平成 年 変異型 イツ ト・ 病 牛海綿状脳症 BSE いわゆ 狂牛病 関連を指摘 た英国政府諮問委員会声明 端を発 たいわゆ 狂牛病問題 発生 始ま ト乾燥硬膜移植 由来 考え イツ ト・ 病 発生 そ 平成 13年 月 牛海綿状脳症 BSE を発症 た牛 わ 国 おい 発見さ
たこ 狂牛病問題 再燃 等 わ 国 おい イツ ト・ 病 ン病 対 関心 高ま い
イツ ト・ 病 異常 ン 呼 タン 質 伝達さ
ト ン病 あ そ 本態解明を目指 研究 進歩 著 い あ
わ 国 おい 昭和51 年 旧厚生省 特定疾患調査研究事業 おい ー イ 感染 難病発症機序 関 研究班 設置さ 以来 現在 遅発性 イ 感 染調査研究班 至 ま ン病等 いわゆ 遅発性 イ 感染 原因 考え
いた疾患 関 調査研究 行わ た イツ ト・ 病 研究
い 着実 推進 図 い
こ 研究 成果を医療 現場 還元 イツ ト・ 病 患者 対 適正 医療を提供 た 平成 年 月 イツ ト・ 病診療 ニ
作成さ 医療機関等 活用さ た こ あ 近年 イツ ト・ 病を た ン病解明 飛躍的 進歩を踏まえ 今般 そ 内容を 最新 知見 基 い 改訂を行うこ た
本改訂 ニ イツ ト・ 病を た ン病 治療
検査 感染因子 滅菌法 感染防御等 い 現在把握 得 最大限 情報を基 構成さ い
こ ニ イツ ト・ 病等 対 診断・治療 向上や医療機 関 おけ 院内感染 伝達 防止策 徹底 さ 患者 適正 資さ こ を期 待
平成 14年 月24日
目
次
第 章 プ オン病 つい ………9
概 念………9
最近のトピック ………10 第 章 プ オン病の分類 ………13 孤発性プ オン病 ………13 家族性プ オン病 ………14 感染性プ オン病 ………16 第 章 プ オン病の臨床 病理 ………17 孤発性プ オン病 ………17 家族性プ オン病 ………21 感染性プ オン病 ………28
第 章 プ オン病の治療 ………43
第 章 プ オン病の検査 ………44
臨床検査 ………44
特殊検査 (異常プ オン蛋白の検出 ………45
第 章 プ オン病感染因子の滅菌法 ………48
完全 滅菌法 ………48
不完全 が 有効 処理 (感染性を0.1%以下 するもの ………48
無効 従来の滅菌法 ………49
滅菌物別の具体例 ………49
略 語
プ オン病 関
CJD, Creutzfeldt-Jakob disease イツ ト・ 病
vCJD, variant form of Creutzfeldt-Jakob disease ント型CJD
GSS, Gerstmann-Sträussler-Scheinker disease ト ン・ ト イ ー・ イン ー病
FFI, fatal familial insomnia 致死性家族性不眠症
BSE, bovine spongiform encephalopathy 牛海綿状脳症
プ オン蛋白 関
PrP, prion protein ン蛋白
PrPSc, scrapie form of prion protein 異常型 ン蛋白
PrPC, normal cellular form of prion protein 正常型 ン蛋白
アミノ酸の略号 Amino acid symbols
Amino acid Three-letter symbol One-letter symbol Japanese
alanine Ala A ニン
arginine Arg R ニン
目
次
第 章 プ オン病の感染防御 ………50
臓器別感染性 つい ………50
感染 ート 関 ………51 患者の看護 感染防止策 ………52
手術時の感染防御の基本的注意事項 ………52
検査時の感染防御の基本的注意事項 ………53
剖検時・病理標本作製時の感染防御の基本的注意事項 ………53
家庭内 の介護 ………55
死後の遺体の感染防御 関 ………55
感染 関わる ま ま 要因 (補足 ………55
第 章 プ オン病患者の看護 介護 ケア 医療福祉 ………57
看 護 ………57
看護 感染防止策 ………57
病気の説明 家族の指導 告知 ………58
胃瘻の増設お び外科治療 ………58
歯科治療 外科治療 ………58
在宅療養 介護施設への移行 ………58
守秘義務 ………58
医療福祉 ………59
第 章 プ オン病のサーベイ ンス ………60
資 料 ………63
aspartic acid Asp D ン酸
cysteine Cys C テイン
glutamine Gln Q タ ン
glutamic acid Glu E タ ン酸
glycine Gly G ン
histidine His H チ ン
isoleucine Ile I イソ イ ン
leucine Leu L イ ン
lysine Lys K ン
methionine Met M メチ ニン
phenylalanine Phe F ニ ニン
proline Pro P ン
serine Ser S セ ン
threonine Thr T ト ニン
tryptophan Trp W ト ト ン
tyrosine Tyr Y チ ン
valine Val V ン
その他
A .発病初期の脳波。いまだ同期性の放電は認めら れていないが、基礎律動は障害されており、徐 波化が著明である。
図2 脳波
P S D の経過を示す。
W ester n blot検査の結果を示している。右の数字は 分 子 量 を 示 し た も の で 、そ れ ぞ れ 32.5K D 、25K D 、 16.5K D の分子量である。W est er n blot のバンドのな かで、タイピングに役立つのは16.5K D と25K D の間に 存在するバンドである。このバンドは、糖鎖のない プリオン蛋白の分子量を示し、タイプ1では21K D に 相当し、タイプ2では19K D に相当する。このバンド の高さの違いによって、異常プリオン蛋白はタイプ1 とタイプ2に分類される。25K D 近くの真中のバンド は、一箇所糖鎖のあるプリオン蛋白であり、この図 では最も量の少ない32.5K D 近くの分子量の大きいバ ンドは2箇所に糖鎖のついたプリオン蛋白である。 図1 異常プリオン蛋白のタイピング
B .典 型 的 な 、P S D 。基 礎 律 動 は 制 御 さ れ 、周 期 的 に、全ての部位で同期的に放電が認められる。
M R IのD iffusionが有効である。大脳皮質では、局所的に(すべての頭葉で見られるのではなく、前頭葉の一部) 高信号が認められる。また、基底核にも高信号が認められることが多い。症例は左から、W ild(M M 1の古典的 C J D )、200(コドン200の変異のある家族性C J D )、232(コドン232の変異のある家族性C J D )で異常な高信号を 検出している。
図4 v C J D のMR I画像 図3 脳の画像
T 2強 調 画 像(a)及 び 拡 散 強 調 (diffusion-w eig ht )画 像(b)に お い て 、両 側 の 視 床 の 枕 (pulv inar )と 背 内 側 核 (dor somedial nuclei)に高信号領域を認める。
A .孤発性プリオン病の古典型C J D (M M 1の症例)。大脳皮質のH E 染色。著明な神経細胞脱落とグリオーシスを 認める。
B . A と同じ症例。プリオン蛋白抗体による免疫染色。異常プリオン蛋白は大脳皮質全体にびまん性に分布して いる。いわゆる、シナプス型の沈着である。
C .挿入変異を有する症例(168bpの挿入変異)。小脳皮質のプリオン蛋白抗体による免疫染色。小脳皮質の分子 層にあわい斑状の異常プリオン蛋白の沈着が見られる。
D .コドン102の変異のGS S 症例。小脳皮質のプリオン蛋白抗体による免疫染色。小脳では分子層と顆粒細胞層に シナプス型の沈着とともに、大きな斑状の沈着(いわゆるアミロイド斑)が認められる。
E . v C J D 症例。H E 染色。軽度の海綿状態が見られる。海綿状態に囲まれたアミロイド斑が認められ 、これが F lor id P laqueと呼ばれるものである。
第 1 章 プリオン病について
概
念
ン病 新 い概念 感染性疾患 あ ン病 理解 役立 う
ン病 概念 確立 歴史 述
1960 年代 ー 高地民族 多発 い kuruー ー いう神経疾患 調査 行わ
疾患 感染 引 起 明 根拠
次 二 報告 あ 一 kuru 死亡 患者 脳乳剤 ン ン ー 接種
同 病気 発症 あ 他 高地民族 儀式的食人習慣 禁止
kuru 発生 終焉 神経病理学的 kuru 同 う 海綿状脳症
示 Creutzfeldt-Jakob・ 病(CJD)や羊 Scrapieー 同様 病気 可能性 推測 CJD
ン ン ー 感染 間 証明
kuruやCJD 動物 Scrapie 伝達性海綿状脳症 総称 う
伝達性海綿状脳症 う 既知 感染因子 発見
従来 原因不明 神経変性疾患 い 感染性疾患 位置付
功績 Gajdusek博士 1976年 ー 賞 受賞 後 家族性
ン・ ー・ ン ー
Gerstmann-Sträussler-Scheinker病(GSS) 同様 感染性 証明 至 い
伝達性海綿状脳症 解明 次 テ 感染因子 精製 あ 精製 感染因
子 核酸 破壊 処理 感染性 低下 質 破壊 処理 行 初
感染性 低下 感染因子 質 構成 い い
う ン仮説 提唱
ン(Prion) Proteinaceous Infectious Paticles 略 あ ン 構成
主 質 ン蛋白(Prion Protein, PrP) 証明 ン蛋白 重
合 線維 性質 示 1983年 報告 時点 ン
蛋白 感染因子 中心 あ 説 疑う研究者 多 ン蛋白 遺伝子 見
遺伝子 正常 動物 脳 存在 ン蛋白 発現 い
感染因子 別 あ い いう 反論 根拠 あ 頃
正常 動物 い ン蛋白 PrP (Cellular) 呼 病気 異常型
PrPSc Scrapie 呼 う ン蛋白 検出 際 使用
い 蛋白分解酵素 あ Proteinase Kテ ー PrPC 完全 分解 う 対
PrPSc 方 少 分子量 低下 検出 可能 あ 異常型 検
出 い いう 現在明 い
ン仮説 認 う 1989年 1993年 二 研究報告
あ 一 家族性GSS ン蛋白遺伝子 ン 102 酸置換
蛋白 原因 考え あ 原因遺伝子 探索 始 ン仮
説 場合 発見 い ン蛋白 中 家族性GSS 遺伝子変異 あ
見出 あ 後 次々 家族性GSS い 新 い遺伝子変異 明
契機 CJDやGSS ン病 いう名称 代わ あ
う一 重要 報告 ン蛋白 欠損 Scrapie由来
感染性蛋白 接種 発病 感染性 ン 増幅 生 い 示
あ
現在 至 正常 ン蛋白 う 異常型 ン蛋白
あ い ン蛋白 異常化 い テ 関与 多 不明
点 残 い 解明 向 活発 研究 進 い
表 プ オン病研究 歴史
Creutzfeldt: 症例報告
Jakob: 症例報告
Gerstmann-Sträussler-Scheinker: 遺伝性症例 報告
Zigas-Gajdusek: Kuru 報告
Gajdusek: Kuru 実験的伝播
Gibbs: CJD 実験的伝播
Gajdusek: ノ ベ 医学生理学賞受賞
Prusiner: プ オン仮説
Oesch: プ オン蛋白遺伝子 ロ ン
Hsiao: GSS症例 プ オン蛋白遺伝子変異 見
Bueler: プ オン蛋白ノッ アウ ウ 作成
Pan: プ オン蛋白 立体構 変化 指摘
Will: new variant CJD 報告
Prusiner: ノ ベ 医学生理学賞受賞
最近
ピック
ン病 最近 大 話題 1996年 起 牛海綿状脳症 bovine
spongiform encephalopathy, BSE いわ 狂牛病騒 あ 従来 動物
種差 羊 Scrapie 感染 う 思わ い
1980年初 羊 ン 羊や牛 肉 脊髄や脾臓 含 作製 肉骨
粉 Meat and bone meals 牛 感染 種 壁 乗 越え 1986年初 BSE
報告 1993年 年間 万頭 及 発生 ー 徐々 沈静化 向 い
あ 1995年 1996年 英国 中心 新 い CJD
10例報告 あ new variant CJD (nvCJD 略 現在 vCJD 略
い ) 10代 含 若年者 認 従来 い臨床・病理 呈 CJD あ
発生CJD 特徴的 脳波 periodic synchronous discharges: PSD 認 い
病理像 斑 多発 異常 ン蛋白 特殊 あ
呼 全身 ン 装置 扁桃 ン 節 脾臓 異常
ン蛋白 沈着 い う vCJD 従来 CJD 異 新 い
CJD あ 現時点 英国 中心 100名以上 vCJD 発生 あ 今後 増
え 可能性 否定 い
BSE 感染 考え vCJD あ 決定的 証拠
証明 わ い BSE vCJD 異常 ン蛋白 同 う 異常型
ン蛋白 動物 野生型 や ン 感染性 類
似 い BSE 多発 い 国 vCJD 認 い 学問的
BSE vCJD 因果関係 確実 あ 考え い
経口的接種 kuru 感染可能 あ 報告 vCJD BSE
感染 牛組織 経口接種 原因 考え い 牛組織 英国
SBO specific bovine offals 年齢 ヶ月以上 牛 脳 脊髄 扁桃 胸腺
腸管 食材 禁止 い 1989年 実際 自然発病 BSE
牛 脳 脊髄 網膜 感染性 証明 実験的 感染 BSE 小腸遠位
部 後根神経節 骨髄 感染性 証明 い 感染実験
感度 問題 あ 1990 年 牛 SBO 哺乳類 鳥類 餌
禁 う WHO Manuals 1998
文 献
) Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a Kuru-like
syndrome to chimpanzees. Nature. 1966, 209:794-796.
) Gibbs CJ Jr, Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, Matthews WB.
Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the
chimpanzee. Science. 1968, 161:388-389.
) Tateishi J, Ohta M, Koga M, Sato Y, Kuroiwa Y. Transmission of chronic
spongiform encephalopathy with kuru plaques from humans to small rodents. Ann
Neurol. 1979, 5:581-584.
) Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982,
216:136-144.
) Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, Barry RA,
Tempst P, Teplow DB, Hood LE, et al. A cellular gene encodes scrapie PrP 27-30
protein. Cell. 1985, 40:735-746.
) Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott
syndrome. Nature. 1989, 338:342-345.
) Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C.
Mice devoid of PrP are resistant to scrapie. Cell. 1993, 73:1339-1347.
) Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S,
Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the
第 章 プリオン病の分類
ン病 原因 三 分類 原因不明 孤発性(sporadic)
ン病 ン蛋白遺伝子 変異 起 家族性(familial)
動物 ン病 感染 考え 感染性(infectious) ン病 あ
孤発性 ン病 現時点 原因不明 あ ン病 大部分 範疇 入
明 感染 病歴 い 遺伝子変異 い症例 相当 家族性 ン
病 ン蛋白遺伝子 変異 起 変異 種類 多様 病態
示 診断 遺伝子解析 容易 あ 同 変異 病像
症例 存在 ン蛋白遺伝子変異 説明 い症例 存在 具体例
後 詳細 記述 最後 感染性 食人習慣 伴 報告
い ー kuru 英国 牛海綿状脳症 いわ 狂牛病 伴う vCJD 硬膜移
植後 CJD 脳下垂体 ン製剤投与後 CJD 分類
三 分類 ン病 分類 細分化
い あ 例え 孤発性 ン病 い 以前 ン
蛋白遺伝子 正常多型 分類 い 現在 ン蛋白遺伝子 正常多型
加え 異常 ン蛋白 ン 分類 い 本
分類 用い 異常 ン蛋白 ン 関 検査項
目 詳細 記述 参照 い
孤発性プ オン病
孤発性 ン病 ン蛋白遺伝子 正常多型 以下 う 分類
ン 129 Met あ Val あ 分類 異常 ン蛋白
分子量 違い いう う 分類 い 図
Proteinase K処理後 異常 ン蛋白 分子量 異 利用 分類方法
あ 糖鎖 い ン蛋白 21KD 19KD 分子量 示
あ ン蛋白 遺伝子型 異常 ン蛋白 ン 合わ 分類
MM1 MV1 MM2 MV2 VV2 呼 MM1 ン 129Met/Met
型 異常 ン蛋白 有 症例 理論的 VV1 存在 あ
実際上 わ 国 う 症例 報告 い 加え 最近異常 ン
蛋白 小 ン 化 ン蛋白(fragmented PrP) 存在
.古典的CJD
MM1 症例 MV1 症例 存在 い 場合 fragmented
PrP 陽性 あ 注意 い点 ン 129Val 有 い 異常
ン蛋白 あ 古典的CJD特有 臨床・病理像 呈 点 あ 意
味 遺伝子型 ン病 分類 不完全 いわ い
.視床型CJD
MM2 症例 相当 fragmented PrP 陽性 あ MM2 症例 皮
質型 呼 ン病 報告 い わ 国 現時点 MM2 症例
視床型 CJD 分類可能 あ 視床型 CJD 呼 わ SFI (sporadic fatal
insomnia:孤発性致死性不眠症) いう命名 FFI fatal familial
insomnia:致死性家族性不眠症 sporadic form 考え 命名 あ
.アミ イ 斑を有す CJD
MV2 VV2 相当 fragmented PrP 認 い 従来 ン
129Val 症例 多 斑 CJD 分類
表 孤発性CJD 分類
孤 性CJD 病型 コ ン 129 遺伝子型 異常プ オン蛋白 タイプ
典的CJD MM1 MV1
視床型CJD MM2
大脳皮質型CJD MM2
ア ロイ 斑 つCJD MV2 た VV2
家族性プ オン病
家族性 ン病 孤発性 ン病 古典的CJD 似 家族性CJD 分類
斑 特 著明 あ GSS 特殊型 FFI あ
い 家族性 ン病 遺伝子変異 位置 分類 家族性
ン病 浸透率 低 家族歴 認 い孤発例 発病 症例
家族性 ン病 40% 認 注意 必要 あ わ 国 認 家族
性 ン病 N末端 う 列挙
.挿入変異
挿入変異 ン51~91 相当 部分 挿入 受 変異 あ 部位 個
酸 構成 構造 回繰 返 40個 酸 ー ン
回 144bp 回 168bp 繰 返 挿入変異 存在 繰 返 多
短い 海綿状脳症 呈 長い 海綿状脳症 示 い いう特徴 あ
CJD GSS 分類 困難 あ 家族性 ン病 いう言葉 い
.コ ン 102
ン 102 Pro Leu 置換 GSS 代表 あ わ 国 家系 ン
219Lys 同 存在 特殊 家系 あ ン 129Met/ ン
219Glu 変異 存在 小脳変性症型GSS あ
.コ ン 105
ン 105 Pro Leu 置換 GSS Spastic Paraparesis 痙性対麻痺
発病 多い ン 129Val/ ン 219Glu 変異 存在 わ
国特有 変異 あ
.コ ン 145
世界 例 報告例 い ン 145 Tyr 停止 ン 変化 変異 あ
.コ ン 178
ン 178 Asp Asn 置換 変異 あ 変異 ン 129Met/ ン
219Glu 存在 FFI 変異 知 ン 129Val/ ン
219Glu 存在 家族性 CJD 変異 報告 い わ 国
変異 疾患 全 FFI 型 あ 家族性 CJD 家系 い 見 い い
.コ ン 180
ン 180 Val Ile 置換 変異 あ 家族性CJD 属 い 浸透率
低 孤発性CJD 見 多い わ 国特有 変異 あ
.コ ン200
ン200 Glu Lys 置換 変異 あ 家族性CJD わ 国
頻度 高い変異 あ
.コ ン210
ン210 Val Ile 置換 変異 あ わ 国 1 家系 認 い
.コ ン232
ン232 Met Arg 置換 変異 あ 家族性CJD 分類 い
症例 孤発例 認 浸透率 低い わ 国特有 変異 あ
感染性プ オン病
感染性 ン病 わ 国 認 症例 硬膜移植後 CJD
あ 感染性 ン病 関 硬膜移植例 関
感染性 ン病 英国 中心 vCJD 説明 行う
文 献
) Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka
H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti
B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease
based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999,
第 章 プリオン病の臨床と病理
孤発性プ オン病
.古典的CJDコ ン 129 (Met/Metあ い Met/Val) プ テアーゼ抵抗型プ オン蛋白 1 型
有病率 100万人 名前後 あ 地域差 い 発症年齢 平均63.0±10.4歳 25
~85歳 あ 古典的 三徴 痴呆 ー 特徴的 脳波所見 周期性同期
性放電:PSD あ 数ヶ月 無動性無言
臨床症状
前駆症状:時 食欲不振 頭痛 倦怠感 睡眠障害 体重減少 あ い 不安感
~ ヶ月見 あ
初期症状:精神・高次機能障害 記憶・記銘力障害 認知障害 計算力低下 失見当識
無関心 行動異常 幻覚 妄想 運動失調 歩行障害 い感 視覚異常 歪視
発症
進行期症状:発病 数ヶ月以内 精神症状 高次機能障害 急速 悪化 高度 痴呆
陥 会話 不能 自発語 起立・歩行 不能 寝
食事摂取 不能 尿失禁 呈
神経学的診察所見 広範 中枢神経系 障害 示 小脳 錐体路 び錐体外路徴
候 含 い 筋強剛 深部腱反射亢進 病的反射 ン ー反射 口
反射 抵抗症(Gegenhalten) 皮質盲 眼球 異常運動 構音・嚥下障害 流涎
尿失禁 脂漏性顔貌 ー 最 重要 臨床所見 あ 四
肢 共 体幹 顔面 軽度 左右差 認 典型例 律動性 同期
性 ー 認 刺激感受性 ー やび 反射
突然 聴覚 視覚刺激や筋肉 進展刺激 反応 や い
末 期:発病 ~ ヶ月 無動性無言 四肢 自発運動 除皮質
状態 上肢 屈曲 下肢 伸展位 あ い 四肢共 強い屈曲状態 関節拘縮
高度 嚥下不能 経鼻胃管栄養 あ い 胃瘻 造設 多い 予
後 不良 褥瘡 誤嚥性肺炎 尿路感染症 併発 や 1~18ヶ月 平均3.9ヶ
月 死亡 数年 わ 症例 あ
検 査
(1) 血算 血清生化学 免疫・炎症 検査 尿 異常 い
(2) 脳波 発症初期 基礎律動 不規則化 徐波化 ー 出
現 う PSD う PSD 出現率 82.2% あ 末期
(3) 髄液 軽度 増加 認 あ 細胞数 正常 あ 早期
NSE( ー ン特異的 ー neuron specific enolase) び 14-3-3蛋白 増
加 認 診断的価値 高い NSE 単純 脳炎 全脳的病変 増加
あ 14-3-3 方 特異性 高い 14-3-3 神経細胞
由来 あ CJD 髄液 94% 証明 診断的特異性 84% いわ い
14-3-3 CSF検査 陽性 他 疾患 列挙
・ 脳炎
・脳梗塞 脳出血
・低酸素脳障害
・ ー 中毒後 代謝性脳症
・脳腫瘍 ー
・肺 小細胞癌 癌性髄膜炎
・傍腫瘍脳症
・橋本病脳症
・神経変性症 (Corticobasal degeneration) 挙 注意 必要
(4) 画 像
初期診断 有用 MRI あ 基底核部や大脳皮質 T2強調画像 高信号 呈
あ 初期 CTやMRI 異常 見出 い 多い う
時期 FLAIR(fluid attenuated inversion recovery)法や拡散強調(diffusion-weighted)
MRI 基底核 視床や大脳皮質 沿 異常 高信号 高率 見出 特 拡散
強調MRI 有用性 高い 図
鑑別診断
CJD 鑑別 疾患 以下 挙 CJD 前述 三徴候 加え 無動性無言
至 経過 早い 画像 全脳 萎縮 急速 進行 髄液 14-3-3
陽性 診断上 重要 あ
CJDと鑑別すべき疾患
老年痴呆 ー型 脳血管障害型
前頭葉・側頭葉型痴呆 病 痴呆 伴う運動 ー ン疾患
ー ン ・痴呆症候群
び 性 ー小体病
皮質基底核変性症
多系統萎縮症
進行性核上性麻痺
悪性症候群 抗精神病薬
単純 脳炎 性脳炎 脳症 神経梅毒
脳原発性 ン 腫
代謝性脳症 脳症 橋本病脳症 中毒性脳症
低酸素性脳症
他 病因 老年期痴呆性疾患
診断基準
●診断確実例 (definite)
特徴的 病理所見 有 症例 Western blot法や免疫染色法 脳 異常
ン蛋白 検出 得 症例
●診断 確実例 (probable)
病理所見 い例 進行性痴呆 示 脳波 PSD 認 ー
錐体路・錐体外路障害 小脳症状 視覚異常 無動性無言 う 項目以上 示
症例
●診断疑い例 (possible)
診断 確実例 同 臨床像 示 PSD 欠 症例
病 理
肉眼的所見:著明 脳萎縮 あ 重量 1,000g以下 あ 多い 脳回 萎縮
海馬 形態 保 割面 灰白質 白質 萎縮 変色 脳室 拡大
組織学的所見:海綿状態 粗 う化やstatus spongiosus 代わ 大脳皮質や基
底核 中心 認 前者 緊張性 膜 覆わ 小孔 海綿状 見え
融合 不規則 間隙 gliosisー 主 status spongiosus 変わ
*神経細胞脱落 gliosis 大脳皮質 線条体 視床 中心 後頭葉 病変
強いHeidenhain型や小脳顆粒層 強いataxic type 呼 あ 図 A
*白質病変 強い症例 わ 国 多 panencephalopathic type 呼 あ
免疫染色:異常 ン蛋白 灰白質 び 性 沈着 一致
synaptic-type 呼 図 B 小脳顆粒層 大型 大顆粒状 沈着 見
kuru斑 塊状 plaque type 沈着 い( )
.視床型CJD ~ コ ン 129 Met/Met
プ テアーゼ抵抗型プ オン蛋白 型
臨 床
発症年齢:36~71 歳 平均52.3歳
初 発:運動失調 自律神経異常症や認知機能障害 発症
能障害 進行 い 睡眠障害 精神運動興奮や幻視 伴う不眠症 や自律神経
異常 発汗 高体温 血圧変動 認 多い 脳波 非特異的 徐波化
認 PSD 認 い 多い 経過 古典型 CJD 緩徐 ~24
ヶ月 平均 15.6ヶ月 あ
病 理
ン蛋白遺伝子型 MM-2型 孤発性CJD 視床 病変 集中 視床変
性症 呼 臨床症状 致死性家族性不眠症 FFI 似 孤発性致死性不眠
症 SFI 呼 あ
肉眼的所見:脳萎縮 い 前頭葉 軽度 脳重量 減少 い
組織学的所見:海綿状態 大脳皮質 軽度 限局性 大脳皮質 第 層 中心
海綿状態 広 認 全層性 海綿状態 脳回 認 い
あ 限局性 多い 注意 要 小脳 顆粒細胞 保 い
古典型 大 差 あ
*神経細胞脱落 gliosis 視床 下 ー 核 著明 あ 視床 背内側核
MD 前核 AV 背外側核 LD PD 強い 下 ー 核 病変 全例
強い 病変 遺伝子異常 あ FFI 同一 あ SFI 大脳皮質 小
脳皮質 歯状核 脳幹 軽度 病変 見 多い
*白質病変 い
免疫染色:異常 ン蛋白 脳 沈着 い synaptic type perivacuolar type
小 plaque type 認 あ Western blot 型 MM2
糖鎖 あ い 比率 SFI FFI 異
.アミ イ 斑を伴う非典型例 ~10) コ ン 129 Val/Val
プ テアーゼ抵抗型プ オン蛋白2型
臨 床
発症年齢:41~80歳 平均61.3歳
初 発:運動失調 発症 多い
経 過:認知障害 あ 加わ 脳波 非特異的 徐波化 認 PSD
認 少 い 予後 ~18ヶ月 平均6.5ヶ月 あ
コ ン 129 Met/Val
プ テアーゼ抵抗型プ オン蛋白 型
臨 床
発症年齢:40~81 歳 平均59.4歳
経 過:経過 古典型CJD 緩徐 ~72ヶ月 平均 17.1 ヶ月 あ 年以
上 生存例 あ 脳波 PSD 認 少 い
病 理 VV2とMV2に関して
わ 国 少 い M/V 遺伝子多型 V/V 遺伝子多型 異常 ン蛋白
Western blot 2型 MV VV2 孤発性症例 古典的CJD 異 症状 病変
示 塊状 Plaque type ン蛋白沈着 あ 遺伝性 ン病 鑑別
異常 ン蛋白 ン ン蛋白遺伝子解析 必要 あ
肉眼的所見:脳萎縮 やや軽度 脳重量 1,000g以下 少 い
組織学的所見:海綿状態 大脳皮質 広範 皮質深部 強い傾向 あ
*神経細胞脱落 gliosis 大脳皮質 線条体 視床 橋核 小脳顆粒層 強い傾向
あ
*白質病変 二次性 思わ
免疫染色:異常 ン蛋白 沈着 特徴的 大小 plaque型 沈着 小脳皮質 主
大脳皮質 単一 unicentric 塊や 周囲 繊維 放散 kuru斑状
主 あ 多数 小塊 集合 malticentric plaque)
場合 遺伝性 ン病 鑑別 必要 あ
.コ ン219 Glu/Lys いて 解説11
一般正常日本人 約 12% ン219 (Glu/Lys) 多型性 示 知
う 家族性GSS 症例 報告 い 孤発型 CJD ン 219 (Glu/Lys)
多型性 示 症例 報告 い い ン219 Lys PrPC PrPSc 転換 抑
制 い 可能性 考え い
家族性プ オン病
ン蛋白 遺伝子 第20染色体 短腕上 存在 う
翻訳 ORF 単一 ン上 あ 253個 酸 ン51 番
91 番 Pro Gly 富 個 回 個 回 酸 繰 返 配列
あ
家族性 ン病 ン病全体 10~15% 占 ン蛋白 遺伝子変
異 認 い 家族性 ン病 40% 症例 浸透率 低 家
族歴 認 い い 臨床上 注意点 あ 今日 15種類 点変異 種類
異 長 反復(octapeptide repeat) 挿入 報告 い
変異 中 最 頻度 高い ン 102 (Pro→Leu) ン200 (Glu→Lys) 変異
.挿入変異 挿入変異の臨床
1-a.96過剰塩基対挿入 (Four octapeptide repeats) 12)
発症年齢:62歳 男性
初 発:62歳 歩行時 転倒 発症 翌年 不良 気
経 過:65歳 自発性低下 見当識・記銘力低下 前頭葉徴候 軽度 小脳失調
認 い 67 歳 ー 出現 68 歳 時脳波 PSD 出現 CT
前頭・側頭葉萎縮 認 69歳 死亡
コメント:本例 前頭葉型痴呆 鑑別 重要 示唆 ン病 あ わ
国 米国 報告 い
1-b.144塩基対挿入 (Six octapeptide repeats) 13)
発症年齢:22~53歳
初発症状:異常行動 無関心 錯乱 不眠 記憶力低下 見当識障害 構音障害
経 過:緩徐 進行 痴呆 筋強剛 錐体路徴候 小脳失調 呈 ~10年後
死亡 ー 記載 脳波 PSD 認 い い
コメント:若年発症 ー病 鑑別 問題
1-c.168塩基対挿入 (Seven octapeptide repeats) 14)
発症年齢:29歳 女性
初発症状:自発性減退 物忘 計算力低下
経 過:痴呆 徐々 進行 失見当識 構成失行 保続 強 34歳頃 筋
強剛 錐体路徴候 小脳失調 加わ 36歳 約 年 経過 死亡 CT 35歳頃
脳室拡大 著明 脳波 34歳 徐波化 平坦化 PSP 認
コメント:北米 本例 同様 168bp過剰挿入例 家系 報告 い 15 本例 若
年発症 前頭・側頭葉型痴呆 鑑別診断 ン病 重要 あ 示 い
挿入変異の病理
8 反復部位 過剰 反復 ・ ・ 回挿入 症例 わ 国 報告
い ・ 回過剰挿入例 似 病変 示 回挿入例 やや異 病変 示
肉眼的所見:脳萎縮 軽度 あ 、 回挿入例 やや強 、 脳重 900g あ
組織学的所見:海綿状態 い 粗 う化 大脳皮質や小脳分子層 中等度 認
回挿入例
*神経細胞 脱落 gliosis 大脳皮質 線条体 小脳皮質 軽度
*白質病変 回例 やや強い以外 軽度 あ
免疫染色:異常 ン蛋白 沈着 特徴的 粗 塊状 太い毛糸 断片状 網目状 沈
着 、 小脳分子層 多発 、 大脳皮質 認 Congo red染色 複屈析 示 典型
.コ ン 102 変異 Pro→Leu 失調型GSS 16~19
臨 床
発症年齢:平均52±12歳 (30~66歳)
初発症状:起立 歩行時 不安定 廻 い 失調症状 数例
初期 失行 性格変化 記憶障害 眼振 深部腱反射 亢進 伴 い
経 過: ヶ月 数年後 痴呆症状 不安 抑う 精神症状 出現
眼振 構音・嚥下障害 小脳・脳幹症状 深部腱反射亢進 筋強剛 広範
神経症状 加わ 約半数 ー 出現 脳波 末期 PSD
認 症例 あ
コメント:家族性 ン病 中 ン 102 変異 示 失調型GSS 最 頻度 多い
失調 数年経過 若年発症者 脊髄小脳変性症 鑑別 問題
病 理
肉眼的所見:脳萎縮 重量 低下 症例 異 長期 臨床経過 比 軽い傾向
あ
組織学的所見:海綿状態 示 い症例 高度 海綿状態 示 症例 あ 同胞間 異
あ
*神経細胞 脱落 gliosis 症例 差 あ 小脳皮質 大脳皮質 線条体 橋
核 多い
*白質病変 症例 異
免疫染色:異常 ン蛋白 plaque 型沈着 小脳皮質 多発 特徴 あ 。
個 塊 unicentric plaque 周囲 線維 放散 kuru斑
数個 小塊 multicentric plaque あ PAS 染色 ン染色
Congo red染色 陽性 糖蛋白 蛋白 特徴 示 synaptic type
沈着 共存 図 D
附 P102L 変異ア 219 Lys多型 合併し 症例20
臨床経過 約 年 例 報告 い
肉眼的所見:正常 脳重量 1,290g
組織学的所見:海綿状態 い
*神経細胞 脱落や gliosis 軽度 あ
*白質変性 い
免疫染色:異常 ン蛋白 大 い綿花状 沈着 大脳 び小脳皮質 認
PAS Congo red染色 染 P102L単独変異例 plaque 性状
分布 異 Western blot 検索 症例 異常 ン蛋白 界
面活性剤 不溶性 あ テ ー 処理 分解 抵抗性 い
.コ ン 105 変異 Pro→Leu 痙性麻痺型GSS 21 22
臨 床
発症年齢:平均45歳 (40~49歳)
初発症状:歩行障害 多い 痴呆 振戦 ー 初発 症例 あ
失調 伴う症例 あ
経 過:全例 痙性対麻痺 呈 痴呆 初発 例 年以上 わ 他
症状 認 痙性対麻痺 初発 症例 ~ 年後 記憶力低下 自発
性低下 精神症状 仮性球麻痺 強制把握 前頭葉徴候 加わ 寝
脳波 PSD 認 死亡 罹病期間 ~12年 あ
コメント:孤発例 脊髄性痙性対麻痺 鑑別診断 問題 痴呆 高次機能障害
併発 確 大切 あ
病 理
肉眼的所見:前頭葉 中心 軽度 脳萎縮
組織学的所見:海綿状態 い
*神経細胞脱落 gliosis 前中心回 中心 大脳皮質 深部 強い 大脳深部 灰白
質や小脳 変化 軽 下位運動 ー ン 障害 い
*白質 皮質脊髄路 選択的 線維脱落 示 他 白質 変性 軽度 あ
免疫染色:異常 ン蛋白 沈着 unicentric 大型 斑 前中心回 多数認
他 頭頂葉 前頭葉 島葉 皮質深部や第一層 あ
小脳 少 い multicentric plaque あ NFT 神経原繊維変化
存在 報告 い NFT 多数認 症例 全 認 い症例
あ
.コ ン 145 変異 (Tyr→Stop) 23~25)
臨 床
発症年齢:38歳 女性
初発症状:物忘 地誌失認
経 過:徐々 進行 痴呆 易怒性 無関心 精神障害 筋強剛 10年後
ー 口唇傾向 出現 四肢屈曲拘縮 死亡 年前 経管栄養 無
動性無言 全経過21 年
コメント: ー病 鑑別診断 困難 あ 症例 あ
病 理
肉眼的所見:著明 脳萎縮 あ 脳重量 640g 脳回 著明 萎縮 示 約21
年 わ 慢性経過 影響 考え
組織学的所見:海綿状態 大脳皮質 一部 軽度
神経原線維変化 あ 老人斑様 ン蛋白斑 共 ー病 鑑別
困難 あ
*白質変性 前頭葉 頭頂葉 後頭葉 強い
免疫染色:異常 ン蛋白 大脳皮質 小脳皮質 多発 unicentric plaque型 老人
斑 似 抗 ー 蛋白抗体 不染 PrP 末端抗体 染 ン146以
降 末端抗体 染 い 脳内 沈着 異常 ン蛋白
ン145 あ 血管 周囲 異常 ン蛋白 沈着 い
.FFI (致死性家族性不眠症) コ ン 178 変異 (Asp→Asn) +コ ン 129 (Met/Met) 26~28)
臨 床
発症年齢:18~61 歳
初発症状:難治性不眠 発汗過多 心拍亢進 高体温 自律神経症状 発症
経 過:錐体路徴候 小脳症状 痴呆 ー 加わ 脳波 PSD 出現
稀 全経過 ~36ヶ月
コメント:わ 国 不眠や自律神経症状 目立 小脳症状 前景 立 脊髄小脳
変性症 鑑別 難 症例 報告 い 診断上 変異 129M
場合 特徴的 不眠症 視床病変 示 FFI (致死性家族性不眠症) 呼 変異
存在 129Val 場合 古典的CJD 似 家族性CJD 病像 呈 わ
国 報告例 い FFI 異 FFI いう診断 変異 129Met
存在 証明 必要 あ
病 理
肉眼的所見:脳萎縮 重量 正常域 あ
組織学的所見:海綿状態 い 大脳皮質 限局性 軽度 あ
*神経細胞脱落 gliosis 視床 下 ー 核 限局 視床 前核(AV)
背内側核(DM) 背外側核(LD LP) 強い 下 ー 核 症例 神
経細胞 変形 消失 あ 大型 増生 他 小脳 ン
細胞 歯状核 中脳被蓋部 軽い病変 あ SFI 似 後者
大脳皮質 病変 拡大 あ
*白質 病変 い
免疫染色:異常 ン蛋白 免疫組織染色 い 軽度 証明
あ FFI症例 免疫染色 Western blot い 部位 PrPSc
検出 い症例 あ 確定診断 数ヶ所 Western blot 行う必要 あ
.コ ン 180 (Val→Ile) 29~31)
臨 床
初発症状:高齢発症 痴呆あ い 不安 精神症状 失調 初発
経 過:比較的緩徐 経過 予後 孤発性CJD 良好 ~ 年 あ 脳波
検査 PSD 認 い場合 多い
コメント: 例 報告 い ン129 多型 Met/Val 有 場合
ー ン ン症状 呈 知 い
一方 ン232 点変異 Met/Arg 併 持 い 症例 1 例報告 い
例 高齢発症 84歳 あ 臨床経過 孤発性CJD 同様 あ 1 年 死
亡
病 理
肉眼的所見:脳萎縮 脳重量 正常域 あ
組織学的所見:海綿状態 広範 特徴 あ 大脳皮質 高度 あ
粗 う化 い 海馬 線条体 視床内側核 軽度 脳幹や小脳
い
*神経細胞脱落 gliosis 大脳皮質 視床内側部 線条体 大脳皮質 中等度
脳幹 小脳 病変 古典型CJD 比 軽度 あ
*大脳白質 軽度 神経線維 減少 あ 全脳型 い
免疫染色:異常 ン蛋白 沈着 普通認 軽度 認 症例 海馬
synaptic type 認 量 多 い Western blot 異常 ン蛋白
量 少量 あ 脳乳剤 Western blot 検出 い症例
多 必 超遠心操作 濃縮操作 必要 あ
.コ ン200 変異 (Glu→Lys) 家族性CJD 32~36)
臨 床
発症年齢:平均57±11 歳 (44~78歳)
初発症状:不安 不眠 異常行動 幻覚 精神症状 記憶障害 失調 感覚異常 視
覚・眼球運動障害 孤発性CJD 類似 症状 初発 い 初発時 ー
認 症例 14例中 例存在 い
経 過:経過 急速 進行 多 ~ ヶ月以内 約半数 症例 無動性無
言 陥 い 脳波上 PSD 全例 認 死亡 期間 平均 14±0.8 ヶ
月( ~36ヶ月) あ 孤発性CJD 平均 17.5±18.4ヶ月 比較 ン200
変異例 方 短
コメント: ン200 変異 示 家族性CJD ン 102 変異例 次い わ 国
多 患者 認 い 症状 孤発性CJD 似 い 経過 早い 特
徴 あ
病 理
組織学的所見:海綿状態 大脳皮質 中心 著明 あ
*神経細胞脱落 gliosis 大脳皮質 線条体 視床 強い 脳幹 小脳 軽い傾向
あ
*白質変性 全脳型 い
免疫染色:異常 ン蛋白 synaptic type あ 変異 129V 症
例 塊状沈着 小脳 認 Western blot 型 あ ー 症
例 本邦 ン219Lys 野生型 症例 例存在 い 症例
発病 遅延 認
.コ ン210 (Val→Ile) 37~39)
臨 床
発症年齢:60歳
初発症状:不安 不眠 幻覚 ー
経 過:急速 嚥下障害 呈 歩行不能 3ヶ月後 無動無言状態 半
年 死亡 脳波 PSD 認
コメント:わ 国 1 例 報告 い
病 理
わ 国 症例 剖検 い ン 各1症例 孤発性CJD
同様 著明 海綿状態 gliosis 大脳 び小脳 ン 症例 前頭
側頭葉 神経細胞脱落 強い いう
.コ ン232 (Met→Arg) 40 41)
臨 床
発症年齢:平均60.4歳 50~73歳
初発症状:不安 性格変化 行動異常 痴呆 主 初発症状 あ 失調 感覚障
害 視覚障害 呈 例 あ
経 過:数ヶ月後 ー 無動無言状態 呈 脳波 PSD
症例 認 予後 0.3~3.5年 平均 1.6年 あ
コメント:現在 13例 報告 い
病 理
肉眼的所見:脳萎縮 著明 脳重量 1,000g以下 多い
組織学的所見:海綿状態 粗 う化 大脳皮質全体 認
*神経細胞 消失 gliosis 大脳皮質 線条体 視床 小脳顆粒層
*白質病変 大脳 強い 脳幹 小脳白質 軽い
図 硬 膜 移 植 患 者 手 術 0 2 4 6 8 10 12 14 16 18
'79 '80 '81 '82 '83 '84 '85 '86 '87 '88 '89 '90 '91
手 術 患
者 数
外 国 例
不 明
Lyodura
図 硬膜移植患者 手術年
図 潜伏期 移植~ 症
0 1 2 3 4 5 6 7 8
1 2 3 4 5 6 7 8 9 10 1 1 12 13 14 15 16
潜伏期 患
者 数
図 潜伏期 移植~発症
患
者
数
人
感染性プ オン病
.硬膜移植歴を有す CJD概 要
. 乾燥硬膜 移植 時期 1979~1991年、特 1983~1987年 多 (図 )
.移植 発症 期間 16ヶ月~17年(図 )
.硬膜移植後 CJD患者数 2001 年 月現在 73例 中 、68名 B.Braun社
未処理 Lyodura 使用 確認
患
者
数
.発症年齢 平均53歳 15~79歳 若年発症 傾向
.初発症状 精神症状 高次機能障害 共 失調症状 多い
.硬膜移植CJD 2群
Dura-classic CJD:孤発性CJD 同様 症状 経過 病理所見 同一
Dura-variant CJD:緩徐 進行 発症 1 年後 簡単 応答可能 無動性無言
脳波 PSD 認 い 脳病理 Dura-classic CJD 比 軽度 あ
限局性 florid plaque 認
.硬膜CJD 剖検脳 Western blot 古典的CJD 同様 1 呈
硬膜移植CJD患者の多発と背景
1987年 月 米国 CDC 乾燥硬膜 移植 受 CJD患者 第 例 報告
42
わ 国 1991 年 最初 硬膜移植CJD患者 報告 い 43 1997年
月 CJD 緊急全国調査 報告書 脳外科 手術時 硬膜移植 受 患者
43名 CJD 認 発表 44 45 後 新 い発症者 続 2001 年
月 76名 達 い
凍結乾燥硬膜 輸入 1973年 開始 当初 製品 処理
ン 感染性 失活 い い 指摘 1987年5月
1N NaOH処理 加わ 新製品 切 替わ い 1997年 月 厚生省 WHO
乾燥硬膜 使用停止勧告 踏 え 乾燥硬膜 使用停止 緊急命令措置 行 い
硬膜CJD 大部分 患者 使用 い 硬膜 B.Braun社 製造 未処
理 旧Lyodura あ 疫学的 CJD 発症 旧Lyodura 因果関係 深
い 示 い
硬膜CJD患者 孤発性CJD 比 若年発症者 存在 初発症状 小脳失調 多
46
脳波 PSD 欠 緩徐 経過 症例 存在 病理像 脳 florid plaque
認 47 48 い 点 孤発性CJD 異 特徴 指摘
い
移植時期 罹病率
移植時期:硬膜移植 受 時期 1979 年 1991 年 及 い 49
1983 年
1987 年 硬膜 移植 受 多 CJD 患者 発症 傾
向 外国例 同一 あ
罹 病 率:1983~85年 移植 受 発症 患者数 推定 1,500名 名 割合
発病 硬膜 CJD発症 間 何 因果関係 存在 示
い 50
硬膜移植後CJDの発症年齢
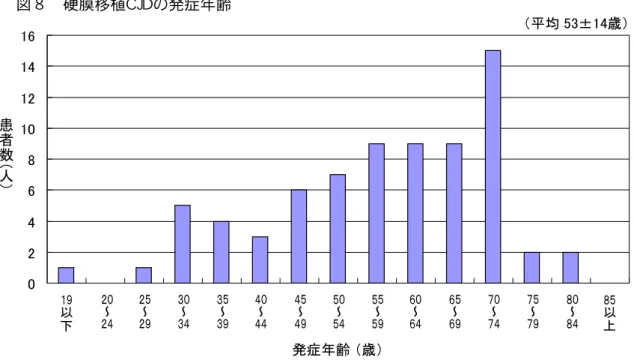
硬膜移植歴 有 CJD患者 発症年齢 15歳 79歳 平均53.0±15.0歳 あ
図 硬膜移植CJD 症 齢 0 2 4 6 8 10 12 14 16
19 20 25 30 35 40 45 50 55 60 65 70 75 80 85
症 齢 歳 患
者 数
均 53±14歳
~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~
以 以
図 硬膜移植CJD 発症年齢
患
者
数
人
臨床的特徴
Dura-classic CJD:硬膜移植CJD 初発症状 歩行時 浮遊感 書字障害
廻 い 小脳失調 記憶力低下 失見当識 計算力低下 方向 場所
分 高次機能障害 不眠 不安 抑う 異常行動 幻覚 精神
症状 眼振 孤発性CJD 比 硬膜移植例 う 初発時 精神症状
共 小脳失調 呈 多 46 発症 3ヶ月後 症状 経過 大半
症例 孤発性CJD 同様 あ 高度 痴呆 全身 筋強剛 振戦 腱反射 亢進
除皮質肢位 両上肢 屈曲位 両下肢 伸展 屈曲 い ー
皮質盲 皮質聾 発語 自発運動 や 無動性無言状態 陥
脳波 PSD 認
Dura-variant CJD:約 10% 患者 発症 年経過 ー
簡単 応答 可能 あ 無動性無言 末期 初 出現 脳波 PSD 観察
い症例 多い
病 理
Dura-classic CJD 51
肉眼的所見:脳萎縮 高度 脳重量 1,000g以下 あ
組織学的所見:海綿状態 粗 う化 強 大脳皮質 線条体 視床 著明
*神経細胞脱落 びgliosis 大脳皮質 小脳顆粒層 線条体 視床 全体 橋底部
高度 あ
*白質病変 大脳や橋一小脳系 強い
免疫染色:異常 ン蛋白 synaptic-type び 性分布 灰白質 病変部位 中
Dura-variant CJD 47 48
肉眼的所見:軽度 脳萎縮 小脳 中心 認 脳重量 1,000g以上 多い
組織学的所見:海綿状態 大脳皮質 基底核 小脳分子層 中等度 認
*神経細胞脱落 gliosis 視床 基底核 小脳 大脳皮質 認
*白質病変 軽度 あ
免疫染色: unicentric plaque 周囲 空胞 囲 florid plaque 大脳 小脳皮質
vCJD 区別 い 他 multicentric plaque や synaptic
type 沈着 認 vCJD 大形 斑状沈着 い ン
節や脾 沈着 証明 い Western blot vCJD B Parchi
分類 Collinge 分類 異常 ン蛋白 分類 い
Dura-variant CJD 異常 ン蛋白 あ
.医原性CJD 医原性CJDの種類
医原性CJD 原因 脳外科手術器具や定位脳深部電極 器具類 あ い
角膜や硬膜移植 う 大脳や小脳実質 感染組織 直接接触 場合 あ 硬膜移
植例 初発時 精神症状 小脳失調 呈 孤発性CJD 多い
下垂体 抽出 成長 ンや ン 投与 受 症例 CJD
発症 い 52 死体 下垂体 抽出 成長 ン 使用 1959
年以降 あ 以来 英国 2,000名 米国 10,000名 投与 い 1985年
例 若年発症 CJD患者 発症 始 約80例 CJD患者 発症 い
1985年以降 組 換えDNA 産生 用い い 感染 危険
い 全国疫学調査 結果 わ 国 例 発見 い い 成長
ン CJD 臨床症状 特徴 小脳失調 初発 あ
血液製剤の安全性
血液製剤 中 CJD患者 献血由来 混入 い 場合 安全対策 深刻
問題 あ 全血 血漿 含 血液製剤 感染 確証 現在 知
い い Créange 肝移植後 発症 例 CJD患者 肝移植 際
投与 い ン提供者 提供 数年後 CJD 発症 判明
ン 感染 可能性 否定 い 報告 い 53
輸血 関 重要 報告 あ BSE 罹患牛 脳 g 羊 経口的 投与
羊 未発症 時期 全血 他 羊 19頭 輸血 頭 羊 神経症状 発現
感染 証明 い 症例 い 報告後 ン投与量
少 ン 起因 発症 い 意見 多 出 い 54
わが国の輸血 臓器移植等における安全性確保
0
10,000
20,000 30,000 40,000
'87 '88 '90 '92 '94 '96 '98 '00
症
0 5 10 15 20 25 30 vCJD BSE S
頭
v
J
患
者
数
人
図 英国 BSEとvCJD 年次発生数
知見 血液 介 古典的CJD 感染 疫学的 証拠 い 可能性
完全 否定 い い 各国 献血時 問診 取扱 参考 わ
国 献血時 問診票 本人及び血縁者 CJD及び類縁疾患 有無 由来成長
ン注射 有無 角膜移植 有無及び硬膜移植 伴う脳外科手術 有無 確認
要因 有 者 献血 念 排除 い
後述 牛海綿状脳症 BSE 関連 あ vCJD 問題 踏 え 英国
ン ン ン ー ン
通算 ヶ月以上 滞在歴 あ 者 献血 念 排除 い
供血者 CJD 発症 供血後 判明 場合 明 古典的CJD
あ 場合 除 関連 血液製剤 念 回収 い
臓器移植 関 CJD 疑い 含 診断 受 い 場合や 以下 う 臓器
提供者 病歴 海外渡航歴及び 血縁者 病歴等 認 場合 当該提供者
臓器提供 行わ い い
成長 ン 投与 受 者
硬膜移植歴 あ 者 角膜移植歴 あ 者
CJD及び 類縁疾患 家族歴 あ 者
CJD及び 類縁疾患 医師 言わ あ 者
1980年以降 英国 ン ン ン
ー ン 10 国 通算 ヶ月以上 滞在歴 有 者
.vCJD バ アン 型CJD 変異型CJD
概 要
(1) 英国 1996年 報告 vCJD BSE 罹患 牛 感染 新
発生 ン病 あ
(2) BSE 減少 い vCJD 最近 急速 患者数 増加 全世界 百余名 達
0 2 4 6 8
10 12 14 16 18
20
19 20 30 40 50 60 70 80
録 時 齢 歳 患
者 数
人
死亡時 均29歳 18~53)
~ ~ ~ ~ ~ ~ 以
以
患
者
数
人
図10 vCJD 登録時年齢 Will et al, 20003)よ
(3) 若年者 多 精神症状 感覚障害 発症 緩徐 進行
(4) 脳波 PSD い
(5) MRI 視床枕 信号異常 見
(6) 脳病理 florid plaque 認
(7) 末梢組織 ン 節 虫垂 扁桃 異常 ン蛋白 証明 血液
介 伝播 危険性 指摘 い
(8) 英国以外 ン ン 香港 vCJD患者 発生 い
vCJDの発生の経緯と背景
1996年3月 英国 海綿状脳症諮問委員会 1985年 爆発的 発生 い BSE
感染 可能性 あ 新 い病型 CJD患者 発生 認 発表 世界 衝
撃 与え 55 BSE 1985年 英国 最初 罹患牛 認 1992年 年
間約3,600頭 発生 後 減少 2000年 千数百頭 い
わ 英国 vCJD 患者 年間23% 増加 2001 年 12月 113
例 達 い 56 (図 10 BSE 英国以外 ー 徐々 発生 認
vCJD患者 ン 例 ン 香港 1 例 報告 い 香
港例 長期間英国 滞在 症例 発症 あ う vCJD 発生国 拡大
世界 新 脅威 投 い
vCJDの特徴
発症年齢 罹病期間:若年発症 経過 長い 特徴 あ い 死亡時 年齢
12~74歳 平均29歳 あ 孤発性CJD 平均63歳 あ 比
罹病期間 平均 18ヶ月 ~38ヶ月 進行 孤発性CJD 緩徐 あ
初発症状:孤発性 CJD 異 潜行性(insidious) 発症 抑う 不安 自閉 異常
行動 精神障害 主 あ 表 記憶障害や持続性 痛 伴う顔面
上・下肢 感覚障害 随伴 認
経過中 症状 全例 失調 舞踏運動 下肢 全身
ー 不随意運動 眼球上方注視障害 多 末期 症状 孤発性CJD 同様
進行性 痴呆 呈 最後 57% 患者 無動性無言 陥 い 表
脳波 基礎律動 異常 全例 認 CJD 特徴 PSD 認
い
髄液 14-3-3蛋白 57% 症例 陽性 あ
MRI 視床枕(pulvinar) 異常信号 認 あ 77% 特 拡散強調
画像(DWI) 検出 や い 所見 特徴的 ー テ 像
呼 い (図 )
58
表 vCJDとCJDと 差異
CJD vCJD
症 齢 44~70 均63 歳 12~74( 均29)歳
現様式 経過 急性 急 進行 insidious onset 緩徐 進行
症状 食欲低 倦怠 抑うつ しび
進行性痴呆 行動異常 性格変化
オ ロ 舞踏運動 小脳失調
オ ロ
脳波 PSD 100% し
MRI 基底核 pulvinar
病理 病変分布 大脳皮質 小脳 基底核 視床 強い
kuru斑 プ 型 び 性 kuru斑 無数 出現
(florid plaque)
表 vCJD 臨床症状 n =35
臨床像 初 時 症状 経過中 症状
精神症状 22
a
34
感覚障害 7 24
四肢 疼痛 4/7 13/24
失調 3 35
健忘 6 29
不随運動 2 33
b
ア 2 12
舞踏運動 0 20
オ ロ 0 25
方注視麻痺 0 14
痴呆 0 35
無動性無言 0 20
a
数例 特別 精神症状 し 無関心や人格変化を示し
b
vCJDの診断基準57)
英国 CJD 諮問委員会 提唱 vCJD 診断基準 確実例(definite) 確実例
(probable) 疑い例(possible) 段階 分 い 確定診断 剖検 生検脳
免疫組織化学 異常 ン蛋白 検出 最 重要 あ
臨床的 若年発症 初発時 精神症状 主体 緩徐 経過 示 脳波 PSD
拡散強調画像 視床枕 高信号 え 重要 あ
表 vCJD 診断基準 WHO 2001
進行性精神 神経障害
経過 6ヶ月以
一般検査 他 疾患 除外
医原性 可能性 い
家族性CJD 否定
症初期 精神症状 抑うつ 不安症 無関心 自閉 錯乱
痛 伴う感覚障害
失調
オ ロ 舞踏運動
痴呆
脳波 PSD陰性
MRI特 拡散強調画像 両側視床枕 高信号
蓋扁桃生検 異常プ オン陽性 扁 桃 生検 通 常 検 査 し 勧
い vCJD 疑う臨床症状 あ 脳波 PSD MRI おい 異常
い 適応 検討
Definite: A 進行性精神 神経障害 神経病理 確認した
Probable: + 4/5項目+ A+ B
又 + A
Possible: + 4/5項目+ A
vCJDの病理59)
肉眼的所見:特別 記載 い
組織学的所見:海綿状態 視床 基底核 著明 小脳や大脳皮質 PrPSc 沈着
部位 強い
*神経細胞脱落 gliosis 視床 小脳皮質 強 大脳皮質 他 認
*白質病変 弱い い
免疫染色:異常 ン蛋白 多量 沈着 特徴 大小 塊状 型沈着 florid
囲 野菊 花 形 塊状沈着 kuru 斑様 や PAS 染色陽性
大形 斑状沈着 小脳分子層 中心 認 synaptic-type び 性沈着 神
経細胞周囲 空胞壁や血管周囲 強い あ 口蓋扁桃 腸管壁 脾
ン 装置 主 濾胞樹状細胞 FDC 異常 ン蛋白 沈着 60 図 F
他 ン病 い 末梢血 異常 ン蛋白 流入
防疫上問題 所以 あ
vCJDとBSE
疫学 び種々 研究結果 vCJD BSE 同一 感染因子 原因 あ
示 い 疫学的 BSE 発生国以外 vCJD 発生
BSE由来 食品 汚染 食品 出回 い 時期(1984~1986) 最初 vCJD患者
発生時期(1994~1996) 10年間あ 考え 潜伏期 一致 い
vCJD ン蛋白 電気泳動 ーン BSE ン蛋白 同一 あ (2
型) 孤発性CJD 型 異 い 61 vCJD患者 脳 接種 潜伏期
症状 経過 孤発性CJD 接種 異 BSE接種 類似
い 病理像 牛 伝達 可能性 裏付
vCJDと血液を介しての感染
最近 vCJD 患者 神経症状 発現 ヶ月前 虫垂炎 虫垂摘出術 受
い ー あ 虫垂 免疫組織化学 ン 濾胞中 樹状濾胞細胞内
ン蛋白 証明 62 未発症者 腸管 ン 組織中 ン蛋白 存在
血液 介 中枢神経系 ン 伝達 考え 同時 血液 感染
危険性 示唆 手術器具 介 感染 拡大 あ い 献血 際 ー
ン 新 問題 投 い
以上 結果 vCJD 患者 血液 介 感染 可能性 完全 否定 い
わ 国 献血時 問診票 英国 ン ン
ン ー ン 通算 ヶ月以上 滞在歴 あ
者 献血 念 排除 い
文 献
) Zerr I, Posshiari M, Collins S, et al. Analysis of EEG and CSF 14-3-3 proteins as aids
to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000, 55:811-815.
) Kitamoto T, Shin RW, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi
J. Abnormal isoform of prion proteins accumulates in the synaptic structures of the
central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol.
1992, 140:1285-1294.
structure of the prion protein influences the distribution of abnormal prion protein in
the central nervous system. Am J Pathol. 1992 141:271-277.
) Parchi P, Capellari S, Chin S, Schwarz HB, Schecter NP, Butts JD, Hudkins P, Burns
DK, Powers JM, Gambetti P. A subtype of sporadic prion disease mimicking fatal
familial insomnia. Neurology. 1999, 52:1757-1763.
) Yamashita M, Yamamoto T, Nishinaka K, Udaka F, Kameyama M, Kitamoto T.
Severe brain atrophy in a case of thalamic variant of sporadic CJD with plaque-like
PrP deposition. Neuropathology. 2001, 21:138-143.
) Kawasaki K, Wakabayashi K, Kawakami A, Higuchi M, Kitamoto T, Tsuji S,
Takahashi H. Thalamic form of Creutzfeldt-Jakob disease or fatal insomnia? Report
of a sporadic case with normal prion protein genotype. Acta Neuropathol (Berl). 1997,
93:317-322
) Kitamoto T, Tateishi J. Human prion diseases with variant prion protein.
Philos Trans R Soc Lond B Biol Sci. 1994, 343:391-398
) Nagashima T, Okawa M, Kitamoto T, Takahashi H, Ishihara Y, Ozaki Y,
Nagashima K. Wernicke encephalopathy-like symptoms as an early manifestation of
Creutzfeldt-Jakob disease in a chronic alcoholic.J Neurol Sci. 1999, 163:192-198.
) Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka
H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti
B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease
based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999,
46:224-233.
10) Miyazono M, Kitamoto T, Doh-ura K, Iwaki T, Tateishi J. Creutzfeldt-Jakob disease
with codon 129 polymorphism (valine): a comparative study of patients with codon
102 point mutation or without mutations. Acta Neuropathol (Berl). 1992, 84:349-354.
11) Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. Codon 219 Lys allele of
PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1998,
43:826-828
12) 磯崎英治 宮本和人 鏡原康裕 前頭葉性痴呆 示 96 bp 過剰塩基挿入 証
明 CJD. Dementia 1994, 8:363-371.
13) Oda T, Kitamoto T, Tateishi J, et al. Prion disease with 144 base pair insertion in a
Japanese family line. Acta Neuropathol 1995, 90: 80-86.
14) 水島節雄 石井 西丸甫夫. 老年痴呆 わ い ン病 168塩基対挿
入例 Dementia 1994, 8:380-390
15) Zeidler M, Gibbs CJ and Meslin F. WHO manual for strengthening diagnosis and
16) Hsiao K, Baker HF, Crow TJ, et al. Linkage of a prion protein missense variant to
Gerstmann-Sträussler syndrome. Nature 1989, 338:342-345.
17) Doh-ura K, Tateishi J, Kitamoto T, et al. Creutzfeldt-Jakob disease patients with
congophilic kuru plaques have the missense variant prion protein common to
Gerstmann-Sträussler syndrome. Ann Neurol 1990, 27: 121-126.
18) Kitamoto T, Yamaguchi K, Doh-ura K, et al. A prion protein missense variant is
integarted in kuru plaque cores in patients with Gerstmann-Strässler syndrome.
Neurology 1991, 41: 306-310.
19) Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y. Pro→Leu change at
position 102 of prion protein is the most common but not the sole mutation related to
Gerstmann-Straussler syndrome. Biochem Biophys Res Commun. 1989 , 1
63:974-979.
20) Tanaka Y, Minematsu K, Moriyasu H, Yamaguchi T, Yutani C, Kitamoto T,
Furukawa H. A Japanese family with a variant of Gerstmann-Straussler-Scheinker
disease. J Neurol Neurosurg Psychiatry. 1997, 62:454-457.
21) Kitamoto T, Amano N, Terao Y et al. A new inherited prion diseaes (PrP-P105L
mutation) showing spastic paraparesis. Ann Neurolo 1993, 34: 808-813.
22) Yamada M, Itoh Y, Fujigasaki H et al. A missense mutation at codon 105 with
codon 129 polymorphism of the prion protein gene in a new variant of
Gerstmann-Sträussler-Scheinker disease. Neurology 1993, 43: 2723-2724.
23) 一宮洋介 飯塚礼二 岩本典彦 老年痴呆 紛 わ い ン病 ン 145
変異症例 Dementia 8: 391-396, 1994
24) Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in
Gerstmann-Straussler syndrome with mutant PrP plaques. Biochem Biophys Res
Commun. 1993, 192:525-531
25)Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T,
Tateishi J, Seiler C, Frangione B, Bugiani O, Giaccone G, Prelli F, Goedert M, Dlouhy
SR, Tagliavini F. Vascular variant of prion protein cerebral amyloidosis with
tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in
PRNP. Proc Natl Acad Sci U S A. 1996, 93:744-748.
26) Nagayama M, Shinohara Y, Furukawa H, Kitamoto T. Fatal familial insomnia with
a mutation at codon 178 of the prion protein gene: first report from Japan. Neurology.
1996, 47:1313-1316.
27) Medori R, Montagna P, Tritschler HJ, LeBlanc A, Cortelli P, Tinuper P, Lugaresi E,
Gambetti P. Fatal familial insomnia: a second kindred with mutation of prion
28) Tateishi J, Brown P, Kitamoto T, Hoque ZM, Roos R, Wollman R, Cervenakova L,
Gajdusek DC. First experimental transmission of fatal familial insomnia. Nature.
1995, 376:434-435.
29) Ishida S, Sugino M, Koizumi N, Shinoda K, Ohsawa N, Ohta T, Kitamoto T, Tateishi
J. Serial MRI in early Creutzfeldt-Jacob disease with a point mutation of prion protein
at codon 180. Neuroradiology. 1995, 37:531-534.
30) Hitoshi S, Nagura H, Yamanouchi H, Kitamoto T. Double mutations at codon 180
and codon 232 of the PRNP gene in an apparently sporadic case of Creutzfeldt-Jakob
disease. J Neurol Sci. 1993, 120:208-212
31) Matsumura T, Kojima S, Kuroiwa Y, Takagi A, Unakami M, Kitamoto T. [An
autopsy-verified case of Creutzfeldt-Jakob disease with codon 129 polymorphism
and codon 180 point mutation]. Rinsho Shinkeigaku. 1995 , 35:282-285.
32) 赤井淳一郎: ・ 病 星和書店 東京 pp102~108 1984
33) 川井 充 高津成美 間宮康喜 . 神経内科 1981, 15:165-171
34) Inoue I, Kitamoto T, Doh-ura K, et al. Japanese family with Creutzfeldt-jakob
disease with codon 200 point mutation of the prion protein gene. Neurology 1994,
44:299-301.
35) 岩淵 潔 遠藤青磁 萩元 浩 ン200 変異(Glu→Lys) ン病
2家系 脳神経 1994, 46:349-354.
36) Hainfellner JA, Parchi P, Kitamoto T, Jarius C, Gambetti P, Budka H. A novel
phenotype in familial Creutzfeldt-Jakob disease: prion protein gene E200K mutation
coupled with valine at codon 129 and type 2 protease-resistant prion protein. Ann
Neurol. 1999, 45:812-816.
37) Furukawa H, Kitamoto T, Hashiguchi H, Tateishi J. A Japanese case of
Creutzfeldt-Jakob disease with a point mutation in the prion protein gene at codon
210. J Neurol Sci. 1996 , 141:120-122.
38) Ripoll L, Laplanche JL, Salzmann M, Jouvet A, Planques B, Dussaucy M, Chatelain
J, Beaudry P, Launay JM. A new point mutation in the prion protein gene at codon
210 in Creutzfeldt-Jakob disease. Neurology. 1993, 43:1934-1938.
39) Pocchiari M, Salvatore M, Cutruzzola F, Genuardi M, Allocatelli CT, Masullo C,
Macchi G, Alema G, Galgani S, Xi YG, et al. A new point mutation of the prion protein
gene in Creutzfeldt-Jakob disease. Ann Neurol. 1993, 34:802-807.
40) Shimizu T, Tanaka K, Tanahashi N, Fukuuchi Y, Kitamoto T. [Creutzfeldt-Jakob
disease with a point mutation at codon 232 of prion protein-a case report]. Rinsho
Shinkeigaku. 1994, 34:590-592.
protein gene at codon 232 in Japanese patients with Creutzfeldt-Jakob disease: a
clinicopathological, immunohistochemical and transmission study. Acta Neuropathol
(Berl). 1996, 92:441-446
42) Prichard J, Thadani V, Kalb R, et al. Rapidly progressive dementia in a patient who
received a cadaveric dura mater graft. MMWR 1987, 36:49-55.
43) Miyashita K, Inuzuka T, Kondo H, et al. Creutzfeldt-Jakob disease in a patient with
a cadaveric dural graft. Neurology 1991, 41: 940-941.
44) 厚生省調査研究 ・ 病等 関 緊急全国調査研究班 研究
報告書 班長 佐藤 猛 1997年 月
45) Sato T, Hoshi K, Yoshino H, et al. Creutzfeldt-Jakob disease associated with
cadaveric dura mater grafts: Japan, January 1979- May 1996. MMWR 1997,
46:1066-1069.
46) Hoshi K, Yoshino H, Urata J, et al: Creutzfeldt-Jakob disease associated with
cadaveric dura mater grafts in Japan. Neurology 2000, 55: 718-721.
47) Takashima S, Tateishi J, Taguti Y et al: Creutzfeldt-Jakob disease with florid
plaque after cadaveric dural graft in a Japanese woman. Lancet 1997, 350:865-867.
48) Schimizu S, Hoschi K, Muramoto T, et al.: Creutzfeldt-Jakob disease with
florid-type plaques after cadaveric dura mater grafting. Arch Neurol 1999, 56: 357-362.
49) 佐藤 猛 ン病:21 世紀 向 課題 順天堂医学 2001 46: 311-321.
50) Nakamura Y, Yanagawa H, Kitamoto T, et al. Epidemiologic features of 65
Creutzfeldt-Jakob disease patients with a history of cadaveric dura mater
transplantation in Japan. Epidemiol Infect 125: 201-205, 2000
51) Yamada S, Aiba T, Endo Y, Hara M, Kitamoto T, Tateishi J. Creutzfeldt-Jakob
disease transmitted by a cadaveric dura mater graft. Neurosurgery. 1994,
34:740-743
52) Brown P, Preece JP, Brandel T, et al. Iatrogenic Creutzfeldt-Jakob disease at the
millenium. Neurology 2000, 55: 1075-1081.
53) Créange A, Gray F, Cesaro P, et al. Creutzfeldt-Jakob disease after liver
transplantation. Ann Neurol 1995, 38: 260-272.
54) Houston F, Foster DJ, Chong A, et al. Transmission of BSE by blood transfusion in
sheep. Lancet 2000, 356:999-1000.
55) Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in
the UK. Lancet 1996, 347:912-925.
56) Department of Health, UK. WWW.doh.gov.uk, 3, December, 2001
57) WHO. www.who.int, 2001
variant Creutzfeldt-Jakob disease. Lancet 2000, 355:1412-1414.
59) Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of
variant Creutzfeldt-Jakob disease. Histopathology. 2000, 37:1-9.
60) Hill AF, Zeidler M, Ironside J, Collinge J. Diagnosis of new variant
Creutzfeldt-Jakob disease by tonsil biopsy. Lancet. 1997, 349:99-100.
61) Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain
variation and the aetiology of 'new variant' CJD. Nature. 1996, 383:685-690.
62) Hilton DA, Fathers E, Edward P, et al. Prion immunoreactivity in appendex before
第
4
章 プ オン病の治療
特異的 治療法 合併症 対 対症療法 主体
ー 著 い や 投与
ー 対 薬剤投与 意識障害 強 あ 薬剤 投
与 控え
嚥下障害 経口摂取 不可能 栄養 補給 経管栄養 行う 多
い
関節拘縮 問題 関節可動域訓練 施行 望 い 四肢 痙直
強 体位交換や清拭 看護 困難 時 ン 投与 あ
褥瘡や気道・尿路感染 合併 注意
動物実験 抗真菌剤 AmphotericinB 抗癌剤 iododoxorubicin 蛋白結合
色素Congo red 感染動物 生存期間 延長 発症 遅 報告 あ
薬剤 い 毒性 強い AmphotericinB CJD症例 投与 報告
あ 有効性 確認
最近 ンあ い ン蛋白 対 抗体 ン感
染細胞系 び ン 動物 用い 系 有効 報告 い ~
看護 治療 際 感染防止 注意 別項 説明
文 献
) Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives
as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A. 2001,
98:9836-9841.
) Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G,
Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB.
Antibodies inhibit prion propagation and clear cell cultures of prion infectivity.
Nature. 2001, 412:739-743.
) Heppner FL, Musahl C, Arrighi I, Klein MA, Rulicke T, Oesch B, Zinkernagel RM,
Kalinke U, Aguzzi A. Prevention of scrapie pathogenesis by transgenic expression of
第
5
章 プ オン病の検査
臨床検査
.髄液検査
髄液検査 神経細胞 破壊 伴 神経細胞 遊離 蛋白 測定
有効 あ Enolase 14-3-3 診断的価値 高い
現時点 ン病 確定診断 い 血液・髄液検査 い
.脳 波
PSD (Periodic synchronous discharges) 存在 有名 あ 古典的CJD
所見 あ 他 脳波所見 重要 PSD 脳 器
質的変化 示唆 う 徐波 存在 上 注意 い い症例
ン 105変異 GSS い 病末期 α波 認 あ
.脳の画像検査
初期診断 有効 MRI検査 あ 基底核部 T2強調画像 High
症例 認 基底核部 異常 FLAIRやDiffusion 強調 多い
基底核部 異常 認 場合 FLAIRやDiffusion 試 有効 あ
. オン蛋白遺伝子検索
ン蛋白遺伝子解析 ン病 診断 重要 検査 あ 遺伝子検索
臨床的 診断 可能 家族性 ン病 限 い 遺伝子変異 存
在 い孤発性 ン病 い ン蛋白 ン 129 ン219 解析
重要 意味 ン 129Met/Met 脳波 PSD 古典的 CJD 経
過 PSD 認 い 視床型CJD 考え い ン 129
Val/Met Val/Val あ 症例 脳波 PSD 認 ン病
取 扱う必要性 あ ン 129 解析 ン病 臨床診断
非常 重要 あ ン219 解析 ン219Lys 検出 孤発性
ン病 診断 可能性 低 他 疾患 考慮 い
ン219Lys 認 硬膜移植後 CJDや家族性 ン病
ン 102 GSS ン200 家族性CJD ン病 発病 症例 存在
特殊検査 異常
オン蛋白の検出
.異常 オン蛋白の検出法 (Western blot法)
異常 ン蛋白 検出法 最 確実 再現性 認 方法 あ 単 脳
ー Proteinase K 蛋白分解酵素 ー 一 あ 処理
検査 十分 あ 異常 ン蛋白 濃度 低い症例 特 孤発性
ン病 視床型CJDや家族性 ン病 FFIや ン 180 変異例 界
面活性剤存在下 不溶性分画 濃縮 必要 あ Western blot 法 異
常 ン蛋白 ン 可能 あ 異常 ン蛋白 Proteinase K処理後
分子量 大 分類 1~3
.異常 オン蛋白の検出法 切片の免疫染色
ー ー 用い 前処理法 導入後 症例 異常 ン蛋白 検
出 成功 い 検出法 利用可能 う 点
記載
)切片のオートクレー 処理
(1) 脱 ン後 切片 水洗 ー ー 処理 行う 切片 種類 応
種々 濃度 1mM 2mM 3mM 塩酸溶液 入 121℃ 10分間 ー
ー 処理 行う ー ー 切片 入 溶液 出 後 30~60分
間 室温 戻 い
(2) 塩酸濃度 切片 異 いう 最 重要 点 あ 剖検後2
週間程度 固定脳 1~3mM 濃度 最良 結果 得 あ 固定期
間 長期 及 症例 30mMや 100mM 塩酸濃度 上 い い い切片
あ 塩酸濃度 組織破壊 起 一歩手前 濃度 最 適当 あ
最適 塩酸濃度 必要 あ
(3) ー ー 処理 行う容器 ン ー 深型容器 600ml 蒸留水
入 濃塩酸 10 濃度 60μl入 1mM塩酸 作製 い 塩
酸溶液中 完全 切片 浸 う 切片 入 切片 染色 用い
同時 何枚 処理可能 あ
) リオン蛋白抗体
(1) 免疫染色 用い 一次抗体 3F4 いう ー 抗体 市販 い Dako
岩井化学 ー 抗体 市販 い IBL) 一次抗体 希釈
5%溶液 効果的 あ
(2) 次抗体系 ン 使用 い系 方 い結果 得 ー ー 処
理 内因性 ン 問題 大脳白質 ン 染色 あ
ン 用い 十分除去 い 多 二次抗体系