2.7.4 臨床的安全性の目次 2.7.4 臨床的安全性の目次 ... 1 2.7.4.1 医薬品への曝露 ... 6 2.7.4.1.1 総括的安全性評価計画及び安全性試験の記述 ... 6 2.7.4.1.1.1 安全性評価に用いた臨床試験の概観 ... 6 2.7.4.1.2 全般的な曝露状況 ... 20 2.7.4.1.2.1 第Ⅰ相・臨床薬理試験 ... 20 2.7.4.1.2.2 第Ⅱ相試験 ... 21 2.7.4.1.2.3 第Ⅲ相試験 ... 22 2.7.4.1.3 治験対象集団の人口統計学的特性及びその他の特性 ... 32 2.7.4.1.3.1 第Ⅰ相・臨床薬理試験 ... 32 2.7.4.1.3.2 第Ⅱ相試験 ... 35 2.7.4.1.3.3 第Ⅲ相試験 ... 36 2.7.4.2 有害事象 ... 51 2.7.4.2.1 有害事象の解析 ... 51 2.7.4.2.1.1 比較的よくみられる有害事象 ... 53 2.7.4.2.1.2 死亡 ... 105 2.7.4.2.1.3 その他の重篤な有害事象 ... 121 2.7.4.2.1.4 その他の重要な有害事象 ... 134 2.7.4.2.1.5 器官別又は症候群別有害事象の解析 ... 241 2.7.4.2.2 個別有害事象の文書による説明 ... 243 2.7.4.2.2.1 死亡、その他の重篤な有害事象 ... 243 2.7.4.3 臨床検査値の評価 ... 243 2.7.4.4 バイタルサイン、身体的所見及び安全性に関連する他の観察項 目 ... 248 2.7.4.4.1 バイタルサイン ... 248 2.7.4.4.2 心電図 ... 248 2.7.4.5 特別な患者集団及び状況下における安全性 ... 253 2.7.4.5.1 内因性要因 ... 253 2.7.4.5.1.1 小児 ... 253 2.7.4.5.1.2 高齢者 ... 253 2.7.4.5.1.3 性別 ... 253
2.7.4.5.1.4 体重 ... 254 2.7.4.5.1.5 人種 ... 254 2.7.4.5.1.6 肝障害 ... 254 2.7.4.5.1.7 腎障害 ... 255 2.7.4.5.2 外因性要因 ... 257 2.7.4.5.3 薬物相互作用 ... 257 2.7.4.5.4 妊娠及び授乳時の使用 ... 257 2.7.4.5.5 過量投与 ... 258 2.7.4.5.6 薬物乱用 ... 258 2.7.4.5.7 離脱症状及び反跳現象 ... 258 2.7.4.5.8 自動車運転及び機械操作に対する影響又は精神機能の障害 ... 259 2.7.4.6 市販後データ ... 260 2.7.4.6.1 (試験 13802;第Ⅳ相製造販売後調査)... 260 2.7.4.6.1.1 試験デザイン ... 260 2.7.4.6.1.2 曝露状況 ... 260 2.7.4.6.1.3 有害事象 ... 261 2.7.4.6.1.4 死亡 ... 264 2.7.4.6.2 自発報告 ... 264 2.7.4.6.2.1 曝露状況 ... 265 2.7.4.6.2.2 総括 ... 265 2.7.4.6.2.3 重篤な有害事象 ... 265 2.7.4.6.2.4 死亡 ... 266 2.7.4.6.2.5 重篤な有害事象 ... 266 2.7.4.6.2.6 出血関連の重篤な有害事象 ... 267
略語一覧
略 語 英 語 名 称 日 本 語 名 称
AE adverse event 有害事象
ALP alkaline phosphatase アルカリホスファターゼ
ALT alanine aminotransferase アラニン・アミノトランスフェラーゼ
ASA aspirin アスピリン
AST aspartate aminotransferase アスパラギン酸アミノトランスフェラーゼ AUC area under the curve 曲線下面積
bid bis in die 1 日 2 回
BILDIR direct bilirubin 直接ビリルビン BMI body mass index 体格指数 BUN blood urea nitrogen 血中尿素窒素 CABG Coronary artery bypass graft 冠状動脈バイパス術 CCUS complete compression ultrasonography 圧迫法による超音波検査 CEC Clinical Endpoint Committee 評価項目判定委員会 CHADS2 An Assessment Score For The Risk Of
Stroke In Patients With Atrial
Fibrillation Incorporating These Risk Factors: Congestive Heart Failure, Hypertension, Age, Diabetes And History Of Stroke Or TIA
(うっ血性)心不全、高血圧症、年齢、糖 尿病及び、脳梗塞又は TIA の既往歴の危険 因子を含む心房細動患者の脳卒中リスクの 評価スコア
CHF congestive heart failure (うっ血性)心不全 CI confidence intervals 信頼区間
CLCR creatinine clearance クレアチニンクリアランス
Cmax maximum observed plasma concentration 最高血漿中濃度
CMV cytomegalovirus サイトメガロウィルス CNS central nervous system 中枢神経系
COX-2 Cyclooxygenase-2 シクロオキシゲナーゼ 2 CSR clinical summary report 治験総括報告書
CYP3A4 cytochrome P450 3A4 シトクローム P450
Den denominator 分母
DVT deep vein thrombosis 深部静脈血栓症
EBV Epstein-Barr Virus エプスタインバーウィルス ECG electrocardiogram 心電図
eDISH evaluation of drug induced severe hepatotoxicity
重篤な薬物肝毒性の評価
EOS end of study 治験薬投与終了
ESMD early study medication discontinuation 治験薬投与早期中止 FDA Food and Drug Administraion (米国)食品医薬品局 FPV first patient visit 最初の被験者の初回来院日
Fu follow up フォローアップ
GGT gamma glutamyl transpeptidase γ-グルタミルトランスフェラーゼ
Hb hemoglobin ヘモグロビン
HEAC Hepatic Event Assessment Committee 肝臓関連事象評価委員会 HIV Human Immunodeficiency Virus ヒト免疫不全ウイルス
HR hazard ratio ハザード比
Ht hematocrit ヘマトクリット
ICH International Conference On Harmonisation
日米 EU 医薬品規制調和国際会議 IDMC Independent Data Monitoring Committee 独立データモニタリング委員会 INR international normalized ratio (プロトロンビン時間の)国際標準比 ITT intent-to-treat 包括解析
J- ROCKET-AF
Japan Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonist For The Prevention Of Stroke And Embolism Trial In Atrial Fibrillation
-
K potassium カリウム
LAP Liver advisory panel 肝臓諮問委員会 LMW low molecular weight 低分子量
LMWH low molecular weight heparin 低分子量ヘパリン
LPV last patient visit 最終投与被験者の最終来院日 MAH marketing authorization holder 医薬品市販承認取得者 MedDRA Medical Dictionary for Regulatory
Activities
ICH 国際医薬用語集 MRI magnetic resonance imaging 磁気共鳴共鳴法 MSSO Maintenance And Support Services
Organization
国際維持管理機関
Na sodium ナトリウム
NSAIDs nonsteroidal anti-inflammatory drugs 非ステロイド性解熱鎮痛消炎薬
Num numerator 分子
NVAF non-valvular atrial fibrillation 非弁膜症性心房細動 NYHA New York Heart Association ニューヨーク心臓協会
od once daily 1 日 1 回
PAI Platelet aggregation inhibitors 血小板凝集阻害薬
PCI percutaneous coronary intervention 経皮的冠動脈インターベンション PD pharmacodynamic 薬力学
PE pulmonary Embolism 肺塞栓症 P-gp p-glycoprotein P-糖たん白 PK pharmacokinetic 薬物動態
POC Point of Care ポイント オブ ケア PPI proton pump inhibitor プロトンポンプ阻害薬 PSUR periodic safety update report 定期的安全性最新報告 PT prothrombin time プロトロンビン時間
PT-INR prothrombin time-international normalized ratio
プロトロンビン時間の国際標準比 QTc corrected QT interval 補正 QT 間隔
RBC red blood cell count 赤血球数 ROCKET
AF
Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonist For The Prevention Of Stroke And Embolism Trial In Atrial Fibrillation
-
SD Standard deviation 標準偏差 SGOT serum glutamic oxaloacetic
transaminase
アスパラギン酸アミノトランスフェラーゼ SGPT serum glutamic pyruvic transaminase アラニン・アミノトランスフェラーゼ SMQ standardized MedDRA queries Meddra 標準検索式
TBL total bilirubin 総ビリルビン TDD total daily dose 1 日用量
TIA transient ischemic attack 一過性脳虚血発作 TTR time in therapeutic range 目標範囲内期間割合 ULN upper limit of normal 正常範囲上限 VKA vitamin K antagonist ビタミン K 拮抗薬 VTE venous thromboembolism 静脈血栓塞栓症 WBC white blood cell 白血球
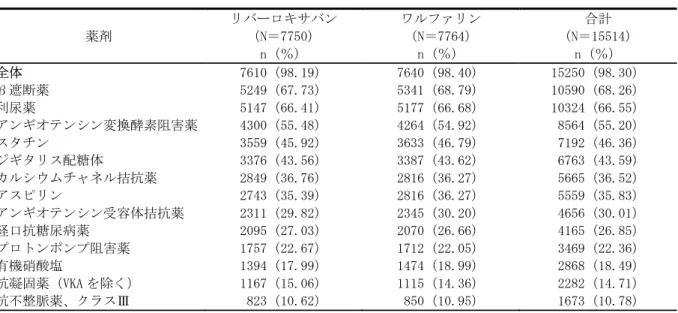
2.7.4.1 医薬品への曝露 2.7.4.1.1 総括的安全性評価計画及び安全性試験の記述 2.7.4.1.1.1 安全性評価に用いた臨床試験の概観 安全性評価には第Ⅰ相・臨床薬理試験 59 試験に対する統合解析(国内実施 5 試験、国外実施 54 試験)、統合解析に含まれない臨床薬理試験 2 試験、第Ⅱ相試験〔日本人非弁膜症性心房細 動(NVAF)患者を対象とした国内第Ⅱ相試験 3 試験、急性症候性深部静脈血栓症(DVT)患者を 対象とした国外第Ⅱ相試験 2 試験、国外第Ⅱ相試験 2 試験の統合解析)、第Ⅲ相試験(日本人 NVAF 患者を対象とした国内第Ⅲ相試験 1 試験、非日本人 NVAF 患者を対象とした国外第Ⅲ相試験 1 試験、国内・国外第Ⅲ相試験の統合解析)、肝臓安全性に関する統合解析を用いた(表 2.7.4.1-1)。 表 2.7.4.1-1 安全性評価に用いた試験及び解析 対象被験者 実施場所 安全性解析対 象被験者数 総括報告書 番号 参照モ ジュール 第Ⅰ相・臨床薬理試験 統合解析 試験によって異なる 国内・ 国外 1,516 例(日 本人 190 例) PH-36320 5.3.5.3.3 試験 14883 日本人健康成人男子 国内 36 例 5.3.3.4.21 試験 12606 非日本人健康成人男子 国外 13 例 PH-36370 5.3.3.4.20 第Ⅱ相試験 試験 11390 日本人 NVAF 患者 国内 36 例 MRR-00199 5.3.5.2.1 試験 12024 日本人 NVAF 患者 国内 100 例 MRR-00267 5.3.5.1.1 試験 11866 日本人 NVAF 患者 国内 102 例 MRR-00297 5.3.5.1.2. 試験 11223 非日本人急性症候性 DVT 患者 国外 604 例 MRR-00150 5.3.5.4.1. 試験 11528 非日本人急性症候性 DVT 患者 国外 542 例 MRR-00223 5.3.5.4.2 試験 11223 と試験 11528 統合解析 非日本人急性症候性 DVT 患者 国外 1,146 例 MRR-00300 5.3.5.3.4 第Ⅲ相試験 試験 12620 日本人 NVAF 患者 国内 1,278 例 A49701 5.3.5.1.3 試験 11630 非日本人 NVAF 患者 国外 14,236 例 R-8570 5.3.5.1.6 試験 12620 と試験 11630 の統合解析 NVAF 患者 国内・ 国外 15,514 例 R-8603 5.3.5.3.1 その他 肝臓安全性統合解析 試験によって異なる 国内・ 国外 - R-8569 5.3.5.3.2 NVAF:非弁膜症性心房細動、DVT:深部静脈血栓
2.7.4.1.1.1.1 第Ⅰ相・臨床薬理試験 国内外で実施された 59 の第Ⅰ相・臨床薬理試験〔試験 10842、10846、10847、10848、10849、 10850、10924、10989、10990、10991、10992、10993、10996、10997、10998、10999、11000、 11001、11002、11003、11032、11123、11124、11125、11126*、11127*、11140、11197、11273、 11275、11279、11321、11322、11325*、11529、11568、11569、11585、11608、11609、11708、 11864、11865、11935、11936、11937、11938、11940、12026*、12089、12090、12359、12362、 12612、12680、13371*、14022、14588、15232(*:国内実施試験)〕の被験者のデータを統合し て安全性の解析を行った(5.3.5.3.3 PH-36320)。1,516 例(内日本人被験者数 190 例)に治験 薬の投与が行われた〔リバーロキサバン:1,335 例(日本人被験者 160 例)、プラセボ:181 例 (日本人被験者 30 例)〕。 治験薬投与下に発現した有害事象及び臨床検査値を評価した。治験薬投与開始日から最終投与 後 30 日目までに発現した有害事象を治験薬投与下に発現した有害事象と定義した。クロスオー バーデザインの試験については、有害事象の発現の直近に投与された治験薬(用量)と関連付け ることとし、最後の治験薬の最終投与後 30 日目までに発現した事象を対象とした。データの欠 落、不完全等で事象発現が治験薬投与下であるか否かが不明の場合は、治験薬投与下に発現した 事象として取り扱うこととした。 上述の統合解析に含まれない以下の 2 つの臨床薬理試験(試験 14883、試験 12606)について も、安全性評価を行った。 試験 14883 は日本人男性健康被験者を対象にワルファリンからリバーロキサバン 15mg に切換 えた際の薬力学的作用、薬物動態、安全性及び忍容性を検討することを目的とした無作為化、単 盲検(A、B 群間のみ)、並行群間比較試験である。ワルファリンを 6~10 日間投与しプロトロ ンビン時間国際標準比(PT-INR 又は INR)2.0~3.0 に達した後、被験者を A:リバーロキサバン 15mg 1 日 1 回 4 日間投与群、B:リバーロキサバンプラセボ 1 日 1 回 4 日間投与群、又は、C:ワ ルファリン前治療なしでリバーロキサバン 15mg 1 日 1 回 4 日間投与群の 3 つの群に無作為に割 り付けた。有害事象、臨床検査値、バイタルサイン、心電図を評価した。治験薬投与開始日から 最終投与後 30 日目までに発現した有害事象を治験薬投与下に発現した有害事象と定義した。A、 B 群については、ワルファリン投与中に発現した有害事象は別途集計した。治験薬が投与された 36 例(各群 12 例)を安全性解析対象集団とした。 試験 12606 は、非日本人男性被験者を対象としたリバーロキサバン 20mg 単回経口投与の安全 性、忍容性、薬物動態へのフルコナゾール 400mg 反復投与の影響を検討する無作為化、非盲検、 2 処置 2 期のクロスオーバー試験である。13 例の被験者が組み入れられ、処置 A としてリバーロ キサバン 20mg 単回投与を行った。処置 B としてリバーロキサバン単回 20mg 投与(Day 0 のみ) とフルコナゾール 1 日 400mg 6 日間投与を行なった。各処置の間には少なくとも 10 日間の wash-out 期間を設けた。有害事象、臨床検査値、バイタルサイン、心電図を評価した。治験薬 投与開始日から最終投与後 30 日目までに発現した有害事象を治験薬投与下に発現した有害事象 と定義した。
表 2.7.4.1-2 国内における第Ⅰ相・臨床薬理試験一覧 試験 デザイン 対象 用法・用量/投与期間 対照 症例数 第Ⅰ相単回投与試験 (試験 11126) 単盲検 用量漸増 健康成人 男子 5、10、20、40mg 空腹時単回投与 プラセボ 40 例 ( 5mg:8 例、 10mg:8 例、 20mg:8 例、 40mg:8 例、 プラセボ:8 例) 第Ⅰ相反復投与試験 (試験 11127) 単盲検 用量漸増 健康成人 男子 10、20、30mg 1 日 2 回 6 日間投与 プラセボ 30 例 (10mg bid:8 例、 20mg bid:8 例、 30mg bid:8 例、 プラセボ :6 例) 臨床薬理単回投与試験 (試験 11325) 単盲検 用量漸増 健康 高齢者 10、20、30、40mg 食後単回投与 プラセボ 64 例 (10mg:12 例、 20mg:12 例、 30mg:12 例、 40mg:12 例 プラセボ:16 例) 臨床薬理反復投与試験 (試験 12026) 非盲検 用量漸増 健康 高齢者 10、15、20mg 1 日 1 回 7 日間投与 なし 36 例 (10mg od:12 例、 15mg od:12 例、 20mg od:12 例) 生物学的同等性試験 (試験 13371) 単盲検 クロス オーバー 健康成人 男子 15mg 単回 なし 20 例 臨床薬理薬物相互作用試 験 (試験 14883) 単盲検 (A 群、B 群のみ) 並行群間 健康成人 男子 A 群:ワルファリン定常状態(PT-INR:2~3)に到達した後、15mg 1 日 1 回 4 日間投与 B 群:ワルファリン定常状態(PT-INR:2~3)に到達した後、プラ セボ 1 日 1 回 4 日間投与 C 群:ワルファリン前投与せず、 15mg 1 日 1 回 4 日間投与 プラセボ 36 例 (A 群:12 例 B 群:12 例 C 群:12 例) od:1 日 1 回、bid:1 日 2 回 2.7.4.1.1.1.2 第Ⅱ相試験 3 つの日本人 NVAF 患者を対象とした国内第Ⅱ相試験及び 2 つの急性症候性 DVT 患者を対象と した国外第Ⅱ相試験を安全性の評価対象とした。 国内第Ⅱ相試験(試験 11390、試験 12024、試験 11866) 日本人 NVAF 患者における、リバーロキサバンの薬物動態、薬力学的効果及び安全性の検討を 目的とした国内第Ⅱ相試験 3 試験(試験 11390、試験 12024 及び試験 11866)を実施した。合計 238 例が治験薬投与を受けた。このうち 185 例に対してリバーロキサバン 2.5~20mg 1 日 2 回及 び 10~20mg 1 日 1 回投与が行われた(表 2.7.4.1-3)。 試験 11390(治験期間:20 年 月~20 年 月)は、日本人の NVAF 患者におけるリバーロ キサバンの用法・用量を段階的に検討する多施設、非盲検試験である。試験は 3 ステップ(ス テップ 1:リバーロキサバン 10mg 1 日 2 回 28 日間投与、ステップ 2:リバーロキサバン 20mg 1
日 2 回 28 日間投与、ステップ 3:リバーロキサバン 30mg 1 日 2 回 28 日間投与)からなり、各 ステップはスクリーニング、治験薬投与期(28±7 日間)、フォローアップ期(28±7 日間)に 分けられる。ステップ 1 から検討を開始し、各ステップでの安全性を確認した上で次のステップ へ移行することとし、次ステップの患者組み入れ開始は、前ステップの目標症例数(25 例)の 組み入れが完了し、過半数例(13 例)のフォローアップが完了し安全性が確認された時点とし た。59 歳以下で少なくとも1つの塞栓症危険因子(高血圧症、糖尿病、冠動脈疾患、うっ血性 心不全)を有する、あるいは 60 歳以上である日本人 NVAF 患者 56 例が組み入れられた。このう ち治験薬が投与された 36 例(リバーロキサバン 10mg 1 日 2 回群 25 例、リバーロキサバン 20mg 1 日 2 回群 11 例)を安全性解析対象集団とした。本試験では、ステップ 2 において最初の 11 例 中 5 例が出血事象あるいは PT-INR 上昇により治験を中止したため、新たな被験者の組み入れを 中止し、ステップ 3 には移行しなかった。 試験 12024(治験期間:20 年 月~20 年 月)は、ワルファリンを対照薬としてリバー ロキサバン(2.5mg、5mg 及び 10mg 1 日 2 回経口投与の 3 用量群)の薬物動態、薬力学的効果及 び安全性を検討する非盲検、無作為化、並行群間比較試験である。59 歳以下で少なくとも1つ の塞栓症危険因子(高血圧症、糖尿病、冠動脈疾患、うっ血性心不全)を有する、あるいは 60 歳以上である日本人 NVAF 患者 121 例が組み入れられた。このうち治験薬が投与された 100 例 (リバーロキサバン 2.5mg 群 24 例、5.0mg 群 26 例、10mg 群 24 例、ワルファリン群 26 例)を安 全性解析対象集団とした。 試験 11866(治験期間:20 年 月~20 年 月)は、ワルファリンを対照薬としてリバー ロキサバン(10mg、15mg 及び 20mg 1 日 1 回の 3 用量群)の安全性、薬物動態及び薬力学的効果 を検討する非盲検、無作為化、並行群間比較試験である。59 歳以下で少なくとも 1 つの塞栓症 危険因子 (高血圧症、糖尿病、冠動脈疾患、うっ血性心不全)を有する、あるいは 60 歳以上で ある日本人 NVAF 患者 114 例が組み入れられた。このうち、治験薬が投与された 102 例(リバー ロキサバン 10mg 群 26 例、15mg 群 25 例、20mg 群 24 例、ワルファリン群 27 例)を安全性解析対 象集団とした。 国内第Ⅱ相試験の概略を示す(表 2.7.4.1-3) 表 2.7.4.1-3 心房細動患者を対象とした国内第Ⅱ相試験(試験 11390、試験 12024、試験 11866):試験デザインの概略 試験 デザイン リバーロキサバンの用量 対照群及び用量 安全性解析対象集団 試験 11390 (MRR-00199) 非盲検、段階的用量 検討、4 週間投与 10、20、(30)mg 1 日 2 回 なし 36 例 R(10mg):25 例 R(20mg):11 例 試験 12024 (MRR-00267) 無作為化、非盲検、 並行群間、4 週間投 与 2.5、5、10mg 1 日 2 回 ワルファリン;70 歳未満には目標 PT-INR 2.0~3.0、70 歳以上には 1.6~ 2.6 で用量調節 100 例 R(2.5mg):24 例 R(5mg):26 例 R(10mg):24 例 W:26 例 試験 11866 (MRR-00297) 無作為化、非盲検、 並行群間、4 週間投 与 10、15、20mg 1 日 1 回 ワルファリン;70 歳未満には目標 PT-INR 2.0~3.0、70 歳以上には 1.6~ 2.6 で用量調節 102 例 R(10mg):26 例 R(15mg):25 例 R(20mg):24 例 W:27 例 PT-INR;プロトロンビン時間の国際標準化比、R;リバーロキサバン、W;ワルファリン 引用元:5.3.5.2.1(MRR-00199)、5.3.5.1.2(MRR-00297)、5.3.5.1.1(MRR-00267)の Table 11-1
これらの 3 試験(試験 11390、試験 12024、試験 11866)では、出血事象、有害事象(出血事 象、脳梗塞、肺塞栓症を含む)、臨床検査値、バイタルサイン、心電図を評価した。同意取得か らフォローアップ期間の終了までに発現した有害事象を収集し、治験薬投与開始日から最終投与 後 7 日目までに発現した事象を治験薬投与下に発現した有害事象と定義した。 出血事象を以下の定義に従って分類した(表 2.7.4.1-4)。試験 11390 では治験担当医師(治 験責任医師又は治験分担医師)から出血事象として報告された事象を、1)「重大な出血事象」、 2)「軽微な出血事象」の 2 つに分類した。試験 12024、試験 11866 では同一の独立した出血事 象判定委員会を設置し、同委員会が、治験担当医師により出血事象として報告された事象を、 1)「重大な出血事象」、2)「重大ではないが臨床的に問題となる出血事象」、3)「軽微な出 血事象」、4)臨床的に明らかでない出血(出血事象としては取り扱わない)の 4 つのカテゴ リー分類した。 表 2.7.4.1-4 出血事象の定義(試験 11390、試験 12024、試験 11866) 試験 11390 試験 12024 試験 11866 「重大な出血事象」 ・臨床的に問題となる出血で、2g/dL 以上のヘモグロビン量の低下を伴うも の ・臨床的に問題となる出血で、2 単位 以上の輸血が必要となるもの ・腹腔内出血、頭蓋内出血、眼出血又 は脊椎内出血 ・死因となった出血 ・治験薬の投与中止を要する出血 ・2g/dL 以上のヘモグロビン量の低下を伴う 出血 ・2 単位以上の輸血(濃厚赤血球又は全血) が必要な出血 ・重要な臓器における出血、頭蓋内出血、後 腹膜出血又は心膜出血 ・死因となった出血 「重大ではないが臨 床的に問題となる出 血事象」 ・血行動態に影響するすべての出血 ・入院を必要とするすべての出血 ・25cm2以上の皮下血腫 ・筋肉内血腫 ・5 分以上継続、24 時間以内に反復して発生 した、又は電気凝固などの処置を必要とした 鼻出血 ・自然発生した(食事や歯磨きに関連のな い)又は 5 分以上継続した歯肉出血 ・肉眼的血尿 ・潜血を伴う下血、吐血などの臨床的に明ら かな肉眼的消化管出血 ・直腸出血(トイレットペーパーに点状を超 える大きさの出血を認めるもの) ・喀血(喀痰中に点状を超える大きさの出血 を認めるもの) ・被験者に臨床的な影響を及ぼすその他の出 血 「軽微な出血事象」 「重大な出血事象」の定義を満たさな い、すべての出血事象 上記の基準を満たさない、すべての臨床的に 明らかな出血事象 臨床的に明らかでな い出血 臨床的に明らかでない出血(出血事象として 扱わない) 引用元:5.3.5.2.1(MRR-00199)、5.3.5.1.2(MRR-00297)、5.3.5.1.1(MRR-00267)
試験 11390、試験 12024、試験 11866 の試験期間は、それぞれ、スクリーニング、治験薬の投与 期(28±7 日間)、フォローアップ期(28±7 日間)からなる。治験実施計画書に定義された安 全性評価スケジュールを表 2.7.4.1-5に示す。
表 2.7.4.1-5 安全性評価スケジュール(試験 11390、試験 11866、試験 12024)
検査・観察日
Pre-Visit Visit 1 Visit 2 Visit 3 Visit 4
投与開始前 投与中 投与終了 フォローアップ 登録前 投与開始1 日前 投与開始 14±7 日目 投与開始 28±7 日目 投与終了 28±7 日目 投与前 投与後 投与前 投与後 治験薬投与 ←---→ 臨床検査 ←---→ X X X 凝固線溶系 パラメータ採血 X X X X X 12 誘導心電図 ←---→ X バイタルサイン X X X X X 有害事象 脳梗塞の臨床徴候 X X X X X 引用元:5.3.5.2.1(MRR-00199)、5.3.5.1.2(MRR-00297)、5.3.5.1.1(MRR-00267) 国外第Ⅱ相試験(試験 11223、試験 11528) 国外第Ⅱ相試験の安全性評価は、急性症候性 DVT 患者を対象とした 2 つの試験(試験 11223 及 び試験 11528)のデータを統合して評価した。個々の試験の安全性成績は治験総括報告書 (5.3.5.4.1 MRR-00150、5.3.5.4.2 MRR00233)を参照。 試験 11223(治験期間:20 年 月~20 年 月)は、非日本人成人急性症候性 DVT 患者を 対象とし、エノキサパリン/ビタミン K 拮抗薬(VKA)併用を対照に用いたリバーロキサバンの有 効性、安全性及び用量‐反応関係を検討するための前向き、多施設共同、無作為化、非盲検(リ バーロキサバンの用量群間は二重盲検)、実薬対照、盲検下評価、並行群間比較試験である。本 試験への適格性が確認された患者は、以下の 5 つの投与群のいずれかに無作為に割り付けられ、 84 日間治験薬を投与された。 リバーロキサバン 10mg 1 日 2 回群 リバーロキサバン 20mg 1 日 2 回群 リバーロキサバン 30mg 1 日 2 回群 リバーロキサバン 40mg 1 日 1 回群 エノキサパリン/VKA 投与群(INR:2.0~3.0) 圧迫法による超音波検査(CCUS)により客観的に確認され、症候性肺塞栓症(PE)を併発して いない急性症候性 DVT 患者 636 例が組み入れられた。このうち治験薬が投与された 604 例(リ バーロキサバン 10mg 1 日 2 回群 119 例、リバーロキサバン 20mg 1 日 2 回群 117 例、リバーロキ サバン 30mg 1 日 2 回群 121 例、リバーロキサバン 40mg 1 日 1 回群 121 例、エノキサパリン/VKA 群 126 例)を安全性解析対象集団とした。
試験 11528(治験期間:20 年 月~20 年 月)は、非日本人成人急性症候性 DVT 患者 を対象とし、低分子量ヘパリン又は未分画ヘパリン/ビタミン K 拮抗薬併用〔(LMW)ヘパリン /VKA〕を対照に用いたリバーロキサバン 1 日 1 回投与の有効性、安全性及び用量‐反応関係を検 討するための前向き、多施設共同、無作為化、非盲検(リバーロキサバンの用量群間は二重盲 検)、実薬対照、盲検下評価、並行群間比較試験である。本試験への適格性が確認された患者は、 以下の 4 つの投与群のいずれかに無作為割り付けされた。 リバーロキサバン 20mg 1 日 1 回群 リバーロキサバン 30mg 1 日 1 回群 リバーロキサバン 40mg 1 日 1 回群 (LMW)ヘパリン/VKA 投与群(INR:2.0~3.0) 症候性肺塞栓症(PE)を併発していない急性症候性 DVT 患者 543 例が組み入れられた。このう ち治験薬が 1 回以上投与された 542 例〔リバーロキサバン 20mg 群:135 例、リバーロキサバン 30mg 群:134 例、リバーロキサバン 40mg 群:136 例、(LMW)ヘパリン/VKA 群:137 例〕を安全 性解析対象集団とした(表 2.7.4.1-6)。 表 2.7.4.1-6 国外第Ⅱ相試験の概略(試験 11223、試験 11528) 試験 治験総括報告書 の添付場所 (報告書番号) FPV – LPV 目的及び デザイン 対象 用法・用量 全体及び用量ごとの 被験者数 (安全性解析対象集団) 主要評価項目 試験 11223 5.3.5.4.1 (MRR-00150) 20 年 月~ 20 年 月 無作為化 一部二重盲検 (本薬用量間の み盲検) 並行群間 症候性の 急性 DVT 613 例 リバーロキサバン: 10mg bid 20mg bid 40mg od 30mg bid VKA/エノキサパリ ン: 12 週間投与 計 604 119 117 121 121 126 ・3 週間後の血栓退縮 ・「重大な出血事象」 試験 11528 5.3.5.4.2 (MRR-00223) 20 年 月~ 20 月 無作為化 一部二重盲検 (本薬用量間の み盲検) 並行群間 症候性の 急性 DVT 543 例 リバーロキサバン: 20mg od 30mg od 40mg od (LMW)ヘパリン /VKA: 12 週間投与 計 542 135 134 136 137 ・12 週間後の DVT 再 発、肺塞栓症の発現、血 栓像の悪化の複合エンド ポイント ・重大な出血事象、重大 ではないが臨床的に問題 となる出血事象 DVT:深部静脈血栓症、LMWH:低分子量ヘパリン(エノキサパリン)、VKA:ビタミン K 拮抗薬、 FPV:最初の被験者来院日、LMW:低分子量、LPV:最終被験者来院日、bid:1 日 2 回、od:1 日 1 回 引用元:5.3.5.4.1(MRR-00150)、5.3.5.4.2(MRR-00223) これらの 2 試験(試験 11528 及び試験 11223)では、出血事象、有害事象、臨床検査値を評価 した。試験 11223 ではバイタルサイン、心電図の評価も行った。同意取得からフォローアップ期
間の終了までに発現した有害事象を収集し、治験薬投与開始日から最終投与後 7 日目までに発現 した事象を治験薬投与下に発現した有害事象と定義した。 試験 11223 の安全性の主要評価項目は治験薬投与下の「重大な出血事象」の発現である。主要 評価項目の解析は出血事象判定委員会による分類に基づく。 出血事象は 2 つのカテゴリーに分類される。 「重大な出血事象」 「重大ではない出血事象」 「重大な出血事象」は以下のように定義される。 死因となった出血 2g/dL 以上のヘモグロビン量の低下を伴う臨床的に明らかな出血 2 単位以上の濃厚赤血球又は全血の輸血を要する臨床的に明らかな出血 重要な臓器(後腹膜、頭蓋内、眼内)における出血 治験薬の投与中止を要する臨床的に明らかな出血 上記以外の出血は「重大ではない出血事象」と判断される。 試験 11528 の安全性の主要評価項目は治験薬投与下の臨床的に重要な出血事象(すなわち「重 大な出血事象」又は「重大ではないが臨床的に問題となる出血事象」)の発現である。主要評価 項目の解析は出血事象判定委員会による分類に基づく。 試験 11528 では(試験 11223 と異なり)出血事象は 3 つのカテゴリーに分類される。 「重大な出血事象」、 「重大ではないが臨床的に問題となる出血事象」、 「軽微な出血事象」 試験 11528 での「重大な出血事象」の定義は、治験薬の投与中止に至った臨床的に明らかな出 血事象が「重大な出血事象」とは判断されないこと以外は試験 11223 と同様である。 「重大ではないが臨床的に問題となる出血事象」は以下のように定義される。(試験 11528 の み) 複数部位からの出血 5 分以上の歯肉の出血 肉眼的血尿(自発的な出血。医学的手技に関連する場合は 24 時間以上継続するもの)
直腸の出血;喀血;吐血 静脈穿刺後の 5 分を超える出血延長 100cm2を超える皮膚の血腫、又は 5 分を超えて継続する又は反復的な鼻出血 試験 11528 では「重大な出血事象」、「重大ではないが臨床的に問題となる出血事象」に該当 しない出血は「軽微な出血事象」に分類された。 試験 11223 及び試験 11528 の試験期間は、いずれもスクリーニング、無作為割り付け日(第 1 日)、治験薬投与期〔第 1 日から第 84(+7)日まで〕及びフォローアップ期〔治験薬最終投与 後 30(+7)日間〕とした。治験実施計画書に定義された安全性評価スケジュールを示す(表 2.7.4.1-7)。 表 2.7.4.1-7 安全性評価スケジュール(試験 11223、試験 11528)
11223 試験 Day 1 Day 2-6 ±2 daysDay 7 ±2 daysDays 21 ±4 daysDay 56 ±4 daysDay 84 F/U Day114±4 days
治験薬服薬状況確認 X X X X X X 有害事象の評価 X X X X X X X バイタルサインの評価 Xa X X X X X ECG Xa X X 臨床検査値の評価 (中央)b Xa X X X X Xc a :臨床検査による評価、ECG、バイタルサイン評価は Day1 で治験薬を投与する前に行う。 b :臨床検査による評価は中央検査によって行われる生化学(肝機能を含む)血液学的検査、凝固パラメータを含む。 c :生化学検査のみ
11528 試験 Day1 Day8 Day15 Day22 Day43 Day84 (D114)F/U
治験薬服薬状況確認 X X X X X X 有害事象の評価 X X X X X X X PK、血液凝固、肝機能 及びクレアチニンa の各検査のための採血 X X X X
a:凝固試験は INR と aPTT を含む。INR は VKA 群に対して 2 から 3 日ごとに行い、その後は少なくとも 1 カ月ごとに行う。肝 機能は ALT と AST、ビリルビン、アルカリホスファターゼ、GGT を含む。これらの試験は割り付け前の Day1 と Day43、 Day84 で行われた。 引用元:5.3.5.4.1(MRR-00150)、5.3.5.4.2(MRR00223) 2.7.4.1.1.1.3 第Ⅲ相試験 2 つの試験〔ROCKET-AF プログラム:国内第Ⅲ相試験(J-ROCKET-AF、試験 12620)及び国外第 Ⅲ相試験(ROCKET-AF、試験 11630)〕を安全性の評価対象とした。試験 11630 及び試験 12620 は共に、NVAF 患者における脳卒中及び非中枢神経系塞栓症の発症抑制に関して、リバーロキサ バン 1 日 1 回経口投与の有効性及び安全性を用量調節ワルファリンと比較評価する、前向き、多 施設共同、無作為化、ダブルダミー法による二重盲検、実薬対照、並行群間比較試験であった。 試験 12620 と試験 11630 の試験デザインは、試験対象の人種、リバーロキサバンの用量、ワル ファリンの目標 PT-INR の範囲、投与期間、症例数を除き、可能な限り同一とすることとした。
2.7.4.1.1.1.3.1 試験方法 国内第Ⅲ相試験(試験 12620、治験期間:20 年 月~20 年 月)は、脳卒中及び非中枢 神経系塞栓症の危険因子を有する日本人 NVAF 患者を対象に、リバーロキサバン 15mg 1 日 1 回 〔クレアチニンクリアランス(CLCR)30~49mL/min の患者には 10mg 1 日 1 回〕の安全性を国内 ガイドライン1)に従って用量調節したワルファリンと比較検討し、安全性の主要評価項目である 「重大な出血事象」又は「重大ではないが臨床的に問題となる出血事象」の複合エンドポイント におけるリバーロキサバンのワルファリンに対する非劣性を検証することを目的とし、合わせて 安全性及び有効性を国外第Ⅲ相試験(試験 11630)と比較検討することにより、外国臨床データ の日本人患者への外挿可能性を評価することも目的とした前向き、多施設共同、無作為化、ダブ ルダミー法による二重盲検、実薬対照、並行群間比較試験である。ワルファリンは国内ガイドラ インの推奨に従い、70 歳未満の被験者に対しては PT-INR2.0~3.0、70 歳以上の被験者には 1.6 ~2.6 を目標にした用量調節を行うこととした。国内 164 施設から脳卒中、一過性脳虚血発作 (TIA)又は非中枢神経系塞栓症の既往を有するか、あるいは心不全、高血圧症、糖尿病、高齢 (75 歳以上)の塞栓症危険因子を 2 つ以上有する日本人 NVAF 患者 1,439 例が組み入れられ、そ のうち 1,280 例がリバーロキサバン群、ワルファリン群のいずれかに無作為に割り付けられた。 このうち、治験薬が 1 回以上投与された 1,278 例(リバーロキサバン群 639 例、ワルファリン群 639 例)を安全性解析対象集団とした。 国外第Ⅲ相試験(試験 11630、治験期間:20 年 月~20 年 月)は、脳卒中と非中枢神 経系塞栓症の発現を有効性主要評価項目として、リバーロキサバン 20mg(CLCR 30~49mL/min の 患者には 15mg)と用量調節ワルファリンの有効性及び安全性を比較検討することを目的とした 前向き、多施設共同、無作為化、ダブルダミー法による二重盲検、実薬対照、イベント主導型、 並行群間比較試験である。ワルファリンは米国・欧州のガイドライン2)の推奨に従い、PT-INR 2.5(2.0~3.0)を目標にした用量調節を行うこととした。国外第Ⅲ相試験は、イベント主導型 試験であり有効性の検証のために必要な、治験実施計画書適合集団における治験薬投与下での主 要有効性評価項目の発現数が 405 に達するまで継続されることとした。有効性の主解析は治験実 施計画書に適合する集団において治験薬投与下に発現した有効性主要評価項目に関する、リバー ロキサバンのワルファリンに対する非劣性の検証であり、非劣性が検証された場合、安全性解析 対象集団における治験薬投与下に発現した事象に関する優位性を検証することとした。45 ヵ国 1,178 施設から 17,232 例の脳卒中、TIA 又は非中枢神経系塞栓症の既往を有するか、あるいは心 不全、高血圧症、糖尿病、高齢(75 歳以上)の塞栓症危険因子を 2 つ以上有する NVAF 患者が組 み入れられた。そのうち 14,264 例がリバーロキサバン群(7,131 例)又はワルファリン群 (7,133 例)のいずれかに無作為に割り付けられた〔Intent-to-Treat(ITT)集団〕。ITT 集団 のうち、治験薬が 1 回以上投与された 14,236 例(リバーロキサバン群 7,111 例、ワルファリン 群 7,125 例)を安全性解析対象集団とした。 2.7.4.1.1.1.3.2 安全性に関わる委員会
独立データモニタリング委員会(Independent Data Monitoring Committee:IDMC)は、試験の 進捗をモニタリングし、被験者の安全が損なわれないよう提言を行った。
評価項目判定委員会(Clinical Endpoint Committee:CEC)は治験実施計画書に記載された定 義に基づき、試験の評価項目を盲検下で評価し分類した。
試験に設定された委員会の詳細は、5.3.5.1.3(A49701)の 4.2 項、5.3.5.1.6 (R-8570)の 3.1.1 項を参照。
肝臓関連事象評価委員会(Hepatic Event Assessment Committee:HEAC): HEAC は外部の肝臓 学の専門家 5 名(3 名は臨床医、2 名は病理医で、病理医は生検又は剖検検体が得られている場 合のみ関与)からなる。同委員会はリバーロキサバンの臨床試験プログラムにおいて肝臓障害に 関する安全性について事前に設定した基準に従って、検討・評価を行う。試験 12620、試験 11630 では基準に合致する全症例が HEAC による評価を受けた。HEAC の評価は、試験実施中に、 各症例に対して各委員が盲検下で行う。評価結果は委員ごとに作成されるもので、委員会として 評価について合意の形成を行うことは目的としていない。HEAC の詳細は 5.3.5.1.3(A49701)の 16.4.2.1 項を参照。 2.7.4.1.1.1.3.3 安全性の評価 安全性は、有害事象、出血事象、肝機能検査を含む臨床検査、心電図、バイタルサイン、理学 所見により評価された。同意取得からフォローアップ期間の終了までに発現した有害事象を収集 し、治験薬投与開始日から最終投与後 2 日目までの期間に発現した事象を治験薬投与下に発現し た事象と定義した。臨床検査は、試験 11630 では中央測定を用い、試験 12620 は各施設又は各施 設が契約する外部検査受託業者で実施した。 2.7.4.1.1.1.3.3.1 特に注目する有害事象 出血事象、肝臓関連有害事象については特に注目する重要な事象として評価した。その他の注 目する有害事象として急性膵炎、血小板減少症、急性腎不全、過敏性反応についても評価した。 当該事象の抽出には、ICH 国際医薬用語集(MedDRA MSSO)の 標準検索式 (SMQ) を用いた。 出血事象(2.7.4.2.1.4.2 参照) リバーロキサバンは直接作用型第Ⅹa 因子阻害剤であり、この薬理作用機序により出血のリス クが増加する。 試験 12620、試験 11630 ともに、安全性の主要評価項目は「重大な出血事象」又は「重大では ないが臨床的に問題となる出血事象」の複合である。 なお、治験責任医師も出血事象を「重大な出血事象」、「重大ではないが臨床的に問題となる 出血事象」、「軽微な出血事象」に分類し症例報告書に記録するが、安全性の評価項目の解析に は CEC により「重大な出血事象」、「重大ではないが臨床的に問題となる出血事象」と判定され た事象を用いた。両試験で同一の CEC を用いた。CEC は、安全性の評価項目(出血事象)に加え て、有効性の評価項目(脳卒中、非中枢神経系塞栓症、心筋梗塞、死亡)についても判定をおこ なった。 「重大な出血事象」は以下の定義を満たす臨床的に明らかな出血である。 ・ 2g/dL 以上のヘモグロビン量の低下を伴う出血 ・ 2 単位以上の輸血(濃厚赤血球又は全血)が必要な出血 ・ 重要な臓器における出血〔頭蓋内、脊髄内、眼内、心嚢(心膜)内、関節内、コンパート メント症候群を伴う筋肉内、後腹膜〕
・ 死因となった出血 頭蓋内出血は CEC により脳卒中及び/又は出血事象の基準に合致するか否かの判定を受けた。 試験 11630 では頭蓋内出血はさらに脳実質内、脳室内、硬膜下、クモ膜下、及び硬膜外出血のい ずれかに分類された。試験 12620 では、出血性脳卒中は実質内及び脳室内のいずれかに分類され たが、出血性脳卒中の定義を満たさない頭蓋内出血についての細分類は、全例には実施されな かった。 「重大ではないが臨床的に問題となる出血事象」は「重大な出血事象」の定義は満たさない以 下の臨床的に明らかな出血である。 ・ 内科的又は外科的処置が必要な出血 ・ 医師との予定外のコンタクト(受診又は電話)が必要となった出血 ・ 治験薬の中断又は中止が必要な出血 ・ 疼痛や日常生活に対する障害などが生じた出血 「軽微な出血事象」は「重大な出血事象」又は「重大ではないが臨床的に問題となる出血事 象」ではない臨床的に明らかな出血と定義した。 CEC 判定のために事前に規定したプログラムによって以下の情報に基づき出血事象の抽出が行 われた。1) 重要な臓器出血、2) 内科的又は外科的介入の有無、3) 医師との予定外のコンタク トの有無、4) 疼痛や日常生活に対する障害の有無、5) 治験薬の変更の有無(治験薬の中止又は 中断後再開)、6) 死亡転帰又は死因となる出血。これらに加えて、ヘモグロビンの低下、輸血 の実施が症例報告書に記載された症例も抽出され、CEC の判定を受けた。CEC は入手可能な臨床 情報に基づいて、各事象を「重大な出血事象」、「重大ではないが臨床的に問題となる出血事 象」、「軽微な出血事象」、又は非出血事象に分類した。 治験担当医師(治験責任医師又は治験分担医師)によって報告された有害事象から SMQ「出血 関連用語(臨床検査用語を除く)」を用いて抽出し、出血に関する安全性の副次的な評価に用い た。 肝臓関連有害事象(2.7.4.2.1.4.2.4 肝臓関連の有害事象 参照) 主に中央検査・各治験施設検査(又は治験施設が契約する外部検査施設)で得られた肝機能検 査値を肝機能の安全性の検討に用いた。試験期間中に重篤な有害事象として報告された肝臓関連 有害事象の観察・モニタリングを行い、肝臓事象を調査した。肝臓関連有害事象の特定は、SMQ 「肝障害」を用いて行った。治験薬の投与中止を必要とする場合、被験者のより頻繁な観察・モ ニタリング調査を必要とする場合の肝機能値に関するガイドラインは治験実施計画書に記載した。 急性膵炎関連有害事象(2.7.4.2.1.4.2.5 急性膵炎関連の有害事象 参照) 試験期間中に重篤な有害事象として報告された膵炎関連有害事象の観察・モニタリングを行い、 急性膵炎事象を調査した。膵炎有害事象の特定は SMQ「急性膵炎」(A 分類)を用いて行った。
血小板減少関連有害事象(2.7.4.2.1.4.2.6 血小板減少関連の有害事象 参照) 試験期間中に重篤な有害事象として報告された血小板減少症に関連した有害事象の観察・モニ タリングを行い、血小板減少症事象を調査した。血小板減少症に関連した有害事象は SMQ「造血 障害による血球減少症」の下位 SMQ「血小板減少症」に基づいた。 急性腎不全(2.7.4.2.1.4.2.7 急性腎不全 参照) 試験期間中に重篤な有害事象として報告された腎不全に関連した有害事象又はクレアチニン値 の増加の観察・モニタリングを行い、腎不全事象を調査した。急性腎不全の有害事象は SMQ「急 性腎不全」に基づいた。 過敏性反応(2.7.4.2.1.4.2.8 過敏性反応 参照) 試験期間中に重篤な有害事象として報告されたアナフィラキシー型有害事象の観察・モニタリ ングを行い、過敏性反応を調査した。過敏性反応は SMQ「アナフィラキシー反応」狭義及び SMQ 「重症皮膚副作用」狭義に基づいた。 2.7.4.1.1.1.3.3.2 安全性評価のスケジュール 試験 12620、試験 11630 は共に、スクリーニング期間、治験薬投与終了時来院で終わる二重盲 検治療期間、及びフォローアップ期間に分かれている。治験薬投与終了時来院、あるいは治験薬 の早期投与中止時来院の際には、被験者の治験薬は治験責任医師の判断で非盲検の VKA あるいは 他の適切な治療薬に変更された。 被験者の来院は、スクリーニング時、二重盲検期間の 2 週目、4 週目、その後 4 週ごとに設定 した。被験者は治験薬投与終了時来院あるいは早期投与中止時来院の 30 日後にフォローアップ のために来院した(フォローアップ来院)。 国内外第Ⅲ相試験(試験 12620、試験 11630)の治験実施計画書に定義された安全性評価スケ ジュールの要約を示す(表 2.7.4.1-8)。詳細については、各治験総括報告書(5.3.5.1.3 A49701 及び 5.3.5.1.6 R-8570)を参照。
表 2.7.4.1-8 安全性評価スケジュール(試験 12620、試験 11630)
安全性の評価 Screen 2 週 4~48 週 52 週 52 週以降 ESMD EOS Visit FU Visit
ECG X 24 週のみ* X 52 週ごと X X 身体検査 X X X バイタルサイン X X X 血液学的検査及び生化 学的検査 a X 24 週のみ X 52 週ごと X X 肝機能検査b X X 4 週ごとc X 12 週ごと X X Xc 有害事象 X X 来院ごと X 来院ごと X X X
ESMD:early study medication discontinuation(治験薬投与早期中止)、ECG:electrocardiogram(心電図)、 EOS:end of study(試験終了時)、FU:follow-up(追跡調査)、Screen:screening visit(スクリー ニング時) *:試験 12620 のみ a:血液学的検査では、ヘモグロビン、ヘマトクリット、白血球分画数、血小板数の検査を実施した。臨床検査 では、ナトリウム、カリウム、アルブミン、血糖、BUN 、クレアチニン、アミラーゼ、リパーゼの検査を実 施した。 b:スクリーニング時の ALT、AST、総ビリルビンと直接ビリルビン、アルカリホスファターゼを含む。ALT、総ビ リルビンと直接ビリルビンは他の測定時点でも測定。AST とアルカリホスファターゼは、ALT が基準値上限 の 3 倍を超える場合のみ実施。 c:試験終了時に治験薬投与を受けている被験者について実施。 引用元 5.3.5.1.3(A49701)、5.3.5.1.6(R-8570) 2.7.4.1.1.1.3.3.3 安全性成績の記述方法 本安全性の概括評価では、試験 12620 及び 11630 の安全性成績をそれぞれ概括し、さらに 2 つ の試験の統合データを用いて投与群間の有害事象の発現頻度を比較する。また、部分集団解析も 実施した。2 試験の統合が可能と判断した根拠を以下に示す。 国内外第Ⅲ相試験は類似したデザインで実施された、多施設共同、無作為化、ダブルダミー 法による二重盲検、並行群間比較試験であり、被験者の選択基準と除外基準は同一のものを 用いた。安全性の評価と臨床検査スケジュールも 2 試験で類似しており、各試験ともに、治 験薬投与中止後の安全性評価を行う期間は 30 日であった。 出血事象の分類(「重大な出血事象」、「重大ではないが臨床的に問題となる出血事象」、 「軽微な出血事象」)と脳卒中を含む心血管事象も両試験で同じ定義が用いられ、同一の CEC により有効性と安全性の評価項目の判定が行われた。 また、有害事象の概括及び安全性主要評価項目については、試験 11630 において東アジア地域 (中国、韓国、香港、台湾)から組み入れられた被験者集団についても検討した。 2.7.4.1.2 全般的な曝露状況 2.7.4.1.2.1 第Ⅰ相・臨床薬理試験 59 の臨床薬理試験が実施され、1,516 例(リバーロキサバン 1,335 例、プラセボ 181 例)に治 験薬が投与され安全性の解析に用いられた。リバーロキサバンの用量と投与期間の要約を表 2.7.4.1-9に示す。
表 2.7.4.1-9 リバーロキサバンの投与期間(第Ⅰ相・臨床薬理 59 試験の統合、安全性解析対 象集団) リバーロキサバン 全被験者 n=1335 <10mg n=77 10mg n=492 15mg n=99 20mg n=325 >20mg n=342 1 日 n(%) 1,075(80.5) 70(90.9) 433(88.0) 81(81.8) 214(65.8) 277(81.0) 2~7 日 n(%) 223(16.7) 7( 9.1) 39( 7.9) 18(18.2) 111(34.2) 48(14.0) 8~10 日 n(%) 37( 2.8) 0( 0.0) 20( 4.1) 0( 0.0) 0( 0.0) 17( 5.0) >10 日 n(%) 0( 0.0) 0( 0.0) 0( 0.0) 0( 0.0) 0( 0.0) 0( 0.0) 引用元:5.3.5.3.3(PH-36320)の Table 14.02.D 上述の解析に含まれない試験 14883 では、すべての被験者が治験薬投与を完了した。24 例は、 ワルファリンを 6~10 日間投与し PT-INR 値が 2.0~3.0 になった後、A 群:リバーロキサバン 15mg 1 日 1 回 4 日間投与(12 例)、B 群:リバーロキサバンプラセボ 1 日 1 回 4 日間投与(12 例)を受けた。C 群に割り付けられた 12 例は、ワルファリンの前投与なしで、リバーロキサバ ン 15mg 1 日 1 回 4 日間の投与を受けた。 試験 12606 では 13 例の被験者がリバーロキサバン 20mg 単回投与を各試験期間(処置 A 及び処 置 B)に受けた。 2.7.4.1.2.2 第Ⅱ相試験 国内第Ⅱ相試験(試験 11390、試験 11866、試験 12024) 試験 11390 ではステップ 1(10mg 1 日 2 回 28 日間)で治験薬を投与された 25 例の被験者が試 験を完了し、ステップ 1 の安全性、忍容性が確認された後にステップ 2 の投与が開始された。ス テップ 2(20mg 1 日 2 回 28 日間)で投薬された 11 例の被験者のうち、6 例(55%)が治験薬投 与を完了し、5 例(45%)が有害事象のために治験薬の投与を中止した。20mg 1 日 2 回群での高 い中止率のためにこの試験への新たな被験者の登録は中止された。 試験 11866 では 102 例の安全性解析対象被験者のうち、100 例(98%)が治験薬投与を完了し た。リバーロキサバン群及びワルファリン群の平均治療期間は 27.2~28.8 日であった。 試験 12024 では 100 例の安全性解析対象被験者のうち、95 例(95%)が治験薬投与を完了し た。リバーロキサバン群及びワルファリン群の平均治療期間は 26.0~28.2 日であった。 国外第Ⅱ相試験(試験 11223、試験 11528) 両試験を統合した場合の、リバーロキサバン群と対照群の安全性解析対象集団の投与量ごとの 症例数を表 2.7.4.1-10に示す。計 883 例がリバーロキサバン 20~60mg/日の投与を受けた。
表 2.7.4.1-10 安全性解析対象の被験者数(試験 11223 と試験 11528 の統合、安全性解析対象 集団) 薬剤及び用量(1 日投与量) 被験者数 リバーロキサバン 20mg 254 リバーロキサバン 30mg 134 リバーロキサバン 40mg 374 リバーロキサバン 60mg 121 リバーロキサバン群の総数 883 (LMW)ヘパリン/VKA 263 (LMW)ヘパリン:低分子量ヘパリン又は未分画ヘパリン VKA:ビタミン K 拮抗薬 引用元:5.3.5.3.4(MRR-00300)の Table 12-1 国外第Ⅱ相 DVT 試験(試験 11223、試験 11528)における治療期間はリバーロキサバン、 (LMW)ヘパリン/VKA 両投与群共に治験薬の平均投与期間は 78~79 日であった。4~12 週間の 治療を受けた被験者の割合は 2 つの投与群で同様であった(表 2.7.4.1-11)。 表 2.7.4.1-11 治験薬投与期間(試験 11223、試験 11528、安全性解析対象集団) リバーロキサバン 合計 ヘパリン/VKA 合計 N 883 263 平均値(日) 78.0 79.0 中央値 84.0 84.0 範囲 1~107 2~102 下記の治療期間の被験者数(%): 4 週間以上 813(92.1%) 246(93.5%) 8 週間以上 788(89.2%) 240(91.3%) 10 週間以上 777(88.0%) 236(89.7%) 12 週間以上 635(71.9%) 166(63.1%) VKA:ビタミン K 拮抗薬 引用元:5.3.5.3.4(MRR-00300)の Table 12-2 2.7.4.1.2.3 第Ⅲ相試験 2.7.4.1.2.3.1 国内第Ⅲ相試験(試験 12620) 試験 12620 では、脳卒中、TIA 又は非中枢神経系塞栓症の既往を有するか、あるいは心不全、 高血圧症、糖尿病、高齢(75 歳以上)の塞栓症危険因子を 2 つ以上有する日本人 NVAF 患者
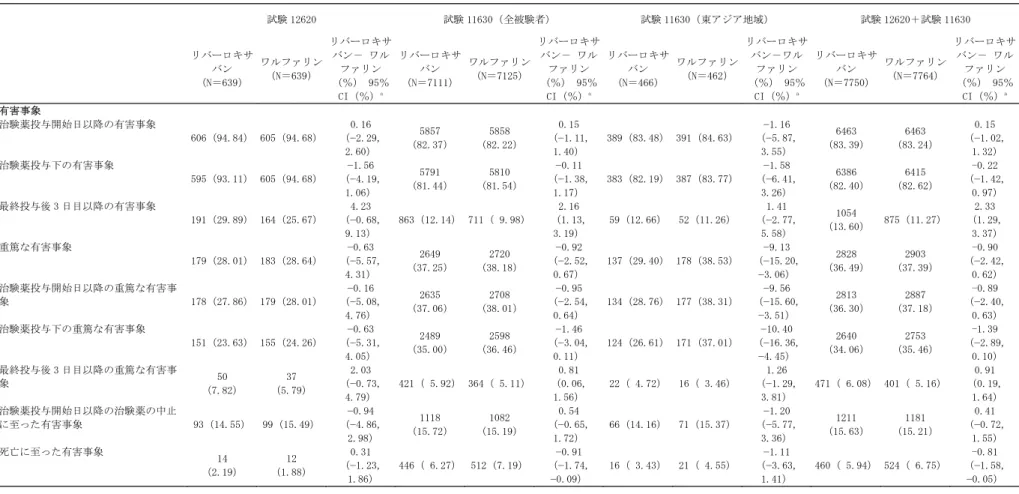
1,280 例がリバーロキサバン群(640 例)、ワルファリン群(640 例)のいずれかに無作為に割 り付けられた。このうち、治験薬が 1 回以上投与された 1,278 例(リバーロキサバン群 639 例、 ワルファリン群 639 例)を安全性解析対象集団とした。 リバーロキサバンの用法用量は 15mg 1 日 1 回夕食後投与であったが、本群でベースラインの CLCRが 30~49mL/min の中等度腎障害を有する被験者には 10mg 1 日 1 回夕食後投与とした。141 例(22.1%)にリバーロキサバン 10mg 1 日 1 回が投与された(表 2.7.4.1-21)。 ワルファリン群の用法はワルファリンカリウムを 1 日 1 回夕食後投与であった。用量は PT-INR が 70 歳未満では 2.0~3.0、70 歳以上では 1.6~2.6 となるよう調節することとした。 二重盲検期終了又は二重盲検期早期中止の 30(±7)日後に設定されるフォローアップ来院を 実施した被験者を治験完了者と定義した。1,226 例〔リバーロキサバン群 610 例(95.5%)、ワ ルファリン群 616 例(96.4%)〕が治験を完了した(図 2.7.4.1-1)。
1,280 無作為割付被験者 / ITT 解析対象集団
リバーロキサバン
ワルファリン
640(無作為割付被験者 /ITT 解析対象集団) 640(無作為割付被験者 /ITT 解析対象集団) 639(安全性解析対象集 団)a 1:治験薬服用せず 639(安全性解析対象集 団)a 1:治験薬服用せず 29:治験中止b 1:有害事象 3:臨床評価項目 の発現c 5:同意撤回 13:死亡 6:追跡不能 1:治験実施計画 書からの逸脱 610:治験完了d 478:二重盲検 治療期を終了し 30 日フォロー アップ来院を実 施 132:二重盲検 治療期を早期中 止後 30 日フォ ローアップ来院 を実施 23:治験中止b 1:有害事象 2:臨床評価項 目の発現c 5:同意撤回 12:死亡 1:医師による 判断 2:追跡不能 616:治験完了d 467:二重盲検治 療期を終了し 30 日フォローアッ プ来院を実施 149:二重盲検治 療期を早期中止 後 30 日フォロー アップ来院を実 施 a:安全性解析対象集団は ITT 解析対象集団のうち少なくとも 1 回の投与を受けたすべての被験者である b:治験中止;30 日フォローアップ来院を実施しなかった被験者 c:臨床評価項目;脳卒中、非中枢神経塞栓症 d:治験完了;30 日フォローアップ来院を実施した被験者 引用元:5.3.5.1.3(A49701)の Table 14.1.1、Table 14.1.4図 2.7.4.1-1 被験者の内訳(試験 12620)
二重盲検治療期を終了した被験者はリバーロキサバン群の 480 例(75.1%)、ワルファリン群 の 468 例(73.2%)であった。治験薬の早期中止に至った最も多い理由は有害事象〔リバーロキ サバン群 73 例(11.4%)、ワルファリン群 70(11.0%)〕で、その他同意撤回〔リバーロキサ バン群 26 例(4.1%)、ワルファリン群 35 例(5.5%)〕、評価項目(脳卒中又は非中枢神経系
塞栓症:治験担当医師判定)の発現〔リバーロキサバン群 18 例(2.8%)、ワルファリン群 28 例(4.4%)〕が主な理由であった(表 2.7.4.1-12)。 表 2.7.4.1-12 二重盲検治療期における治験薬早期中止理由(試験 12620) リバーロキサバン n(%) ワルファリン n(%) 合計 n(%) 治験薬投与終了例 480 ( 75.1) 468 ( 73.2) 948 ( 74.2) 治験薬投与早期中止例 159 ( 24.9) 171 ( 26.8) 330 ( 25.8) 有害事象 73 ( 11.4) 70 ( 11.0) 143 ( 11.2) 臨床評価項目の発現a 18 ( 2.8) 28 ( 4.4) 46 ( 3.6) 同意撤回b 26 ( 4.1) 35 ( 5.5) 61 ( 4.8) 死亡 8 ( 1.3) 3 ( 0.5) 11 ( 0.9) 治験実施計画書に基づかない医師 の判断 4 ( 0.6) 13 ( 2.0) 17 ( 1.3) 追跡不能 4 ( 0.6) 1 ( 0.2) 5 ( 0.4) 治験薬投与不遵守 3 ( 0.5) 1 ( 0.2) 4 ( 0.3) 治験実施計画書に基づく判断 12 ( 1.9) 8 ( 1.3) 20 ( 1.6) 治験実施計画書からの逸脱 9 ( 1.4) 9 ( 1.4) 18 ( 1.4) 医師による施設の打ち切り 1 ( 0.2) 2 ( 0.3) 3 ( 0.2) 治験依頼者による施設の打ち切り 1 ( 0.2) 1 ( 0.2) 2 ( 0.2) 治験担当医師によって報告された主な中止理由の集計であり、それぞれのイベント(例えば、有害事象による中 止例あるいは死亡例)の総数を示すものではない。 a:脳卒中又は非中枢神経塞栓症 b:合計 61 例(リバーロキサバン群 26 例、ワルファリン群 35 例)が同意撤回により治験薬の投与を中止した。 そのうち 53 例(リバーロキサバン群 23 例、ワルファリン群 30 例)は 30 日フォローアップ調査を完了した。 引用元:2.7.6.46 の表 2.7.6.46-1 治験薬の投与期間はリバーロキサバン群で平均 498.85 日(1~868 日)、ワルファリン群で平 均 481.08 日(1~861 日)であった。被験者全体の 80.36%が治験薬投与を少なくとも 12 ヵ月間 受け、21.05%が少なくとも 24 ヵ月投与を受けた(表 2.7.4.1-13)。 服薬率は治験薬服薬日数を全投与期間(休薬期間も含む)で除して 100 を乗じて求めた。服薬 率は両投与群とも 99%以上と服薬コンプライアンスは非常に良好であった(2.7.6.46 A49701 の 表 2.7.6.46-6 参照)。
表 2.7.4.1-13 治験薬(実薬)の投与期間(試験 12620、安全性解析対象集団) 累積投与期間 リバーロキサバン ワルファリン 合計 (N=639) (N=639) (N=1278) n(%) N(%) n(%) ≧ 1 回投与 639(100) 639(100) 1278(100) ≧ 1 ヵ月 614(96.09) 617(96.56) 1231(96.28) ≧ 3 ヵ月 596(93.27) 586(91.71) 1182(92.49) ≧ 6 ヵ月 569(89.05) 556(87.01) 1125(88.03) ≧ 9 ヵ月 547(85.60) 527(82.47) 1074(84.04) ≧12 ヵ月 530(82.94) 497(77.78) 1027(80.36) ≧18 ヵ月 262(41.00) 246(38.50) 508(39.75) ≧24 ヵ月 136(21.28) 133(20.81) 269(21.05) 平均値(日) 498.85 481.08 489.96 標準偏差 218.99 228.18 223.72 最小値(日) 1 1 1 Q1(日) 385 364 370 中央値(日) 483 455 476 Q3(日) 693 679 679 最大値(日) 868 861 868 Q1:最小値側から 25%値、Q3:最小値側から 75%値 各群の被験者数を分母とし発現頻度を算出した。 全投与期間:最終投与日-投与開始日+1 1 ヵ月:30 日
引用元:5.3.5.1.3(A49701)の Table DSUB008PB
PT-INR 値の前後の測定値から直線補完法によって算出した帰属値(imputed)をもとに延べ投 与日数を分母に、INR が目標範囲内に入っていた日数を分子として PT-INR が目標範囲(70 歳以 上:1.6~2.6、70 歳未満:2~3)内に入っていた期間割合(TTR)を算出した。ワルファリン群 全体での TTR は 65.0%であった。70 歳以上の被験者では TTR が 74.0%であったが、70 歳未満の 被験者では TTR は 51.8%であり、43.8%が目標範囲以下であった。参考として 70 歳未満で PT-INR 値が 1.6~2.6 に入っていた期間割合を計算してみると 72.7%で、70 歳以上と同様であった。 また、ワルファリン群全例が 2.0~3.0 に入っていた期間割合は 46.8%であった。PT-INR 値が特 定範囲内に入っていた期間割合を表 2.7.4.1-14に示す。
表 2.7.4.1-14 ワルファリン群における PT-INR 値が特定範囲内にある期間割合(試験 12620、 安全性解析対象集団) ワルファリン群全例 (被験者数:639 例) 曝露日数 INR 目標範囲以下 (%) INR 目標範囲内 (%) INR 目標範囲以上 (%) 全投与期間 327352 28.0 65.0 6.9 70 歳以上 (被験者数:389 例) 曝露日数 INR:<1.6 目標範囲以下 (%) INR:1.6~2.6 目標範囲内 (%) INR:>2.6 目標範囲以上 (%) 全投与期間 195371 17.3 74.0 8.6 70 歳未満 (被験者数:250 例) 曝露日数 INR:<2 目標範囲以下 (%) INR:2~3 目標範囲内 (%) INR:>3 目標範囲以上 (%) 全投与期間 131981 43.8 51.8 4.2 70 歳未満 (被験者数:250 例) 曝露日数 INR:<1.6 (%) INR:1.6~2.6 (%) INR:>2.6 (%) 全投与期間 131981 13.9 72.7 13.3 ワルファリン群全例 (被験者数:639 例) 曝露日数 INR:<2 (%) INR:2~3 (%) INR:>3 (%) 全投与期間 327352 49.7 46.8 3.4
PT-INR(INR):プロトロンビン時間国際標準比(prothrombin time-international normalized ratio) 期間割合:PT-INR 値の前後の測定値から直線補完法によって算出した帰属値(imputed)をもとに延べ投与日数 を分母に、INR が目標範囲内に入っていた日数を分子として算出 引用元:2.7.6.46 の表 2.7.6.46-7、表 2.7.6.46-8、5.3.5.1.3(A49701)の表 14.1.19 2.7.4.1.2.3.2 国外第Ⅲ相試験(試験 11630) 試験 11630 では、脳卒中、TIA 又は非中枢神経系塞栓症の既往を有するか、あるいは心不全、 高血圧症、糖尿病、高齢(75 歳以上)の塞栓症危険因子を 2 つ以上有する非日本人 NVAF 患者 14,264 例がリバーロキサバン群(7,131 例)、ワルファリン群(7,133 例)のいずれかに無作為 に割り付けけられた。このうち、治験薬が 1 回以上投与された 14,236 例(リバーロキサバン群 7,111 例、ワルファリン群 7,125 例)を安全性解析対象集団とした。 リバーロキサバンの用法・用量は、20mg 1 日 1 回夕食後投与であったが、本群でベースライ ンの CLCRが 30~49mL/min の被験者には 15mg 1 日 1 回夕食後投与とした。1,502 例(21.1%)に リバーロキサバン 15mg 1 日 1 回が投与された(表 2.7.4.1-21)。 ワルファリン群の用法はワルファリンナトリウムを 1 日 1 回夕食後投与であった。用量は 2.5 (2.0~3.0)の PT-INR を目標として調節することとした。 試験 11630 はイベント主導型試験であるため、治験実施計画書適合集団における治験薬投与下 の有効性主要評価項目(CEC 判定)の発現数が事前に設定した 405 例になるまで継続された。有 効性評価項目のイベント数が必要数に達した事を施設へ通知した日(施設通知日:20 年 月 日。南アフリカ共和国のみ 20 年 月 日)当日あるいはそれ以降に、最終コンタクトが実
施された被験者(施設通知日に治験薬投与が行われていたか否かを問わない。また最終コンタク トには早期中止者を対象に実施されていた電話等による有効性評価項目に関するイベント評価及 び生存確認のためのコンタクトを含む)を治験完了者と定義した。12,064 例〔リバーロキサバ ン群 6,035 例(84.87%)、ワルファリン群 6,029 例(84.62%)〕が治験を完了した(図 2.7.4.1-2)。 a:施設通知日時点〔南アフリカ(20 / / )を除き 20 / / 〕 b:5 例が 2 回割り付けされたが、初回の割り付けに伴うデータのみが ITT 解析に使用された。 c:安全性解析対象集団は ITT 解析対象集団のうち治験薬の投与を少なくとも 1 回受けたすべての被験者。 d:治験薬投与及び追跡中止;治験薬を早期中止し、かつ最終コンタクトが施設への施設通知日以前に行われた 被験者 e:治験完了;最終コンタクトが施設通知日かそれ以後に行われた被験者(治験薬投与が行われていたか否かを 問わない) f:除外施設の被験者を含む g:除外施設の被験者を含まない h:除外施設;GCP 違反のため治験依頼者が除外した施設 i:閉鎖施設;施設終了通知日以前に閉鎖され、追加の被験者情報が得られない施設 j:有効性評価項目により中止され、最後のコンタクトが施設通知日(20 / / )の 3 日前であったワルファリ ン群の 1 例 引用元:2.7.6.47-2 の図 2.7.6.47-2 図 2.7.4.1-2 被験者の内訳 被験者の内訳a ワルファリン 7,111(安全性解析対象集団)c ・20:治験薬服用せず リバーロキサバン 7,125(安全性解析対象集団)c ・8:治験薬服用せず 14,264b 無作為割付被験者/ ITT 解析対象集団 1,076:治験薬投与及び追 跡中止d ・583:死亡f ・18:追跡不能f ・380:同意撤回g ・89:除外施設の被験者h ・6:閉鎖施設の被験者i 6,035:治験完了e ・4,591:二重盲検治 療期間の終了例 ・1,444:二重盲検治 療期間の早期中止例 1,096:治験薬投与及び追 跡中止d ・638:死亡f ・14:追跡不能f ・354:同意撤回g ・78:除外施設の被験者h ・11:施設退去i ・1:その他j 6,029 治験完了e ・4,657:二重盲検治 療期間の終了例 ・1,372:二重盲検治 療期間の早期中止例 7,133(無作為割付被験者/ITT 解析対象集団) 7,131(無作為割付被験者/ITT 解析対象集団)
二重盲検治療期を終了した被験者はリバーロキサバン群の 4,591 例(64.56%)、ワルファリ ン群の 4,657 例(65.36%)であった。早期中止に至った最も多い理由は有害事象〔リバーロキ サバン群 993 例(13.96%)、ワルファリン群 919 例(12.90%)〕で、その他同意撤回〔リバー ロキサバン群 671 例(9.44%)、ワルファリン群 673 例(9.45%)〕、評価項目(脳卒中又は非 中枢神経系塞栓症:治験担当医師判定)の発現〔リバーロキサバン群 300 例(4.22%)、ワル ファリン群 332 例(4.66%)〕が主な理由であった(2.7.6.47 R-8570 の表 2.7.6.47-1 参照)。 治験薬の投与期間はリバーロキサバン群で平均 572.2 日(1~1,263 日)、ワルファリン群で 平均 579.9 日(1~1,263 日)であった。被験者全体の 78.55%が治験薬投与を少なくとも 12 ヵ 月間、35.71%が少なくとも 24 ヵ月投与を受けた。少なくとも 36 ヵ月の投与を受けた被験者が 2.02%(288 例)存在した。服薬率はいずれの投与群も 98%以上であった(2.7.6.47 R-8570 の表 2.7.6.47-5、表 2.7.6.47-6 参照)。 試験 11630 ではワルファリン群の目標 PT-INR は 2.5(2.0~3.0)とした。PT-INR 値が目標範 囲内であった期間割合(TTR)は、被験者ごとの TTR は平均 55.2%(中央値 57.8%)であった。 試験 12620 と同様に、INR 全測定値の帰属値(imputed)をもとに、延べ投与日数を分母に、INR が目標強度に入っていた日数を分子として算出した TTR は 58.4%であった(表 2.7.4.1-15)。 表 2.7.4.1-15 ワルファリン群における PT-INR 値が目標範囲内にある期間割合(試験 11630、 安全性解析対象集団) ワルファリン群 曝露日数 INR:<2.0 (%) INR:2.0-3.0 (%) INR:>3.0 (%) 全投与期間 4092922 26.4 58.4 15.3
PT-INR(INR):プロトロンビン時間国際標準比(prothrombin time-international normalized ratio) 期間割合:PT-INR 値の前後の測定値から直線補完法によって算出した帰属値(imputed)をもとに延べ投与日数 を分母に、INR が目標範囲内に入っていた日数を分子として算出 引用元:5.3.5.1.6(R-8570)の Table DSUB410IB 2.7.4.1.2.3.3 国内・国外第Ⅲ相試験統合データベースに基づく解析 試験 11630 と試験 12620 で無作為割り付けされた 10,196 例(65.72%)の被験者が治験薬の投 与を受け、二重盲検治療期を終了した。2 試験で 5,318 例(34.28%)が早期に治験薬投与を中 止した〔リバーロキサバン群 2,679 例(34.57%)、ワルファリン群 2,639 例(33.99%)〕。主 な中止理由は有害事象〔リバーロキサバン群 1,066 例(13.75%)、ワルファリン群 989 例 (12.74%)〕であった。中止理由となった有害事象では、非出血関連有害事象が多く、その頻 度は両投与群で同程度であった〔リバーロキサバン群 743 例(9.59%)、ワルファリン群 751 例 (9.67%)〕(表 2.7.4.1-16)。
表 2.7.4.1-16 二重盲検治療期における早期中止理由(試験 12620 と試験 11630 の統合、安全 性解析対象集団) リバーロキサバン (N=7750) n(%) ワルファリン (N=7764) n(%) 合計 (N=15514) n(%) 治験薬投与終了例 5071(65.43) 5125(66.01) 10196(65.72) 治験薬投与早期中止例 2679(34.57) 2639(33.99) 5318(34.28) 有害事象 1066(13.75) 989(12.74) 2055(13.25) ・出血関連 323( 4.17) 237( 3.05) 560( 3.61) ・非出血関連 743( 9.59) 751( 9.67) 1494( 9.63) 同意撤回 697( 8.99) 708( 9.12) 1405( 9.06) 追跡不能 10( 0.13) 9( 0.12) 19( 0.12) 治験実施計画書からの逸脱 151( 1.95) 133( 1.71) 284( 1.83) 臨床評価項目の発現a 318( 4.10) 360( 4.64) 678( 4.37) 治験薬投与不遵守 137( 1.77) 165( 2.13) 302( 1.95) 治験依頼者による施設の打ち切り 83( 1.07) 70( 0.90) 153( 0.99) 治験実施計画書に基づかない医師の判断 195( 2.52) 191( 2.46) 386( 2.49) 欠落/不完全データ 1( 0.01) 1( 0.01) 2( 0.01) 死亡 8(11.82) 3(12.13) 11(11.98) 各群の被験者数を分母とし発現頻度を算出した。 投与完了例:試験 11630 は、被験者の最終投与日が施設通知日より以降であるもの。試験 12620 は症例報告書の 記載に基づく。 死亡のカテゴリーがあるのは試験 12620 のみ a:脳卒中又は非中枢神経系塞栓症 引用元:5.3.5.3.1(R-8603)の Table DSUB004KB4I 平均投与期間は、リバーロキサバン群 566.18 日(中央値 584 日)、ワルファリン群 571.73 日 (中央値 588 日)であった。全被験者中、約 79%が少なくとも 1 年、約 35%が少なくとも 2 年、 約 2%が少なくとも 3 年の治験薬投与を受けた(表 2.7.4.1-17)。