2.7.6.41
【国外試験】外国人健康男性被験者を対象とした第Ⅰ相臨床試験:ワルファリ
ンからリバーロキサバンへの切り替え試験(試験番号 10849)[報告書番号
PH-36332
]
治験の標題: 健康男性被験者を対象にワルファリンから BAY 59-7939/リバーロキサバンに切り替えたときの薬力 学的効果を検討することを目的とした無作為化、プラセボ対照、並行群間比較試験 BAY 59-7939 / 10849 治験責任医師: 実施医療機関: , Germany , Germany 公表(参考文献): 該当せず 治験期間: 20 年 月 日~20 年 月 日 (最初の被験者の初回来院日から最後の被験者の最終来院日まで) 治験終了日:20 年 月 日 開発のフェーズ: 第 I 相臨床薬理試験 目的: 主目的: ワルファリン〔定常状態に達するまで投与、国際標準化比(INR)2.0~3.0〕からリバーロキサ バン(20mg 1 日 1 回)へ切り替えたときの各種薬力学的パラメータを検討すること 副次目的: ワルファリンからリバーロキサバンへ切り替えたときの薬物動態、安全性及び忍容性を検討す ること 方法論(試験デザイン): 多施設 無作為化 並行群間比較(A、B、C 群) プラセボ対照(B 群) 単盲検(A、B 群)被験者数: 計画被験者数:84 名(最低対象被験者数:75 名) 登録被験者数:96 名(実年齢幅:18.0~45.0 歳、平均年齢:32.4 歳) 薬力学的解析対象被験者数:84 名 薬物動態学的解析対象被験者数:84 名 安全性解析対象被験者数:91 名 診断及び主な組み入れ基準 健康男性被験者 年齢 18 歳~45 歳 BMI:18~29 kg/m2
被験者は、野生型であるCYP2C9*1をホモで有し、VKORC1遺伝子の 6484 位及び 7566 位に C-allele を有する者とした。 被験薬、用法及び用量、バッチ番号: BAY 59-7939(リバーロキサバン)錠剤 20mg 開発番号: バッチ番号:BX02J3F 対照薬、用法及び用量、バッチ番号: プラセボ バッチ番号:BXA13KJ 併用薬剤、用法及び用量、バッチ番号: ワルファリン(CoumadinⓇ 、BSM)5mg 錠 バッチ番号:8F73 中和薬、用法及び用量、バッチ番号: ビタミン K(KonakionⓇ、Hoffmann-LaRoche) バッチ番号:F013211 治療期間: A 群(ワルファリン+リバーロキサバン):
ワルファリンを 1 日 1 回 6 日間(Day 6~Day 1)、PTINR が 2.0~3.0 になるように、Day -6 及び Day -5 は 10mg 以下を投与し、Day -4~Day -1 は 2.5~15mg の範囲を 1 日 1 回投与し た。
リバーロキサバン 20mg を 1 日 1 回 4 日間(Day 0~Day 3)投与した。
リバーロキサバン投与後 6 日目(Day 5)にビタミン K 10mg を 1 日 1 回投与した。 B 群(ワルファリン+プラセボ):
ワルファリンを 1 日 1 回 6 日間(Day 6~Day 1)、PTINR が 2.0~3.0 になるように、Day -6 及び Day -5 は 10mg 以下を投与し、Day -4~Day -1 は 2.5~15mg の範囲を 1 日 1 回投与し た。
プラセボを 1 日 1 回 4 日間(Day 0~Day 3)投与した。
プラセボ投与後 6 日目(Day 5)にビタミン K 10mg を 1 日 1 回投与した。 C 群(リバーロキサバン単独):
評価項目: 薬力学: 以下の項目の AUCτ及び Emax PT、PT-INR、aPPT、抗第Xa因子活性、第Xa因子活性阻害、第Ⅱa因子活性、第Ⅶa因子活 性、HepTestⓇ、ETP(ラグタイム、ピーク、ピークまでの時間、AUC)、PiCT (主要な薬力学的パラメータ:PT の Emax) 薬物動態: 主要評価項目: R-ワルファリン及び S-ワルファリン:最終投与後の t1/2 リバーロキサバン:初回投与後の AUC(0-24)及び Cmax 副次評価項目: R-ワルファリン及び S-ワルファリン:最終投与時及び最終投与後の Ctrough,ss及び Ctrough,ss/D リバーロキサバン:初回投与後の AUC(0-24)norm、Cmax、norm及び tmax;2~4 回目の投与における Cpeak及び Ctrough;最終投与後の t1/2 その他の評価項目:Points terminal 安全性: バイタルサイン、心電図、臨床検査項目、治験薬投与後の有害事象の発現 統計学的手法: 薬力学的パラメータ PT 延長の AUC(0-tn)、AUCabs(0-tn)、Emax,abs及び Emaxは探索的に解析した。処置群間の動態を比較す るためにこれらのパラメータを対数変換し、処置効果を組み込んだ分散分析(ANOVA)を実施した。 処置群間の比に関する点推定値及び探索的な片側 90%信頼区間は、ANOVA により得られた対数結果を 再変換して算出した。 薬物動態学的パラメータ リバーロキサバンの初回投与後の AUC(0-24)及び Cmax並びにワルファリンの最終投与後の t1/2は、 データが対数正規分布に従うと仮定して解析した。これらのパラメータの対数値は、処置効果を組 み込んだ ANOVA を用いて解析した。処置群間の比の点推定値(最小二乗幾何平均値)及び探索的な 95%信頼区間は、ANOVA の被験者間の標準偏差を用いて対数結果を再変換して算出した。 安全性パラメータは記述統計量を用いて表した。 要約及び結論: 薬力学: 本治験の主目的として、ワルファリンの効果が十分に認められている状態(PT-INR 2.0~3.0)で、 リバーロキサバン(20mg 1 日 1 回、心房細動における脳卒中予防で使用予定の用量)へ切り替えた ときの各種薬力学的パラメータの変化を検討した。 測定した薬力学的パラメータは、第Xa因子活性の阻害、抗第Xa因子活性、PT、PT-INR、aPTT、 HepTestⓇ、第Ⅶa因子活性、第Ⅱa因子活性、ETP(ラグタイム、ピーク、ピークまでの時間、 AUC)及び PiCT であった。 本治験は、3 群の並行群間比較試験であった。A 群では、PT-INR が 2.0~3.0 となるまでワルファリ ンを漸増し、ワルファリンの投与完了 24 時間後にリバーロキサバンの投与を開始した。B 群では、 PT-INR が 2.0~3.0 となるまでワルファリンを漸増し、ワルファリンの投与完了 24 時間後にプラセ ボの投与を開始した。C 群では、ワルファリンは投与せず、リバーロキサバンのみ投与した。 リバーロキサバン 20mg を 1 日 1 回 4 日間(Day 0~Day 3)投与した。ワルファリン投与群では、各 被験者の PT-INR を 2.0~3.0 へと増加させるため、リバーロキサバン又はプラセボの投与に先立ち ワルファリン 2.5~15mg を 1 日 1 回約 6 日間投与することとした。
薬力学的解析対象及び薬物動態学的解析対象はいずれも 84 名、1 群あたりの被験者数は 28 名で あった。 薬力学的パラメータの評価では、各測定時点におけるベースライン値からの変化率の中央値を統計 学的評価により求め、AUC 及び Emax(相対変化率及び絶対変化率)を効果-時間曲線から求めた。 ベースライン値の定義は、A 群(ワルファリン/リバーロキサバン)及び B 群(ワルファリン/プラ セボ)では、ワルファリン投与開始直前の測定値とし、C 群(リバーロキサバン単独投与)では、 リバーロキサバンの初回投与直前の測定値とした。 主要な薬力学的パラメータである PT の結果は以下のとおりであった。 プロトロンビン時間測定は、ビタミン K 拮抗薬を測定するようにデザインされた。リバーロキサバ ンも PT に影響を及ぼすが、変化の程度は試験の種類により異なる。Neoplastinでは、血漿中濃度 に対する PT の傾きが、Innovinでの傾きに比べ大きかった。したがって、Neoplastinを用いて PT を測定した。 ・PT の測定には Neoplastinのみを用いた: ワルファリン単独投与により、PT はベースライン値の 1.87 倍に延長(11.2 秒の延長)した。B 群 では、ワルファリンからプラセボへの切り替え後、ワルファリンの効果が減少し、Day 3 の朝(ワ ルファリン最終投与 4 日後)にベースライン値まで戻った。 リバーロキサバン単独投与(C 群)により、最大 7 秒の PT 延長(ベースライン値の 1.6 倍)が認め られたが、リバーロキサバン 20mg の投与で予測された範囲内であった(効果-時間曲線の Emax)。PT はリバーロキサバンの投与 24 時間後にベースライン値まで戻った。 ワルファリンからリバーロキサバンへの切り替え後、最大で 45 秒の PT 延長(ベースライン値の 4.4 倍、効果-時間曲線の Emax幾何平均値)が Day 0 に認められた。被験者ごとの値では、Day 0 の 投与 1 時間後に約 90 秒の PT 延長が認められた。ワルファリンの効果の減少により、PT は Day 4 の 朝、リバーロキサバン投与前にベースライン値まで戻った。リバーロキサバンの効果は、最終投与 24 時間後にはベースライン値まで戻った。この時点では、ワルファリンの効果が完全に消失してい た。
・PT-INR:
PT-INR の測定には、次の 3 種類の試験を用いた。(i) Day -1 及び Day 0~Day 5 には、Neoplastin を、(ii) ワルファリン投与期間中は、Innovinを、(iii) A 群の Day 0 での被験者の部分集団に は、Hemosenseを用いた。Innovinと Neoplastin並びに Hemosenseと Neoplastinの全体的な相 関性は、いずれも高かった(ピアソンの相関係数は、それぞれ 0.94 及び 0.89、p<0.0001)。相関 プロットで PT-INR が 2.0 以下と 2.0 以上の場合で比較すると、2.0 以下でより厳密な相関性が示さ れた。 Neoplastinの結果を以下に示す。 ワルファリンからリバーロキサバン又はプラセボへの切り替え直前の PT-INR(中央値)は、A 群で は 2.2(範囲:1.92~2.55)、B 群では 2.3(範囲:1.90~2.70)であった。PT-INR がわずかに 2.0 以下(0d00h00m)であった被験者 6 名も、リバーロキサバン又はプラセボ投与群に組み入れた。こ れらの被験者の組み入れ時点における PT-INR 最小値は 1.9 であり、リバーロキサバン又はプラセボ 投与開始に適切な値と判断された。 B 群の PT-INR(中央値)は、4 日間のプラセボ投与期間で次第に減少し、Day 4 の朝にはベースライ ン値まで戻った。時間-効果曲線の Emaxでは、ワルファリン投与後に PT-INR がベースライン値の 2.25 倍に増加した。 C 群では、リバーロキサバン 20mg を投与した過去の試験から予測されたとおり、PT-INR はベースラ イン値の最大 1.8 倍まで増加した(時間-効果曲線の Emax)。投与後 24 時間でベースライン値まで減 少した。
ワルファリンからリバーロキサバンへの切り替えで、PT-INR はワルファリン投与終了時の 2.2 か ら、Day 0 の投与後 3 時間で通常時のリバーロキサバン投与前値の 6.7 倍にまで増加した(時間-効 果曲線の Emax)。被験者ごとの PT-INR で 4.19~10.29 の範囲が得られたのは、ワルファリン最終投 与後 27 時間及びリバーロキサバン初回投与後 3 時間であった。Day 0 の PT-INR は 28 名の被験者す べてで 3.0 以上であり、平均 12.6 時間持続した。Day 1 の投与後、PT-INR が 3.0 以上の被験者は 25 名で、平均 7.5 時間持続した。Day 2 で PT-INR が 3.0 以上の被験者は 8 名であった。Day 2 以降 では、PT-INR が 3.0 以上の被験者は認められなかった。ワルファリン/リバーロキサバンの投与で は、相加作用を上回る効果が認められたが、ワルファリン/プラセボ及びリバーロキサバン単独投与 での効果を考慮すると指数的な変化ではなかった。投与期間中、PT-INR(中央値)は減少し、切り 替え後にベースライン値に戻ったのは Day 4 の投与前であった。 予測されたとおり、PT ではワルファリン及びリバーロキサバンのいずれの効果も検出された。 主要な薬力学的パラメータである PT の統計解析を Table 2-2 に示す。
副次的な薬力学的パラメータについて以下に示す: ・第Xa因子活性阻害 ワルファリン投与により、第Xa因子活性は中央値としてベースライン値(0d00h00m)の 41~42% に減少した。ワルファリンからプラセボへの切り替え後、このワルファリンの効果は 4 日間のプラ セボ投与の間に減少した。リバーロキサバン 20mg の投与により、第Xa因子活性は 50%阻害され (効果-時間曲線の Emax)、この効果は投与後約 24 時間持続した。ワルファリンからリバーロキサバ ンへの切り替えにより、最大 76%の阻害(Emax)が認められた。リバーロキサバンとワルファリンの 最大効果は、ワルファリンとプラセボ及びリバーロキサバン単独での効果を考慮するとおおむね相 加的であった。リバーロキサバン投与前のトラフ時の第Xa因子活性の阻害は、リバーロキサバン 投与期間中、ワルファリンの効果が減少するにつれて減少した。 予測されたとおり、第Xa因子活性の阻害は、ワルファリン及びリバーロキサバンのいずれの効果 も検出された。 ・抗第Xa因子活性 ワルファリン投与及びワルファリンからプラセボへの切り替えによる抗第Xa因子活性への影響は 認められなかった。 ワルファリンからリバーロキサバンへの切り替え(A 群)による効果は、リバーロキサバン単独投 与(C 群)の効果と同程度であった。リバーロキサバン投与開始後、抗第Xa因子活性はベースラ イン値の最大 15.8 倍に増加した(時間-効果曲線の Emax)。この増加は、投与後 3~4 時間で一時的 に認められた。24 時間以内に、抗第Xa因子活性はベースライン値の 1.5 倍に減少した。 抗第Xa因子活性測定ではワルファリンの効果が検出されないため、リバーロキサバン単独の効果 を測定できる。 ・aPTT: ワルファリン単独投与により、aPTT は中央値として 1.24~1.25 倍に延長した。 ワルファリンからプラセボへの切り替えによる相加作用は認められず、aPTT 延長はプラセボ投与期 間中に減少した。 リバーロキサバン単独投与により、aPTT はベースライン値の 1.41 倍に延長した(効果-時間曲線の Emax)。この効果は、投与後 24 時間以降にベースライン値に戻った。 ワルファリンからリバーロキサバンへの切り替え後、aPTT はベースライン値の最大 1.84 倍に延長 した(時間-効果曲線の Emax)。ワルファリンとプラセボ及びリバーロキサバン単独での効果を考慮 すると、この効果は相加的と考えられた。リバーロキサバン投与前のトラフ時の aPTT 延長は、リ バーロキサバン投与期間の Day 3 までに、ワルファリンの効果の減少に伴い減少した。 ・HepTest: ワルファリン投与及びワルファリンからプラセボへの切り替えによる HepTestへの影響は認められ なかった。 リバーロキサバン単独投与後の最大効果は、ベースライン値の 2.01 倍の延長で(時間-効果曲線の Emax)、これは過去の試験結果と一致していた。この効果は、投与後 24 時間で消失した。 ワルファリンからリバーロキサバンへの切り替えにより、ベースライン値の 2.15 倍の延長が認めら れた(時間-効果曲線の Emax)。最大延長は、投与後 3~4 時間で認められ、24 時間で投与前のベー スライン値に戻った。 HepTestではワルファリンの効果が検出されないため、リバーロキサバン単独の効果が測定でき る。
・PiCT: ワルファリン投与及びワルファリンからプラセボへの切り替えによる PiCT への影響は認められな かった。リバーロキサバン単独投与により、PiCT はベースライン値の最大 2.7 倍に延長し、ワル ファリンからリバーロキサバンへの切り替えでは 3.2 倍に延長した(時間-効果曲線の Emax)。最大 効果は、投与後 3 時間で認められた。投与後 24 時間及び次のリバーロキサバン投与前に、PiCT は ほぼベースライン値に戻った。 PiCT ではワルファリンの効果が検出されないため、リバーロキサバン単独の効果が測定できる。 ・ETP:
ワルファリンの 6 日間投与により、ETP の AUC はベースライン値の 0.44 倍に減少し、ETP のピーク はベースライン値の 0.45 倍に減少した。また、ETP のラグタイムは 1.59 倍に延長し、ピークまで の時間は 1.30 倍に延長した(ベースライン値からの変化率の中央値)。
ワルファリン投与により生じた変化は、ワルファリンからプラセボへの切り替え後、5 日間のプラ セボ投与で回復した。
リバーロキサバン単独投与により、ETP の AUC は最大でベースライン値の 0.55 倍に減少し、ETP の ピークは最大でベースライン値の 0.15 倍に減少した(時間-効果曲線の 1/Emax)。 リバーロキサバン単独投与により、ETP のラグタイムはベースライン値の 2.57 倍に延長し、その結 果としてピークまでの時間もベースライン値の 3.79 倍に延長した(時間-効果曲線の Emax)。リバー ロキサバン単独投与の効果は短時間であったが、投与後 24 時間でもベースライン値には戻らなかっ た。 ワルファリンからリバーロキサバンへの切り替え後、ETP の AUC は顕著に減少し、最大でベースラ イン値の 0.24 倍に減少した(時間-効果曲線の 1/Emax)。同様の結果が ETP のピークでも認められ、 ワルファリンからリバーロキサバンへの切り替えによる相加作用で、最大でベースライン値の 0.05 倍に減少した。ワルファリン投与による ETP のラグタイム及びピークまでの時間の延長は、リバー ロキサバン単独投与後に比べて顕著であり、これらの最大効果はそれぞれベースライン値の 4.0 倍 及び 4.2 倍であった。これらの四つのパラメータのトラフ時の結果は、Day 5、すなわちリバーロキ サバンの最終投与後 48 時間及びワルファリンの最終投与後 6 日目でベースライン値に戻った。 ・第Ⅶa因子活性: 6 日間のワルファリン投与により、第Ⅶa因子活性はベースライン値の 0.15 倍に減少した。 ワルファリンからプラセボへの切り替えにより、第Ⅶa因子活性は 5 日間の観察期間の間に回復し た。 リバーロキサバン単独投与でも、第Ⅶa因子活性は投与後 8 時間までにベースライン値の 0.74 倍に 減少した(時間-効果曲線の 1/Emax)。リバーロキサバン投与期間中、投与後にベースライン値の 0.81~0.87 倍へのわずかな減少が認められた。これらの減少は、投与後 24 時間でベースライン値 に戻った。 ワルファリンからリバーロキサバンへの切り替えにより、第Ⅶa因子活性は更に減少し、ベースラ イン値の 0.08 倍となった(時間-効果曲線の 1/Emax)。全般的に、ワルファリンにより生じた第Ⅶa 因子活性の変化は、リバーロキサバン投与期間中に回復した。各投与後、第Ⅶa因子活性の中等度 かつ短時間の減少が認められた。ベースライン値に達したのは Day 4 であった。リバーロキサバン 単独投与で認められた効果は、必ずしも第Ⅶ因子活性への直接的な影響を示すものではない。本試 験は、第Ⅶ因子活性を直接的に測定する系ではなく、むしろ第Ⅶ因子の下流のすべての因子が関与 する凝固時間を通して間接的に測定する系である。そのため、本試験が第X因子の活性に依存して いることから、認められる効果は凝固試験の感度を反映していると考えられる。
・第Ⅱa因子活性: ワルファリンの最終投与後、第Ⅱa因子活性はベースライン値の 0.36 倍に減少した。 ワルファリンからプラセボへの切り替え後、第Ⅱa因子活性は回復し、Day 5 にベースライン値に 達した。 リバーロキサバン単独投与による第Ⅱa因子活性への影響は認められなかった。 ワルファリンからリバーロキサバンへの切り替え及びワルファリンからプラセボへの切り替えは、 第Ⅱa因子活性に関して同様なプロファイルを示した。投与前の状態に達したのは、Day 5、すなわ ちワルファリンの最終投与後 6 日目であった。 本治験では、ワルファリンからリバーロキサバンへ切り替えたときの、種々の血液凝固パラメータ に及ぼすワルファリン及びリバーロキサバンの影響を評価した。両剤が関与するアゴニスト作用は PT で認められ、ワルファリンからプラセボへの切り替え(B 群)及びリバーロキサバン単独投与(C 群)の結果から予測される相加作用を上回る効果がワルファリンからリバーロキサバンへの切り替 えで認められた。第Xa因子活性阻害、aPTT 延長及び ETP(AUC 及びピークの高さの減少、ラグタ イム及びピークまでの時間の延長)など他の薬力学的パラメータでは、ワルファリンからリバーロ キサバンへの切り替えで相加作用が認められた。リバーロキサバン単独の効果の測定には、ワル ファリンの効果を検出せず、リバーロキサバンの効果を確実に検出できる試験系である抗第Xa因 子活性、PiCT 及び HepTest®が推奨される。 以上より、ワルファリンからリバーロキサバンへの切り替えにおいて、薬力学的パラメータには、 使用した測定系に依存し、顕著な影響を受けたもの(PT-INR)、相加的な影響を受けたもの(第X a因子活性阻害、aPTT 及び ETP)、相加的な影響を受けなかったものが認められた。 薬物動態: 84 名の被験者を薬物動態解析対象とし、このうち 56 名にワルファリン 2.5~15mg を 6 日間投与し た。ワルファリンの最終投与 24 時間後に、リバーロキサバン 20mg(28 名、A 群)又はプラセボ (28 名、B 群)の 1 日 1 回の投与を開始し、以後 4 日間継続した(Day 0~Day 3)。また、ワル ファリンの先行投与無しでリバーロキサバンを投与した群を C 群とした。 リバーロキサバンの薬物動態学的パラメータでは、A 群と C 群の間で差が認められなかった。ま た、AUC(0-24)及び Cmaxについて A 群と B 群とを比較したところ、最小二乗幾何平均値の比とその 95%信頼区間は生物学的同等性の基準(0.80、1.25)の範囲内であった。 リバーロキサバンの主要な薬物動態学的パラメータの幾何平均値、幾何 CV%及び範囲を Table 2-3 に示す。
ワルファリン最終投与後の R-ワルファリン及び S-ワルファリンの t1/2について A 群と B 群とを比較 したところ、最小二乗幾何平均値の比とその 95%信頼区間は生物学的同等性の基準(0.80、1.25) の範囲内であった。 R-ワルファリン及び S-ワルファリンの主要な薬物動態学的パラメータを Table 2-4 に示す。 安全性: 91 名の健康男性被験者を安全性解析対象とし、A 群、B 群及び C 群の 3 群(各群 28 名)に無作為化 した。 A 群(ワルファリン/リバーロキサバン):ワルファリン 2.5~15mg を 6 日間投与(Day -6~ Day -1)した後、リバーロキサバン 20mg を 4 日間投与(Day 0~Day 3)した。ワルファリンの 効果を除くため、Day 5 にビタミン K 10mg を投与した。 B 群(ワルファリン/プラセボ):ワルファリン 2.5~15mg を 6 日間投与(Day -6~Day -1)し た後、プラセボを 4 日間投与(Day 0~Day 3)した。 C 群(リバーロキサバン単独投与):ワルファリンの先行投与無しで、リバーロキサバン 20mg を 4 日間投与した。 無作為化した 3 群のほか、ワルファリン投与期間中に投与中止した被験者(7 名)を、別の群とし て評価した。この群の薬物曝露量は、用量及び投与期間により異なった。 安全性データに関して、A 群、B 群及び C 群それぞれの結果に加え、ワルファリンを投与した被験者 のデータを集計した結果(63 名)も示した。 健康被験者 91 名のうち、治験薬投与期間中に発現した 1 件以上の有害事象を報告したのは 42 名 (46%)であった。発現した有害事象のうち、軽度は 89 件、中等度は 7 件であった。 治験薬に関連する有害事象は、91 名中 12 名(13%)に発現した。このうち各薬剤の投与期間別で は、ワルファリン投与期間中に発現した有害事象が 8%(5/63)、ワルファリンからリバーロキサ バンへの切り替え後が 21%(6/28)、リバーロキサバン単独投与後が 11%(3/28)であった。な お、ワルファリンからプラセボへの切り替え後では有害事象は発現しなかった。 重篤及び重度の有害事象は発現せず、すべての事象が治験終了までに回復した。 ワルファリンと関連する有害事象は、腹部不快感、下痢及び消化不良(各 1 件)で、いずれも軽度 かつ短期間の発現であった。頭痛を伴う多汗症及びワルファリン投与開始後の注射部位血腫が、そ れぞれ 1 名に発現した。臨床的に問題となる臨床検査値の変動は認められなかった。被験者 1 名 (被験者番号:010849-131)は、PT-INR が 3.0 以上で持続したため投与を中止した。ワルファリン 投与は 5 日後に中止したが、PT-INR は減少しなかった。3 日後に、治験責任医師はビタミン K を投 与した。
ワルファリンからリバーロキサバンへの切り替え後、治験薬に関連する ALT 増加が発現し、その結 果、リバーロキサバン投与は早期に中止された。
本症例では、被験者はリバーロキサバンの初回投与前に ALT 増加を示していた。投与後、ALT 値が更に増加したため、リバーロキサバンの投与を Day 3 に中止した。ALT 値は、Day 5 に施設 基準値上限の 4.12 倍に達し、その後は減少し最終投与後 18 日目には正常範囲に戻った。この 増加は軽度であり、治験薬に関連する有害事象として報告された。 また、ワルファリンからリバーロキサバン切り替え後、治験薬に関連する頭痛を伴う軽度の浮動性 めまい、多発性舌びらん及びリパーゼ増加がいずれも 1 件発現した。治験薬に関連しないリパーゼ 増加も 1 件報告された。軽度の出血が 2 名に発現し、いずれもリバーロキサバンの初回投与後約 1 日目に発現した治験薬に関連する歯肉出血として報告された。このうち 1 名は、注射部位血腫も発 現した。すべての出血事象は、治験薬に関連する有害事象であった。 リバーロキサバン単独投与で発現した治験薬に関連する有害事象は、軽度の鼻出血、注射部位血腫 及びリパーゼ増加で、いずれも発現は 1 件であった。また、治験薬に関連しないリパーゼ増加も 1 件報告された。治験薬に関連する出血事象は鼻出血及び注射部位血腫で、いずれも軽度かつ短期間 の発現であった。 バイタルサイン及び心電図検査では、いずれの治験薬の影響も認められなかった。 治験薬に関連する有害事象を Table 2-5 に示す。計算上の相対開始時点はリバーロキサバン又はプ ラセボの投与初日とし、この日を Day 0(00d00h00min)と表した。
結論: リバーロキサバンは安全で、忍容性も良好であった。 PT-INR が 2.0~3.0 で、ワルファリンの定常状態からリバーロキサバン 20mg へ切り替えたとこ ろ、PT 及び PT-INR で顕著なアゴニスト作用が認められた。 切り替え後の PT はベースライン値の 4.4 倍(45 秒)に延長し、一方リバーロキサバン単独投 与ではベースライン値の 1.6 倍(7 秒)に延長した(効果-時間曲線の Emax)。 ワルファリンからリバーロキサバンへの切り替えにより PT-INR(NeoplastinⓇを用いて測定) はベースライン値の 6.1 倍になり、一方リバーロキサバン単独投与ではベースライン値の 1.6 倍になった(0d03h00m での中央値)。被験者ごとの PT-INR は、ワルファリンの最終投与後 27 時間及びリバーロキサバンの初回投与後 3 時間で、4.19~10.29 の範囲に達した Day 0 の PT-INR は 28 名の被験者すべてで 3.0 以上であり、平均 12.6 時間持続した。Day 1 の投与後、PT-INR が 3.0 以上の被験者は 25 名で、平均 7.5 時間持続した。Day 2 で PT-の投与後、PT-INR が 3.0 以上の被験 者は 8 名であった。Day 2 以降では、PT-INR が 3.0 以上の被験者は認められなかった。 リバーロキサバンは、第Xa因子活性、aPTT 及び ETP においてワルファリンの効果を増強させ た。両剤による効果は、第Xa因子活性では相加的で、aPTT 及び ETP でもおおむね相加的で あった。リバーロキサバン投与により生じた変化は、24 時間以内は可逆的であった。また、ワ ルファリン投与により生じた変化は、ワルファリンの最終投与後約 4 日間で回復した。 抗第Xa因子活性、HepTestⓇ及び PiCT では、ワルファリンの影響が認められなかった。ワル ファリンからリバーロキサバンへの切り替え後に認められた最大効果は、抗第Xa因子活性が ベースライン値の 16~23 倍の増加(相対変化率の中央値)、HepTestⓇがベースライン値の 2.15 倍の延長(Emax)、PiCT がベースライン値の 3.16 倍の延長(Emax)であった。抗第Xa因 子活性、HepTestⓇ及び PiCT の各試験は、ワルファリンの効果に影響されず、リバーロキサバ ン単独の効果を検出できる。 ワルファリン投与により、第Ⅶa因子活性はベースライン値の 0.15 倍に減少した。リバーロキ サバン単独投与でも、第Ⅶa因子活性はベースライン値の 0.74 倍に減少した(1/Emax)。ワル ファリン投与による活性減少に対するリバーロキサバンの相加作用は小さいが、これは試験に よる影響と考えられ、真の効果ではないと考えられた。 ワルファリン投与による第Ⅱa因子活性の変化に対するリバーロキサバンの相加作用は認めら れなかった。 以上より、ワルファリンからリバーロキサバンへの切り替えにおいて、薬力学的パラメータは 使用した評価法の種類に依存し、顕著な影響を受けたもの、相加的な影響を受けたもの、相加 的な影響を受けなかったものが認められた。 ワルファリンからリバーロキサバンへの切り替え後、先行投与したワルファリンは、リバーロ キサバンの薬物動態に影響しなかった。薬物動態学的パラメータのうち、特に AUC(0-24)及び Cmaxは、リバーロキサバン単独投与での投与初日と、ワルファリンから切り替え後のリバーロ キサバンの投与初日の間で同程度であった。 ワルファリンからリバーロキサバンへの切り替え後、リバーロキサバンは、ワルファリンの t1/2に影響しなかった。すなわち、R-ワルファリン及び S-ワルファリンの t1/2は、リバーロキ サバンへ切り替えた場合とプラセボへ切り替えた場合とで同程度であった。
2.7.6.42
【国外試験】健康男性被験者 12 名を対象とした第 I 相臨床試験(試験 11140)
[報告書番号 PH-33444]
治験の標題: 健康男性被験者 12 名を対象に BAY 59-7939 5mg 及び 30mg 投与した際のトロンビン生成に対する影響を 検討する無作為非盲検 2 期クロスオーバーパイロット試験 BAY 59-7939/Impact 011140 治験責任医師: 実施医療機関: , Germany 公表(参考文献): 報告書作成時においては該当なし 治験期間: 初回スクリーニング:20 年 月 日 最終来院日:20 年 月 日 開発のフェーズ: 第 I 相臨床薬理試験 目的: 血小板により誘導されるトロンビン生成及び乏血小板血漿(PPP)でのトロンビン生成に対して、第 Xa 因子活性阻害薬となる経口による BAY 59-7939 の影響に関するデータの作成 方法論(試験デザイン): 単施設 2 期クロスオーバー 無作為化 非盲検 被験者は BAY 59-7939 又はプラセボを投与されるよう無作為に割り付けられた。各治験薬は DAY 1 に 5mg 及び Day 14 に 30mg 又はその逆に投与した。 被験者数: 予定被験者数:12 名(最低 6 名の被験者に実薬を投与する) 登録被験者数:12 名 薬物動態解析の対象とされた被験者数:実薬を投与した 8 名 安全性解析の対象とされた被験者数:12 名(男性 実年齢幅:27~37 歳 年齢中央値:30 歳) 診断及び主な組み入れ基準: 健康男性被験者 年齢 18~45 歳 BMI 18~32kg/m2被験薬、用法及び用量、バッチ番号: BAY 59-7939 5.00mg 微粒剤 微結晶性セルロース 欧州薬局方、米国国民医薬品集、日本薬局方収載 40.0mg クロスカルメロースナトリウム 欧州薬局方、米国国民医薬品集収載 3.00mg ハイプロメルロース 欧州薬局方、米国薬局方収載 3.00mg ラクトース一水和物 欧州薬局方、米国国民医薬品集、日本薬局方収載 33.1mg ラウリル硫酸ナトリウム 欧州薬局方、米国国民医薬品集、日本薬局方収載 0.30mg ステアリン酸マグネシウム 欧州薬局方、米国国民医薬品集、日本薬局方収載 0.60mg ハイプロメルロース 15cP 欧州薬局方、米国薬局方、日本薬局方収載 マクロゴール/PEG 3.350 欧州薬局方、米国国民医薬品集、日本薬局方収載 二酸化チタン 欧州薬局方、米国薬局方、日本薬局方収載* E171 規格、投与単位:各錠剤 5mg 投与経路:経口 開発番号: 品目番号: ロット/バッチ番号:BX0003J 治療期間: 無作為割付に従い、Day 1 に 5mg 及び Day 14 に 30mg 又はその逆に投与。被験者は実薬又はプラセボの 投与を受けるように無作為割付された。 対照、用法及び用量、バッチ番号: プラセボ錠 微結晶性セルロース 欧州薬局方、米国国民医薬品集、日本薬局方収載 67,575mg ラクトース一水和物 欧州薬局方、米国国民医薬品集、日本薬局方収載 17.0mg ステアリン酸マグネシウム 欧州薬局方、米国国民医薬品集、日本薬局方収載 0.425mg ハイプロメルロース 15cP 欧州薬局方、米国薬局方、日本薬局方収載 マクロゴール/PEG 3.350 欧州薬局方、米国国民医薬品集、日本薬局方収載 二酸化チタン 欧州薬局方、米国薬局方、日本薬局方収載 E171 規格、投与単位:BAY 59-7939 5mg と同様 投与経路:経口 開発番号: 品目番号: バッチ番号:BX00042
評価項目:
薬力学的パラメータ:
第 Xa 因子活性阻害率、PT、aPTT、HepTest®、選択した時点における第 IIa 因子活性阻害率、アンチト ロンビン多血小板血漿(PRP)及びアンチトロンビン乏血小板血漿(PPP)により誘導される内因性トロ ンビン生成量(ETP)、血小板誘導トロンビン生成時間(PITT)及び Russel のマムシ毒による血液凝固 試験(Russel Viper Venom time)(プロトロンビナーゼ誘導凝固試験:PICT 試験)。BAY 59-7939 が 第 VII 因子活性により中和されるかどうかを検討するためin vitro試験を実施した。任意で抗第 Xa 因 子活性阻害試験を実施した。 薬物動態学的パラメータ: 主要評価項目:AUC、AUCnorm 副次評価項目:AUC/D、Cmax、Cmax,norm、Cmax/D、tmax、t1/2、MRT、CL/f、Vz/f、AUC(0-tn)、AUC(0-tn)norm その他の評価項目:AUC(tn-∞)、points terminal 安全性パラメータ: 有害事象、血圧、心拍数、心電図、臨床検査項目の評価 統計学的手法: 薬力学的解析の結果は適切な要約統計量及び図を用いて示した。所見がある場合は、更に探索的に検討 することとした。 薬物動態学的パラメータである AUC は、対数正規分布を仮定して解析した。このパラメータの対数値に ついて、シーケンス、被験者(シーケンス)、期間及び投与効果を含めて分散分析法(ANOVA)を用い て解析した。これらの解析に基づき、AUC 比に関する推定値(最小二乗平均)及び探索的な 90%信頼区 間は、ANOVA により得られた被験者内標準偏差を用いて、対数値を再変換して求めた。検証的な統計学 的解析は計画しなかった。 要約及び結論: 安全性: 全体で被験者 12 名が安全性解析の対象とされた。本治験において重篤な有害事象は発現しなかった。 被験者 1 名のみが 1 件の有害事象を報告し、それは治験薬と関連するかもしれないと考えられた軽度の 頭痛であった。当該事象は 30mg の投与の約 30 分後に発現し、8 時間後に回復した。 臨床検査項目の評価において、問題となる異常は認められなかった。 血圧、心拍数及び心電図パラメータは BAY 59-7939 の影響を受けなかった。
薬力学に関する概要: 血液凝固パラメータ(hemostaseologic parameters)の解析で、健康被験者において、BAY 59-7939 が 第 Xa 因子活性を阻害することにより、その結果としてトロンビンの生成が抑制されることが示されて いる。 他の試験で用いた方法により測定した第 Xa 因子活性阻害率は、先行する試験の結果よりも若干高かっ た。血液凝固試験(PT、aPTT、HepTest®)では、先行する試験で認められたのと同様の変動が認められ た。これらのパラメータと血漿中濃度の相関性は PT が最も高く(相関係数:0.97)、次いで第 Xa 因子 活性阻害率及び HepTest® (相関係数:0.93)、aPTT(相関係数:0.89)の順であった。 第 Xa 因子活性について更に調べるため、2 つの代替方法、すなわち PICT 及び抗第 Xa 因子を測定し た。予測どおり、いずれの方法でも第 Xa 因子活性の用量依存的な阻害が認められた。しかしながら、3 つの方法の間に血漿中 BAY 59-7939 濃度との相関性の差が認められた:相関性が最も高かったのは抗第 Xa 因子(相関係数:0.98)、次いで第 Xa 因子活性(相関係数:0.93)、若干精度の低いのが PICT(相 関係数 0.86)であった。 BAY 59-7939 は第 Xa 因子活性の直接阻害薬であるため、血漿中の遊離トロンビン(第 IIa 因子)の全 体的な活性又はアンチトロンビンレベルのいずれも変化が認められなかった。したがって、これらのパ ラメータと血漿中濃度には相関性がない。 しかしながら、本治験の目的は、コラーゲンなどの血小板活性化因子や組織因子が存在する場合に、血 小板により誘導されるトロンビン生成のうち、内因性トロンビン生成能(ETP)を測定することであっ た。 全体的な血液凝固系の能力指標としての内因性トロンビン生成量の AUC(ETP AUC)は、BAY 59-7939 が 存在する場合に生成されるトロンビンの総量に対して、用量依存的に低下した。この効果は、外因性経 路及び内因性経路を介したいずれの活性化の後にも認められた。影響は、組織因子がある場合に血漿中 BAY 59-7939 濃度とはっきりと相関しており(相関係数:-0.8)、コラーゲンが存在する場合には関連 はそれほど厳密なものではなかった(相関係数:0.6)。 内因性トロンビン生成量(ETP)のピークも、コラーゲン又は組織因子による活性化の後に BAY 59-7939 を投与すると、用量依存的に低下した。しかしながら、内因性トロンビン生成量(ETP) のピークと血漿中濃度の相関性は活性化因子に依存していた(相関係数 組織因子:-0.8、コラーゲ ン:-0.6)。内因性トロンビン生成量(ETP)のピーク最大効果は、組織因子の場合と比較すると、コ ラーゲンが使用された場合に大きく低下した。コラーゲンにより活性化された内因性トロンビン生成量 (ETP)のピーク効果が BAY 59-7939 30mg の投与後 24 時間目にベースライン値に戻らず、そのことが 抗血栓症効果の可能性をいくらか示唆していることに注目すべきである。 血小板により誘導されるトロンビン生成量の低下は、血小板誘導トロンビン生成時間(PITT)の評価に より更に示すことができ、その結果は血小板凝集開始までの時間及び血液凝固までの時間を測定した際 に用量依存的な延長が認められたというものであった。凝集までの PITT は、治験薬投与後 24 時間まで 延長したが、血液凝固までの PITT はそのときにはベースライン値に戻っていた。このことは、血小板 凝集力に対する BAY 59-7939 30mg の影響が延長する可能性を示しているかもしれない。凝集までの PITT 及び血液凝固までの PITT と血漿中 BAY 59-7939 濃度の相関性はむしろ低いものであったが、最も 顕著な延長は高い血漿中濃度と関連があった。
総じて、PITT 試験及び ETP 試験の結果は、BAY 59-7939 が血液凝固反応までの時間を遅延させ、トロン ビン生成のピーク効果、並びに凝固系が内因性及び外因性な血液凝固経路を介して活性化される際に生 成されるトロンビンの総量を減少させることを示している。すべての影響は用量依存的であったが、血 漿中濃度と厳密には相関していなかったことから、それらは BAY 59-7939 の影響をモニターできる可能 性を持つ最適なパラメータではない。内因性トロンビン生成量(ETP)ピーク及び凝集までの血小板誘 導トロンビン生成時間(PITT)は BAY 59-7939 30mg 投与後 24 時間目においてもまだ変動を示している が、その他のパラメータはすべてその時点までにベースライン値(又は生理学的範囲)に戻っていた。 これらの変動が BAY 59-7939 30mg の投与によって、24 時間を超えて影響を示すものかどうか、臨床試 験において更に検討する必要がある。 薬物動態に関する概要:
30mg 用量の AUC 及び Cmaxは、5mg 用量と比較して比例的より小さい程度に増加した。AUC/D
「30mg/5mg」の平均比は 74.5%で、90%信頼区間は 66.0~84.0%であった。Cmax/D に関して用量間の差 はより著しく、tmax中央値はいずれの投与量でも 1.5 時間であった。 結論: BAY 59-7939 の 5mg 及び 30mg の単回投与について良好な忍容性が認められた。 BAY 59-7939 は、測定方法(第 Xa 因子活性、プロトロンビナーゼ誘導凝固試験(PICT)、抗第 Xa 因子活性)にかかわらず、第 Xa 因子活性を用量依存的に阻害した。 BAY 59-7939 は、用量依存的に血液凝固パラメータ(PT、aPTT、HepTest®)を延長した。 BAY 59-7939 は、トロンビンの生成に関するラグタイムを遅延させ、総トロンビン生成量を減少さ せ、トロンビン生成のピーク(血小板誘導トロンビン生成時間(PITT)、内因性トロンビン生成量 の AUC(ETP AUC)、内因性トロンビン生成量(ETP)のピーク)を低下させた。血漿中のトロンビ ン活性及びアンチトロンビンは影響を受けなかった。 抗第 Xa 因子活性と血漿中濃度の相関が最も相関係数が高く、次が若干低い値を示した PT であっ た。 30mg 用量の AUC 及び Cmaxは、5mg と比較すると、比例的よりも小さい程度で増加した。
2.7.6.43
【国外試験】QT/QTc 評価試験(試験番号 11275)[報告書番号 PH-34050]
治験の標題: 健康男女被験者を対象に BAY 59-7939 15 及び 45mg を単回投与した際の QTc 間隔に対する影響を検討す るための、第Ⅰ相、無作為化、ダブルダミー法による二重盲検、プラセボ及び陽性対照、4 期クロス オーバー試験 BAY 59-7939/11275 治験責任医師: 実施医療機関: , Germany 公表(参考文献): 該当なし 治験期間: 20 年 月 日~20 年 月 日 開発のフェーズ: 第 I 相臨床薬理試験 目的: 主要目的: プラセボと比較して手動で読み取った QTc 間隔に対する BAY 59-7939 45mg の単回経口投与の影響 を除外すること(すなわち、影響がないことを証明すること) 副次目的: QTc 間隔評価の感受性を評価/検証するため、モキシフロキサシン 400mg の単回経口投与の QTc 間 隔に対する影響をプラセボと比較して特徴付けること BAY 59-7939 15mg の単回経口投与の QTc 間隔に対する影響をプラセボと比較して特徴付けること モキシフロキサシン 400mg、BAY 59-7939 15 及び 45mg の単回経口投与の QTc 間隔及び心拍数 (HR)に対する影響をプラセボと比較して特徴付けること BAY 59-7939 及びモキシフロキサシンの血漿中での曝露作用をそれぞれ特徴付けること BAY 59-7939 及びモキシフロキサシンへの曝露と心電図パラメータ(QTc 及び心拍数)の関連を調 べること 方法論(試験デザイン): 単施設 無作為化 二重盲検 ダブルダミー 4 期クロスオーバー プラセボ及び陽性対照(モキシフロキサシン 400mg)の単回投与及び 2 種類の BAY 59-7939(15 及 び 45mg)の単回投与 本治験は投与期間 1 日及び観察期間 2 日(0~2d)からなる各期の 4 期で計画され、投与と投与の 間には 7 日間以上の休薬期間を設けた。 年齢 50 歳以上の白人女性(出産不可能な人)及び白人男性被験者被験者数: 合計 145 名の被験者にスクリーニングを実施し、91 名が不適格又は待機被験者となったため 54 名が登 録された。男女被験者各 27 名が本治験に参加し、50 名が予定通り治験を完了した。被験者 3 名が治験 薬に関連しない重篤な有害事象のため、また 1 名が治験実施計画書からの逸脱のため早期に治験を中止 した。 診断及び主な組み入れ基準: 健康男女白人被験者、年齢 50 歳以上、BMI が 20~32kg/m2の標準体重、12 誘導心電図に臨床的に問題 となる所見がないこと 女性は出産不可能であること。 被験薬、用法及び用量、バッチ番号: BAY 59-7939: 経口投与 5mg 白色コーティング錠 バッチ番号:BX0060S 経口投与 プラセボ白色コーティング錠 バッチ番号:BX00042
BAY 59-7939 として 0、15 又は 45mg(BAY 59-7939 5mg 錠を 0、3 又は 9 錠)及び BAY 59-7939 5mg 錠 のプラセボを 9、6 又は 9 錠を、モキシフロキサシンのプラセボ 1 カプセルと共に、食後単回経口投与 する。 治療期間: 各被験者の試験期間は約 8~9 週間であった。 スクリーニング:治験薬の初回投与の前 14 日間以内 各期の投与:3 日間(0~2d) 投与と投与の間に 1 週間以上の休薬期間を設ける 試験終了時検査:治験薬の最終投与後 1~2 週間目 対照、用法及び用量、バッチ番号: モキシフロキサシン: 経口投与 赤褐色硬ゼラチンカプセル 400mg サイズ:DB AA バッチ番号:BX01G55 経口投与 プラセボ赤褐色硬ゼラチンカプセル サイズ:DB AA バッチ番号:BX004E7 モキシフロキサシンとして 400mg(1 カプセル)を BAY 59-7939 5mg 錠のプラセボ 9 錠と共に、食後単 回経口投与する。モキシフロキサシンのプラセボは 1 カプセルを BAY 59-7939 5mg 錠の実薬又はプラセ ボ合計 9 錠と共に、食後単回経口投与する。 評価項目: 薬力学的パラメータ: QT、QTcF(Fridericia の補正式で補正)、QTcI 薬物動態学的パラメータ: Cmax、Cmax,norm、tmax データがある場合、AUC、AUCnorm、AUC0-tn、t1/2、AUCtn-∞% その他の評価項目:AUCtn-∞%,points terminal 安全性及び血液凝固パラメータ: PT(INR)、aPTT、12 誘導心電図、バイタルサイン(血圧、PR)、臨床検査所見(尿検査含む)及び有 害事象
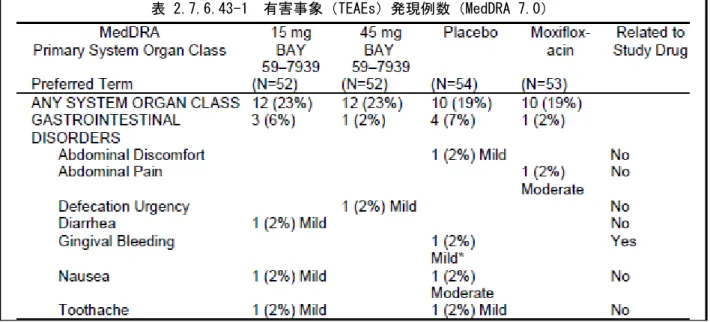
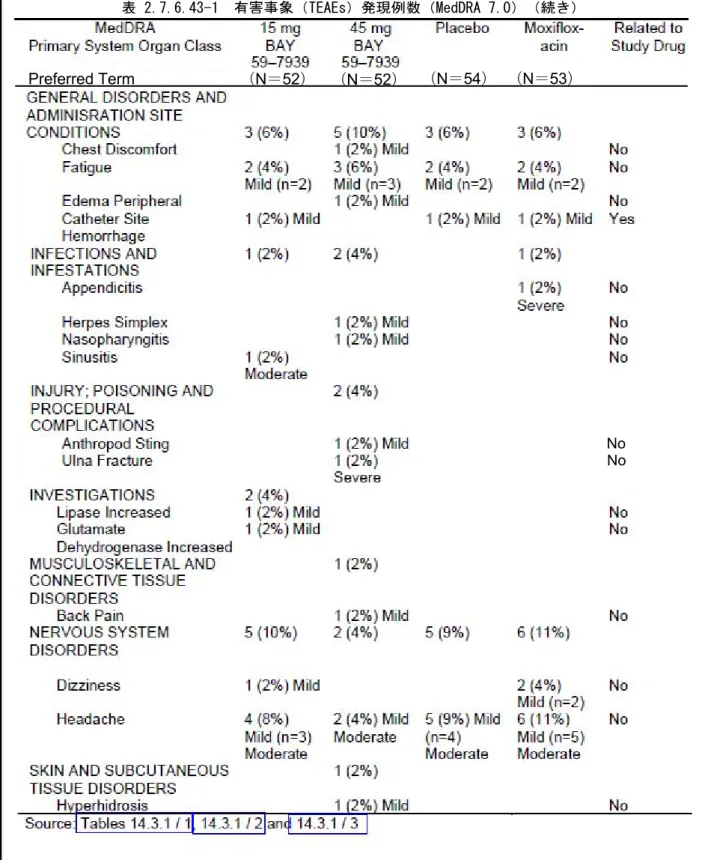
統計学的手法: 安全性及び血液凝固パラメータ: 安全性及び血液凝固パラメータは記述統計量を用いて表にまとめた。 薬力学(PD): 本治験は、主要評価項目に対する BAY 59-7939 45mg の影響を除外するようにデザインされた。 検討した帰無仮説及び対立仮説は、H0: µΤ-µЅ≧δ 及び H1: µΤ-µЅ<δ (ただし、µΤは 45mg 投与 後 3 時間目の QTc 変動の絶対値、µЅ はプラセボ投与後 3 時間目の QTc 変動の絶対値、δ は 10 ミリ秒に おける臨床的に問題となる差)であった。Fridericia の QT 補正式(QTcF=QT/RR1/3 )、すなわち QTcF を主要解析の基本とした。更に、QTc に関する解析も被験者別に補正した QT、すなわち QTcI を用いて 実施した。QTc 解析は、QT 及び RR の対数線形回帰モデルに基づいて実施した:各被験者について、プ ラセボのすべてのベースライン値及び投与後のすべての値を被験者別の対数線形回帰モデル(1n(QT)= a+b*1n(RR) ただし、RR=60/心拍数)に適用した。その場合、QTcI=QT/RRbで表した。 主要評価項目は、BAY 59-7939 45mg とプラセボの投与後 3 時間目における QTcF 変動の差、すなわち 「BAY 59-7939 45mg-プラセボ」の値であった。副次評価項目は、(i)BAY 59-7939 15mg とプラセ ボ、(ii)モキシフロキサシン 400mg とプラセボ、(iii)BAY 59-7939 45mg とモキシフロキサシン 400mg の QTcF 変動の差であった。BAY 59-7939 及びモキシフロキサシンの血漿中濃度と心電図パラメー タ(QTc 及び必要に応じてその他)の関係をグラフを用いて検討した。 薬物動態(PK): BAY 59-7939 又はモキシフロキサシンに関する濃度-時間推移を用量ごとにまとめた。標準統計量は血 液採取時点ごとに求めた。被験者別及び平均の濃度-時間推移は、線形及び片対数目盛りの両方を用い て用量ごとにプロットした。薬物動態学的パラメータ(tmaxを除く)は上記の統計量を用いてまとめ た。tmaxは最小値、最大値、中央値及び度数を表した。 要約及び結論: 安全性: 男女被験者各 27 名で実施した 4 期クロスオーバー試験において、BAY 59-7939 15 又は 45mg 及びモキ シフロキサシン 400mg を単回経口投与した際に、安全性及び良好な忍容性が認められた。 被験者 3 名が有害事象のため、1 名が計画書からの重大な逸脱のため脱落した。合計で被験者 54 名が 安全性の評価に組み入れられた。被験者 2 名から 2 件の重篤な有害事象が報告された:被験者 011275-10 は肘頭骨折、被験者 011275-51 は壊疽性虫垂炎を発現した。治験薬に関連すると考えられた事象は なく、すべて治験終了時までに回復した。 全体で被験者 25 名(46%)が 56 件の治験薬投与期間中に発現した有害事象を報告し、それらは各投与 期間に均一に分布していた(表 2.7.6.43-1 参照)。頭痛が最も高頻度に報告され、ほとんどの有害事 象は軽度であった。2 件の重篤な有害事象(虫垂炎及び肘頭骨折)及び 1 件の有意な有害事象(静脈洞 炎)のため、該当患者は治験を早期に中止したが、治験薬投与に関連する事象とは考えられなかった。 BAY 59-7939 投与後に有害事象として報告された 2 件の臨床検査異常値(リパーゼの 95U/L から 670U/L への上昇及び GLDH 活性の 5.0U/L から 18.3U/L への上昇。いずれも治験薬投与に関連なし)及び血液凝 固パラメータの予測された変動を除き、臨床検査項目又はバイタルサインに関して臨床的に問題となる 変動は認められなかった。
表 2.7.6.43-1 有害事象(TEAEs)発現例数(MedDRA 7.0)(続き)
(N=52) (N=52) (N=54) (N=53) Preferred Term
No No
薬力学: 主要な解析結果から、投与後 3 時間目の QTcF に対する BAY 59-7939 45mg の影響が認められないことが はっきりと示された。実際の試験データでは、プラセボとの QTcF の差の平均(95%信頼区間)は-0.91 ミリ秒(-3.33~1.52)で、ICH E14 のガイダンスに記載されているとおり薬剤の QTc 延長効果をはっ きりと除外しており、BAY 59-7939 による影響は認められない。 副次的な薬力学的評価項目は、「BAY 59-7939 15mg-プラセボ」、「モキシフロキサシン 400mg-プラ セボ」、「BAY 59-7939 45mg-モキシフロキサシン 400mg」、「BAY 59-7939 45mg 対 BAY 59-7939 15mg」で求めた投与後 3 時間の値、tmax、投与後平均値及び投与後最大値の変動であった。QTcF に加え て、被験者別の QTcI 及び未補正の QT も評価した。 BAY 59-7939 の用量 45 及び 15mg に関して、プラセボと比較した QTcF、QTcI 及び QT の全変動平均は、 95%信頼区間を含めて 5 ミリ秒未満であった。これらの副次的評価項目はすべてプラセボを投与した場 合と差が認められず、用量相関的な変動は認められなかった。対照的に、陽性対照のモキシフロキサシ ン 400mg に対する反応が、同じ被験者において一貫して認められた。投与後 3 時間の値、tmax、投与後 平均値及び投与後最大値における評価で得られた QTcF、QTcI 及び QT の平均をプラセボと比較すると、 モキシフロキサシンの投与後ですべて 5 ミリ秒を超えていた。更に、QTcF 及び QTcI の投与後の平均 (7.64 ミリ秒及び 7.67 ミリ秒)を除き、ほとんどすべての 95%信頼区間の上限値が 8 ミリ秒を超えて いた(表 2.7.6.43-2 参照)。 いずれの投与に関しても 500 ミリ秒を超えた QT、QTcF 又は QTcI 間隔はなかった。QT、QTcF 及び QTcI 間隔の絶対値は、BAY 59-7939 及びプラセボで同等であるか、又は延長は治験薬投与前に認められた。 モキシフロキサシン投与後の QT、QTcF 又は QTcI 間隔の延長は、BAY 59-7939 投与後と比較すると明ら かに大きかったが、先行する試験で認められた範囲内の程度であった(表 2.7.6.43-3 参照)。いずれ の投与に関しても、ベースライン値からの QT、QTcF 又は QTcI 間隔の変動は 60 ミリ秒以下であった。 BAY 59-7939 及びプラセボの投与では、ベースライン値からの QT、QTcF 又は QTcI 間隔の変動は 30 ミ リ秒未満であった(表 2.7.6.43-4 参照)。 モキシフロキサシン投与後の QT 変動に関してよく反応が認められた被験者の結果は、BAY 59-7939 の 15 又は 45mg の単回投与により QT/QTc が変動しないことを示している。モキシフロキサシン投与後で は QT/QTc の濃度依存的な変動が認められたが、薬物動態/薬力学的解析において BAY 59-7939 投与後 では QT/QC が用量比例的に変動しないことが、QT/QTc が濃度依存的に変動しないことにより、実証 された(図 2.7.6.43-1 参照)。
表 2.7.6.43-2 主要及び副次的解析による QTcF における調整済平均に基づいた ANCOVA 群間比較
表 2.7.6.43-4 Fridericia の QT 補正式―分類されたベースラインからの相対変化量[全被験者] 図 2.7.6.43-1 BAY 59-7939 の濃度[µg/L]及び Fridericia の QT 補正式[ms]間の相関関係 薬物動態: 薬物動態学的パラメータである tmax、Cmax及び AUC の値は、先行する試験で認められた値の範囲内によ く収まっていた。BAY 59-7939 の薬物動態評価項目に関する幾何平均/幾何 CV%(最小値―最大値)を 表 2.7.6.43-5 に示す。
表 2.7.6.43-5 BAY 59-7939 15mg、45mg 経口単回投与時及びモキシフロキサシン 400mg 経口単回投与時の血漿中薬物動態学的パラメータ
結論: BAY 59-7939 の 15 又は 45mg 及びモキシフロキサシンの単回投与について安全性及び良好な忍容性 が認められた。 BAY 59-7939 及びモキシフロキサシンの薬物動態学的パラメータは先行する試験の結果と同等で あった。 本治験の主要目的に関して、BAY 59-7939 45mg の単回投与は QTcF 間隔に影響を及ぼさなかった。 投与後 3 時間目におけるプラセボとの QTcF の差は-0.91 ミリ秒で、95%信頼区間最大値の差は 2 ミリ秒未満であった。 本治験の副次目的に関しては、薬物動態評価項目の tmaxにおける QTcF、QTcI 及び QT 解析、並びに QTcF の平均値及び最大値に関する投与後の変動は BAY 59-7939 15mg 及び 45mg のいずれの投与に おいても用量依存的な QTcF 延長を示さなかった。95%信頼区間を含む最小二乗平均の変動はすべ て 5 ミリ秒未満であった。 QT 変動によく反応が認められたことが、陽性対照であるモキシフロキサシンの投与後に、QTcF、 QTcI 及び未補正の QT に関する平均変動が 5 ミリ秒超、95%信頼区間最大値が 10 ミリ秒超であっ たことによりはっきりと示された。モキシフロキサシンについて認められた全体的な変動は、先行 する試験で認められた結果と同等であった。 いずれの投与に関しても QT、QTcF 又は QTcI 間隔は 500 ミリ秒以下であった。QT、QTcF 及び QTcI 間隔のその他の項目は BAY 59-7939 及びプラセボのいずれの投与量についても同等か、又は治験薬 投与前に認められた。モキシフロキサシン投与後の QT、QTcF 又は QTcI 間隔の延長は、 BAY 59-7939 投与後と比較すると明らかに大きかったが、先行する試験で認められた範囲内の程度 であった。 いずれの投与に関しても、ベースライン値からの QT、QTcF 又は QTcI 間隔の変動は 60 ミリ秒以下 であった。BAY 59-7939 及びプラセボの投与では、ベースライン値からの QT、QTcF 又は QTcI 間隔 の変動は 30 ミリ秒未満であった。 BAY 59-7939 投与の QTcF に対する濃度依存的な影響は認められなかった。 したがって、BAY 59-7939 15mg 又は 45mg の単回投与は、QT/QTc に影響を及ぼさなかった。実薬 対照のモキシフロキサシンは、5 ミリ秒超の QTc 変動及び 8 ミリ秒超の各信頼区間を検出すること により、試験の感受性を証明した。
2.7.6.44
【国内試験】心房細動患者を対象とした第Ⅱ相臨床試験(試験番号 12024)
[報告書番号 MRR-00267]
2.7.6.44.1
試験計画
治験の標題:リバーロキサバン(第Ⅹa 因子阻害薬)の心房細動に対する低用量第Ⅱ相臨床試験 治験調整医師:該当なし 治験責任医師: 、 、 、 、 、 、 、 、 実施医療機関:本治験は国内の 10 施設において実施された。 公表論文:なし 治験期間: 最初の患者の組み入れ日:20 年 月 日 最後の患者の最終来院日:20 年 月 日 開発のフェーズ:第Ⅱ相 目的:心房細動患者を対象に、リバーロキサバン(BAY 59-7939)2.5mg、5mg、10mg の 1 日 2 回投 与における薬物動態と薬力学的効果との関係を探索的に検討すること、及び安全性を予備的に検討 すること。 試験方法: 本治験は、ワルファリンを対照としてリバーロキサバン(2.5mg、5mg 及び 10mg 1 日 2 回反復経口 投与の 3 用量群)の薬物動態、薬力学的効果及び安全性を検討する非盲検、無作為化、並行群間比 較試験である。薬物動態及び薬力学的効果の検討はリバーロキサバン投与群のみで行った。 ワルファリンは 1 日 1 回経口投与とし、その投与量は心房細動治療(薬物)ガイドライン(2001 年)に従って、プロトロンビン時間国際標準比(PT-INR)により調整した。すなわち、69 歳以下で 塞栓症の危険因子(高血圧、糖尿病、冠動脈疾患、うっ血性心不全)を少なくとも 1 つ有している 患者は PT-INR の 2.0~3.0 を目標に調整し、それ以外(70 歳以上で塞栓症の危険因子を少なくとも 1つ有している患者、又は 60 歳以上で塞栓症の危険因子を有さない患者)の場合は PT-INR の 1.6 ~2.6 を目標に調整した。 治験薬投与期間は 28 日間で、各投与群 25 例、合計 100 例の被験者について検討を行うこととし た。 診断と選択基準: 非弁膜症性心房細動患者 選択基準 過去 2 回以上の心房細動発作があり、少なくとも 1 回は無作為割り付け前 4 週間以内に心電図によ り心房細動が確認された、発作性あるいは慢性の非弁膜症性心房細動患者。 (1) 塞栓症発症リスクが高く抗凝固療法が必要と考えられる以下の患者 塞栓症の危険因子(高血圧、糖尿病、冠動脈疾患、うっ血性心不全)を少なくとも 1 つ 有している患者 60 歳以上の患者(上記の危険因子の有無を問わない) (2) 本治験への参加について文書による同意が得られた成人男性及び閉経後の女性。 除外基準(1) 脳梗塞あるいは一過性脳虚血発作(TIA)の既往を有する患者、又は合併している患者。 (2) 出血の既往又は合併:無作為割り付け前 6 ヵ月以内に脳出血、眼内出血あるいは消化管出血 の既往を有する患者、又は合併している患者。 (3) 無作為割り付け時に以下の出血を有する患者 血小板減少性紫斑病 喀血 肉眼的血尿 便潜血(便ヘモグロビン)検査陽性、など (4) 無作為割り付け時に以下の出血リスクを有する患者 無作為割り付け前 1 ヵ月以内の中枢神経系の外科手術又は外傷 高い出血リスクがある消化器疾患(炎症性腸疾患、びらん性胃炎など) 細菌性心内膜炎 コントロール不良の高血圧〔収縮期血圧(SBP)>180mmHg 又は拡張期血圧(DBP)> 95mmHg〕 コントロール不良の糖尿病(HbA1c≧8.0%) 先天性又は後天性出血性素因〔プロトロンビン時間(PT)及び活性化部分トロンボプラ スチン時間(aPTT)がいずれも正常範囲上限を超える〕 血小板数が 100,000/μL 未満、など (5) 治験薬の吸収が損なわれる可能性がある胃腸疾患(例:重度活動性炎症性腸疾患、短腸症候 群)を合併している患者。 (6) 肝障害(トランスアミナーゼ値が正常範囲上限の 2 倍を超える)、又は腎障害(血清クレア チニン値が正常範囲上限の 1.5 倍を超える)を合併している患者。 (7) 悪性腫瘍(現在の病変)を合併している患者。 (8) 体重が 45kg 未満の患者。 (9) 薬物又はアルコール依存症と診断された患者。 (10) 無作為割り付け時に PT-INR が 1.5 を超えるワルファリン治療を行っている患者(ワルファ リン投与を受けている場合はワルファリン投与の中断又は減量を行い、PT-INR が 1.5 以下 になった時点で無作為割り付けを行う)。 (11) 無作為割り付け前 1 週間以内にワルファリン以外の抗凝固薬(ヘパリン製剤など)を投与し た患者、あるいは線維素溶解療法(アルテプラーゼ、ウロキナーゼなど)を受けた患者。 (12) 無作為割り付け前 1 週間以内にアスピリン又はその他の抗血小板薬(チクロピジン、ジピリ ダモールなど)を投与していた患者。 (13) 無作為割り付け時にその他血液凝固系に影響を及ぼす可能性のある薬剤を使用していた患者 (半減期が 17 時間以下の NSAIDs は使用可能)。 (14) 無作為割り付け前 3 日以内に全身又は局所投与によりアゾール系薬剤(ケトコナゾール、フ ルコナゾール及びイトラコナゾールなど)の投与を受けた患者。 (15) 上記以外にワルファリン投与が禁忌とされる患者。 (16) 電気的除細動あるいは大規模な手術が予定されている患者。 (17) 無作為割り付け前 1 ヵ月以内に他の薬剤の治験に参加した患者。 (18) 他の治験に参加している患者。 (19) 治験責任医師又は治験分担医師が不適格と判断した患者。 有効性の評価項目: 有効性の評価は行わなかった。 なお、血栓形成の抑制に関する代替マーカーを検討する目的で、凝固線溶系検査(D-ダイマー、ト ロンビン・アンチトロンビンⅢ複合体、プロトロンビンフラグメント F1+2 及びトロンビン生成量) を実施した。 安全性の評価項目: 安全性の評価項目は以下のとおりとした。
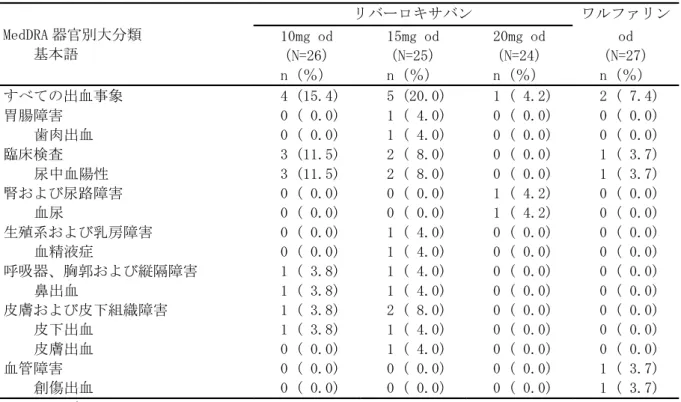
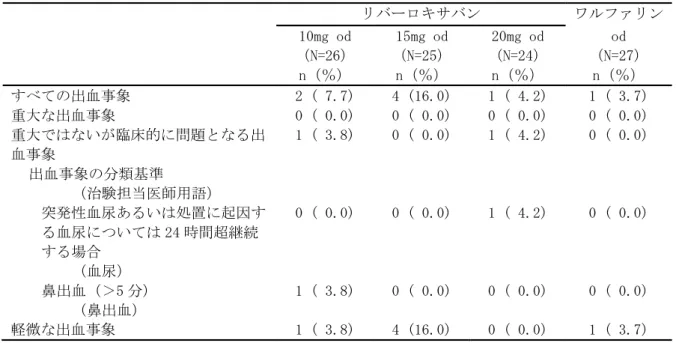
• 出血事象 • 有害事象(出血事象、脳梗塞、肺塞栓症を含む) • 重篤な有害事象(出血事象、脳梗塞、肺塞栓症を含む) • 臨床検査値 臨床的に明らかな出血事象を、1)重大な出血事象、2)重大ではないが臨床的に問題となる出血事 象、及び 3)軽微な出血事象の 3 つに分類した。その基準は以下のとおりとした。 (1) 重大な出血事象:臨床的に明らかな以下の出血 ・ 2g/dL 以上のヘモグロビン量の低下を伴う出血 ・ 2 単位以上の輸血(濃厚赤血球又は全血)が必要な出血 ・ 重要な臓器における出血、頭蓋内出血、後腹膜出血又は心膜出血 ・ 死亡に至った出血 (2) 重大ではないが臨床的に問題となる出血事象:臨床的に明らかな以下の出血 ・ 血行動態に影響するすべての出血 ・ 入院を必要とするすべての出血 ・ 25cm2以上の皮下血腫 ・ 筋肉内血腫 ・ 5 分以上継続、24 時間以内に反復して発生した、又は電気凝固などの処置を必要とした 鼻出血 ・ 自然発生した(食事や歯磨きに関連のない)又は 5 分以上継続した歯肉出血 ・ 肉眼的血尿 ・ 潜血を伴う下血、吐血などの臨床的に明らかな肉眼的消化管出血 ・ 直腸出血(トイレットペーパーに点状を超える大きさの出血を認めるもの) ・ 喀血(喀痰中に点状を超える大きさの出血を認めるもの) ・ 被験者に臨床的な影響を及ぼすその他の出血 (3) 軽微な出血事象 ・ 上記の基準を満たさない、すべての出血事象 なお、これらの基準による出血事象の分類は、出血事象判定委員会が実施した。 安全性主要評価項目は、治験薬投与開始後から投与終了後 7 日目までの間に認められた「臨床的に 問題となる出血事象」(「重大な出血事象」と「重大ではないが臨床的に問題となる出血事象」) の頻度とした。なお、この期間の後に発現した「臨床的に問題となる出血事象」は別途評価を行っ た。 薬物動態及び薬力学的効果の評価項目: 薬物動態: ・薬物濃度より、薬物動態パラメータとして全身クリアランス(CL/f)等を算出することとした。 薬力学的効果: ・薬力学的効果パラメータとして、第Ⅹa 因子活性、PT、PT-INR、aPTT、Heptest を測定した。 薬物動態と薬力学的効果に関する検討: ・薬物動態と薬力学的効果との関連を明らかにするために、NONMEM 等の母集団解析による手法を用 い検討することとした。 症例数: 計画時:100 例 症例数の設定根拠 本治験では、統計学的考察に基づいた被験者数の算出は行わなかった。日本人の心房細動患者に対 して、本薬を 1 日 2 回投与した際の薬物動態と薬力学的効果との関係を探索的に検討し、安全性を 予備的に評価するために、安全性解析対象例として各投与群で少なくとも 25 例、計 100 例程度が必 要と判断した。 解析時:121 例の被験者が組み入れられ、100 例が治験薬の投与を受けた。
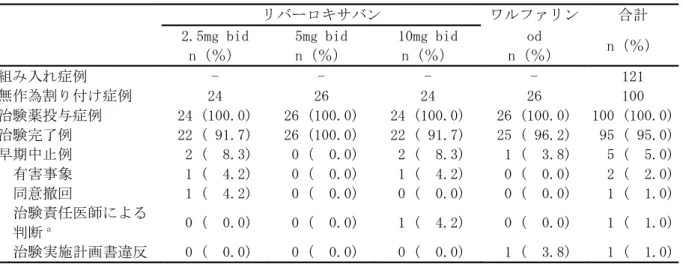
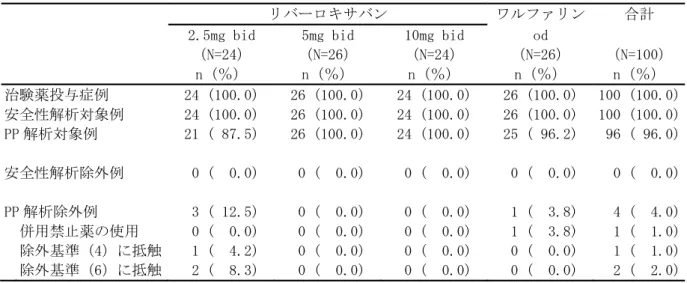

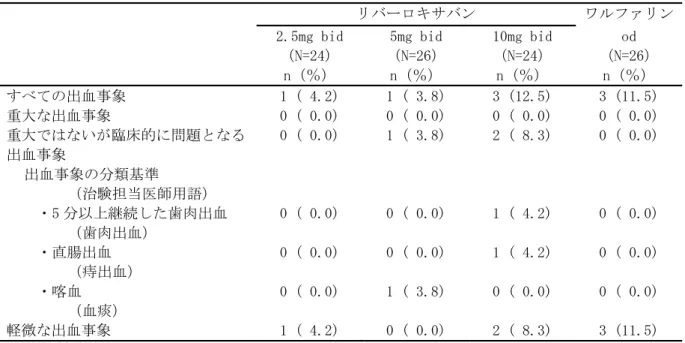
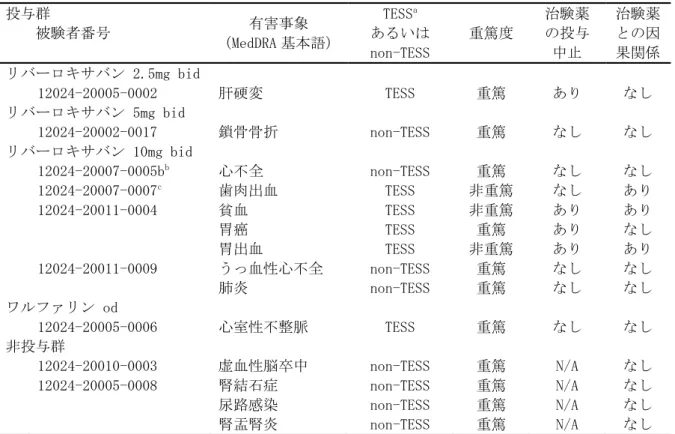
 n 中央値 範囲 リバーロキサバン 2.5mg bid 来院 1 ベースライン 21 0.410 0.190 ~ 9.710 来院 2 投与前 21 0.440 0.210 ~ 3.160 来院 2 投与 2 時間後 21 0.350 0.160 ~ 1.150 来院 3 投与前 20 0.415 0.120 ~ 1.090 来院 3 投与 2 時間後 20 0.355 0.1](https://thumb-ap.123doks.com/thumbv2/123deta/8704248.959424/49.892.102.789.191.613/プロトロンビンフラグメント中央値リバーロキサバンベースライン.webp)