = 14.4, 7.6 Hz), 3.72 (3H,s), 5.31 (1H, m), 5.47 (1H, m); 13 C NMR (100 MHz, CDCl3) δ 1.7 (3C), 6.8 (3C), 7.4, 18.0, 31.2, 40.2, 52.5, 86.8, 124.2, 129.8, 171.0, 203.7; EIMS: m/z 314 [M]+ , 283, 258 (base), 115; HREIMS: m/z 314.1899 [M]+
(314.1913 calcd. for C16H30O4Si).
Methyl 2-propanoyl-2-(triethylsilyloxy)hexanoate (36) の合成
35 (100.0 mg, 0.32 mmol) を EtOH (2.0 mL) に溶かし,palladium hydroxide on carbon (7.6 mg, 0.01mmol) を加え,水素雰囲気下室温で 5 時間攪拌した.反応液を濾過して,減 圧下溶媒を留去し.残渣をシリカゲルクロマトグラフィーに付し,n-hexane-EtOAc (100: 1) 溶出画分より 36 (70.9 mg, 70%) を得た.36: colorless oil. 1 H NMR (400 MHz , CDCl3) δ 0.65 (6H, t, J = 8.0 Hz), 0.90 (9H, q, J = 8.0 Hz), 0.96 (3H, t, J = 7.6 Hz), 1.18 (3H, t, J = 7.2 Hz), 1.25 (2H, m), 1.33 (1H, s), 1.80 (1H, m), 2.07 (1H, m), 2.30 (1H, m), 2.65 (2H, q, J = 7.2 Hz), 3.76 (3H, s); 13 C NMR (100 MHz, CDCl3) δ 1.7 (3C), 6.8 (3C), 7.4, 13.8, 21.7, 22.5, 30.6, 37.6, 52.6, 83.3, 171.2, 207.2; EIMS: m/z 316 [M]+ , 285, 258 (base), 115; HREIMS: m/z 316.2117 [M]+
(316.2070 calcd. for C16H32O4Si).
Methyl 2-butyl-2,5-dihydroxy-4-methyl-3-oxohexanoate (37) の合成
trimethylamine (10.0 µL, 0.14 mmol),化合物 36 (18.2 mg, 0.06 mmol) を順次加え,反応 液を 0 ºC で 2 時間攪拌した後.続いて,-78 ºC で acetaldehyde (16.0µL, 0.29 mmol) を 加え, 0 ºC で 15 時間攪拌した後, 0 ºC で反応液に MeOH (1.0 mL), pH 7 buffer (1.0 mL), H2O2 (30 %) 水溶液 (1.0 mL),チオ硫酸ナトリウム (10 %) 水溶液 (1.0 mL) を順次加 えた後,反応液に水を加え EtOAc で 3 回抽出した.得られた有機層を飽和食塩水で 洗浄し,無水硫酸ナトリウムで乾燥後,減圧下溶媒を留去した.残渣をシリカゲルクロ マトグラフィーに付し,n-hexane-EtOAc (39:1~19:1) 溶出画分より 37 (2.1 mg, 20%) を 得た.37: colorless oil. 1 H NMR (400 MHz, CDCl3) δ 0.90 (3H, t, J = 7.0 Hz), 1.00 (3H, d, J = 6.8 Hz), 1.14 (3H, d, J = 6.0 Hz), 1.20~1.36 (4H, m), 1.67~1.74 (1H, m), 2,00 (1H, br), 2.10 (1H, m), 3.26 (1H, m), 3.59 (1H,m), 3.77 (3H, s), 4.78 (1H, s); EIMS: m/z 245 [M+H]+ , 146 (base), 103; HREIMS: m/z 245.1332 [M+H]+ (245.1467 calcd. for C12H23O5).
Methyl 2-butyl -4,5-dimethyl-3-oxo-2,3-dihydrofuran-2-carboxylate (rac-18) の合成
化合物 37 (2.1 mg, 0.01 mmol) を CH2Cl2 (1.0 mL) に溶かし,0 ºC で Dess-Martin
periodinane (15 wt.% CH2Cl2 溶液) (40 µl) を加え,室温で 2 時間攪拌した.反応液に飽和
チオ硫酸ナトリウム水溶液と 1M NaOH を順次加え EtOAc で 3 回抽出した.得られ た有機層を水・飽和食塩水で洗浄し,無水硫酸ナトリウムで乾燥後,減圧下溶媒を留去 した.残渣をシリカゲルクロマトグラフィーに付し,n-hexane-EtOAc (9:1) 溶出画分よ り rac-18 (0.6 mg, 31%) を得た.rac-18: colorless oil. 1
H NMR (400 MHz, CDCl3) δ 0.88 (3H, t, J = 7.2 Hz), 1.21~1.34 (4H, m), 1.70 (3H, s), 1.94 (1H, m), 2.19 (1H, m), 2.27 (3H, s) , 3.76 (3H, s); EIMS: m/z 226 [M]+ , 170 (base), 138, 125; HREIMS: m/z 226.1205 [M]+ (226.1190 calcd. for C12H18O4)
(S)-Ethyl 2-(tert-butyldiphenylsilyloxy)propanoate (39) の合成
L-(-)-乳酸エチル (5.0 g, 42.32 mmol) を DMF (10.0 mL) に溶かし,butylchlorodiphenyl-silane (11.3 mL, 41.6 mmol), imidazole (5.8 g, 84.64 mmol) を加え室温で 5 時間攪拌した. 反応液を 1M HCl で酸性化したのち,水を加え EtOAc で 3 回抽出した.得られた有 機層を水・飽和食塩水で洗浄し,無水硫酸ナトリウムで乾燥後,減圧下溶媒を留去した. 残渣をシリカゲルクロマトグラフィーに付し,n-hexane-EtOAc (19:1) 溶出画分より 39 (12.1 g, 80%) を得た.39: colorless oil. 1 H NMR (400 MHz, CDCl3) δ 1.10 (9H, s), 1.15 (3H, t, J = 7.2 Hz ), 1.37 (3H,d, J = 6.8 Hz), 4.27 (2H, q, J = 6.8 Hz), 7.35~7.44 (6H, m), 7.66~7.70 (4H, m); 13 C NMR (100 MHz, CDCl3) δ 14.3, 19.2, 21.3, 26.8 (3C), 60.5, 69.0, 127.5 (2C), 127.6 (2C), 129.7 (2C), 130.6 (2C), 131.7 (2C), 135.8 (2C), 170.0; EIMS: m/z 299 [M-t-butyl]+ ; HREIMS: m/z 299.0114 [M-t-butyl]+
(299.1090 calcd. for C17H19O3Si).
(S)-(3-Buten-2-yloxy)-(tert-butyl)-diphenylsilane (40) の合成
-78ºC で化合物39 (1.78 g, 5.0 mmol) を CH2Cl2 (10.0 mL) に溶かし,diisobutylaluminium
hydride (5.0 mL, 5.0 mmol)を加えて,2 時間攪拌した後.反応液に MeOH (10.0 mL) を 加え,30 分間攪拌したのち,飽和 (+) 酒石酸ナトリウムカリウム四水和物 (20.0 mL) を 加え,1 時間攪拌した.反応液に水を加え EtOAc で 3 回抽出した.得られた有機層 を飽和食塩水で洗浄し,無水硫酸ナトリウムで乾燥後,減圧下溶媒を留去した.続いて, -20 ºC で methyltriphenylphosphonium (1.83 g, 7.13 mmol) を THF (15.0 mL) に溶かし, lithium hexamethyldisilazide (7.1 mL, 7.13 mmol) を加え,30 分間攪拌した後,残渣を反 応液に加え,2 時間攪拌した.反応液に飽和塩化アンモニウム水溶液を加え EtOAc で 3
回抽出した.得られた有機層を飽和食塩水で洗浄し,無水硫酸ナトリウムで乾燥後,減 圧下溶媒を留去した.残渣をシリカゲルクロマトグラフィーに付し,n-hexane-EtOAc (49:1) 溶出画分より 40 (69.7 mg, 5%) を得た.40: colorless oil. 1 H NMR (400 MHz CDCl3) δ 1.22 (3H, d, J = 7.2 Hz), 3.73 (1H, m), 4.94 (1H, dt, J = 10.6, 1.8 Hz), 5.09 (1H, dt, J = 17.2, 1.8 Hz), 5.87 (1H, ddd, J = 17.2, 10.6, 6.0 Hz), 7.34~7.44 (6H, m), 7.68~7.75 (4H, J = 7.8 Hz); 13 C NMR (100 MHz, CDCl3) δ 19.3, 24.3, 27.0 (3C), 69.3, 115.8, 127.9 (2C), 128.0 (2C),130.1 (2C), 130.8, (2C), 131.2 (2C), 135.9 (2C), 136.1; EIMS: m/z 253 [M-t-butyl]+ ; HREIMS: m/z 253.1078 [M-t-butyl]+
謝辞
本研究に際し,始終御懇篤なる御指導,御鞭撻を賜りました東北大学大学院薬学研究科 教授 大島吉輝先生に謹んで感謝致します. 副査として本論文の審査にあたり, 有益なる御助言を賜りました東北大学大学院薬学研 究科准教授 叶 直樹先生に厚く御礼申し上げます. 細部に至る御指導と御助言を賜りました東北大学大学院薬学研究科准教授 菊地晴久先 生に謹んで御礼申し上げます. 様々な面でお力添えをいただきました東北大学大学院薬学研究科助教 加藤泰弘先生に 心より御礼申し上げます. また,研究室生活全般にて公私共にご支援頂いた東北大学大学院薬学研究科助教 浅井禎吾先生に御礼申し上げます. NMR スペクトル,質量スペクトルの測定の労をとられました東北大学大学院薬学研究 科中央機器分析室の諸氏に御礼申し上げます. 本研究を続けるにあたり公私共に御支援頂いた東北大学大学院薬学研究科反応制御科学 分野 三原 健修士;医薬資源化学分野 熊谷 史由修士, 壁谷 尚宏修士, 佐藤 雄一修士, 中村 哲也修士, 阿部 由布子修士,江 宇修士,藤村 信平修士,松尾 祐介修士,星川 毅修士,山本 崇史修士,羅 丹修士,コウ スイマン修士,並びに教室員の方々に心より 感謝致します. 最後に,これまで様々な面において御世話になりました家族に心から感謝致します.参考文献
1. Kodaira, Y. Agric. Biol. Chem. 1962, 26, 36-42.
2. Myokei, R.; Sakurai, A.; Chang, C.; Kodaira, Y.; Takahashi, N.; Tamura, S. Tetrahedron Lett.
1969, 9, 695-698.
3. Hamill, R. L.; Higgens, C. E.; Boaz, H. E.; Gorman, M. Tetrahedron Lett. 1969, 49, 4255-4258.
4. Suzuki, A.; Kanaoka, M.; Isogai, A.; Murakoshi, S.; Ichinoe, M.; Tamura, S. Tetrahedron Lett.
1977, 25, 2167-2170.
5. Miyaka, Y.; Kozutsumi, Y.; Nakamura, S.; Fujita, T.; Kawasaki, T. Biochem. Biophys. Res.
Commun. 1995, 211, 396-403.
6. Kaczka, E. A.; Trenner, N. R.; Arison, B.; Walker, R. W. and Folkers, K. Biochem. Biophys.
Res. Commun. 1964, 14, 456-457.
7. a) Stuart, B. K.; Ivan, K.; Richard, E. G.; Doletha, M. E. S.; Bruno, G. G. D.; Alice, C. L. C.; Donna, M. G. J. Nat. Prod. 2007, 70, 1919–1924. b) Miwa, A.; Ishidoh, K.; Kinoshita, H.; Nihira, T.; Ihara, F.; Fujita, T.; Igarashi, Y. J. Nat. Prod. 2008, 71, 278–280. c ) Stuartb, K.; Christopher, H.; Yong, S, M.; Bruno, G. G. D.; Jonh, D. V.; Alice, C. L. C.; Donna, M. G. J.
Agric. Food Chem. 2006, 54, 7083-7088. d) Patrick, S.; Mathias, M.; Dirk, H. Mol. Pharmaceutics. 2008, 5 (2), 234–242.
8. Carlos, A. C.; Ana, L. A. C.; Letcia, A. S.; Thais, G.; Donald, W. R.; Norberto, P. L.; Gilberto, U. L. B. Fungal Biol. 2010, 114 , 473- 480.
9. Schneider, I. J. Embryol. Exp. Morph. 1972, 27, 353.
10. 喜多山実,修士論文,東北大学大学院薬学研究科,2004.
11. Kagamizono, T.; Maejima, A.; Kawashima, A.; Akiyama, T.; Ando, T.; Isogai, K.; Morimoto, S. WO 006646, September 3rd, 1995.
12. 星朋子,修士論文,東北大学大学院薬学研究科, 2008.
Chem. 1993, 58, 1062-1067.
14. Kikuchi, H.; Hoshi, T.: Kitayam, M.; Sekiya, M.; Katou, Y.; Ueda, K.; Kubohara, Y.; Sato, H.; Shimazu, M.; Kurata, S.; Oshima, Y. Tetrahedron. 2009, 65, 469-477
15. John, L. W.; Brian, M. S.; Hans-Ju1rgen, D.; Derek, A. P.; Dejah, T. P. J. Am. Chem. Soc.
1997, 119, 9641-965.
16. Aoki, S.; Oi, T.; Shimizu, K.; Shiraki, R.; Takao, K.; Tadano, K. Bull. Chem. Soc. Jpn. 2004,
77, 1703-1716.17.
17. Li, P.; Li, J.; Arikan, F.; Ahlbrecht, W.; Dieckmann, M.; Menche, D. J. Org. Chem. 2010, 75, 2429-2444. b) Ganesan, K.; Brown, H. C. J. Org. Chem. 1994, 59, 2336-2340. c)Evans, D. A.; Nelson, J. V.; Taber, T. R. J. Am. Chem. Soc. 1981, 103, 3099-3111.
18. Helen, A. C.; Karim, H.; William, B. M. Synlett. 2010, 4, 595-598. 19. Hirama, M.; Shigemoto, T. J. Org. Chem. 1987, 52, 3342-3346.