学位論文
Coherent Anti-Stokes Raman Spectroscopy
を
用いた高分子の
Coherent
振動緩和過程の研究
香川大学大学院 工学研究科
博士後期課程 材料創造工学専攻
指導教官
:
中西 俊介 教授
12D551
香西 貴典
平成
27
年
1
月
3
目 次
第 1 章 序章 7 1.1 分子振動に関する研究. . . 7 1.1.1 Raman 散乱研究の歴史 . . . 7 1.1.2 Raman 分光法 . . . 9 1.1.3 赤外分光法 . . . 10 1.1.4 Raman 分光と赤外分光における選択則 . . . 13 1.2 分子振動ダイナミクスと CARS 法 . . . 14 1.2.1 時間分解振動分光法 . . . 15 1.2.2 フェムト秒時間分解分光法 . . . 16 1.2.3 誘導 Raman 分光法 . . . 161.2.4 Coherent Anti-Stokes Raman Spectroscopy について. . . 17
1.2.5 時間分解 CARS 法 . . . 19 1.3 研究目的 . . . 23 第 2 章 Raman 分光とその理論的背景 29 2.1 Raman 散乱の古典的モデル . . . 29 2.2 Raman 散乱の量子論的描像 . . . 32 2.3 非線形光学効果について . . . 34
2.4 Coheretn Anti-Stokes Raman Spectroscopy の量子力学的理論 . . . 37
2.4.1 時間に依存する摂動論 . . . 37 2.4.2 無摂動系における時間に依存する Schr¨odinger 方程式 . . . 37 2.4.3 摂動がある場合の時間に依存する Schr¨odinger 方程式 . . . 40 2.4.4 展開係数 an(t) の時間変化 . . . 40 2.4.5 密度行列の導入 . . . 41 2.4.6 密度行列の運動方程式 . . . 43
2.5 密度行列を用いた Coherent Anti-Stokes Raman Spectroscopy の理論. . . 45
2.5.1 マスター方程式の導出 . . . 47
2.5.2 密度行列に対する摂動論 . . . 52
2.5.4 CARS 信号光の理論的表現 . . . 60 第 3 章 実験方法 67 3.1 試料と実験条件 . . . 67 3.1.1 Polyethylene の特徴 . . . 67 3.1.2 Polyvinyl alcohol の特徴 . . . 69 3.1.3 Polymethyl methacrylate の特徴 . . . 70 3.1.4 試料作製 . . . 70 3.2 実験装置 . . . 73 3.2.1 Raman 分光装置 . . . 73 3.2.2 レーザーシステム . . . 74 3.2.3 CARS 光学系 . . . 77 3.2.4 CARS 測定用の励起パルスの特性 . . . 78 3.3 CARS 測定の励起配置と結果の解析手法について . . . 80 3.3.1 CARS 信号の励起とそのスペクトル測定 . . . 80 3.3.2 時間分解 CARS 法 . . . 81 3.3.3 CARS 信号の温度変化の測定 . . . 83 3.3.4 コヒーレント振動緩和時間の導出について . . . 84 3.3.5 Fourier 変換 . . . 86 第 4 章 実験結果と考察 89 4.1 Polyvinyl alcohol についての測定結果と解析 . . . 89 4.1.1 Raman スペクトル結果 . . . 89 4.1.2 CARS スペクトルの測定結果 . . . 91 4.1.3 時間分解 CARS 法の実験結果 . . . 92 4.1.4 時間分解 CARS 信号の Fourier 変換結果 . . . 93 4.1.5 CH2基の Coherent 振動緩和についての解析 . . . 95 4.1.6 CH2基についてのまとめ . . . 97 4.1.7 OH 基の Coherent 振動緩和についての解析 . . . 99 4.1.8 OH 基についてのまとめ . . . 100 4.2 Polymethyl Methacrylate についての測定結果と解析 . . . 101 4.2.1 Raman スペクトル結果 . . . 101 4.2.2 CARS スペクトルの測定結果 . . . 102 4.2.3 時間分解 CARS 法の実験結果 . . . 103 4.2.4 時間分解 CARS 信号の Fourier 変換結果 . . . 104
5 4.2.5 PMMA 中の CH2基のコヒーレント振動緩和時間 . . . 105 4.3 Polyethylene について . . . 106 4.3.1 Raman スペクトル結果 . . . 106 4.3.2 CARS 分光の測定結果 . . . 108 4.3.3 時間分解 CARS 法の実験結果 . . . 109 4.3.4 PE 中の CH2基のコヒーレント振動緩和時間 . . . 110 4.4 Polyethylene の CARS 信号の低温での測定について . . . 112 4.4.1 低温 (6 K) での CARS 分光の測定結果 . . . 112 4.4.2 低温での時間分解 CARS 法の実験結果 . . . 113 4.4.3 Coherent 分子振動の温度変化に関する考察 . . . 115 第 5 章 まとめ 121 謝辞 125
7
第
1
章
序章
この章では、本研究のテーマである「Coherent Anti-Stokes Raman Spectroscopy を用 いた高分子の Coherent 振動緩和過程の研究」に関系する Raman 散乱や赤外分光によ る分子振動の研究の歴史と現状、レーザーの発展の経緯と Coherent Anti-Stokes Raman Spectroscopy の確立にいたる過程について説明する。その後、本研究の研究目的つい ての概要をまとめる。
1.1
分子振動に関する研究
1.1.1
Raman
散乱研究の歴史
Raman 散乱は、1928 年に C. V. Raman と K. S. Krishnan によって発見された [1]。彼 らはランプの光を液体に照射したとき、散乱される一部の光の波長が大きく変化する ことを観測した。この現象は、物質をいくら純粋にしても、照射した光の波長と異な る波長の散乱光が現われるというものであった [2–5]。このことから Raman らは、す べての物質が蛍光を出すのではないかと考えたが、現在ではこの現象は Raman 効果 として知られている新しい現象であることが明らかになった。その後、この現象の研 究者たちは光源を線スペクトルの光源に変え、また散乱スペクトルの測定に分光器を 使って、物質によって散乱した一部の光の波長が変化するということが今までに知ら れていなかった新しい効果であることを確かめた [6]。 Raman 散乱は、図 1.1 に示すように分子の固有振動数ν1、ν2· · · νnが入射線ν と結合 してν ± ν1、ν ± ν2· · ·ν ± νn のスペクトル線としてあらわれる。初期の Raman 分光 法ではそのスペクトル線を写真の乾板上に映し出し [7]、散乱光のスペクトルを観測 するというものであった。ここで観測される入射光と異なった波長を持つ光の振動数 変化が分子の固有振動数になっていることが分かった。この固有振動数は官能基の種 類によって決まっており、得られた Raman スペクトルから物質の分子の構造や結晶構 造を調べることが可能となった。この Raman 散乱光の特性を応用し、その後さまざ まな化合物の Raman スペクトル測定が盛んに行われ、Raman 分光実験の対象とされ た試料は膨大な数に上ったが [8, 9]、一般的な分析方法として分析機器の形で市販さ
入射線 ν 固有振動数 νn 入射線 ν 固有振動数 νn スペクトル線 ν − νn スペクトル線 ν + νn 図 1.1: 光散乱遷移 れるまでには至らなかった。その原因として、散乱光の強度が入射光(励起光)に対 して非常に微弱なため測定が難しかったことが挙げられる。また、赤外吸収分光法が フーリエ変換赤外分光 [10] の登場で著しく進歩した結果、測定の難しい Raman 分光 法はあまり利用されない時期が続いた。しかし、1960 年に Raman 分光にとって理想 的な光源であるレーザーが発明されたことで、それを励起光源にすることにより今ま で微弱であった Raman 信号光の強度を飛躍的に上げることに成功し、分析法としての 地位を確かなものとした。1960 年代には、波長 694.3nm のルビーレーザー、632.8nm の He-Ne レーザーを用いたレーザーラマン分光法が登場した。しかし、レーザーの出 力は数十 mW と低かった。 1970 年代には、現在でも用いられている Ar+レーザーが 登場し、充分強いレーザー出力が可能になった [11]。それに伴って光学顕微鏡との組 合せにより顕微レーザーラマン分光光度計が誕生し、数µm という局所的な領域に対 する Raman 分光法による分析が可能となった。1980 年代後半には、多数の測定点 (波 数) を一度に測定できる、マルチチャンネル検出器を装備した Raman 分光光度計が登 場し、その後マルチチャンネル検出器の 1 つである CCD 検出器の性能が飛躍的に向 上したため、 CCD 検出器を用いる Raman 分光光度計が一般的になった。1990 年代 後半には、高性能なレイリー光除去フィルターが出現し、ダブルモノクロメータを用 いる大型 Raman 分光光度計に代わって、 小型シングルモノクロメータでも高感度な Raman 分光光度計が誕生した。こうした分光機器の発達によって測定精度が飛躍的に 向上し、物質中の分子の挙動、特に分子振動スペクトルを調べる手法として現在も研 究に広く用いられている。
1.1. 分子振動に関する研究 9
1.1.2
Raman
分光法
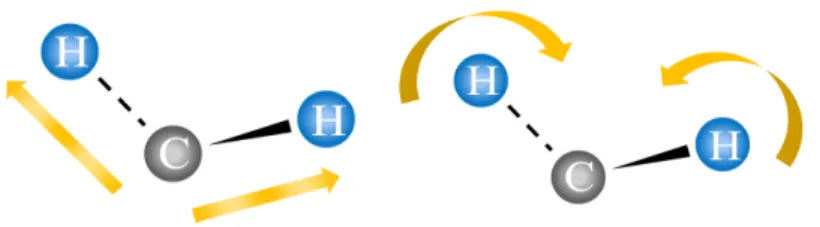
Raman 分光法における測定はレーザーを光源として試料を照射し、試料からの散乱 光のスペクトルを検出器で観測するというものである。レーザーが物質に入射するこ とによって発生する散乱光の多くはフォトンエネルギー (波長) が変化しない Rayleigh 散乱と呼ばれる弾性散乱光である。しかしながら、物質から散乱する光の中に非弾性 散乱を起こしているものも存在する。その非弾性散乱光は入射したフォトンの波長と は異なる波長を持ったフォトンである。発生したフォトンの中で、波長が長く (レッ ドシフト) なったものを Stokes 散乱と呼び、波長が短く (ブルーシフト) なったものを Anti-Stokes 散乱と呼ぶ [6, 12]。これらの光散乱における物質系の状態間の遷移を図 1.2 に示す。点線は仮想準位を表しており、散乱の過程で仮想準位に実際に系が遷移 するわけではない。この散乱の中で、Anti-Stokes 散乱は物質が高温の状況に置かれて いる場合、強く観測することができる。これはボルツマン分布が大きく関係している と考えられる。詳しい関係は節 2.2 で述べる Raman 散乱の量子力学的モデルの所で説 明するが、図 1.2 でわかるように Anti-Stokes 散乱の出発状態は固有振動の励起状態で あり、その状態に系が存在する確率に依存しているために生じる現象である。また、 全エネルギーは散乱過程の間に保存されるので、フォトンが得たエネルギー、あるい は失ったエネルギーは、分子のエネルギー変化に等しくなる。これを利用することに より分子振動の振動数を特定することができ [13, 14]、その測定値から分子の化学組 成、分子構造などを調べることができる。 (a) Stokes Scattering (b) Rayleigh Scattering (c) Anti-Stokes Scattering 図 1.2: 光散乱における遷移1.1.3
赤外分光法
(a) 赤外分光法の概略 赤外分光法は、物質に赤外光を照射し、透過または反射した光の強度変化、スペク トル変化を測定することで、試料の構造解析や定量を行う分析手法である [15–17]。 赤外光は、分子の振動や回転運動によって吸収される。それは赤外光(λ = 2.5∼ 25µm)が、紫外・可視光(λ = 0.2∼0.78µm)よりもエネルギーが小さく、そのエネ ルギーが電子遷移には足りず、分子振動のエネルギーに当るためである。また、分子 の振動や回転の状態を変化させるのに必要なエネルギー(赤外光の波長)は、物質の 化学構造によって異なるので、物質に吸収される赤外光を測定すれば、化学構造や状 態に関する情報を得ることができる。すでに述べたように、赤外分光法によって分子 の振動スペクトルを得ることができる。 赤外分光光度計には、分散型とフーリエ変換型 (FTIR) があり、横軸に波数(または 波長)、縦軸に透過率(または吸光度)をプロットしたグラフを出力する。このグラ フを赤外吸収スペクトル(IR スペクトル)という。 IR スペクトルは、物質固有のパ ターンを示すことから、構造解析や定性分析に有効である。IR スペクトルの縦軸であ る吸光度は物質の濃度や厚みに比例するため、ピークの高さや面積から定量分析を行 うことも可能となる。しかし、赤外分光法にはデメリットも存在する。赤外分光法で はもちろん赤外光を用いることになるが、その赤外光を発生させることが可能な光源 が少ない。また、それを観測する測定器も高価な場合が多い。そのほかに、赤外光は 水による吸収 (3300 cm−1付近) が大きいため水溶液中の物質を測定することは難しい 点などがあげられる。 近年では、予めデータベースに記録された標準試料のスペクトルと、測定したスペ クトルを照合することにより未知試料を同定することができる。 特に、IR スペクト ルは数十万という膨大なデータが存在するため、未知試料分析に非常に有効である。 特に、有機物質を中心に多くのデータが蓄積されており、薬学、農学、生物学、ガス 分析、鑑識など広い分野で利用されている。[18–20] 図 1.3 は赤外分光法で標準物質として知られているポリスチレンの IR スペクトル である。現在では、フェムト秒赤外光パルスを発生させ分子振動のダイナミクスを Pump-Probe 法で調べる手法も研究されている。[21–23] (b) 赤外分光法の原理 分子に関するエネルギー値は、一般的に量子化された飛び飛びの値をとる。このた め表 1.1 に示すように、分子にさまざまなエネルギーを持つ電磁波を当てることによ1.1. 分子振動に関する研究 11
透
過
光
強
度
透
過
光
強
度
透
過
光
強
度
透
過
光
強
度
波数
波数
波数
波数 (cm
-1))))
ポリスチレン ポリスチレンポリスチレン ポリスチレン 図 1.3: スチレンの IR スペクトル [18] り、分子に関する多様な情報を得ることができる。赤外線の場合、図 1.4 に示すよう に振動準位や回転準位の遷移エネルギーに相当するため、分子振動、回転準位に関す る情報を得ることが可能である。 表 1.1: 分光法によって得られる分子の情報 [18] 電磁波 電磁波 電磁波 電磁波ののの種類の種類種類種類 スペクトルのスペクトルの種類スペクトルのスペクトルの種類種類種類 スペクトルからわかるスペクトルからわかるスペクトルからわかるスペクトルからわかる情報情報情報情報 XXXX線線線線 回折像回折像を回折像回折像を利用をを利用利用利用したしたしたした結晶構造結晶構造結晶構造結晶構造 真空紫外線 真空紫外線 真空紫外線 真空紫外線 ((((遠紫外線遠紫外線遠紫外線))))遠紫外線 真空紫外線真空紫外線真空紫外線真空紫外線スペクトル((((遠紫外遠紫外遠紫外スペクトル遠紫外スペクトルスペクトル))))スペクトルスペクトルスペクトルスペクトル 内殻および内殻内殻内殻およびおよびおよびssss電子電子電子電子のののの遷移遷移遷移エネルギー遷移エネルギーエネルギーエネルギー 紫外線 紫外線紫外線 紫外線 紫外紫外紫外スペクトル紫外スペクトルスペクトルスペクトル nnnnおよびおよびおよびおよびpppp電子電子の電子電子のの遷移の遷移遷移遷移エネルギーエネルギーエネルギーエネルギー 可視光線 可視光線可視光線 可視光線 可視可視可視可視スペクトルスペクトルスペクトルスペクトル 近赤外線 近赤外線近赤外線 近赤外線 近赤外近赤外スペクトル近赤外近赤外スペクトルスペクトルスペクトル 振動振動振動振動のpppp電子電子の電子電子ののの倍音倍音倍音、、、、結合音倍音の遷移のの遷移エネルギー遷移遷移結合音と結合音結合音エネルギーエネルギーエネルギーと高共役とと高共役高共役高共役 赤外線 赤外線赤外線 赤外線 赤外赤外赤外赤外スペクトルスペクトルスペクトルスペクトル 振動準位の振動準位振動準位振動準位ののの遷移遷移遷移エネルギー遷移エネルギーエネルギーエネルギー 遠赤外線 遠赤外線遠赤外線 遠赤外線 遠赤外遠赤外スペクトル遠赤外遠赤外スペクトルスペクトルスペクトル 振動振動と振動振動と回転準位とと回転準位回転準位回転準位のののの遷移遷移遷移エネルギー遷移エネルギーエネルギーエネルギー マイクロ マイクロマイクロ マイクロ波波波波 マイクロマイクロマイクロマイクロ波分光波分光波分光波分光スペクトルスペクトルスペクトルスペクトル 電磁波 電磁波電磁波 電磁波((((ラジオラジオラジオラジオ波波波))))波 核磁気共鳴核磁気共鳴核磁気共鳴核磁気共鳴スペクトルスペクトルスペクトルスペクトル 核核核核スピンのスピンのスピンのスピンの遷移遷移遷移遷移エネルギーエネルギーエネルギーエネルギー 分子中で化学結合をしている原子の運動様式は、ばねで結ばれた原子の動きとして 考えることができる (図 1.5: 古典モデル)。たとえば、図 1.6 に示すように原子および 官能基の伸縮振動や結合角度の変わる変角振動としてあらわすことができる。この分 子振動は赤外光 (光エネルギー) が分子に吸収されるときに励起され、吸収された赤外 光のエネルギーは振動エネルギーに変換される。この振動の周期は、結合が強いほど、 また原子が軽いほど短くなる。すなわち、振動数が高くなる。すでに述べたように、 分子振動も量子力学に従って運動するため、そのエネルギー準位は離散的である。そ のため、吸収される赤外光エネルギーは振動準位間のエネルギーに対応するもののみ である。その結果、赤外吸収スペクトルで吸収が増加する波長が振動エネルギー準位 間のエネルギーに対応し、分子振動の振動数を得ることができる。図 1.4: 赤外吸収のモデル
図 1.5: 分子の運動様式 (古典モデル)
1.1. 分子振動に関する研究 13
1.1.4
Raman
分光と赤外分光における選択則
Raman 分光法と赤外分光法はお互い分子振動に関する情報を得られる手法として用 いられているが、検出されるピークの位置や強度、形状は両者で異なる場合がある。 場合よっては、分子振動のうち一方の分光法で測定したときに得られるスペクトルに 現れない振動モードも存在する。つまり、赤外光を吸収しない振動モードでも Raman 散乱が起こる場合やその逆の場合がある。このことは光学遷移の選択則と呼ばれる規 則によって決まっている [6]。 分子の基準振動のうち選択則によって許されるものだけがスペクトルとして観測さ れる。赤外線吸収の場合、振動数が振動モードの振動と一致しなければならない。さ らに、吸収に関与する振動準位間の遷移双極子モーメントが 0 でない必要がある。つ まり、ある振動モードが赤外線吸収スペクトルに現れるためには、分子振動準位間の 遷移双極子モーメントが有限である必要がある。 次に、Raman 散乱の場合は項 1.1.2 で述べたように、入射光に対して散乱した光の 周波数が変化した光が観測されるが、この原因は分子の振動と入射光との直接的な相 互作用によって発生するものではないことに由来する。これは 分子内の電子が入射 光の影響を受けて強制振動を起こすことに由来する。これらの電子が、入射光と同じ 振動数ν で強制振動を起こすと分子からは同じく振動数 ν の光が発生する (Rayleigh 散 乱)。このとき分子がν0の固有振動数で振動していれば、入射光はこの分子の電子状態 に周期的な変化を受ける。つまり、ν±ν0の振動数の光が発生する。それゆえ、Raman 効果が表れるためには、分子の固有振動によって電子状態の変化、具体的にいうと分 極率の変化が生じることが重要である。量子力学的にいうと Raman 散乱の場合の振動 準位間の遷移は、遷移双極子モーメントが 0 で光吸収で遷移できないが、エネルギー の大きな電子遷移を 2 回経ることで、振動励起状態への遷移が生じることに対応する。 以上をまとめると赤外線吸収と Raman 効果の選択則は光遷移の仕方によって決まっ ており、分子の対称性に大きく影響を受ける。たとえば、図 1.7 に示すような対称中 心を持つような分子の場合、その赤外吸収および Raman 効果の選択則は、対称中心 に対称の振動は Raman 活性で赤外不活性である。これに対して、対称中心に逆対称 の振動は Raman 不活性で赤外活性である。そのため、対称中心のある分子の基準振 動のうち Raman 活性の者は赤外吸収に現れず、また赤外活性のものは、Raman 散乱 としては現れない。これを交互禁制律という。一般的な分子の場合、対称中心と同時 に他の対称要素を持つので、それを考慮したうえで活性・不活性の振動であるのか決 定する必要がある。また、Raman 散乱と赤外吸収の両者を生じるような振動も一般的 に存在するが、その強度比率は分子の対称性に依存する。図 1.7: 対称中心を持つ分子振動
1.2
分子振動ダイナミクスと
CARS
法
物質内の分子振動に関する研究は物理・化学の分野において重要な役割を担ってい る。物質中の分子は絶えず振動し、気体、液体や固体中で振動、回転や並進の運動を している。これら分子の振動は、レーザーを用いた Raman 分光法や赤外吸収といった 手法で分子の同定、特定官能基の同定、分子振動の解析などに応用されてきた。例え ば、ポリエチレンなどでは Raman 光や赤外吸収から得られたスペクトルの形状から分 子構造が推測されたり [24]、分子内相互作用や分子構造の解明 [25] などが行われた。 その他に、分子振動のスペクトルに加えて分子振動ダイナミクスにも注目が集まり だした。たとえば、振動状態、あるいは回転状態が高いエネルギー状態に励起された 分子は熱 (Phonon) などとの相互作用によって緩和し平衡状態となる。そのほかに、外 部からエネルギーを加えることにより、振動が誘起された分子は化学結合の切断や生 成を行う。これらの緩和が生じる時間領域を時間軸で示したのが図 1.8 である。この ように分子振動は化学反応において重要な働きをしていると予測されたが、制御され た安定な短パルスの発生が困難であったため、分子振動ダイナミクスといった現象を 研究することは非常に難しい実験を遂行する必要があり、困難であった。現在では比 較的容易に制御されたフェムト秒パルス [26, 27] を得ることができるため、分子振動 ダイナミクスに関する研究が盛んにおこなわれている。 10 fs 1 ps 100 ps 10-14 10-13 10-12 10-11 10-10 10-9 秒 原子核の振動 回転運動 振動位相緩和 振動エネルギー緩和 電子 位相緩和 電子エネルギー緩和 図 1.8: 分子における様々な超高速緩和過程が生じる時間領域1.2. 分子振動ダイナミクスと CARS 法 15
1.2.1
時間分解振動分光法
時間分解 Raman 分光法 分子振動ダイナミクスを時間分解 Raman 分光法を用いて測定した研究は、1970 年 代後半のマイクロ秒時間分解 Raman 分光の実験の成功がはじめである [28]。1980 年 代に入り、安定なナノ秒 Q-スイッチ Nd:YAG レーザーが市販されるようになると、ナ ノ秒領域の時間分解 Raman 分光が急速に発展した。数多くの短寿命過渡種の Raman スペクトルが測定され、それまで得ることのできなかった過渡種の貴重な構造情報が もたらされた。1980 年代の後半までには、ナノ秒時間分解 Raman 分光の測定手法が 確立された [29]。1990 年代に入ると、モード同期ピコ秒レーザの増幅技術が進歩し、 ピコ秒領域での時間分解 Raman 分光の試みが本格的化した。1993 年には Nd3+:YAG 再生増幅器出力を励起光源とする色素増幅器を用いた、フーリエ限界のピコ秒時間分 解 Raman 分光装置 [30] の製作が報告された。現在では、Ti3+:Al2O3結晶を用いたモー ドロックレーザー [26, 27] と再生増幅技術により、安定で強力な 5fs のパルス幅を有 するフェムト秒パルスを発生させる技術が確立されており、容易にフェムト秒パルス を得ることができるので、高度で精緻な実験を行うことができるようになっている。 多くのグループが再生増幅器を用いた方式を踏襲し、時間分解能がフェムト秒領域ま で拡張されて現在に至っている。 時間分解赤外分光法 一方、時間分解赤外分光の発展は、時間分解 Raman 分光法に対してやや遅れをとっ た。その原因は、赤外領域の光パルス発生と検出が困難であること、またその開発が 時間分解 Raman 分光法に比べると遅れていたからである。1980 年代にいくつかのグ ループにより先駆的研究が行われていたが、論文数が Raman 分光法と肩を並べるよ うになったのは、1990 年代に入ってからのことである。時間分解赤外分光法の研究に は、おおまかにいって2つの流れがある。第一の流れは、レーザーではない通常光源 を用い、電気的なゲート検出によりマイクロ秒からナノ秒の時間分解能を得るもので ある。現在では、分散型の分光器を用いる方式 [31] と、ステップ掃引 FT 方式 [10] の 装置がすでに市販されている。両方式の特徴として、FT 法は広い波数範囲で時間分解 スペクトルを観測するときに有力であり、分散法は狭い波数範囲のキーバンドに注目 してダイナミクスを調べる場合に有利となる [32]。第二の流れは、超高速パルスレー ザーを用いてピコ秒の時間分解能を達成するものである。ピコ秒の赤外パルスを検索 光として用いる方式 [33] と、ピコ秒の紫外・可視パルスにより連続発振の赤外検索光 にゲートをかける方式が行われている。最近の研究では、10 fs 以下の中赤外光パルス
を発生させることに成功し、それを超高速の赤外吸収分光に応用する試みが進んでい る。この場合、通常、パルス幅と光電場の波長が同程度となるため、発生が極めて困 難である。Ti:Sapphire レーザー出力を光パラメトリック増幅によって波長変換する方 法が代表的であるが、中赤外光と Ti:Sapphire レーザーの波長である 800nm の光を両 方透過する結晶の種類は限られ、目的の位相整合を満たすことのできる結晶はいまだ 開発段階である。そういった中、気体を非線形媒質としてフィラメンテーションを発 生させ中赤外パルスを発生させる方法が編み出された。この手法で発生した中赤外光 パルスの中心波長は 3.3µm であり、スペクトル幅は 2.5∼5.5µm 程度まで広がってい て、中赤外と呼ばれる領域をほぼ覆っている。またこの中赤外光パルスは気相の分光 にも使用可能なエネルギー領域であり、パルス幅は 13fs である。この手法により発生 した中赤外パルスを用いて高分子の IR スペクトルの観測に成功している [22]。この パルスを用いた時間分解振動分光への応用も期待されている。
1.2.2
フェムト秒時間分解分光法
フェムト秒パルスレーザーの登場により、分子にエネルギーと位相のそろった電子 励起状態や振動状態 (コヒーレント振動状態) を作り出すような実験も行うことが可能 となった。衝突の少ない希薄気相状態では、比較的長時間にわたるコヒーレント振動 状態の観測や分子回転コヒーレンスの観測 [34] が 1980 年代になされている。その他 に 1990 年代にはサブ 5 fs ポンプ・プローブ実時間分解法を用いて、ポリジアセチレ ンの一種である PDA-4BCMU4A と呼ばれる共役高分子の伸縮振動とねじれあるいは 変角振動との結合の実時間的観測を C=C 二重結合や C≡C 三重結合において測定した 研究 [35, 36] や、Mokhtari ら [37] は NaI の光解離における分子振動の様子を実時間分 光で観測している。Zewail はこれら一連のフェムト秒分光の業績により、1999 年の ノーベル化学賞を受賞している。このように分子振動の動的振舞を正確にとらえる方 法として、ピコ秒やフェムト秒の実時間領域で分子振動の動きを直接検出する手法が 多く考えだされている。このようにパルスレーザーを用いてコヒーレントな振動状態 を生成し、その分子振動の時間変化を理解することは重要な課題になってきている。 このようなフェムト秒領域の現象への興味とその応用性がフェムト秒分光の推進力に なっており、多くの研究が現在も行われている。1.2.3
誘導
Raman
分光法
Raman 散乱は自発 Raman 散乱と誘導 Raman 散乱に大別される。自発 Raman 散乱 において、光は一般に周波数が変化し、全立体角方向に散乱される。そのため、自発
1.2. 分子振動ダイナミクスと CARS 法 17 Raman 散乱の強度は弱いものとなる。一方、誘導 Raman 散乱は、非常に強いレーザー 光を物質に入射した場合に、入射レーザーの周波数ω から物質の振動数 ωνだけずれ た周波数ω − ωνのコヒーレントな光が散乱 (放射) される現象で、1962 年に Woodbury と Ng [38] によりニトロベンゼンで観測された。誘導 Raman 散乱の特徴は、非常にコ ヒーレントで強い光であること、入射レーザー光強度にある閾値が存在し、それ以下 では生じないこと、入射光と誘導 Raman 散乱光の周波数差が分子などの振動数となっ ていることなどである。この誘導 Raman 散乱が生じるのは、強力な励起レーザー光の 入射により物質内で Raman 散乱光 (ω − ων) に対する利得が大きくなり、ある方向 (位 相整合方向) に進む散乱光が増幅されることによる。この場合、物質内では分子振動が コヒーレントに誘起されていることによる。そのため、入射レーザー光の Anti-Stokes 散乱に対応する周波数 (ω + ων) も増幅され、コヒーレントで強力な散乱を示すことも ある。 この誘導 Raman 散乱を用いた分光法を誘導 Raman 分光法という。この分光法の利 点は誘導 Raman 散乱光が強いため、スペクトルを容易に得ることができる点である。 欠点としては、非常に強力なレーザー光が必要であること、増幅される振動モードは 少数であり、全ての Raman 活性スペクトルを得ることができないことである。誘導 Raman 散乱光をレーザー発振させた Raman レーザーも実用化されている。 誘導 Raman 散乱を制御して生じさせる方法として、2 つの強いレーザー光ω1とω2 を入射する方法がある。特に、ω1とω2 = ω1− ωνの 2 波長のレーザーを使って、ωνの 固有振動をコヒーレントに強く励起することで強い Raman 散乱光を観測することがで きる。この誘導 Raman 分光法の代表的なものにと Coherent Anti-Stokes Raman Spec-troscopy (CARS) がある。もし周波数の役割が入れ替わると、Coherent Stokes Raman Spectroscopy (CSRS) となる。信号強度が最大となるためには波数ベクトルの整合条件 を満たす必要がある。この 2 つの方法では、スペクトル的にも空間的にも信号光 (誘 導 Raman 散乱光) を入射光から分離することができるので、バックグラウンド雑音を 除去できる。
1.2.4
Coherent Anti-Stokes Raman Spectroscopy
について
Coherent Anti-Stokes Raman Spectroscopy(CARS) に関する実験は 1965 年に P. D. Maker と R. W. Terhune の二人によって初めて報告された [39]。この実験ではルビー パルスレーザーを励起光源として、物質の 3 次の非線形応答を調べる手法として用い られた。彼らは、周波数ω のパルスレーザーを、ωνの Raman シフトが起こる物質に 照射し、ω − ωνの散乱光を得た。それらを測定試料へ同時に照射することで CARS 信 号 (ω + ων) を観察した。この信号は、測定試料の Raman シフト量がωνに一致すると
きに著しく強度が増加することを実証した。通常の Raman 散乱では、散乱強度が弱 く測定が難しいという難点があったが、レーザーの登場により信号レベルが飛躍的に 強くなり、容易に Raman 散乱と同じ情報が得られるようになった。大きく改善された 理由は、2 つの異なる周波数のレーザーがその差の周波数で分子を強制振動させるこ とができる点である。これにより、コヒーレントな Raman 信号振幅を大幅に増強し て発生することが可能になった。その約 10 年後の 1974 年に R. F. Begley らによって、 CARS によるガス、固体や液体の非線形特性が調べられた [40, 41]。ガスのような気 体の場合、分子の回転状態も知ることができる。CARS では、ヨウ素や水素の回転ダ イナミクスの報告も数多くある [42, 43]。その他に CARS 法で特筆すべきなのは、生 体物質の構造を調査する手法として注目されるようになったことである。CARS 法に より生体物質中の化学構造の研究や分子振動の研究が盛んに行われ、見事な研究成果 につながっている [44]。また、この分子の振動モードの違いを利用したイメージング 装置の開発も盛んに行われている。例えば、サンプルとして生きた細胞にレーザーを 照射し、イメージングを行う研究がなされている。サンプルに存在する振動モードを CARS により誘起し、フィルターなどで特定の狙った波長を取り出し CCD カメラで 検出する手法がとられている [45, 46]。こういった細胞のイメージングは通常のレー ザー Raman でも行われていたが、CARS の場合、微弱な Raman 信号を増幅して検出 ができ、サンプルから指向性のよい信号光が発生するなど、従来の手法では得られな い利点がいくつかあり、イメージングの制度を向上することに貢献している。さらに、 CARS の場合、照射するパルスを制御することにより時間発展を観測することも可能 となる。実際に、図 1.9 に示すように、細胞の時間変化を観測した実験結果も報告さ れている [47]。 図 1.9: 出芽酵母細胞の時間分解 CARS イメージ [47]
1.2. 分子振動ダイナミクスと CARS 法 19
1.2.5
時間分解
CARS
法
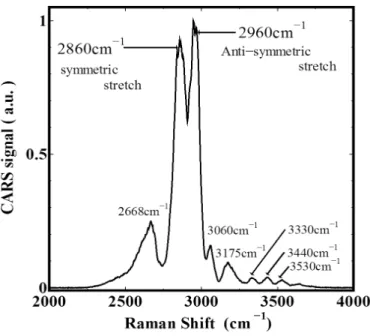
分子の振動や緩和といったダイナミクスはフェムト秒からピコ秒の時間領域で生じ る現象である。この超高速な時間領域で生じる分子内のエネルギー移動や分子内相互 作用といった現象を解明するためにレーザー分光法を用いた研究が盛んにおこなわれ るようになった。超高速時間分解 CARS 法は超短レーザーパルスの開発とともに発 展してきたもので、1970 年代後半から現在に至るまでよく用いられている手法であ る [48–58]。CARS 法の時間分解能はフェムト秒パルスレーザーを利用することによ り、10 fs 以下まで向上してきている [59–61]。そのため、フェムト秒レーザーを用い ることで超高速な分子振動のダイナミクスを観測することができる。たとえば、メタ ノール水溶液の分子振動ダイナミクスに関する研究報告がある [62]。この研究ではメ タノールに含まれている CH 基のコヒーレント緩和時間をメタノールと水の濃度比を 変化させて測定を行っている。図 1.10、1.11 はコヒーレント振動緩和測定結果である。 CARS 信号に周期的な強度変化 (beat) がみられる。これは、振動エネルギーの近い振 動モードがある場合に起こる現象として知られており、今回の場合 CH3基の対称伸縮 振動と反対称伸縮振動に由来する CARS 信号の干渉によって beat がみられる。この ビートの周期から関係する振動モードの特定がおこなえる。また、図 1.11 から明らか なように、メタノール濃度によってコヒーレント緩和時間が大きく変化していること が分かる。この研究結果はメタノールと水との分子間相互作用の変化によるものでは ないかと予測されている。このようにコヒーレント分子振動緩和から、分子のおかれ る状況により分子振動ダイナミクスがどのように変化するのか予測する研究が行われ ている。 その他に、結晶のポリジアセチレンを低温に冷やしたときの分子振動ダイナミクス に関する報告もある [63]。図 1.12 はコヒーレント分子振動緩和の様子を 2 次元と 3 次 元でプロットしたものである。この結果にも CARS 信号に周期的な強度変化 (beat) が みられる。ポリジアセチレンでは C-C 結合の曲げ振動と伸縮振動や C-C 結合と C=C 結合の伸縮振動がお互いに結合し振動しているため beat が見られると報告されてい る。その他に C-C 結合と C=C 結合の振動による信号が予想よりも強く観測されてい るため、図 1.13 のようにポリジアセチレン中の炭素鎖の結合状態が変化していると 予測している。このように、分子振動の動的振舞を正確にとらえる方法として、時間 分解 CARS 法は用いられている。こういった手法を用いて分子振動の本質を解明し ようとする研究が行われている一方で、それを踏まえフェムト秒パルスを用いて分子 振動を制御し、これまでにない新しい化学反応や制御を目指す研究もおこなわれてい る [64]。これは複数のパルスを用いてコヒーレントな分子振動状態を誘起し、反応に 有利な状態を作り出し、一つの反応経路を選択的に増進する、いわゆるコヒーレント制御を実現した研究である。時間分解 CARS 法では、特定の分子振動を選択的に励起 して化学反応を制御することなども可能である。時間分解 CARS 法はフェムト秒時間 領域で分子振動を制御する FemtoChemistry と同種の方法であり、分子振動のダイナ ミクスを知り、制御する手法として有力な手段となている。 本研究においても、高分子の振動ダイナミクスを研究する方法として時間分解 CARS 法を用いている。そこでは、CARS 信号光を誘起するために 3 つのフェムト秒パルス を用いるが、それ等のパルス間の時間遅延の制御、試料中における 3 つのパルスの空 間的重なりの確認など技術的に難しい調整を行う必要がある。そのような困難さのた め、時間分解 CARS 法を採用した研究はそれほど多くない。第 4 章で示す高分子にお ける時間分解 CARS 法による結果は新奇で斬新な結果である。特に、常温において高 分子の CH2基の分子振動の緩和が時間領域で測定されたことは、特筆に値すると考え ている。
1.2. 分子振動ダイナミクスと CARS 法 21
図 1.10: メタノールのコヒーレント緩和時間測定結果 (2 次元マッピング)
図 1.12: ポリジアセチレンのコヒーレント振動緩和測定結果
1.3. 研究目的 23
1.3
研究目的
前節までに述べたように分子振動ダイナミクスに関する研究は広汎に行われてきて いる。本研究では物質の分子振動ダイナミクスの研究を進めるという方針の下で、時 間分解 CARS 分光の対象とされていない高分子の基本骨格を構成する CH 基や OH 基 に着目し、分子振動のコヒーレント分子振動緩和の測定を目的とした。高分子におけ るコヒーレント分子振動緩和が測定されていないのは高分子がアモルファス固体であ るため液体に比べて分子振動の理解が難しく、また結晶のように純然たる Phonon モー ドもないと考えられたためである。そのため、時間分解 CARS を用いてコヒーレント 分子振動緩和を測定することは、高分子中の分子振動ダイナミクスの理解につながる と期待した。 今回、高分子の基本骨格に注目した理由は以下のようなものである。タンパク質な どの生体高分子は水素結合やファンデルワールス力、疎水性相互作用など比較的弱い 相互作用を媒介として、機能発現に必要な固有構造を形成あるいは変化させており、 また生体分子は室温における熱揺らぎ程度の大きさのエネルギーを巧みに利用して機 能発現あるいは構造変化を行っていると考えられている。これらは主に CH 基や OH 基などの官能基を含んだ分子であり、生体高分子の機能発現に主要な役割を果たして いると考えられる。生体高分子中での分子振動ダイナミクスを調べる前段として、CH 基や OH 基を含むより単純な系である高分子に対象を定めた。 時間分解 CARS 法では物質中の分子をコヒーレントに振動励起するが、CARS 信 号強度はこのコヒーレント分子振動状態を正確に反映する。コヒーレント分子振動状 態は、Phonon はもちろん高分子中の相互作用 (分子・分子相互作用、分子・Two level system 相互作用など) に敏感に影響を受けてコヒーレンスが壊れると予測される。我々 は高分子鎖中のコヒーレント分子振動時間を求め、物質中のダイナミクスについて の詳しい知見を得たいと考えた。今回用いた試料は分子の構造が比較的簡単な高分子 Polyvinyl alcohol、Polymethyl methacrylate、Polyethylene である。これら高分子のコ ヒーレント分子振動緩和に関する報告はほとんどないため、基本的な振動モード (伸 縮振動の対称振動、反対称振動など) のコヒーレント分子振動状態のダイナミクスを 調べた。具体的には、分子構造の違いによるコヒーレント緩和への影響、Phonon に よるコヒーレント緩和への影響である。この結果から、コヒーレント分子振動緩和の メカニズム、振動エネルギー移動のメカニズムや相互作用のタイプの予測が可能とな る。そして、我々の高分子におけるダイナミクスの基礎的な研究の成果が、より構造 が複雑で分子量も大きいたんぱく質などの生体高分子の特性の理解へとつながってい くと期待している。25
参考文献
[1] C. V. Raman and K. S. Krishnan, Nature, 121, 501 (1928). [2] G. Landsberg and L. Mandelstam, Naturwiss., 16, 557 (1928). [3] C. V. Raman, Indian J. Phys., 2, 387 (1928).
[4] C. V. Raman, Nature, 121, 619 (1928). [5] A. Smekal, Naturewiss., 11, 873 (1923).
[6] 島内武彦, 水島三一郎 共著 ”赤外線吸収とラマン効果 ”, (共立出版株式会社, 1976).
[7] 水島三一郎,“ Raman effect ”, Handbuch der Physik XXVI, Springer, Berlin (1957). [8] J. H. Hibben,“The Raman Effect ant its Chemical Applications”,Reinhold Publishing
Corporation, New York (1939).
[9] H. Pajenkamp, Fortschr. Chem. Forsch., 1, 417 (1950). [10] O. Widlich and F. Siebert, Appl. Spectrosc., 47, 1394 (1993).
[11] J. P. Russell, R. Loudon, Proceedings of the Physical Society, 85, 321, pp. 1029 (1965).
[12] キャリー 著, 伊藤 紘一, 尾崎 幸洋 訳,“ ラマン分光学 ”(共立出版株式会社 1984). [13] A. D. Buckingham, Transactions of the Faraday Society, 56, 753 (1960).
[14] G. Eckhardt, D. P. Bortfeld, M. Geller, Appl. Phys. Lett. 3, 137, (1960). [15] V. H. Segtnan, S. Sasic, T. Isaksson, Y. Ozaki, Anal. Chem., 73, 3153 (2001). [16] Q. Li, G. Wu and Z. Yu, J. Am. Chem. Soc., 128, 1438 (2006).
[18] 宇野 英満, 築部 浩 編“ はじめての有機スペクトル解析 IR, NMR, MS データを 読む ”(丸善株式会社, 2008).
[19] 中西 香爾, P. H. ソロモン, 古館 信生 共著“ 赤外線吸収スペクトル ‐定性と演習 ‐ 改訂版 (南江堂 1987).
[20] E. Pretsch, T. Clerc, J. Seibl, W. Simon 著, 中西 香爾, 梶原 正宏, 堤 健太郎 訳“ 有 機化合物スペクトルデータ集 ”(講談社サイエンティフィック 1982).
[21] Y. Nomura, Y. T. Wang, T. Kozai, H. Shirai, A. Yabushita, C. W. Luo, S. Nakanishi, T. Fuji, Optics Express, 21, 18249 (2013).
[22] T. Fuji, T. Suzuki, Opt. Lett. 32, 3330 (2007).
[23] Y. Nomura, H. Shirai, K. Ishii, N. Tsurumachi, A. A. Voronin, A. M. Zheltikov, T. Fuji, Opt. Express, 20, 24741―24747 (2012).
[24] S. Mohan and A. R. Prbakaran, Asian J. Chem., 1, 162 (1989).
[25] Du Xin, He Xing, Liu Yu-Qiang, Wang Ying-Hui, Yang-Qiang, Chin. Phys. B, 21, 34210 (2012).
[26] D. E. Spence, P. N. Kean, W. Sibbett, Opt. Lett., 16, 42 (1991). [27] 三沢一彦, 竹内 佐年, 小林 孝嘉, 日本物理学会誌, 50, 463 (1995).
[28] P. Pagsberg, R. Wilbrandt, K. B. Hansen, K. V. Weisberg, Chem. Phys. Lett., 39, 538 (1976).
[29] J. Herrmann, B. Wilhelmi 著, 小林孝嘉訳, ”超短光パルスレーザー”, 共立出版 (1991).
[30] K. Iwata, H. Hamaguchi, J. Phys. Chem., 101, 632 (1997).
[31] T. Yuzawa, C. Kato, M. W. George, H. Hamaguchi, Appl. Spectrosc., 48, 684 (1994). [32] 濱口宏夫, 生物物理, 214, 259 (1997).
[33] P. Hama, M. Lim, R. M. Hochstrasser, J. Phys. Chem. B, 102, 6123 (1998). [34] J. S. Baskin, P. M. Felker, A. H. Zewail, J. Chem. Phys., 84, 4708 (1986).
[35] K. Yamashita, K. Morokuma, F. LeQuere, C. Leforestier, Chem. Phys. Lett., 191, 515 (1992).
1.3. 研究目的 27 [36] C. Leforestier, F. LeQuere, K. Yamashita, K. Morokuma, J. Chem. Phys., 101, 3806
(1994).
[37] A. Mokhtari, P. Cong, J. L. Herek, A. H. Zewail, Nature, 348, 225 (1990). [38] E. J. Woodbury, W. K. Ng, Proc. IRE, 50, 2347 (1962).
[39] P. D. Maker, R. W. Terhune, Phys. Rev., 137, A801 (1965).
[40] R. F. Begley, A. B. Harvey, R. L. Byer, Appl. Phys. Lett., 25, 387 (1974). [41] W. B. Roh, P. W. Schreiber, J. P. E. Taran, Appl. Phys. Lett., 29, 74 (1976). [42] M. Schmitt, G. Knopp, A.Materny, W. Kiefer, Chem. Phys. Lett., 270, 9 (1997). [43] T. Lang, K. -L. Kompa, M. Motzkus, Chem. Phys. Lett., 310, 65 (1999).
[44] L. A. Carreira, T. C. Maguire, T. B. Malloy Jr., J. Chem. Phys. 66, 2621 (1976). [45] N. Dudovich, D. Oron, Y. Silberberg, Nature, 418, 512 (2002).
[46] M. M¨uller J. M. Schins, J. Phys. Chem. B, 106, 3715 (2002). [47] Y-S. Huang, et. al., Biochemistory, 44, 10009 (2005).
[48] W. M. Tolles, J. W. Nibler, J. R. McDonald, A. B. Harvey, Appl. Spectrosc., 31, 253 (1977).
[49] S. A. J. Druet, B. Attal, T. K. Gustafson,J. P. Taran, Phys. Rev., 18, 1529 (1978). [50] L. A. Rahn, R. L. Farrow, M. L. Koszykowski, P. L. Mattern, Phys. Rev. Lett., 45, 620
(1980).
[51] R. Leonhardt, W. Holzapfel, W. Zinth, W. Kaiser, Chem. Phys. Lett., 133 , 373 (1987). [52] T. Joo, A. C. Albrecht, J. Chem. Phys., 99 , 3244. (1993).
[53] T. Lang, K.-L. Kompa, M. Motzkus, Chem. Phys. Lett., 310 , 65 (1999).
[54] M. Heid, S. Schlucker, U. Schmitt, T. Chen, R. Schweitzer-Stenner, V. Engel, W. Kiefer, J. Raman Spectrosc., 32 , 771 (2001).
[55] Y. R. Shen,“ The Principles of Nonlinear Optics ”, A Wiley-Interscience Publication, (2002).
[56] T. Siebert, M. Schmitt, S. Grafe, V. Engel, J. Raman Spectrosc. 37, 397 (2006). [57] Kozai, S. Yamashita, K. Hirochi, H. Miyagawa, N. Tsurumachi, S. Koshiba, S.
Nakan-ishi, H. Itoh, Chem. Phys. Lett., 553 , 26 (2012).
[58] Y. Zhao, S. Zhang, B. Zhou, Z. Dong, D. Chen, Z. Zhang, Y. Xia, J. Raman Spectrosc.,
45, 826 (2014).
[59] R. L. Fork, C. H. Brito Cruz, P. C. Becker, C. V. Shank, Opt. Lett., 12, 483 (1987). [60] M. Spshenichnikov. W. P. de Boeji, D. A. Wiersma, Opt. Lett., 19, 572 (1994). [61] M. Nisoli, S. de Silverstri, O. Svelto, Appl. Phys. Lett., 68, 2793 (1996).
[62] D. Pestov, M. Zhi, Z. -E. Sariyanni, N. G. Kalugin, A. Kolomenskii, R. Murawski, Y. V. Rostovtsev, V. A. Sautenkov, A. V. Sokolov, M. O. Scully, J. Raman Spectrosc., 37, 392 (2006).
[63] A. Vierheiling, T. Chen, P. Waltner, W. Kiefer, A. Materny, A. H. Zewail, Chem. Phys. Lett., 312, 349 (1999).
[64] A. Assion, T. Baumert, M. Bergt, T. Brixner, B.Kiefer, V. Sayfried, M. Strehle, G. Gerber, Science, 282, 919 (1998).
29
第
2
章
Raman
分光とその理論的背景
この章では、本研究で用いた Raman 分光と Coherent Anti-Stokes Raman Spectroscopy の理論的背景について説明する。Coherent Anti-Stokes Raman Spectroscopy はレーザー の出現と発展に伴って観測が可能になった非線形光学効果を用いた代表的な分光法で あり、四光波混合法と呼ばれる非線形分光法の範疇に入いる。非線形光学効果の理論 的取り扱いは古典的な理論をベースにする場合も多いが、分光の対象とする物質系は 基本的には量子力学系であるため、量子力学的取り扱いをする必要がある。この章で は測定信号の物理的理解を深めることを目的に、本研究で測定される CARS 信号の量 子力学的理論による表式を得ることである。
2.1
Raman
散乱の古典的モデル
Raman 散乱とは、入射光と分子との相互作用の結果、入射光と異なる振動数の光が 散乱される現象である。古典的な描像では、光波は電磁場からなる進行波であるが、 電場成分のみが電子雲と原子核からなる分子と相互作用して、ラマン散乱を引き起こ す。分子の大きさを 1nm 程度とすると、その大きさは光の波長に比べて十分に小さ い。したがって、電場の大きさは分子のどの部分においても、常に同じであると考え てよい。すなわち電場は分子中のすべての電子に同じ力を及ぼす。そして正電荷を帯 びた原子核の周りに存在する電子を、その平均的位置から変位させる。この電子の変 異によって、分極 P が分子中に生じる。このことは、Raman 散乱過程において、まず 最初に重要ことである。分極は、十分良い近似で電場 E に比例すると考えてよい。 P= ϵ0χE(t) = αE (2.1) ここで比例定数α は分子の電子分極率と呼ばれる。一般にベクトル P の方向はベク トル E の方向とは異なっているので、α は簡単なスカラー量ではない。P を定義する 三つの成分、すなわち Px、Py、Pzの大きさは電場 E の大きさと次のような関係が ある。Px = αxxEx+ αxyEy+ αxzEz (2.2) Py = αyxEx+ αyyEy+ αyzEz (2.3) Pz = αzxEx+ αzyEy+ αzzEz (2.4) 上の式から E の三つの成分すべてが P に寄与する。9 個の係数αi jは、分極率α テ ンソルの成分である。ある固定された分子に対して振動数ν0の単色光が照射されたと すると、z 方向に偏光した入射光の電場 Ezは時間の関数として次のようになる。 Ez(t) = E cos(2πν0t) (2.5) ここで E は Ezの最大値であり、t は任意の開始時間からの時間 (秒) である。よって P の z 成分は次のようになる。 Pz(t) =αzzE cos(2πν0t) (2.6) Pzはαzzと Ezによって決まるので、分子の性質はαを通して Pzに反映される。電 子は電場の影響によって変位するが、その変位しやすさは電子がどの程度、原子核に 結びつけられているかにより決まる。一方、電子は原子核間距離が変化すると、その 影響を必然的に受ける。したがってα は分子が振動するとき、時間とともに変化する。 ここで、原子核 m1、m2の平衡位置からの変位の差をあらわす座標を q(t)= r2− r1で 表すと、時間依存性を、次のようにあらわすことができる。 q(t) = A cos(2πνvibt+ ϕ) (2.7) ここで、νvibは分子振動の振動数をヘルツで表したものである。q は時間 t における 原子核間距離の平衡状態からのずれであり、q(t) = r2− r1=∆r(t) と定義する。また、 原子間振動の振幅 A は原子核間距離∆rmaxとなり、これは原子核間距離の平衡値から のずれの最大値である。したがって、式 (2.7) は書き換えると次のようになる。
2.1. Raman 散乱の古典的モデル 31 さらに、時間 t の出発点を、光波の式 (2.5) における時間軸と等しくすることで位相 ϕ を 0 とすることができる。二原子分子の分極率が ∆r(t) によって変化し、その依存性 が∆r(t) に比例するとすれば、α の αzz成分は次のようになる。 αzz(t) =α(equivvib .)+ dαzz dr ∆r(t) (2.9) = α(equiv.) vib + dαzz
dr ∆rmaxcos( 2πνvibt )
定数α(equiv.) vib は平衡位置ある分子の分極率成分である。(dαzz)/ dr は r の変化に伴う 分極率の変動率を表している。αzzを表す式 (2.9) を式 (2.6) に代入すると、次のように P がα と E の時間的揺らぎに依存する様子を示すことができる。 Pz(t) =α (equiv.) vib E cos( 2πν0t )+ dαzz
dr rmaxE cos(2πν0t) cos(2πνvibt) (2.10)
ここで、cosθ cos ϕ=1 2[cos(θ+ ϕ ) + cos( θ − ϕ )] を用いると Pz(t) =α (equiv.) vib E cos( 2πν0t ) +1 2 dαzz
dr ∆rmaxE cos{2π( ν0+ νvib)}t
+1 2
dαzz
dr ∆rmaxE cos{2π( ν0− νvib)}t (2.11)
式 (2.11) は光波が振動する二原子分子と相互作用するとき、分極が振動数の異なる 三つの成分からなることを示している。式 (2.11) の第一項は入射光の振動数と同じ振 動数で振動する成分を表す。この成分の大きさはα(equiv.) vib と E によって決まる。した がって、入射光と同じ振動数をもつ光が放射される。この場合、散乱光は入射光の方 向とは異なった方向に観測される。これが Rayleigh 散乱である。第二項によって生じ る散乱光は振動数がν0+ νvibであり Anti-Stokes Raman 散乱、第三項はν0-νvibであり
Stokes Raman 散乱として知られている。この第二項と第三項から生じる振動数 (ν0+
νvib) と (ν0-νvib) の Raman 散乱光を解析することによって分子内振動 (振動数νvib ) を
2.2
Raman
散乱の量子論的描像
前節では古典力学モデルにより散乱過程を記述したが、量子力学的取り扱いはこの 古典的モデルとは異なる。量子力学では、分子のエネルギー準位は量子化されている と考えられる。まず、気体の二原子分子の分子エネルギー Emolは、十分に良い近似
で、次のような調和振動子モデルで書くことができる。
Emol= Eelec+ Evib (2.12)
Emol:全エネルギー、Eelec:電子状態のエネルギー、Evib :振動の状態エネルギーを表
す。ここでは、分子の回転あるいは並進のエネルギーの寄与は無視する。電子遷移エ ネルギーは、振動遷移エネルギーに比べてはるかに大きなエネルギーを必要とする。 すなわち、前者の場合は、10000∼50000 cm−1のエネルギーを必要とするが、後者の 場合は 10∼4000 cm−1のエネルギーで十分である。電子基底状態と電子励起状態の間 には大きなエネルギーギャップがあるが、振動エネルギー準位間の間隔はそれに比べ て小さい。その二原子分子の振動エネルギー Evibは調和振動子モデルが十分によい近 似であるため、つぎのように書くことができる。 Evib= ( n +1 2 ) hνvib (n = 0,1,2…) (2.13) ここで振動量子数 n は整数値のみをとる。したがって電子基底状態において振動 エネルギー準位は、等間隔 hνvibで並んでいる。量子力学モデルは光散乱を二光子過 程として取り扱う。二光子過程ではじめにフォトンと分子との衝突が起こり、その結 果、極端に短い時間の間、分子は高いエネルギー状態に仮想的に励起される。きわめ て短い時間の後、第 2 の過程としてフォトンの放出がおこる。図 2.1 に示すように、 Rayleigh 散乱の場合、上矢印と下矢印の長さは等しい。すなわち、向きを、別にすれ ば二つの遷移は同じエネルギーをもつ。したがって、Rayleigh 散乱過程では散乱され るフォトンの振動数は変化しない。次に、ラマン散乱過程では下向き矢印が最初の振 動エネルギー準位よりも高い振動エネルギー準位に到達する場合には、Stokes 過程が 起こる。この場合、放出されるフォトンは式 (2.6) の第三項に示した振動数ν0−νvibを 持つ。Stokes 過程とは逆に、下向きの矢印が最初の振動エネルギー準位よりも低い振 動エネルギー準位に到達する場合、Anti-Stokes 過程によって放出されるフォトンの振 動数はν0+νvibとなる。この振動数は式 (2.11) の第二項と等しい。どちらの過程も全エ ネルギーは保存されるが Stokes 散乱の場合には分子は量子化されたエネルギー hνvibを
2.2. Raman 散乱の量子論的描像 33 得ることになり (振動励起状態に遷移し)、Anti-Stokes 散乱の場合にはその逆となる。 (a) Stokes Scattering (b) Rayleigh Scattering (c) Anti-Stokes Scattering 図 2.1: 光散乱における遷移 このように古典論によるモデルと量子力学によるモデルはお互いに矛盾することは ない。しかし、古典論モデルでは、Stokes 遷移と Anti-Stokes 遷移の相違について何ら 説明することができない。一方、量子力学モデルでは 2 つのラマン散乱光の間の強度 の違いを説明することができる。まず、Anti-Stokes 遷移が起こるためには分子は電子 基底状態において高い振動状態にいる必要がある。高い振動エネルギー準位に存在す る分子の数はごくわずかである。電子基底状態において高い振動エネルギー状態の分 子数を N1、低いエネルギー状態の分子数を N0とするとその比は熱平衡状態において 次のようになる。 N1 N0 = exp ( −hνvib kT ) (2.14) ここで h はプランク定数、T は絶対温度、k はボルツマン定数である。このような、 Stokes 光と Anti-Stokes 光の強度比を利用すると、物質の温度を測定することが可能 であり、実際に応用されている [6]。
2.3
非線形光学効果について
光と物質は、光電場によって誘起される分極を介して相互作用する。通常の光学で は、分極は光電場に比例すると仮定して議論される。しかし、レーザーを用いた場合、 光電場が非常に強くなるため、分極は光電場に比例せず非線形性を持つようになる。 そして光と物質との相互作用にも非線形性を考えなければ説明することができない現 象が発現するようになる。このような現象を扱う分野を非線形光学と呼ぶ [2–5]。物 質に光 (電場) を照射した場合、分極は電場や磁場の作用によって電子や原子核のよう な電荷粒子が分極するモデルを用いる (図 2.2)。 電子 原子核 E ==== 0000 : 電場なし 電子 原子核 E ≠ 00 : 00: : : 電場あり 図 2.2: 分子の分極 まず電場がない場合、原子核と電子の重心はずれることなく一致する。一方、電場 がある場合、電子と原子核は電場の影響を受けるので、重心がずれる。特に、電子は 質量が原子核に比べてとても軽いので図 2.2 のように大きくずれることになる。 この状態での分極を計算すると、 P(t) = (−e)r = ϵ0χE(t) (2.15) と表すことができる。P(t) は分極、ϵ0は真空の誘電率、χ は分極率、E(t) は電場であ る。しかし、レーザー光を用いることで分極と電場が比例関係におさまらない場合を 生じさせることができる。この場合、線形応答の限界を超えてしまう。このように非 線形性を無視できないとき、 分極 P(t) は非線形分極を含めて、光電場のべき級数展 開で表される。この展開のうち電場の 2 次の項に起因する現象を 2 次の非線形光学効 果、3 次の項のものを 3 次の非線形光学効果と呼ぶ。P(t)= Plin+ Pnon= ϵ0χE(t) + ϵ0χ(2)E : E(t)+ ϵ0χ(3)E : E : E(t)+ · · · (2.16)
2.3. 非線形光学効果について 35 の非線形光学効果は、光学の分野では図 2.3 で示すように、第 2 高調波発生 (Second harmonic generation)、和周波発生 (Sum frequency generation)、差周波発生 (Difference frequency generation) として知られており、波長変換の手法としてよく用いられてい る。これらはすべて 2 次の非線形光学効果である。また、4 光波混合、縮退 4 光波混 合、誘導散乱 (Coherent Raman 散乱) などは 3 次の非線形光学効果として知られてい る。本研究で用いる CARS は 3 次の非線形光学効果の一種であり、図 2.4 で示した 3 つの励起光ω1、ω2、ω3によってω4の光が放出される現象である。これらの励起光が すべて同一の場合には、放出される光の周波数も励起光と同じになる。この場合には 縮退 4 光波混合と呼ばれる。
第 第 第 第2高調波発生高調波発生高調波発生高調波発生 和周波発生和周波発生和周波発生和周波発生 差周波発生差周波発生差周波発生差周波発生
ħ
ω
ħ
ω
2ħ
ω
ħ
ω
2ħ
ω
1ħ
ω
3ħ
ω
3ħ
ω
2ħ
ω
1 図 2.3: 2次の非線形光学効果ħ
ω
1ħ
ω
2ħ
ω
3ħ
ω
4 図 2.4: 3次の非線形光学効果 (4光波混合、CARS)2.4. Coheretn Anti-Stokes Raman Spectroscopy の量子力学的理論 37
2.4
Coheretn Anti-Stokes Raman Spectroscopy
の量子力
学的理論
2.4.1
時間に依存する摂動論
時間変動する摂動(たとえば、電磁波やパルス的電場など)が入ったときに、考え ている非摂動系がどのようになるのかは重要である。実際に、時間変動する摂動が入 ると、非摂動系が起こす現象として、電磁波の吸収や放出などが起こる。また、分散 などの現象も生じる。このとき、系では非摂動の状態からの変化が起きている。この ような時間変動する摂動が入ったときの系の振る舞いを考えるには、時間に依存する Schr¨odinger 方程式を解く必要がある。 時間に依存する Schr¨odinger 方程式は次のように表される。 iℏ∂Ψ(r, t) ∂t = ˆHΨ(r, t) (2.17) ここで、Ψ(r, t) は波動関数、 ˆH = ˆH0+ ˆH′であり、 ˆH0、ˆH′はそれぞれ無摂動のハミル トニアン、摂動との相互作用ハミルトニアンである。後者が摂動と考えている系の間 の相互作用を表している。しかし、摂動がある場合、これを解析的に解くことは非常 に難しいため、摂動展開を用いる摂動論を援用する必要がある。 そこで、まずはじめに無摂動系における時間に依存する Schr¨odinger 方程式の解に ついて考え、次に摂動がある場合の時間に依存する Schr¨odinger 方程式の解を求める 方法を示す。2.4.2
無摂動系における時間に依存する
Schr¨odinger
方程式
無摂動系ではハミルトニアン演算子は時間変化を示さない。すなわち、露わには時 間変数を含まない。 ˆ H0 = ˆH0(r, ˆP) (2.18) また、時間に依存しない Schr¨odinger 方程式は解がわかっており、その結果として 物質の定常状態についての情報はわかっていると仮定する。すなわち、時間的に依存 しない Schr¨odinger 方程式は次のようになる。 ˆ H0ψn(r)= Enψn(r) (2.19)ここで、Enはエネルギー値、ψn(r) は固有関数 である。 さらに、ψn(r) は正規完全直交系をなすので、以下の関係を満足する。 < ψm(r)|ψn(r)> = ∫ ψ∗ m(r)ψn(r)dr = δnm (2.20) この系に摂動が入らない場合の時間に依存する Schr¨odinger 方程式を解いてみる。 iℏ∂Ψ(r, t) ∂t = ˆH0Ψ(r, t) (2.21) となる。 波動関数Ψ(r, t) を Ψ(r, t) = ψ(r)T(t) と仮定し、式 (2.21) に代入すると、 iℏψ(r, t)∂T(t) ∂t = ˆH0ψ(r)T(t) = T(t) ˆH0ψ(r) (2.22) となる。 両辺をΨ(r, t) = ψ(r)T(t) で割ると、 iℏ ∂T(t) ∂t T (t) = ˆ H0ψ(r) ψ(r) = E (2.23) 式 (2.23) では、左辺は t のみの関数、右辺は r のみの関数なので、定数でなければ ならない。 ゆえに、 iℏ∂T(t) ∂t = ET(t) (2.24) ˆ H0ψ(r) = Eψ(r) (2.25) 式 (2.24) は時間に依存する関数 T に対する方程式である。また、式 (2.25) は時間に 依存しない Schr¨odinger 方程式そのものであるので、解は求まっていて、ψ(r) = ψn(r), E = Enである。したがって式 (2.24) の解は、
2.4. Coheretn Anti-Stokes Raman Spectroscopy の量子力学的理論 39 iℏ∂T(t) ∂t = ET(t) ∂T(t) T (t) = E iℏ∂ ∫ dT (t) T (t) = E iℏ ∫ dt log T (t) = (E iℏ ) t+ C ∴ T(t) = A exp[E iℏt ] (2.26) となる。 以上より、波動関数Ψn(r, t) は、 Ψn(r, t) = ψn(r) exp [E n iℏt ] (2.27) となる。 この関数系Ψn(r, t) も正規完全直交系になっている。 < Ψm(r)|Ψm(r)> = ∫ Ψ∗ m(r)Ψn(r)dr = ∫ exp [ −Em iℏ ] tψ∗m(r) exp [E n iℏ ] tψn(r)dr = exp[En− Em iℏ t ] ∫ ψ∗ m(r)ψn(r)dr (2.28) = exp[En− Em iℏ t ] δnm = δnm