Studies on the Application of Second
Generation Palladium-Catalyzed
Cycloalkenylation to the Synthesis of
Biologically Active Natural Products
著者
Takeda Kazutaka
内容記述
学位授与大学: Osaka Prefecture University(大阪
府立大学), 学位の種類: 博士(理学), 学位記番号:
論理第98号, 学位授与年月日: 2012-03-31, 指導教
員: 豊田真弘.
Studies on the Application of Second Generation
Palladium-Catalyzed Cycloalkenylation to the Synthesis of
Biologically Active Natural Products
(第二世代型触媒的環化アルケニル化反応を利用した
生物活性天然物の合成研究)
竹 田 一 貴
Osaka Prefecture University
2012
i
Table of Contents
General Discussion
1
Detailed Discussion
Chapter 1 Concise Construction of Bicyclo[3.2.1]octane Framework
14
Chapter 2 Diastereoselective Total Syntheses of Isoiridomyrmecin and
Isodihydronepetalactone
16
Chapter 3
Efficient Synthesis of 3-Azabicyclo[3.3.0]octanes and
3-Azabicyclo[4.3.0]nonanes
18
Chapter 4 Diastereoselective Total Synthesis of α-Skytanthine
23
Conclusion
25
Acknowledgment
26
Experimental Section
1. General
27
2. Experiments in Chapter 1
28
3. Experiments in Chapter 2
32
4. Experiments in Chapter 3
36
5. Experiments in Chapter 4
49
References
53
ii
Abbreviations
Å
angstrom(s)
Ac
acetyl
acac
acetylacetonate
atm
atmosphere(s)
Bn
benzyl
Boc
tert-butoxycarbonyl
br
broad (spectral)
Bu, n-Bu
normal (primary) butyl
t-Bu
tert-butyl
°C
degrees Celsius
calcd.
calculated
CAN
ceric ammonium nitrate
cat.
catalytic amount
cm
-1wavenumber(s)
CMBP
cyanomethylenetributylphosphorane
COSY
correlation spectroscopy
DEAD
diethyl azodicarboxylate
DMAP
4-(N,N-dimethylamino)pyridine
DMF
dimethylformamide
DMSO
dimethyl sulfoxide
EI
electron ionization
equiv.
equivalent
FAB
fast atom bombardment
Et
ethyl
h
hour(s)
HMPA
hexamethylphosphoramide
HPLC
high performance liquid chromatography
HRMS
high resolution mass spectrum
iii
IBX
2-iodoxybenzoic acid
IR
infrared
J
coupling constant (in NMR spectrometry)
L
liter(s)
LDA
lithium diisopropylamide
LHMDS
lithium hexamethyldisilazide, lithium bis(trimethylsilyl)amide
LRMS
low resolution mass spectrum
M
molar (moles per liter)
M
+parent molecular ion
Me
methyl
min
minute(s)
mol
mole(s)
MOM
methoxymethyl
MS 3
A
molecular sieves, 3
Å
N
normal (equivalents per liter)
NMR
nuclear magnetic resonance
NOE
nuclear Overhauser effect
Ns
2-nitrobenzenesulfonyl
Pd-C
palladium on charcoal
Ph
phenyl
i-Pr
isopropyl
rt
room temperature
TBAI
tetrabutylammonium iodide
TBS
tert-butyldimethylsilyl
THF
tetrahydrofuran
TLC
thin layer chromatography
TMANO
trimethylamine N-oxide
TMS
trimethylsilyl
1
General Discussion
Intellectual curiosity is the primary motivation underlying human progress. Interest in nature
has impelled the development of modern science, which is divided into various branches. Chemistry
is the branch of science concerned with the substances of which matter is composed, the
investigation of their properties and reactions, and the use of such reactions to form new substances.
Transition metals have gained an indispensable role in modern synthetic chemistry; palladium is
being one of the most versatile and widely used of these metals in organic synthesis. The extensive
utility of palladium stems from its effects on an extraordinary number of very difficult reactions,
including carbon–carbon bond forming reactions, under relatively mild reaction conditions.
Furthermore, palladium is generally required in only catalytic amounts, and tolerates a wide variety
of functional groups.
Palladium(0) catalysis has dominated the landscape of catalyst development for the past several
decades. Specifically, palladium-catalyzed cross-coupling has led to a number of synthetically
useful products and provides organometallic chemists with a platform for investigating fundamental
chemical processes.
1)In 2010, this research area was awarded a Nobel Prize.
In contrast, palladium(II)-catalyzed oxidation reactions
2)have developed at a much slower pace
during this same period. The mechanistic differences between Pd(0)- and Pd(II)-catalytic cycles are
summarized in Scheme 1. In Pd(0) chemistry, the catalytic cycle is generally initiated by oxidative
addition of an organic electrophile, often organic halides, to the Pd(0) center, followed by a
Pd(II)-mediated process such as transmetallation or migratory insertion/β-hydride elimination. The
cycle is completed by reductive elimination to form the desired organic product and regenerate the
Pd(0) catalyst.
Pd(II) oxidation catalysis, on the other hand, is initiated by Lewis acid activation of a substrate,
which undergoes deprotonation, often followed by β-hydride elimination. This process is referred to
as dehydrogenation (Scheme 1). The resulting Pd-hydride can undergo reductive elimination to
form a Pd(0) species. However, because Pd(0) catalysis is not the desired process in this case, the
use of an organic electrophile is generally not acceptable except in a few select cases. Instead, an
alternative oxidant must be used to generate the Pd(II) species from the Pd(0) species. This has
classically been accomplished with the use of stoichiometric Cu(II) salts or benzoquinone. Efficient
reoxidation of the catalyst is important for the success of palladium(II)-catalyzed oxidation
reactions. The reduced attention to oxidation reactions possibly reflects the added complexities that
arise from the need for a stoichiometric oxidant to regenerate the active catalyst.
2
LnPd0 X LnPdII X R LnPdII X O R X H LnPdII H LnPdII X R LnPdII R' R R' HX Pd(0) cycle Pd(II) cycle HX HO R R X R' M X M O R H oxidant Oxidation Oxidative Addition -Hydride Elimination Reductive Elimination Oxidation Cross-Coupling reduced oxidant (Dehydrogenation) TransmetallationScheme 1
Olefins are widely employed substrates in Pd(II) chemistry. Generally, olefins are considered to
be nucleophiles, where they react with various electrophiles. The complexation of an olefin moiety
with a metal can reverse this reactivity, thereby promoting subsequent nucleophilic attack. This
reaction motif is most commonly demonstrated by the Wacker oxidation where water attacks the
olefin. The efficiency of the activation of olefins with Pd(II) species is relevant in the process
whereby stabilized nucleophiles such as enolates and related species can undergo a carbometalation
reaction onto unactivated unsaturated systems. In 1980, Hegedus et al. reported the intermolecular
palladium-mediated alkylation of terminal olefins with the sodium or lithium anion of
1,3-dicarbonyl compounds (Scheme 2).
3)In 2003, Widenhoefer and co-workers reported
palladium-catalyzed intramolecular oxidative alkylation of unactivated olefins (Scheme 3).
4)PdCl2(MeCN)2 (1 equiv.) Et3N (2 equiv.) THF, –78 °C to rt 90% EtO O OEt O Na EtO2C CO2Et +
Scheme 2
3
PdCl2(MeCN)2 (5 mol %) CuCl2 (2.5 equiv) 1,2-dichloroethane 96% O O O OScheme 3
Likewise, Toyota developed the palladium-catalyzed cycloalkenylation of olefinic silyl enol
ethers,
5)and this method has been successfully adapted to the stereoselective syntheses of
polycyclic natural products (Scheme 4).
6)Me H H Me CO2Me O R Me H H Me CO2Me Me H H Me CO2Me Me H Me H CO2Me MeO2C R1 R2 (±)-GA12: R1=H, R2=H (±)-GA111: R1=OH, R2=H (±)-GA112: R1=H, R2=OH (–)-Methyl atis-16-en-19-oate (–)-Methyl kaur-16-en-19-oate (–)-Methyl trachyloban-19-oate H HO H HO Aphidicolin OH OH OH S O H H HO2C
Serofendic Acids A and B
OTBS MOMO O MOMO Pd(OAc)2 (3 mol %) O2 (1 atm) DMSO, 45 °C 81%
Scheme 4
4
In efforts to expand the synthetic utility of the palladium-catalyzed cycloalkenylation
methodology, the synthesis of 6-oxatricyclo[6.3.0.0
1,5]undecane, which is part of the overall
structure of ginkgolide C, was undertaken (Scheme 5).
7)Using ketene silyl acetal 1 as the starting
material, the desired tricyclic compound 2 could be produced under palladium-catalyzed
cycloalkenylation conditions with limited yield of less than 9%; this low yield may be due to the
instability of ketene silyl acetal 1.
O OTMS H O O H H Pd(OAc)2 (10 mol %) O2 (1 atm) DMSO, 45 °C 1 2 (9%) O O O O O O O HO Me HO HO t-Bu OH H O O H H
Ginkgolide C 6-oxatricyclo[6.3.0.01,5]undecane O O H 3 (72%) +
Scheme 5
To circumvent this limitation, the palladium-catalyzed oxidative cyclization of olefinic keto and
lactone esters (second generation palladium-catalyzed cycloalkenylation) was developed as an
alternative palladium-catalyzed cycloalkenylation method for the construction of polycyclic
compounds (Scheme 6).
8) X O CO2Me Pd(OAc)2 (cat.) X H O 4: (X = O, CH2; n = 0,1) 5 CO2Me ( )n O2 (1 atm), DMSO ( )nScheme 6
5
The postulated mechanism for the second generation palladium-catalyzed cycloalkenylation is
outlined in Scheme 7. Initially, Pd(OAc)
2acts as a Lewis acid, activating the terminal alkene;
subsequent intramolecular attack of the nucleophilic enol then occurs (step A). The resulting
alkyl-Pd(II) species undergoes β-hydride elimination (step B) to form a cyclized product and a
Pd(II)-hydride complex. Reductive elimination of acetic acid from the Pd(II)-hydride complex
generates the Pd(0) species. Finally, the catalytic cycle is completed by the regeneration of active
Pd(II) species from Pd(0) using molecular oxygen as the oxidant. The use of molecular oxygen,
instead of traditional CuCl
2or benzoquinone, as the sole stoichiometric oxidant has emerged as a
simple and environmentally favorable method for reoxidation of Pd(0) to Pd(II) species.
2d)CO2Me O Pd O H O Pd–OAc O CO2Me H H O CO2Me H O O2, AcOH H2O2 Pd(OAc)2 O O CO2Me Pd(H)OAc Pd(0) AcOH AcOH A B OAc OAc
Scheme 7
On the basis of this hypothesized mechanism, second generation palladium-catalyzed
cycloalkenylation may be applied to compounds containing both unactivated olefins and enolizable
activated moieties in the construction of new carbon–carbon bonds. Consequentry, the author
undertook the investigations outlined as follows, with the objective of expanding the scope and
limitation of reaction substrates and achieving the total synthesis of bioactive natural products by
using second generation palladium-catalyzed cycloalkenylation;
1. Concise Construction of Bicyclo[3.2.1]octane Framework
2. Diastereoselective Total Syntheses of Isoiridomyrmecin and Isodihydronepetalactone
3. Efficient Synthesis of 3-Azabicyclo[3.3.0]octanes and 3-Azabicyclo[4.3.0]nonanes
4. Diastereoselective Total Synthesis of α-Skytanthine
6
Concise Construction of Bicyclo[3.2.1]octane Framework
Because the bicyclo[3.2.1]octane ring system is a structural subunit of bioactive natural
products such as gibberellins
9)and aphidicolin,
10)the second generation palladium-catalyzed
cycloalkenylation was modified here for the construction of the bicyclo[3.2.1]octane ring systems
(Scheme 8).
O R O R Pd(OAc)2 (cat.) O2 (1 atm), DMSO 6 7 CO2Me CO2Me O R CO2Me + 8Scheme 8
On the basis of the above scheme, the second generation palladium-catalyzed cycloalkenylation
of 6 was examined. The reaction of compound 6a produced the desired cyclization product 7a,
along with small amounts of the phenol derivative 9 (Scheme 9). The details of the above
examination are expounded in the first chapter of the ensuing discussion.
OH CO2Me O O H CO2Me H CO2Me 7a (58%) 9 (12%) 6a Pd(OAc)2 (10 mol %) O2 (1 atm) DMSO, 60 h +
Scheme 9
Diastereoselective Total Syntheses of Isoiridomyrmecin and Isodihydronepetalactone
Iridoids are a large class of naturally occurring compounds. They comprise a highly oxygenated
monoterpenoid skeleton characterized by a functionalized cyclopentane ring cis-fused to a
dihydropyran, δ-lactol, or δ-lactone. These cyclopentanoid monoterpenes have attracted
considerable attention because of their diverse biological activity and interesting structure. The
name “iridoid” originated from the fact that the first members of this natural product were isolated
from the secretion of ants belonging to the genus Iridomyrmex; isoiridomyrmecin (10) was isolated
from
Iridomyrmex nitidus in 1956 (Figure 1).
11)This lactone is used by ants as an agent of defense
against preying insects and as possible means of communication. This iridoid exhibits insecticidal
7
activity, a factor that ensures their efficiency in defense.
12)Apart from isoiridomyrmecin,
isodihydronepetalactone (11) represents another type of cyclopentapyranone isolated from the
volatile oils of the cat-attracting plant Actinidia polygama in 1965 (Figure 1).
13)It is well known
that this lactone shows highly excitatory activity toward cats and animals of the family Felidae and
exhibits ant repellent effects.
14)The stereochemistry at the four contiguous, asymmetric centers in
isoiridomyrmecin
15)and isodihydronepetalactone
16)has attracted the attention of synthetic chemists.
O H H O Isoiridomyrmecin (10) O H H O Isodihydronepetalactone (11)
Figure 1
In 1994, Tsunoda reported the total synthesis of (–)-isoiridomyrmecin (Scheme 10).
15j)The
optically active tosylamide was converted to the aza-Claisen precursor by the Birch reduction of the
tosylamide followed by acylation with propanoic anhydride. Stereospecific aza-Claisen
rearrangement was successfully carried out. The hydroboration/oxidation of alkene gave alcohol.
However, the yield remained rather low because of the concurrent reduction of the amide part. The
acid hydrolysis of amide alcohol afforded (–)-isoiridomyrmecin.
N O Ph H i) LHMDS, –78 °C 77% ii) 100 °C ii) NaOH, H2O2 i) H H O H H benzene, reflux O BH O OH 90% N Ph Ts 1) Na/NH3 2) (EtCO)2O, Et3N 85% for 2 steps O TsOH 46% N H O Ph N H Ph O Isoiridomyrmecin
Scheme 10
Recently, Hofferberth synthesized isoiridomyrmecin and isodihydronepetalactone using a
divergent approach.
15a)A common aminal intermediate that was converted from citronellal was
subjected to catalytic hydrogenation followed by hydrolysis of the aniline residue to provide lactol.
8
The oxidation of lactol with Fetizon’s reagent yielded isodihydronepetalactone (Scheme 11).
O O O Citronellal 1) SeO2, t-BuOOH salicylic acid 2) IBX, DMSO (30–45%) O H H NPhMe NHPhMe Et2O 84% 1) H2, Pd-C 2) TsOH, THF EtOH, 90% 67% AgCO3, Celite benzene, 80 °C 88% O H H Isodihydronepetalactone O O H H OH
Scheme 11
In addition, the common aminal intermediate was subjected to a functional group transformation
involving eight steps such as protection, reduction, deprotection, and oxidation to provide
isoiridomyrmecin (Scheme 12). This is because the translocation of carbonyl in aminal requires a
multi-step transformation.
O H H NPhMe H H OBn S S H H OBn OAc CO2H H H OAc O H H O Isoiridomyrmecin 1) H2, Pd-C 2) 1,2-ethanedithiol 3) BnBr, NaH, TBAI BF3•OEt2 80% for 3 steps 1) CAN, MeCN; NaBH4 2) Ac2O, pyridine DMAP 66% for 2 steps 1) H2, Pd-C 2) RuCl3, NaIO4 62% for 2 steps TsOH CCl4-MeCN-H2O EtOH EtOH quant.Scheme 12
The second generation palladium-catalyzed cycloalkenylation of δ-lactone ester has been
recognized as an important method for constructing the iridoid carbon skeleton. Therefore, the total
syntheses of isoiridomyrmecin and isodihydronepetalactone were undertaken by using the second
9
generation palladium-catalyzed cycloalkenylation.
The retrosynthetic analysis of these iridoids is outlined in Scheme 13. Both natural products 10
and 11 could be synthesized by functional group transformations of the common intermediate 12,
which could be constructed from the trans-substituted olefinic lactone ester 13 by employing
second generation palladium-catalyzed cycloalkenylation. Lactone ester 13 could be constructed by
the stereoselective conjugate addition of a Grignard reagent to α,β-unsaturated lactone 15,
17)followed by methoxycarbonylation. Because the optically active form of 15 is a known
compound,
18)asymmetric synthesis could be possible by means of the same route.
O H H O (±)-Isoiridomyrmecin (10) + O H H O (±)-Isodihydronepetalactone (11) O H O 12 O O 13 CO2Me CO2Me O O Second Generation Palladium-Catalyzed Cycloalkenylation 15 O O 14
Scheme 13
On the basis of this retrosynthetic analysis, the syntheses of isoiridomyrmecin and
isodihydronepetalactone were carried out. Both iridoids were synthesized from the racemic
compound 15 by using second generation palladium-catalyzed cycloalkenylation. The details of the
synthesis are expounded in the second chapter of ensuing discussion.
Efficient Synthesis of 3-Azabicyclo[3.3.0]octanes and 3-Azabicyclo[4.3.0]nonanes
Second generation palladium-catalyzed cycloalkenylations are primarily synthetically useful in
the preparation of bicyclic ring systems containing heterocyclic units. The cyclization of olefinic
lactone esters provides cis-fused 3-oxabicyclo[3.3.0]octanes and 3-oxabicyclo[4.3.0]nonanes.
8a)To expand the synthetic utility of this method, the applicability of olefinic lactam esters 16 and
17 to this method for the construction of the corresponding 3-azabicyclo[3.3.0]octanes 18 and
10
R1N O CO2R2 R1N H O 16 (n = 0) 18 (n = 0) CO2R2 Pd(OAc)2 (cat.) ( )n ( )n 17 (n = 1) 19 (n = 1) O2 (1 atm)Scheme 14
3-Azabicyclo[3.3.0]octanes and 3-azabicyclo[4.3.0]nonanes represent major classes of
nitrogen-containing heterocycles, which continue to play an increasingly important role in drug
discovery. These compounds are structural units of a vast range of natural products such as
dendrobine
19)and skytanthines
20)(Figure 2).
Dendrobine MeN H H -Skytanthine MeN O O H H H
Figure 2
On the basis of the above scheme, the second generation palladium-catalyzed
cycloalkenylations of 16 and 17 were investigated. The reaction of compounds 16a and 17a
produced the desired cyclization products 18a and 19a, as a mixture of olefin isomers (Scheme
15). The details of the investigation are expounded in the third chapter of the ensuing discussion.
CO2Me MeN O MeN O CO2Me H 18a 16a CO2Me CO2Me H 19a 17a MeN MeN O O Pd(OAc)2 (10 mol %)
DMSO (5 equiv.), O2 (1 atm)
toluene, 45 °C, 94 h 76%
(exo/endo = 2.6/1)
Pd(OAc)2 (10 mol %)
DMSO (5 equiv.), O2 (1 atm)
toluene, 45 °C, 94 h 68%
(exo/endo = 1/1.6)
11
Diastereoselective Total Synthesis of α-Skytanthine
α-Skytanthine, which has a unique 3-azabicyclo[4.3.0]nonane core, was isolated from the
Chilean endemic plant Skytanthus acutus (Apocynaceae) as a monoterpene piperidine alkaloid in
1961.
20)The structure and absolute stereochemistry of this compound have been determined on the
basis of partial and total syntheses.
21)This alkaloid exhibits hypotensive activity.
22)Because many
natural products and pharmaceutically active agents possess substituted piperidine ring systems, the
synthesis of this class of functionalized heterocyclic compounds remains of interest to synthetic
chemists.
23)The total synthesis of (+)-α-skytanthine was first achieved by Oppolzer in 1986 by using the
magnesium-ene reaction (Scheme 16).
21a)The magnesium-ene reaction on the 2,7-dienylchloride,
which was synthesized from (S)-3-methyl-1-penten-5-ol, and oxidative trapping afforded cyclized
alcohol as the major component, although with poor yield and moderate stereoselectivity.
Hydroboration/oxidation afforded a diol, which was treated with tosyl chloride/pyridine and
methylamine to give enantiomerically pure (+)-α-skytanthine.
(+)--Skytanthine OH 1) MsCl, Et3N 2) LiBr Br i) Mg turnings CHO 67% OH SOCl2 i) Mg powder; ii) MoOPh OH H H 49% 79% reflux i) BH3, THF ii) NaOH, H2O2 OH H H OH 1) TsCl 2) MeNH2 H H NMe 68% (dr = 4.2:1) 67% ii) (three isomers: 9%) pyridine Cl
Scheme 16
In 2008, Honda reported the diastereoselective synthesis of (+)-α-skytanthine based on the
intramolecular Pauson–Khand reaction (Scheme 17).
21e)The intramolecular Pauson–Khand reaction
of optically active enyne that was obtained by the condensation of three components—alkenyl
alcohol, alkynyl alcohol, and nosylamide—generated cyclized enone, although the undesired
reduction of the nitro group was observed. Catalytic hydrogenation and epimerization afforded
ketone with the desired stereochemistry. The conversion of ketone to the target molecule was
achieved by carrying out Wolff–Kishner reduction, deprotection, and N-methylation.
12
NH H H NMe H H (+)--Skytanthine 2) AcCl, NaH, 81% 3) Na-Hg, Na2PHO4, 66% 1) H2NNH2•H2O, 130 °C; HCHO N H SO2 O NNs OH 1) NsNHBoc, DEAD, PPh3, 98% 2) DMF, 120 °C, 97% 3) DEAD, PPh3, 100% Co2(CO)8 THF-H2O (3:1) 71% N H SO2 O + + MeOH, reflux 95% 6% (separation) 8% (separation) 2) NaOMe PtO2 (3 mol%) 1) H2 (1 atm) 86% (:=1:1) N H SO2 O H2N H2N O2N TMANO•2H2O N H SO2 O H NH2 KOH, 170 °C, 94% OH HCO2HScheme 17
In the previous investigation, the second generation palladium-catalyzed cycloalkenylation of
lactam esters was demonstrated to be synthetically useful as a construction method for generating
the 3-azabicyclo[3.3.0]octane and 3-azabicyclo[4.3.0]nonane structures. In order to illustrate the
additional versatility of the second generation palladium-catalyzed cycloalkenylations, the
stereoselective total synthesis of (±)-α-skytanthine (20) was undertaken. Owing to the structural
similarity between α-skytanthine and isoiridomyrmecin, α-skytanthine may be synthesized from
alkenyl lactone 14, which is a synthetic intermediate of iridoid syntheses. Scheme 18 shows the
retrosynthetic analysis of α-skytanthine. The target molecule could be obtained through a series of
functional group manipulations starting from 3-azabicyclo[4.3.0]nonane 21, which could be
constructed from 22 via the second generation palladium-catalyzed cycloalkenylation process.
Substrate 22 could be synthesized from lactone 14 by lactam formation followed by
methoxycarbonylation.
13
Second Generation Palladium-Catalyzed Cycloalkenylation MeN H H (±)--Skytanthine (20) MeN O CO2Me 21 O O 22 14 MeN O H CO2MeScheme 18
On the basis of this retrosynthetic analysis, the synthesis of α-skytanthine was carried out.
α-Skytanthine was synthesized diastereoselectively from the racemic compound 14 by using second
generation palladium-catalyzed cycloalkenylation. The details of the synthesis are expounded in the
fourth chapter of the ensuing discussion.
14
Detailed Discussion
Chapter 1
Concise Construction of Bicyclo[3.2.1]octane Framework
On the basis of synthetic plan described in the previous discussion, the second generation
palladium-catalyzed cycloalkenylation of 6 was examined. The feasibility of this protocol was
demonstrated in the synthesis of the requisite keto esters 6a–d from starting materials 23a–d, as
depicted in Scheme 19. The protocol was executed by treating compound 23a with methyl
cyanoformate
24)at –78 °C in THF, in the presence of lithium hexamethyldisilazide (LHMDS), to
provide keto ester 6a in moderate yield.
O O R R CO2Me LHMDS; NCCO2Me 23a (R=H) THF, –78 °C to rt 23b (R=Me) 23c (R=CH2OMOM) 23d (R=CH2CH2OMOM) 6a (55%) 6b (38%) 6c (86%) 6d (57%)
Scheme 19
With substrates 6 in hand, the second generation palladium-catalyzed cycloalkenylation of these
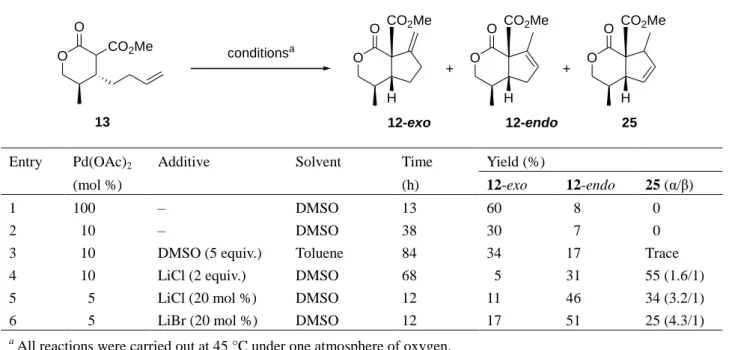
substrates was examined. The results of these cyclizations are summarized in Table 1. Subjection of
compound 6a to the second generation palladium-catalyzed cycloalkenylation produced the desired
cyclization product 7, along with small amounts of phenol derivative 9 (entries 1–3). It should be
noted that the use of 1 atm oxygen may not be required for the cyclization process given that the
reaction was successful under ambient atmosphere (entry 3). Under “open-flask” conditions, longer
reaction times were required; nonetheless, the yields remained at levels comparable to those of the
reactions performed under one atmosphere of oxygen. Superior yields were obtained with substrate
6b, having a methyl group at the angular position (entry 4). Heating the reaction mixture to 80 °C
increased the yields of cyclized products (entry 5). When the reaction was carried out in the
presence of 5 mol % of Pd(OAc)
2, the yield of 7b improved (entry 6). Moreover, further reducing
the amount of catalyst to 2 mol % produced no evidence of compromised yield; nonetheless, a
longer reaction time was necessitated (entry 7). In the case of substrates 6c and 6d, which contain a
15
methoxymethoxy (MOMO) group in the terminal position of the alkyl chains, the yield of the
corresponding cyclization products was decreased relative to 6b (entries 9–14).
Table 1. Preparation of bicyclo[3.2.1]octanes by second generation palladium-catalyzed cycloalkenylation
OH CO2Me O O O R CO2Me R CO2Me R CO2Me conditionsa + + 7 8 9 6
Entry Substrate Pd(OAc)2 Atmosphere Temperature Time Yield (%)
(mol %) (°C) (h) 7 8 9 1 6a 10 O2 rt 60 58 0 12 2 (R = H) 5 O2 rt 72 48 0 12 3 10 Air rt 70 52 0 6 4 6b 10 O2 rt 67 63 Trace – 5 (R = Me) 10 O2 80 13 72 7 – 6 5 O2 80 11 76 3 – 7 2 O2 80 26 72 6 – 8 10 Air 80 20 70 5 – 9 6c 10 O2 rt 67 50 Trace – 10 (R = CH2OMOM) 10 O2 80 6 66 5 – 11 10 Air 80 12 67 3 – 12 6d 10 O2 rt 84 53 6 – 13 (R = (CH2)2OMOM) 10 O2 80 5 55 6 – 14 10 Air rt 132 43 6 – a
All reactions were run in DMSO.
16
Chapter 2
Diastereoselective Total Syntheses of Isoiridomyrmecin and
Isodihydronepetalactone
O O MgBr HMPA, TMSCl THF, –78 °C 94% 15 O O 14 THF, –78 °C 96% O O 13 CO2Me Table 2. O H O 12 CO2Me 86% (2 steps) O H O (±)-11 H 0 °C to rt 95% H H HO HO 24 Pd(OAc)2 ( 5 mol %) pyridine (20 mol %) toluene, 60 °C 90% (±)-11 O H H O (±)-10 + (1 : 1) LiAlH4 THF LDA; ClCO2Me CuBr•SMe2 10% Pd-C, MeOH (1) H2 (1 atm) DMSO-H2O, 160 °C (2) LiCl (10 equiv.) MS 3A, O2 (1 atm)Scheme 20
On the basis of the retrosynthetic analysis described in the previous discussion, the syntheses of
isoiridomyrmecin (10) and isodihydronepetalactone (11) were undertaken. Scheme 20 provides a
detailed outline of the total syntheses of isoiridomyrmecin and isodihydronepetalactone. Conjugate
addition
25)of homoallyl magnesium bromide to 15 provided trans-substituted lactone 14 in 94%
yield; lactone 14, was subsequently subjected to methoxycarbonylation to afford lactone ester 13 in
96% yield as a 3:1 mixture of diastereoisomers. The second generation palladium-catalyzed
cycloalkenylation of 13 was performed under a variety of conditions, as shown in Table 2. Initially,
13 was treated with a stoichiometric amount of Pd(OAc)
2in DMSO as a solvent, which gave the
desired product 12 in 68% isolated yield, as a mixture of olefin isomers (entry 1). Subjection of 13
to catalytic cyclization at 45 °C in DMSO, in the presence of 10 mol % Pd(OAc)
2, gave 12 in 37%
yield (entry 2). The use of another solvent system such as toluene-DMSO (5 equiv.), was
satisfactory for this catalytic cyclization (entry 3). The use of lithium halide additives increased the
reaction rate, and further olefin isomerization occurred (entries 4–6). It should be noted that these
17
latter reaction conditions gave rise to compound 25.
1H-
1H COSY studies of 25a and 25b enabled
all protons of each compound to be assigned, and the relative stereochemistries were established on
the basis of NOE correlations, as depicted in Figure 1. Subjection of 13 to catalytic cyclization at
45 °C, in DMSO, in the presence of 5 mol % Pd(OAc)
2and 20 mol % LiBr, furnished the desired
cyclization product 12 in 68% yield (entry 6). Investigations into the role of lithium halides in this
reaction sequence are underway. Without separation of the exo and endo isomers, 12 was reduced in
the presence of 10% Pd-C, under a hydrogen atmosphere (1 atm), to provide the corresponding
lactone ester, which was subjected to the Krapcho reaction
26)to yield (±)-isodihydronepetalactone
(11) in 86% total yield. The
1H and
13C NMR spectra of synthesized 11 were identical to those
previously reported.
16)(±)-Isoiridomyrmecin (10) was assembled by reducing compound 11 with LiAlH
4to generate
diol 24,
16h)which was subsequently oxidized with catalytic Pd(OAc)
2in the presence of pyridine
and 3 Å molecular sieves
27)to produce a 1:1 mixture of the desired products, 10 and 11, in 90%
yield. The natural products were separated by HPLC; the
1H and
13C NMR spectra of synthesized
10 were identical to those provided by Professor Tsunoda.
15jTable 2. Second generation palladium-catalyzed cycloalkenylation of olefinic lactone ester 13
O CO2Me H O O O CO2Me conditionsa O CO2Me H O O CO2Me H O + + 12-exo 12-endo 25 13
Entry Pd(OAc)2 Additive Solvent Time Yield (%)
(mol %) (h) 12-exo 12-endo 25 (α/β)
1 100 – DMSO 13 60 8 0
2 10 – DMSO 38 30 7 0
3 10 DMSO (5 equiv.) Toluene 84 34 17 Trace
4 10 LiCl (2 equiv.) DMSO 68 5 31 55 (1.6/1)
5 5 LiCl (20 mol %) DMSO 12 11 46 34 (3.2/1)
6 5 LiBr (20 mol %) DMSO 12 17 51 25 (4.3/1)
a All reactions were carried out at 45 °C under one atmosphere of oxygen.
O O H 25a (-methyl) CO2Me O O H CO2Me 25b (-methyl) H NOE NOE
Figure 3
18
Chapter 3
Efficient Synthesis of 3-Azabicyclo[3.3.0]octanes and
3-Azabicyclo[4.3.0]nonanes
On the basis of synthetic plan described in the previous discussion, the second generation
palladium-catalyzed cycloalkenylations of 16 and 17 were investigated. As the initial step in the
evaluation of the possibility of the transformation of 16→18 and 17→19, the requisite substrates 16
and 17 were prepared. Heating γ-lactone 26 with methylamine at 220 °C in dioxane in the presence
of TsOH, using a stainless autoclave, produced γ-lactam 27 directly in 69% yield.
Methoxycarbonylation of 23 was next conducted using methyl chloroformate in THF in the
presence of LDA to give the desired γ-lactam ester 16a in 86% yield (Scheme 21).
O O MeNH2, TsOH dioxane, 220 °C 69% MeN O LDA; ClCO2Me THF, –78 °C to rt 86% MeN O CO2Me 26 27 16a (stainless autoclave)
Scheme 21
In contrast with the direct conversion of the lactone to the lactam achieved in Scheme 21, the
reaction of δ-lactone 28 with methylamine at 200 °C in a stainless autoclave afforded the
corresponding amide alcohol 29 (63%), which was then converted to δ-lactam 30 in 76% yield
using POCl
3. Finally, methoxycarbonylation of 30 was performed in the same manner described in
Scheme 21 to furnish δ-lactam ester 17a (Scheme 22).
28 O O 63% POCl3, pyridine CH2Cl2, –78 °C to rt 76% MeN O 30 MeN O CO2Me 17a 29 MeHN O HO dioxane, 200 °C MeNH2 (stainless autoclave) LDA; ClCO2Me THF, –78 °C to rt 89%
Scheme 22
19
At this stage, the transformation of the amide alcohol to the corresponding δ-lactam under a
variety of conditions was examined and selected results are summarized in Table 3. Generally,
amide alcohol could be converted to the corresponding δ-lactam under the Mitsunobu conditions
(DEAD-PPh
3)
28)or modified Mitsunobu conditions, developed by Tsunoda.
29)Because the
Mitsunobu conditions were not suitable for 29, compound 29 was subjected to modified Mitsunobu
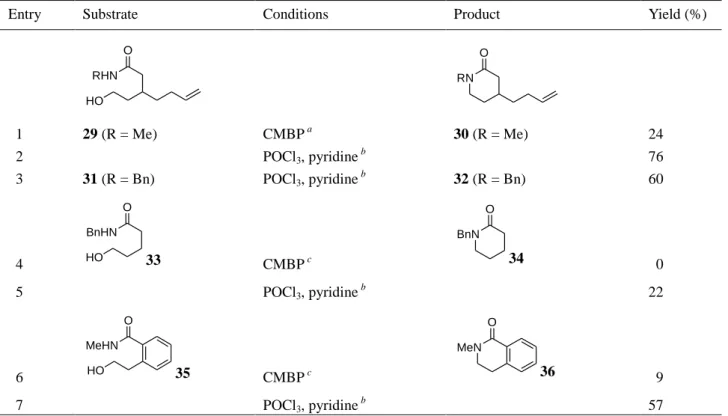
conditions using cyanomethylenetributylphosphorane (CMBP) to provide 30 in 24% yield (entry 1).
On the other hand, the reaction of 29 with POCl
3at temperatures ranging from –78 °C to room
temperature gave rise to 30 in 76% yield (entry 2). The use of substrate 31, in which a benzyl
substituent is attached to the nitrogen, furnished 32 in 60% yield (entry 3), even though the
cyclization product 34 was obtained in only 22% yield using POCl
3(entry 5). It should be noted
that the reaction of the simple amide alcohol 33 with CMBP did not give the cyclization product 34.
This procedure proved to be effective for the synthesis of 3,4-dihydroisoquinolinone 36, and the
desired product 36 was obtained in 57% yield using POCl
3(entry 7). To the best of our knowledge,
the use of POCl
3in a lactam synthesis, as described above, is rare.
30)Table 3. Lactam formation from amide alcohols
Entry Substrate Conditions Product Yield (%)
RHN O HO RN O 1 29 (R = Me) CMBP a 30 (R = Me) 24 2 POCl3, pyridine b 76 3 31 (R = Bn) POCl3, pyridine b 32 (R = Bn) 60 4 BnHN O HO 33 CMBP c BnN O 34 0 5 POCl3, pyridine b 22 6 MeHN O HO 35 CMBP c MeN O 36 9 7 POCl3, pyridine b 57 a Reaction was run at 100 °C in benzene.
b Reactions were run from –78 °C to rt in CH 2Cl2. c Reactions were run at rt in benzene.
20
Having successfully synthesized γ-lactam esters 16 and δ-lactam esters 17, the second
generation palladium-catalyzed cycloalkenylations of 16 and 17 were investigated. Initially,
γ-lactam substrates 16a and 16c were subjected to a treatment in which the palladium source,
solvent, additive, and reaction temperature were varied as shown in Table 4. The use of N-methyl
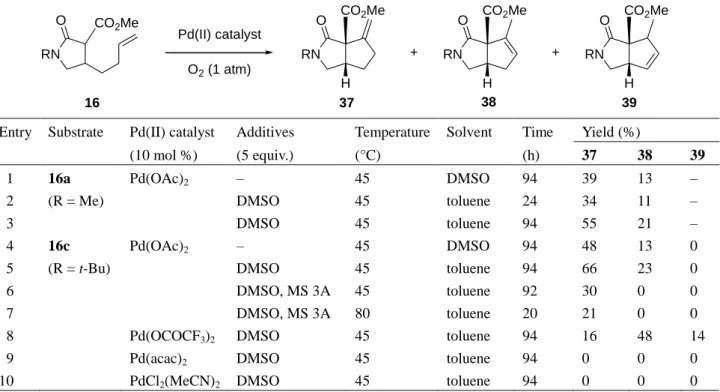
substituted lactam 16a as a substrate furnished the desired cyclization products 37a and 38a in 52%
yield under the standard conditions (entry 1). Compared to the γ-lactone esters, γ-lactam ester 16a
was less reactive and necessitated a much longer reaction time for conversion. Unlike the
palladium-catalyzed cycloalkenylation of the olefinic keto and lactone esters, the reaction of 16a
proceeded at 45 °C in toluene in the presence of 5 equivalents of DMSO, leading to 37a and 38a in
76% yield as a 2.6:1 mixture (entry 3). N-tert-Butyl substituted lactam 16c was amenable to this
reaction (entries 4 and 5). Notably, no olefin isomerization product was observed when 3 Å
molecular sieves were used as an additive (entries 6 and 7).
The catalyst efficiency of various Pd(II) complexes was examined in the subsequent evaluation
by using N-tert-butyl substituted lactam 16c as a substrate. The catalytic reaction with Pd(OAc)
2,
which was found to be the most suitable catalyst, proceeded to furnish the desired cyclization
products 37c and 38c in 89% yield as a 2.9:1 mixture (entry 5). Pd(OCOCF
3)
2gave rise to a
mixture of 37c and 38c in 64% yield (entry 8), however, the desired compounds were not generated
when either Pd(acac)
2or PdCl
2(MeCN)
2were used as catalysts (entries 9 and 10).
Table 4. Second generation palladium-catalyzed cycloalkenylation of γ-lactam esters 16
CO2Me RN O RN O CO2Me H RN O CO2Me H RN O CO2Me H 37 38 39 + + Pd(II) catalyst O2 (1 atm) 16
Entry Substrate Pd(II) catalyst Additives Temperature Solvent Time Yield (%)
(10 mol %) (5 equiv.) (°C) (h) 37 38 39
1 16a Pd(OAc)2 – 45 DMSO 94 39 13 –
2 (R = Me) DMSO 45 toluene 24 34 11 –
3 DMSO 45 toluene 94 55 21 –
4 16c Pd(OAc)2 – 45 DMSO 94 48 13 0
5 (R = t-Bu) DMSO 45 toluene 94 66 23 0
6 DMSO, MS 3A 45 toluene 92 30 0 0
7 DMSO, MS 3A 80 toluene 20 21 0 0
8 Pd(OCOCF3)2 DMSO 45 toluene 94 16 48 14
9 Pd(acac)2 DMSO 45 toluene 94 0 0 0
21
Having optimized the reaction conditions, examination of the scope of the reaction was
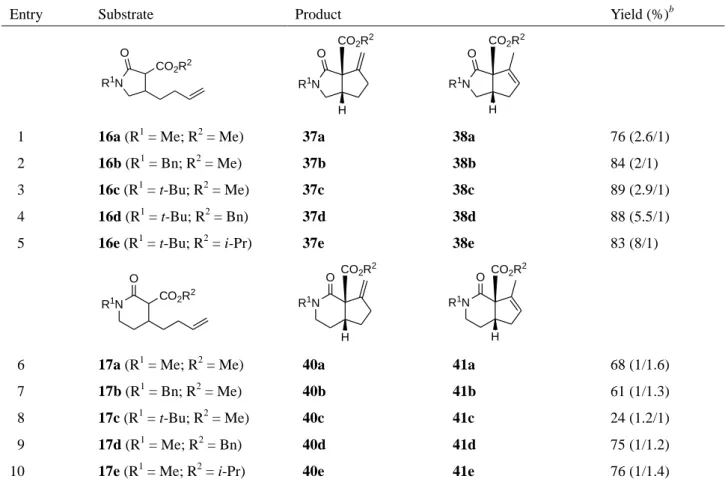
continued using various substrates (Table 5). N-Benzyl substrate 16b was converted to 37b and 38b
in 84% yield as a 2:1 mixture (entry 2). The highest yield was achieved with substrate 16c (entry 3),
indicating that the tertiary butyl group was an effective protecting group for the lactam moiety in 16.
The bulkiness of the ester moiety in 16 did not affect the cyclization yield but rather exerted an
effect on the olefin isomerization (entries 4 and 5).
The reaction of olefinic δ-lactam 17 was subsequently investigated. N-Methyl substrate 17a
gave the desired cyclization products 40a and 41a in moderate yield as a 1:1.6 mixture (entry 6);
however, the yield decreased slightly in the case of the reaction employing the N-benzyl substrate
17b (entry 7). Compared to the γ-lactam 16, the N-tert-butyl substituent proved to be ineffective for
furnishing the desired products in good yield (entry 8). Changing the ester group from the methyl to
the benzyl substituent improved the yield (entry 9). N-Methyl isopropyl ester 17e provided 40e and
41e in good yield (entry 10). The relative stereochemistries of compounds 37e and 40e were
established on the basis of NOE correlations, as shown in Figure 4.
Table 5. Second generation palladium-catalyzed cycloalkenylations of olefinic lactam esters 16 and 17a
Entry Substrate Product Yield (%)b
R1N O CO2R2 R1N O H CO2R2 R1N O H CO2R2
1 16a (R1 = Me; R2 = Me) 37a 38a 76 (2.6/1) 2 16b (R1 = Bn; R2 = Me) 37b 38b 84 (2/1) 3 16c (R1 = t-Bu; R2 = Me) 37c 38c 89 (2.9/1) 4 16d (R1 = t-Bu; R2 = Bn) 37d 38d 88 (5.5/1) 5 16e (R1 = t-Bu; R2 = i-Pr) 37e 38e 83 (8/1)
R1N O CO2R2 R1N O H CO2R2 R1N O H CO2R2
6 17a (R1 = Me; R2 = Me) 40a 41a 68 (1/1.6) 7 17b (R1 = Bn; R2 = Me) 40b 41b 61 (1/1.3) 8 17c (R1 = t-Bu; R2 = Me) 40c 41c 24 (1.2/1) 9 17d (R1 = Me; R2 = Bn) 40d 41d 75 (1/1.2) 10 17e (R1 = Me; R2 = i-Pr) 40e 41e 76 (1/1.4) a All reactions were run at 45 °C in toluene, in the presence of 10 mol % of Pd(OAc)
2 and 5 equivalents of DMSO, for 94 h, under one atmosphere of oxygen.
22
t-BuN MeN O H H O O O H O O 37e 40e NOE NOEFigure 4
23
Chapter 4
Diastereoselective Total Synthesis of α-Skytanthine
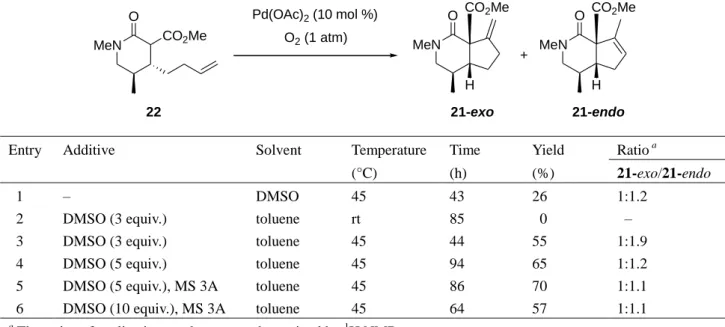
O O 14 aq. MeNH2 dioxane, 200 °C (stainless autoclave) MeHN O HO 42 99% POCl3, pyridine CH2Cl2, –78 °C to rt 73% MeN O 43 LDA; ClCO2Me 91% THF, –78 °C MeN O CO2Me 22 21 MeN O H CO2Me H2 (1 atm) 10% Pd-C MeN O H CO2Me 44 LiCl (10 equiv.) 160 °C 99% 91% MeN O H H 45 0 °C to rt 88% (±)-20 Table 6. MeN H H EtOH DMSO-H2O LiAlH4 THF
Scheme 23
On the basis of the retrosynthetic analysis described in the previous discussion, the synthesis of
α-skytanthine was undertaken (Scheme 23). Compound 14 was transformed into the lactam 43 in
good overall yield, with formation of amide alcohol 42 as a transitional step, as described in
Chapter 3. After introduction of a methoxycarbonyl moiety on 43, the desired 22 was obtained in
91% yield. The second generation palladium-catalyzed cycloalkenylation of lactam ester 22 was
then investigated under the various conditions listed in Table 6. The catalytic cyclization under the
standard conditions used in the case of the olefinic keto and lactone esters was not suitable in this
case (entry 1). Although the reaction did not proceed at all at room temperature (entry 2), the
desired cyclization products 21-exo and 21-endo were obtained in good to moderate yield when the
reaction was performed at 45 °C in toluene in the presence of 10 mol % Pd(OAc)
2with additives
(entries 3–6). The best result, a 70% total yield, was obtained under the reaction conditions listed in
entry 5. Without separating the exo and endo isomers, 21 was subjected to hydrogenation in the
presence of 10% Pd-C to furnish saturated lactam ester 44 as a single stereoisomer. In this case,
reduction occurred from the side opposite the angular ester group (Scheme 17). The relative
24
stereochemistry of the newly formed methyl group of compound 44 was established on the basis of
NOE correlations, as shown in Scheme 10. The Krapcho reaction
26)of lactam ester 44 led to lactam
45 as the sole product. Finally, 45 was converted to (±)-α-skytanthine (20) in 88% yield via LiAlH
4reduction. The spectral data acquired for synthesized 20 were identical to those of an authentic
sample provided by Professor Honda.
21e)Table 6. Second generation palladium-catalyzed cycloalkenylation of olefinic lactam ester 22
MeN O CO2Me MeN CO2Me H O Pd(OAc)2 (10 mol %) O2 (1 atm) MeN CO2Me H O + 22 21-exo 21-endo
Entry Additive Solvent Temperature Time Yield Ratio a
(°C) (h) (%) 21-exo/21-endo
1 – DMSO 45 43 26 1:1.2
2 DMSO (3 equiv.) toluene rt 85 0 –
3 DMSO (3 equiv.) toluene 45 44 55 1:1.9
4 DMSO (5 equiv.) toluene 45 94 65 1:1.2
5 DMSO (5 equiv.), MS 3A toluene 45 86 70 1:1.1
6 DMSO (10 equiv.), MS 3A toluene 45 64 57 1:1.1
a
The ratios of cyclization products were determined by 1H NMR.
MeN CO2Me H O H2, Pd-C 44 MeN CO2Me H O 21 MeN O O OMe disfavored favored NOE
Scheme 24
25
Conclusion
The author demonstrated the construction of bicyclic carbocycles and heterocycles, and total
syntheses of natural products by means of second generation palladium-catalyzed cycloalkenylation.
The transformations employed the Pd(OAc)
2(catalyst)/DMSO (solvent or additive) system with
molecular oxygen as the sole oxidant. Functionalized bicyclo[3.2.1]octanes, 3-oxabicyclo-
[4.3.0]nonanes,
3-azabicyclo[3.3.0]octanes,
and 3-azabicyclo[4.3.0]nonanes
were readily
synthesized by second generation palladium-catalyzed cycloalkenylation. In addition, two types of
iridoids, isoiridomyrmecin and isodihydronepetalactone, and α-skytanthine, a typical
3-azabicyclo[4.3.0]nonane alkaloid, were stereoselectively constructed by applying the
palladium-catalyzed cyclization protocol as a key step.
26
Acknowledgment
My heartfelt appreciation goes to Professor Masahiro Toyota whose comments and suggestions
were of inestimable value for my study. I would like to express my gratitude to Professors Ilhyong
Ryu and Hiroyuki Matsuzaka whose comments made enormous contribution to my work. I would
also like to thank Dr. Hideo Kojima who provided meticulous comments. I am indebted to
Professors Toshio Honda (Hoshi University) and Tetsuto Tsunoda (Tokushima Bunri University) for
providing copies of the
1H and
13C spectra of α-skytanthine and isoiridomyrmecin, respectively. I
would also like to thank Dr. Yasushi Makino and laboratory members. Finally, I would like to
express my gratitude to my family for their moral support and warm encouragements.
27
Experimental section
1. General
Unless otherwise noted, all reactions were performed in oven-dried glassware, sealed with a
rubber septum under an atmosphere of argon. Anhydrous THF and CH
2Cl
2were purchased from
Kanto Chemical Co., Inc. Pyridine, i-Pr
2NH and TMSCl were distilled from CaH
2prior to use.
Toluene and benzene were distilled from P
2O
5. DMSO and HMPA were distilled from CaH
2under
reduced pressure. Unless otherwise mentioned, materials were obtained from commercial suppliers
and used without further purification. Flash column chromatography was carried out using Cica 60
N (spherical, neutral / 40-50 μm) silica gel. Reactions and chromatography fractions were analyzed
employing precoated silica gel 60 F
254plates (Merck). Compounds were visualized using ultraviolet
lamp (254 nm) and by staining with p-anisaldehyde (in EtOH), and KMn
2O
7(in water). IR spectra
were measured on a SHIMADZU FT-IR 8300 spectrophotometer.
1H NMR spectra were recorded
on Varian 400 MR (400 MHz) spectrometers with CHCl
3(δ 7.26) as an internal standard.
13C NMR
spectra were recorded on Varian 400 MR (100 MHz) spectrometer with CHCl
3(δ 77.16) as an
internal standard. Mass spectra were recorded on JEOL JMS-700 spectrometers. Analytical and
semi-preparative HPLC separation were performed by using a HPLC system composed of Jasco
PU-2086 pump, Jasco UV-2075 detector, and Jasco RI-2031 detector.
28
2. Experiments in Chapter 1
2.1. O CO2Me H 6aMethyl 5-(2-propenyl)-2-oxo-3-cyclohexene-1-carboxylate (6a)
A solution of 23a (1.00 g, 7.34 mmol) in THF (10 mL) was added dropwise to a solution of LHMDS, prepared from hexamethyldisilazane (1.60 mL, 7.63 mmol) and n-butyllithium (1.69 M in hexane, 4.4 mL, 7.4 mmol) in THF (20 mL), at –78 °C. After 1 h, methyl cyanoformate (0.60 mL, 7.5 mmol) was added dropwise. After 1 h, the mixture was treated with water and extracted with hexane-EtOAc (1:1 v/v, three times). The combined organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, and concentrated. The
residue was subjected to flash chromatography (hexane-EtOAc 4:1 v/v) to afford 6a (786.2 mg, 55%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 6.92-6.85 (1H, m), 6.07-6.02 (1H, m), 5.82-5.72 (1H, m), 5.16-5.09
(2H, m), 3.78 (2H, s), 3.73 (1H, s), 3.47 (0.33H, dd, J= 5.6, 5.6 Hz), 3.43 (0.67H, dd, J= 14.4, 4.8 Hz), 2.75-2.45 (1.67H, m), 2.28-2.22 (2H, m), 2.06 (0.67H, ddd, J= 14.4, 13.6, 11.6 Hz), 1.94 (0.33H, ddd, J= 13.6, 8.4, 5.2 Hz); IR (neat) 1743, 1682, 1237, 1165 cm-1; HRMS (EI) m/z 194 (M)+ calcd for C11H14O3 194.0943, found 194.0941.
2.2 O CO2Me Me 6b Methyl 5-methyl-2-oxo-5-(2-propenyl)-3-cyclohexene-1-carboxylate (6b) 1 H NMR (400 MHz, CDCl3) δ 11.87 (0.6H, br s), 6.71 (0.2H, dd, J= 10.0, 2.0 Hz), 6.68 (0.2H, dd, J= 10.2, 2.0 Hz), 6.05 (0.6H, d, J= 10.0 Hz), 5.95 (0.2H, d, J= 10.2 Hz), 5.93 (0.2H, d, J= 10.0 Hz), 5.89 (0.6H, d, J= 10.0 Hz), 5.84-5.71 (1H, m), 5.19-5.00 (2H, m), 3.79-3.76 (3H, m), 3.60 (0.2H, dd, J= 4.8, 2.4 Hz), 3.57 (0.2H, dd, J= 5.2, 2.0 Hz), 2.46-2.04 (4H, m), 1.20 (0.6H, s), 1.16 (0.6H, s), 1.03 (1.8H, s); IR (neat) 1746, 1684, 1659, 1443, 1361, 1304, 1239 cm-1; HRMS (EI) m/z 208 (M)+ calcd for C12H16O3 208.1099, found 208.1100.
2.3 O CO2Me MOMO 6c Methyl 5-(methoxymethoxymethyl)-2-oxo-5-(2-propenyl)-3-cyclohexene-1-carboxylate (6c) 1 H NMR (400 MHz, CDCl3) δ 11.87 (0.4H, br s), 6.82 (0.3H, dd, J= 10.0, 2.0 Hz), 6.71 (0.3H, dd, J= 10.0, 2.0 Hz), 6.13-5.98 (1.6H, m), 5.84-5.69 (1H, m), 5.20-5.02 (2H, m), 4.63-4.58 (2H, m), 3.82-3.76 (3.3H, m), 3.66-3.45 (1.7H, m), 3.40-3.33 (3.6H, m), 2.48-2.03 (4H, m); IR (neat) 1744, 1661, 1240, 1153, 1109, 1042 cm-1; HRMS (EI) m/z 268 (M)+ calcd for C14H20O5 268.1311, found 268.1309.
29
2.4 O
CO2Me MOMO
6d
Methyl 5-(2-methoxymethoxyethyl)-2-oxo-5-(2-propenyl)- 3-cyclohexene-1-carboxylate (6d) 1
H NMR (400 MHz, CDCl3) δ 11.85 (0.6H, br s), 6.80 (0.2H, dd, J= 10.0, 2.0 Hz), 6.77 (0.2H, dd, J= 10.0, 2.0
Hz), 6.11-5.92 (1.6H, m), 5.82-5.69 (1H, m), 5.20-5.01 (2H, m), 4.61-4.56 (2H, m), 3.79-3.76 (3H, m), 3.69-3.51 (2H, m), 3.38-3.34 (3.4H, m), 2.45-2.00 (4H, m), 1.94-1.64 (2H, m); IR (neat) 1745, 1683, 1238, 1152, 1109, 1045 cm-1; HRMS (EI) m/z 282 (M)+ calcd for C15H22O5 282.1467, found 282.1468.
2.5 O
CO2Me H
7a
Methyl (1S*,5R*)-7-methylene-2-oxobicyclo[3.2.1]oct-3-ene-1-carboxylate (7a)
To a solution of 6a (34.6 mg, 0.178 mmol) in DMSO (3 mL) was added Pd(OAc)2 (4.0 mg, 0.018 mmol). The
mixture was stirred under one atmosphere of oxygen. After 60 h, the mixture was treated with aqueous NaCl solution, extracted with hexane-EtOAc (1:1 v/v, three times). The combined organic layers were dried over MgSO4, filtered and concentrated. The residue was subjected to flash chromatography (hexane-EtOAc 4:1 v/v) to
provide 7a (20.0 mg, 58%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.17(1H, ddd, J= 9.6, 6.8, 1.6 Hz),
5.89-5.87 (1H, m), 5.85 (1H, d, J= 9.6 Hz), 5.37-5.35(1H, m), 3.79 (3H, s), 2.95 (1H, ddd, J= 6.0, 6.0, 4.0 Hz), 2.79 (1H, dddd, J= 15.2, 5.6, 2.8, 2.8 Hz), 2.51 (1H, dd, J= 11.6, 2.8 Hz), 2.39 (1H, ddd, J= 15.2, 1.2, 1.2 Hz), 2.21 (1H, ddd, J= 11.6, 4.0, 1.6 Hz); 13C NMR (100 MHz, CDCl3) δ 193.3, 169.8, 154.4, 141.2, 126.5, 117.2,
67.2, 52.4, 44.2, 37.7, 34.8; IR (neat) 1748, 1683, 1260, 1069 cm-1; HRMS (EI) m/z 192 (M)+ calcd for C11H12O3
192.0786, found 192.0790. 2.6 OH CO2Me 9 Methyl 5-(2-propenyl)-2-hydroxybenzoate (9) 1 H NMR (400 MHz, CDCl3) δ 10.62 (1H, s), 7.64 (1H, d, J= 2.4 Hz), 7.29 (1H, dd, J= 8.4, 2.4 Hz), 6.92 (1H, d, J= 8.4 Hz), 5.93 (1H, ddt, J= 16.8, 10.4, 6.4 Hz), 5.10-5.03 (2H, m), 3.94 (3H, s), 3.32 (2H, d, J= 6.4 Hz); 13C NMR (100 MHz, CDCl3) δ 170.7, 160.2, 137.4, 136.4, 130.8, 129.5, 117.7, 116.2, 112.2, 52.4, 39.3; IR (neat)
30
2.7 O CO2Me Me 7b Methyl (1S*,5R*)-5-methyl-7-methylene-2-oxobicyclo[3.2.1]oct-3-ene-1-carboxylate (7b) 1 H NMR (400 MHz, CDCl3) δ 6.98 (1H, dd, J= 9.6, 2.4 Hz), 5.83 (1H, d, J= 9.6 Hz), 5.82-5.80 (1H, m), 5.30-5.28 (1H, m), 3.79 (3H, s), 2.54 (1H, ddd, J= 15.6, 2.4, 2.4 Hz), 2.44-2.37 (2H, m), 2.10 (1H, dd, J= 11.6, 2.0 Hz), 1.36 (3H, s); 13C NMR (100 MHz, CDCl3) δ 193.4, 169.7, 158.9, 142.5, 125.6, 116.6, 67.8, 52.4, 50.3,45.1, 40.8, 23.4; IR (neat) 1749, 1686, 1281, 1240, 1049 cm-1; HRMS (EI) m/z 206 (M)+ calcd for C12H14O3
206.0943, found 206.0940. 2.8 O CO2Me Me 8b Methyl (1S*,5R*)-5,7-dimethyl-2-oxobicyclo[3.2.1]octa-3,6-diene-1-carboxylate (8b) 1 H NMR (400 MHz, CDCl3) δ 7.11 (1H, dd, J= 9.6, 2.0 Hz), 6.08-6.06 (1H, m), 5.38 (1H, d, J= 9.6 Hz), 3.77 (3H, s), 2.89 (1H, d, J 10.0 Hz), 2.58 (1H, dd, J 10.0, 2.0 Hz), 1.09 (3H, d, J 1.6 Hz), 1.33 (3H, s); 13C NMR (100 MHz, CDCl3) δ 194.3, 170.0, 161.2, 143.8, 141.3, 121.3, 71.7, 59.8, 52.1, 46.3, 21.7, 14.8; IR (neat) 1747, 1682, 1280,
1170, 1049 cm-1; HRMS (EI) m/z 206 (M)+ calcd for C12H14O3 206.0943, found 206.0935.
2.9 O CO2Me MOMO 7c Methyl (1S*,5S*)-5-(methoxymethoxymethyl)-7-methylene-2-oxobicyclo[3.2.1]oct-3-ene-1-carboxylate (7c) 1 H NMR (400 MHz, CDCl3) δ 7.09 (1H, dd, J 9.6, 2.4 Hz), 5.91 (1H, d, J= 9.6 Hz), 5.87-5.85 (1H, m), 5.34-5.31 (1H, m), 4.67 (2H, s), 3.80 (3H, s), 3.67 (1H, d, J= 9.6 Hz), 3.64 (1H, d, J 9.6 Hz), 3.39 (3H, s), 2.66 (1H, ddd, J= 15.2, 2.4, 2.4 Hz), 2.50-2.40 (2H, m), 2.16 (1H, dd, J= 11.6, 2.0 Hz); 13C NMR (100 MHz, CDCl3) δ 193.2, 169.6, 155.2, 141.3, 126.4, 117.1, 96.8, 71.6, 67.3, 55.6, 52.5, 46.2, 45.2, 40.9; IR (neat) 1748, 1687, 1234, 1151, 1110, 1040 cm-1; HRMS (EI) m/z 266 (M)+ calcd for C14H18O5 266.1154, found 266.1154.
2.10 O CO2Me MOMO 8c Methyl (1S*,5S*)-5-(methoxymethoxymethyl)-7-methyl-2-oxobicyclo[3.2.1]octa-3,6-diene-1-carboxylate (8c) 1 H NMR (400 MHz, CDCl3) δ 7.27 (1H, dd, J= 9.6, 1.6 Hz), 6.16-6.13 (1H, m), 5.46 (1H, d, J= 9.6 Hz), 4.67 (2H, s), 3.77 (3H, s), 3.66 (1H, d, J= 9.6 Hz), 3.63 (1H, d, J= 9.6 Hz), 3.39 (3H, s), 2.91 (1H, d, J= 10.0 Hz), 2.68 (1H,
31
dd, J= 10.0, 1.6 Hz), 1.93 (3H, d, J= 1.6 Hz); 13C NMR (100 MHz, CDCl3) δ 194.1, 169.7, 157.0, 142.4, 139.8,
122.0, 96.8, 71.2, 69.8, 55.5, 52.2, 50.9, 15.0; IR (neat) 1747, 1683, 1111, 1042 cm-1; HRMS (EI) m/z 266 (M)+ calcd for C14H18O5 266.1154, found 266.1154.
2.11 O CO2Me MOMO 7d Methyl (1S*,5R*)-5-(2-methoxymethoxyethyl)-7-ethylene-2-oxobicyclo[3.2.1]oct-3-ene-1-carboxylate (7d) 1 H NMR (400 MHz, CDCl3) δ 7.09 (1H, dd, J= 9.6, 2.0 Hz), 5.85 (1H, d, J= 9.6 Hz), 5.84-5.82 (1H, m), 5.31-5.29 (1H, m), 4.61 (2H, s), 3.79 (3H, s), 3.67 (2H, td, J= 6.4, 1.6 Hz), 3.35 (3H, s), 2.61 (1H, ddd, J= 15.2, 2.4, 2.4 Hz), 2.52-2.42 (2H, m), 2.09 (1H, dd, J= 11.6, 2.0 Hz), 2.01 (1H, dt, J= 14.4, 6.4 Hz), 1.91 (1H, dt, J= 14.4, 6.4 Hz); 13C NMR (100 MHz, CDCl3) δ 193.3, 169.7, 157.6, 141.7, 125.8, 116.8, 96.7, 67.5, 64.9, 55.6, 52.5, 48.5, 44.0, 43.4, 37.2; IR (neat) 2950, 1747, 1683, 1281, 1232, 1150, 1107, 1047 cm-1; HRMS (EI) m/z 280 (M)+ calcd for C15H20O5 280.1311, found 280.1315.
2.12 O CO2Me MOMO 8d Methyl (1S*,5S*)-5-(2-methoxymethoxyethyl)-7-methyl-2-oxobicyclo[3.2.1]octa-3,6-diene-1-carboxylate (8d) 1 H NMR (400 MHz, CDCl3) δ 7.26 (1H, dd, J= 9.6, 2.0 Hz), 6.14-6.12 (1H, m), 5.39 (1H, d, J= 9.6 Hz), 4.61 (2H, s), 3.77 (3H, s), 3.66 (2H, t, J= 6.4 Hz), 3.35 (3H, s), 2.91 (1H, d, J= 10.0 Hz), 2.64 (1H, dd, J= 10.0, 2.0 Hz), 2.01 (1H, dt, J= 14.4, 6.4 Hz), 1.92 (1H, dt, J= 14.4, 6.4 Hz), 1.91 (3H, d, J= 1.6 Hz); 13C NMR (100 MHz, CDCl3) δ 194.3, 169.9, 160.0, 142.4, 141.4, 121.2, 96.7, 71.5, 64.6, 57.6, 55.6, 52.1, 48.9, 35.0, 14.9; IR (neat)
32
3. Experiments in Chapter 2
3.1 O O 14 (4S*,5R*)-4-(3-Butenyl)-tetrahydro-5-methylpyran-2-one (14)Copper(I) bromide-dimethyl sulfide complex (172 mg, 0.837 mmol) and HMPA (3.5 mL) were added to a solution of 3-butenylmagnesium bromide, prepared from magnesium turnings (299 mg, 12.3 mmol) and 4-bromo-1-butene (1.25 mL, 12.3 mmol) in THF (25 mL), at –78 °C. After 30 min, a mixture of 15 (889 mg, 7.93 mmol) and TMSCl (2.2 mL, 17 mmol) in THF (8 mL) was added dropwise. After 1.5 h, the mixture was treated with 10% aqueous NH4Cl solution, and extracted with hexane-EtOAc (3:1 v/v, three times). The combined
organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, and concentrated.
The residue was subjected to flash chromatography (hexane-EtOAc 3:1 v/v) to provide 14 (1.259 g, 94%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.77 (1H, ddt, J= 17.2, 10.0, 6.8 Hz), 5.04 (1H, ddt, J= 17.2, 1.6, 1.6
Hz), 5.00 (1H, ddt, J= 10.0, 1.6, 1.6 Hz), 4.26 (1H, dd, J= 11.2, 4.8 Hz), 3.88 (1H, dd, J= 11.2, 9.2 Hz), 2.70 (1H, dd, J= 17.2, 6.0 Hz), 2.20-2.10 (1H, m), 2.19 (1H, dd, J= 17.2, 8.8 Hz), 2.07-1.96 (1H, m), 1.78-1.57 (3H, m), 1.36-1.28 (1H, m), 1.00 (3H, d, J= 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 172.2, 137.7, 115.5, 73.5, 37.3, 34.6,
33.58, 33.56, 30.3, 15.7; IR (neat) 2974, 2917, 1746, 1213, 1051 cm-1; HRMS (EI) m/z 168 (M)+ calcd for C10H16O2 1168.1150, found 168.1146. 3.2 O O CO2Me 13 Methyl (4R*,5R*)-4-(3-butenyl)-tetrahydro-5-methyl-2-pyrone-3-carboxylate (13)
A solution of 14 (263.1 mg, 1.56 mmol) in THF (3 mL) was added dropwise to a solution of LDA, prepared from diisopropylamine (0.50 mL, 3.6 mmol) and n-butyllithium (1.65 M in hexane, 2.0 mL, 3.3 mmol) in THF (8 mL), at –78 °C. After 1 h, methyl chloroformate (0.12 mL, 1.6 mmol) was added dropwise. After 1 h, the mixture was treated with 10% aqueous NH4Cl solution, and extracted with hexane-EtOAc (3:1 v/v, three times). The combined
organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, and concentrated.
The residue was subjected to flash chromatography (hexane-EtOAc 4:1 v/v) to afford 13 (337.7 mg, 96%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.77 (1H, ddt, J= 17.2, 10.4, 6.4 Hz), 5.03 (1H, ddt, J= 17.2, 1.6, 1.6
Hz), 5.00 (1H, ddt, J 10.4, 1.6, 1.6 Hz), 4.22 (1H, dd, J 11.2, 4.0 Hz), 3.99 (1H, dd, J 11.2, 8.4 Hz), 3.80 (3H, s), 3.36 (1H, d, J= 8.0 Hz), 2.15-2.04 (3H, m), 1.89-1.78 (1H, m), 1.60-1.45 (2H, m), 1.06 (3H, d, J= 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 169.5, 168.5, 137.4, 115.7, 72.6, 53.1, 52.5, 40.8, 33.3, 33.0, 30.0, 16.6; IR (neat)
33
3.3. Typical procedure of second generation palladium catalyzed cycloalkenylation of 13
(Table 2, entry 6) To a solution of 13 (100.8 mg, 0.445 mmol) in DMSO (3 mL) were added Pd(OAc)2 (5.0 mg,
0.022 mmol) and LiCl (7.7 mg, 0.089 mmol). The mixture was stirred at 45 °C under one atmosphere of oxygen. After 12 h, the mixture was treated with aqueous NaCl solution, extracted with hexane-EtOAc (1:1 v/v, three times). The combined organic layers were dried over MgSO4, filtered and concentrated. The residue was subjected
to flash chromatography (hexane-EtOAc 3:1 v/v) to provide a mixture of 12 and 25 (92.6 mg, 93%) as a colorless oil. Each compound was isolated by HPLC (hexane-EtOAc 3:1 v/v).
3.4 O
H O CO2Me
12-exo
Methyl (1S*,5R*,6R*)-5-methyl-9-methylene-2-oxo-3-oxabicyclo[4.3.0]nonane-1-carboxylate (12-exo) 1
H NMR (400 MHz, CDCl3) δ 5.42 (1H, dd, J= 2.4, 2.4 Hz), 5.31 (1H, dd, J= 2.0, 2.0 Hz), 4.20 (1H, dd, J= 11.2,
4.0 Hz), 3.89 (1H, dd, J= 11.2, 11.2 Hz), 3.78 (3H, s), 2.62 (1H, ddd, J= 9.2, 6.4, 6.4 Hz), 2.54-2.41 (2H, m), 1.98 (1H, ddq, J= 8.8, 6.8, 6.8 Hz), 1.84-1.72 (1H, m), 1.57 (1H, dddd, J= 13.2, 7.6, 7.6, 6.0 Hz), 1.04 (3H, d, J= 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 170.9, 168.6, 146.7, 113.5, 73.2, 63.0, 53.5, 50.5, 32.3, 31.1, 28.8, 15.4; IR
(neat) 2957, 1732, 1236 cm-1; HRMS (EI) m/z 224 (M)+ calcd for C12H16O4 224.1049, found 224.1048.
3.5 O
H O CO2Me
12-endo
Methyl (1S*,5R*,6R*)-5,9-dimethyl-2-oxo-3-oxabicyclo[4.3.0]nona-8-ene-1-carboxylate (12-endo) 1
H NMR (400 MHz, CDCl3) δ 5.70-5.60 (1H, m), 4.19 (1H, dd, J= 11.2, 3.6 Hz), 3.95 (1H, dd, J= 11.2, 9.6 Hz),
3.78 (3H, s), 2.74 (1H, dddd, J= 16.4, 8.8, 2.4, 2.4 Hz), 2.66 (1H, ddd, J= 8.8, 8.8, 4.0 Hz), 2.11 (1H, dddq, J= 16.4, 4.0, 2.4, 2.4 Hz), 1.89-1.77 (4H, m), 1.05 (3H, d, J= 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 171.8, 168.9,
137.3, 130.5, 72.4, 70.0, 53.3, 49.2, 37.4, 35.4, 15.8, 15.1; IR (neat) 2957, 1730, 1244 cm-1; HRMS (EI) m/z 224 (M)+ calcd for C12H16O4 224.1049, found 224.1049.
34
3.6 O H O CO2Me 25aMethyl (1R*,5R*,6R*,9R*)-5,9-dimethyl-2-oxo-3-oxabicyclo[4.3.0]nona-7-ene-1-carboxylate (25a) 1
H NMR (400 MHz, CDCl3) δ 5.85 (1H, ddd, J= 6.0, 2.4, 1.6 Hz), 5.63 (1H, ddd, J= 6.0, 2.4, 1.6 Hz), 4.26 (1H,
dd, J= 10.8, 3.6 Hz), 4.07 (1H, dd, J= 10.8, 9.6 Hz), 3.76-3.72 (4H, m), 2.81 (1H, ddd, J= 9.6, 2.4, 1.6 Hz), 1.84-1.72 (1H, m), 1.08-1.04 (6H, m); 13C NMR (100 MHz, CDCl3) δ 172.1, 169.5, 137.5, 127.8, 73.7, 61.4, 56.0,
53.4, 46.4, 35.0, 18.2, 15.0; IR (neat) 1747, 1729, 1235 cm-1; HRMS (EI) m/z 224 (M)+ calcd for C12H16O4
224.1049, found 224.1037. 3.7 O H O CO2Me 25b Methyl (1R*,5R*,6R*,9S*)-5,9-dimethyl-2-oxo-3-oxabicyclo[4.3.0]nona-7-ene-1-carboxylate (25b) 1 H NMR (400 MHz, CDCl3) δ 5.63-5.57 (2H, m), 4.07 (1H, dd, J= 10.8, 4.0 Hz), 3.84-3.78 (2H, m), 3.78 (3H, s), 3.51 (1H, dddd, J= 10.0, 2.0, 2.0, 2.0 Hz), 1.80-1.68 (1H, m), 1.10 (3H, d, J= 6.8 Hz), 1.02 (3H, d, J= 7.6 Hz); 13 C NMR (100 MHz, CDCl3) δ 170.7, 170.2, 134.9, 128.8, 71.7, 62.4, 54.4, 53.2, 47.3, 45.0, 16.2, 16.1; IR (neat)
2964, 1732, 1242, 1191, 1161 cm-1; HRMS (EI) m/z 224 (M)+ calcd for C12H16O4 224.1049, found 224.1039.
3.8 O H O H 11 (±)-Isodihydronepetalactone (11)15a)
A solution of 12 (89.1 mg, 0.397 mmol) in MeOH (3 ml) in the presence of 10% Pd/C (20 mg) was stirred under one atmosphere of hydrogen. After 42 h, the reaction mixture was filtered through Celite® and concentrated under reduced pressure. The residual oil was dissolved in DMSO (2.0 mL) and H2O (0.4 mL). LiCl (181 mg, 4.27
mmol) was added, and the resulting mixture was heated at 160 °C for 8 h. After the mixture was cooled to room temperature, aqueous NaCl solution was added, and extracted with hexane-EtOAc (3:1 v/v, three times). The combined organic layers were dried over MgSO4, filtered, and concentrated. The residue was subjected to flash
chromatography (hexane-EtOAc 3:1 v/v) to afford 11 with its diastereomer (9:1) (57.2 mg, 86% for 2 steps) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.15 (1H, dd, J= 10.8, 3.2 Hz), 3.87 (1H, dd, J= 10.8, 10.8 Hz), 2.34
(1H, dd, J= 10.8, 8.8 Hz), 2.28 (1H, ddd, J= 10.0, 6.4, 6.4 Hz), 2.13-1.98 (2H, m), 1.86 (1H, dddd, J= 12.0, 6.0, 6.0, 2.0 Hz), 1.60 (1H, ddqd, J= 9.6, 9.6, 6.8, 3.2 Hz), 1.23-1.10 (2H, m), 1.20 (3H, d, J= 6.8 Hz), 0.99 (3H, d,
![Table 1. Preparation of bicyclo[3.2.1]octanes by second generation palladium-catalyzed cycloalkenylation](https://thumb-ap.123doks.com/thumbv2/123deta/8513569.1805686/22.892.80.817.233.705/table-preparation-bicyclo-octanes-generation-palladium-catalyzed-cycloalkenylation.webp)