筋収縮最小運動系(ナノ筋収縮系)と 顕微技術の開発
Development of a minimum contractile system of muscle (Nanomuscle)
and microscopic techniques
平成 17 年(2005 年) 3 月
早稲田大学大学院 理工学研究科 生命理工学専攻 実験生物物理学研究
鈴木 団
2004年度 博士論文 鈴木 団
【目 次】
【省略語一覧】 ・・・・・4
第 1 章 序論
1-1) 筋収縮系の概要 ・・・・・ 5 1-2) 骨格筋収縮系の階層構造 ・・・・・ 6 1-3) 本論文の概要 ・・・・・ 7
第1章 図 ・・・・・12
第 2 章 本研究で用いたタンパク質の調製法と顕微解析装置、
実験法について
2-1) ウサギ骨格筋筋原線維の調製
2-1-1) グリセリン筋の調製 ・・・・・15
2-2) タンパク質の調製
2-2-1) G-アクチン調製法 ・・・・・17
2-2-2) nTm調製法 ・・・・・17
2-2-3) プラズマゲルゾリン調製法 ・・・・・21
2-2-4) G1-G3調製法 ・・・・・25
2-3) 顕微解析装置について
2-3-1) ミクロ熱励起・ミクロ温度計測法で用いた装置について
・・・・・29
2-3-2) ナノ筋収縮系で用いた装置について ・・・・・30
第2章 図 ・・・・・31
第 3 章 新しいミクロ熱励起・ミクロ温度計測法の開発について
3-1) 要約 ・・・・・35
3-2) 研究の背景 ・・・・・35
3-3) 材料と方法
3-3-1) 試薬 ・・・・・37
3-4) 実験結果
3-4-1) 温度計ピペットの作成 ・・・・・38
3-4-2) ヒーターピペットの作成 ・・・・・38
1
2004年度 博士論文 鈴木 団
3-4-3) 温度の測定 ・・・・・39
3-4-4) レーザー焦点位置の調整 ・・・・・39
3-4-5) 局所熱励起・温度計測 ・・・・・40
3-5) 考察 ・・・・・41
第3章 図 ・・・・・43
第4章 アクチンフィラメントを選択的に除去する分子道具と しての、アクチンフィラメント切断・キャップタンパク 質ゲルゾリンの性質
4-1) 要約 ・・・・・49
4-2) 研究の背景 ・・・・・49
4-3) 材料と方法
4-3-1) タンパク質 ・・・・・51
4-3-2) 筋線維中でのF-アクチン除去過程の観察 ・・・・・51
4-3-3) in vitroでのF-アクチン及び再構成した細いフィラメント 切断・除去過程の観察 ・・・・・53
4-3-4) 顕微解析の方法 ・・・・・53
4-4) 実験結果と考察
4-4-1) 筋線維中でのG1-G3による細いフィラメント除去
・・・・・55 4-4-2) G1-G3によるF-アクチン切断・除去に対するCa2+の寄与
・・・・・56 4-4-3) G1-G3とnTmのF-アクチンへの競合的結合
・・・・・57
第4章 図・表 ・・・・・59
第5章 ナノ筋収縮系の開発とその特性の顕微解析
5-1) 要約 ・・・・・64
5-2) 研究の背景 ・・・・・64
5-3) 材料と方法
5-3-1) 筋原線維とタンパク質 ・・・・・66
5-3-2) 実験装置 ・・・・・66
5-3-3) ナノ筋収縮系の調製 ・・・・・67
5-4) 実験結果
5-4-1) ナノ筋収縮系 ・・・・・69
5-4-2) ナノ筋収縮系における発生張力の測定 ・・・・・70
2
2004年度 博士論文 鈴木 団
5-4-3) 長さ−力関係 ・・・・・71
5-4-4) 負荷−速度(P-V)関係 ・・・・・72
5-5) 考察
5-5-1) ミオシン頭部1個の平均発生力 ・・・・・74
5-5-2) 線形な長さ−力関係 ・・・・・76
5-5-3) 筋(原)線維におけるP-V関係との相似 ・・・・・77
5-5-4) 張力ゆらぎ ・・・・・77
5-6) 結論 ・・・・・78
第5章 図・表 ・・・・・79
第6章 まとめ
6-1) 本論文のまとめ ・・・・・86
6-2) 展望 ・・・・・87
【参考文献】 ・・・・・89
【研究業績】 ・・・・・96
【 謝 辞 】 ・・・・・101
3
2004年度 博士論文 鈴木 団
【省略語一覧】
AMS 硫酸アンモニウム
A-PMSF:(p-amidinophenyl) methanesulfonyl fluoride ATP アデノシン三リン酸
BDM 2,3-butanedione 2-monoxime DFP:diisopropyl fluorophosphate DMSO dimethyl sulfoxide
DTT dithiothreitol
EGTA ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid F-アクチン アクチンフィラメント
G-アクチン アクチンモノマー
G1-G3 ヒトゲルゾリンN末端断片。アミノ酸1-406 G.S. Guba-Straub solution
HMM heavy meromyosin
IPTG isopropyl β-D-thiogalactoside IR infra red
MOPS 3-(N-morpholino)propanesulfonic acid MQ 超純水
nTm native tropomyosin,トロポミオシン−トロポニン複合体 P-V関係 load-velocity relationship, 負荷−速度関係
PMSF phenylmethanesulfonyl fluoride Tm tropomyosin
Tn troponin
4
2004年度 博士論文 鈴木 団
第 1 章 序論
本研究の対象である筋収縮系と、本論文の概要について述べる。
1-1) 筋収縮系の概要
歩く、走る、食べる、話す、といった人間の自発的な運動は、筋肉と骨で構成され る機構が共同的に働くことにより可能となる。この機構において能動的なのは、両 端が骨あるいは弾性のある臓器に腱を介してつながれている骨格筋である。この随 意筋の収縮あるいは弛緩状態は神経から伝えられる電気信号により、オン−オフの 二値化された状態として制御されている。
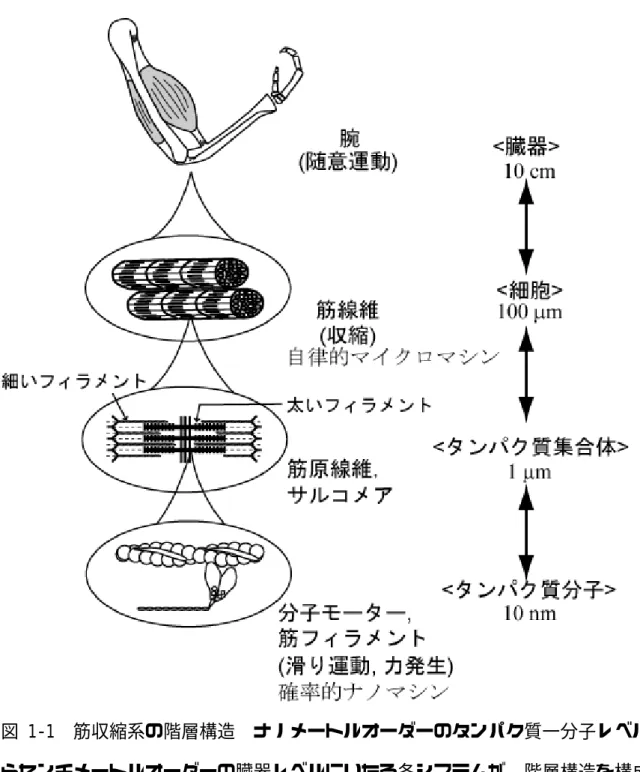
筋収縮系は、一分子(タンパク質)から臓器に渡る階層構造を構成している(図 1-1)。筋収縮系は生理的機能を達成できるよう、高度に統制のとれた構造をしてお り、各階層の大きさは10 nmから約1 cmの範囲に及ぶ。10 nmともなると、それ らを我々が見るためには電子顕微鏡や原子間力顕微鏡(AFM)のような道具が必要 である。一方 1 cm という大きさならば、目で見ることができ、手で触れる。即ち この階層における巨視的な筋収縮は、アクチン−ミオシン複合体(力学的酵素)、ト ロポミオシン−トロポニン複合体(制御系タンパク質)、コネクチン/タイチン(弾 性タンパク質)など数種の筋タンパク質の微視的な動きが集約された結果なのであ る。
運動する装置を実現する必要十分条件は、生物機械と人工機械とで共通している
(図1-2)。しかしながら運動装置とその枠組みを与える材料とを比べたとき、二つ の機械の間には大きな違いが存在する。例えば、人工的な機械の動作原理は完全に 理解されている。自動車は熱力学・機械工学といった物理的原理に基づき動作する。
その一方で我々は、筋肉の分子モーターが動作する際の化学−力学エネルギー変換
5
2004年度 博士論文 鈴木 団
の物理的原理を未だ完全には理解できていない。また生物機械を構成する全てのタ ンパク質は環境変化に素早く応答できるような柔軟性(自律的フィードバック機構)
を備えているのに対し、人工機械では応答と制御は予め設計・プログラムされてい なくてはならない。
1-2) 骨格筋収縮系の階層構造
骨格筋の構造的・機能的な単位は筋線維(muscle fiber)である(図1-3)。筋線維は 長く(例えばキリンでは1 mに及ぶ)細い(直径10~100 µm)線維で、円筒状をし、
細胞融合で生み出される多核細胞である。筋線維の周囲は細胞膜に囲まれ、神経末 端が結合している。筋線維同士は互いに平行に寄り添い、束となって筋組織を構成 している(Engel and Franzini-Armstrong, 1994)。
このような筋線維は、収縮要素である筋原線維(myofibril)が平行に多数集ま ったものである。筋原線維には縞模様があり、さらに隣り合う筋原線維同士では位 相がそろっているため筋線維もまた縞模様をしている。以上のような縞模様は骨格 筋と心筋に共通で、規則正しい周期構造を持たない平滑筋とは対照的である。筋原 線維の縞模様はA帯とI帯との周期的配列に起因する。A帯は約1.6 µmの長さ一定 の構造だが、I帯の長さは変動する。各々のI帯の中央を通る太い線はZ線あるはZ 盤と呼ばれている。縞模様の構造単位は Z 線で区切られ、筋節(sarcomere)と呼 ばれており、通常2~3 µmの長さである。
A帯は太いフィラメントで構成されており、フィラメントの中央部がM線でま とめられている。またI帯は直径約8 nm、長さ1 µmの細いフィラメントが平行に 配列したものである。細いフィラメントはZ線からA帯中央に向かって伸び、その 一部はA帯で太いフィラメントと重なっている。細いフィラメントの無いA帯の中
6
2004年度 博士論文 鈴木 団
央部はHゾーンと呼ばれている。これら太いフィラメントと細いフィラメントとは 格子構造を成しており、例えば哺乳類の骨格筋では、正三角形の頂点に位置する3 本の太いフィラメントに三角形の重心に位置する1本の細いフィラメントが囲まれ るようにして、六角形に納められている(図1-3、右上)。
ミオシンとアクチンはそれぞれ太いフィラメント、細いフィラメントの主な構 成分子である。これらタンパク質は、全筋タンパク質の約70%を占める。太いフィ ラメントは二極性の分子集合体で、約300個のミオシン分子からなる。その構造は 中央を境にして対称的であり、中央では太いフィラメントに結合したMタンパク質 が M 線を形成している。細いフィラメントは極性のあるらせん状の分子集合体で、
アクチン分子からなる。細いフィラメントの自由端はP端と呼ばれ、トロポモジュ リン(tropomodulin)と呼ばれるキャップタンパク質が結合している(Littlefield et al.,
2001)。反対側(B端)は Z 線に結合し、細いフィラメントの格子構造がZ 線で固
定されて、Z線で対称な構造を持つI-Z-Iブラシを形作っている。
1-3) 本論文の概要
本論文は、筋収縮分子機構の解明を目指して新規に開発したナノ筋収縮系と、
ナノ筋収縮系を開発する過程で得られたタンパク質間相互作用の研究の成果、及び ナノ筋収縮系のみでなく、細胞機能を探る上でも強力で新たな道具となるミクロ熱 励起・ミクロ温度計測法の開発の成果についてまとめたものである。
筋収縮分子機構は生体機能における最大の謎の1つであり、この解明は他の生 体システムの多様な動作メカニズムを理解するための基礎となるものである。なぜ なら、主要な筋タンパク質であるミオシンファミリーのタンパク質や、細胞骨格で あるアクチンは筋肉以外の細胞でも発現し、そのアクチン−ミオシン相互作用は、
7
2004年度 博士論文 鈴木 団
細胞分裂を始めとした細胞活動の根幹に関わっているからである。また、筋タンパ ク質の動作機構、制御機構の研究は、キネシン−微小管系といった他のモータータ ンパク質群によるシステム制御メカニズムに関する研究と共に、人工的なナノシス テムを構築する上での基礎となりうる。
第一章では、筋収縮分子機構に関する研究の背景、そして未解明な問題をまと めた。
第二章では、本研究で用いた生体試料の調製法と顕微解析装置、実験法につい てまとめる。生体試料は、大腸菌で発現・精製を行ったタンパク質、動物から単離 したタンパク質や筋肉試料と多岐に渡るので、そのそれぞれについて解説する。ま た顕微解析装置、実験法は各研究において異なるため、それらについても詳細に述 べる。
第三章では、新しいミクロ熱励起・ミクロ温度計測法の開発に関する研究につ いて述べる。本研究において申請者は、マイクロメートルサイズの微小領域を対象 に、顕微鏡下の任意の箇所で水温(室温)から水の沸点に渡る広い範囲で温度上昇 と下降を行うことができる、新しい技術を開発した。温度上昇・下降の速さは約10 msで、最高温度は任意に変えられる。さらにこれに併せて開発したマイクロメート ルの空間分解能を持つミクロ温度センサーを用いれば、0.1°C の精度で局所的な温 度変化を検出できる。
局所熱励起は、マイクロマニピュレーターにより操作されるガラスマイクロピ ペット先端(直径1〜2 µm)に固着した金属微粒子に、赤外レーザー(波長1064 nm) を照射して行う。赤外レーザーは、レーザー通過領域に影響を与えずに金属微粒子
8
2004年度 博士論文 鈴木 団
のみが熱源となるよう、開口数の大きな対物レンズ(N.A.1.3〜1.4)を通して集光 する。
ミクロ温度センサーには、水溶液から隔離された微小領域に閉じ込めた蛍光色
素 Europium-TTA の、蛍光強度変化を利用する。蛍光色素を外部環境から隔離する
ことにより、色素が熱以外の環境変化にも応答してしまうという従来技術の欠点を 克服した。温度変化による蛍光色素の蛍光強度変化は充分速く、時間分解能はビデ オレート(毎秒30フレーム)を超える。
この技術は、筋収縮系や第五章で述べるナノ筋収縮系と併せて利用することが でき、筋収縮時における熱発生の検知や、収縮装置が温度により受ける影響につい て詳細に調べる手段となる。また例えば標準的な電気生理学や、蛍光イメージング の技術と互換性が良い。そのため、生きた単一細胞の局所熱刺激に対する応答性や、
細胞内小器官からのイオンの流出に伴う局所温度変化を検出するなどといった、こ れまでにない全く新しい細胞生物学研究への応用が直ちに可能である。
第四章では、アクチンフィラメント(F-アクチン)を選択的に除去する分子道 具としての、F-アクチン切断・キャップタンパク質ゲルゾリンの性質を調べた研究 について述べる。ゲルゾリンからCa2+感受性の切断抑制部位を遺伝子操作により取 り除いたN末断片(G1-G3)を用いて、心筋筋線維での細いフィラメント切断の様 子を観察し、次にin vitro系(精製タンパク質系)で再構成フィラメント切断速度を 計測した。
グリセリン処理ラット心筋筋線維では、蛍光色素Alexa488-ファロイジンでラベ ルした細いフィラメントがG1-G3により除去されていく様子を、共焦点蛍光顕微鏡 を用いて観察した。その結果、細いフィラメントはI帯部分から先に除去されること が分かった。またCa2+の無い条件では、Ca2+のある時と比べて、G1-G3による細い
9
2004年度 博士論文 鈴木 団
フィラメントの除去速度は極端に遅いことが分かった。
次にin vitro系で、F-アクチン、およびトロポミオシン・トロポニン複合体(nTm)
を再構成した細いフィラメントを用いて、筋線維で見られた現象を詳しく調べた。
このin vitro系ではまず、ビオチン化したアクチンを含むF-アクチンを蛍光色素ロー ダミンファロイジンでラベルし、再構成した細いフィラメントの場合はさらにウサ ギ骨格筋から精製したnTmを混ぜ氷上で一晩放置する。それをガラス面に吸着して いるビオチン化したBSAにアビジンを介して固定して、蛍光顕微鏡で観察した。切 断・除去の時間経過は蛍光強度の減少から定量化した。その結果、Ca2+非存在下で のG1-G3がF-アクチンを切断する速度は、Ca2+存在下と比べて遅かった。さらにnTm を再構成した細いフィラメントについても同様の実験を行ったところ、Ca2+非存在 下でのフィラメント除去速度はさらにずっと遅いことが分かった。一方Ca2+存在下 では、nTmの結合していないF-アクチンと比べてわずかな差が見られた。
以上の結果を、G1-G3のF-アクチン切断能に対するCa2+の寄与、及びF-アクチ ン結合タンパク質により誘起されるF-アクチンの構造変化の可能性と、その生理的 意義について議論する。
第五章では、筋収縮最小運動系(ナノ筋収縮系,Nanomuscle)の開発と、その 特性の顕微解析について述べる。本研究では、筋(原)線維収縮系とin vitro滑り運 動系の中間に位置し、ミオシンフィラメントが生体と同じ格子構造を保持している 全く新しいナノ筋収縮系を開発し、その力発生・運動特性に関する研究を行った。
本研究の目的は、アクトミオシン分子モーターの運動特性を生体内の筋肉に近 い条件で、規則的に配列した数〜数十個の分子モーター(筋収縮系ではナノ分子機 械であるタンパク質が立体格子を形成している)と相互作用する単一F-アクチンレ ベルの顕微解析により、明らかにすることである。そこで申請者は、ミオシン分子
10
2004年度 博士論文 鈴木 団
モーターの3次元集合体と、再構成した1本のF-アクチンとで構成される、ナノ筋 収縮系を開発した。アクトミオシン相互作用は、生体内と同じ立体配置で行なわれ る。また溶液条件も、極度に人工的なin vitro滑り運動系と比べてより生理的な条件 にできる。この実験系は、筋原線維から直接調製される分子モーター集合体(A帯)
と、光ピンセット技術・蛍光観察技術を用いて操作・計測される単一F-アクチンに より構成される。よって、筋収縮系を模倣した最小システムでのアクトミオシン相 互作用を計測すると言える。
ナノ筋収縮系の調製にはまず、グリセリン処理したウサギ腸腰筋をホモジナイ ズして筋原線維を用意する。そして筋原線維内の細いフィラメントだけをゲルゾリ ンを用いて選択的に除去し、露出したA帯を得た。そこでは太いフィラメントのフ ィラメント格子が、生体内と同じ三次元構造を保っている。次に B 端に直径 1 µm のポリスチレンビーズを結合した単一F-アクチンを光ピンセットで操作し、A帯の 表面と内部における張力発生の様子を位相差像と蛍光像で観察・解析した。その結 果、長さ−張力関係はA帯表面では太いフィラメントの端から0〜300 nmの範囲で、
A帯内部では0〜150 nmの範囲で線形である事が確認された。なお、それ以上の範 囲については張力が光ピンセットの捕捉力を越えるため計測できなかった。この長 さ−張力関係から、A帯内部におけるミオシン頭部 1 個当たりの平均発生力を、約 1 pN と見積もった。また収縮に伴い負荷の上昇する増張力性収縮条件(auxotonic
condition)下での負荷−収縮速度関係を求めたところ、上に凸の曲線であった。以
上の結果はいずれも、筋線維や筋原線維で得られた結果を再現していた。
第六章では、本研究のまとめと、関連する研究における本研究の位置づけ、そ して今後の研究のあり方について述べる。
11
2004年度 博士論文 鈴木 団
図 1-1 筋収縮系の階層構造 ナノメートルオーダーのタンパク質一分子レベルか らセンチメートルオーダーの臓器レベルにいたる各システムが、階層構造を構成し ている。この階層構造は横紋筋(骨格筋・心筋)に共通している。
12
2004年度 博士論文 鈴木 団
図 1-2 運動装置を構成するための必要十分条件 自然の創った運動装置(筋肉)
と、ヒトの作ったそれ(自動車)との比較。自然物と人工的産物とで、運動装置を 構成する必要十分条件は共通しており、各機能は1:1に対応している。しかし少 なくとも二つの大きな違いがある。それは、動作(力発生)原理と、材料である。
13
2004年度 博士論文 鈴木 団
図1-3 筋収縮系 構造・機能上の最小単位は筋節(sarcomere)で、そこでは細 いフィラメントと太いフィラメントとが、フィラメント格子を構成している。細 いフィラメントと太いフィラメントはどちらもタンパク質分子がらせん状に結合 したもので、前者は一極性、後者は二極性のフィラメントである。ミオシンはア クチンと結合してクロスブリッジを結合し、またATP加水分解部位でもある二つ の頭部(head)を持つ長いタンパク質である。もう一つの尾部(rod)と呼ばれる
部位は、coiled-coil構造をしている。アクチンは馬蹄形の小さな分子で、らせん状
に重合する。そのZ線に結合する方をB端、逆側の自由端をP端と呼ぶ。
14
2004年度 博士論文 鈴木 団
第 2 章 本研究で用いたタンパク質の調製法と顕微解析装置、
実験法について
本章では、本研究で用いた生体試料の調製法と実験装置、実験方法について述べる。
手順は全て、早稲田大学の動物実験に関する規定にのっとって行なわれた。
2-1) ウサギ骨格筋筋原線維の調製
筋原線維はグリセリン処理したウサギ腸腰筋筋線維をRigor溶液(60 mM KCl, 5 mM MgCl2, 10 mM 3-(N-morpholino)propanesulfonic acid (MOPS), pH 7.0, and 1 mM EGTA)中でホモジナイズして調製した。グリセリン筋の調製は、Ishiwata and Funatsu(1985)の方法に従った。
2-1-1) グリセリン筋の調製
<ウサギ屠殺1週間前>
ウサギ雄2.5 kgコンベンショナルを注文する。
<ウサギ屠殺2日前>
1.直径3 mmのパイレックスのガラス棒を用意する
−ガラス切りを使って長さ10 cm(試験管より5 mmほど長く)切断し、その 両端をガスバーナーで熱して丸める。
−よく洗い乾燥棚で乾かしておく。
2.糸を用意する
−ガラス棒×2本分の長さのたこ糸より少し太めの糸をガラス棒に巻く
−1%NaHCO3で煮沸消毒 5 min.
−MQで煮沸消毒 5 min.
−MQで洗い、はさみで約5 cmに切り乾燥棚へ。長く置くと焦げるので注意。
15
2004年度 博士論文 鈴木 団
3.試験管を用意する
−細長いものを使用
−ガラス棒×2本+予備分を洗浄し、乾燥棚へ。
<ウサギ屠殺前日>
1.糸
−低温室に置いておく 2.溶液作成
(i) 51%(v/v)グリセリン溶液 7 ml×試験管の本数×2セット
51% グリセリン、0.5 mM NaHCO3、5 mM EGTA (ii) リンゲル 100 ml
154 mM NaCl、0.1g Glucose、1 mM EGTA、5.6 mM KCl、2.4 mM NaHCO3
3.試験管
−51%グリセリン溶液を6.86 mlずつ入れる。
−1から通し番号をふったラベルを貼る。
−氷中で低温室に放置。
4.低温室に実体顕微鏡を置き、場所の用意をする
<ウサギ屠殺当日>
−試験管1セットに入った51%グリセリン溶液に0.1 M Leupeptinを0.14 ml 加える(final 2 mM)。
−ウサギ腸腰筋から太さ数mmの筋線維束を切り出し、その両端を用意した糸 でガラス棒に固定する。その際ピンセット・ハサミでは筋線維束の両端の みを持つように気をつける。
−試験管に入れよく攪拌し、氷中放置。
−6時間後に攪拌する。
16
2004年度 博士論文 鈴木 団
<ウサギ屠殺翌日>
−新鮮な0.1 M Leupeptinを0.14 ml加えた、新しい51%グリセリン溶液入り試験 管にガラス棒ごと筋線維束を移し、よく撹拌する。
−-20°C冷凍庫で保存
2-2) タンパク質の調製
G-アクチンは、トロポミオシン−トロポニン複合体(nTm)をあらかじめ取り除い てあるウサギ骨格筋のアセトンパウダーから精製した(Kondo and Ishiwata, 1976)。 nTmはEbashiらの方法(Ebashi et al., 1968)でウサギ骨格筋から調製した。ゲルゾ リンはKurokawa et al.の方法に倣いウシ血清から調製した(Kurokawa et al., 1990)。 ゲルゾリンは低Ca2+の溶液にさらすと機能を失いやすいため、低Ca2+条件に置く時 間はなるべく短くした。またKurokawaらはプロテアーゼインヒビター 0.1 mM DFP を全ての溶液に混ぜているが、我々は一番初めにプラズマに添加した以外は利用し ないようにした。それでも活性・収量はほぼ変わらない。ヒトゲルゾリンcDNAのN 末端断片(アミノ酸 1-406, G1-G3)は、pET3dベクターにより発現、調製した
(Granzier et al., 1997; Trombitas and Granzier, 1997)。
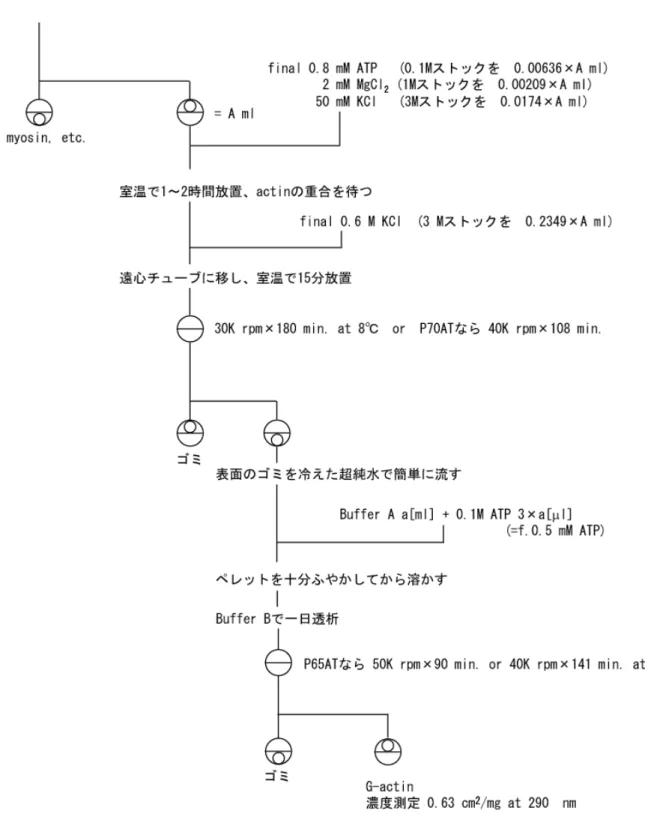
2-2-1) G-アクチン調製法
アセトンパウダーからのG-アクチン調製法は、図2-1にまとめた。
2-2-2) nTm調製法
太字で斜体はウサギ2羽用
[Guba Straub Solution:G.S.] イオン強度〜0.5 M 2リットル、3リットル 0.3 M KCl 粉44.7 g、67.05 g
0.15 M KH2PO4 粉42.1 g、63.15 g
17
2004年度 博士論文 鈴木 団
室温にて、KOHでpH6.4に調製
[4 %(w/v) NaHCO3溶液] 筋肉から脂肪を除くため。 1リットル、2リットル 粉末NaHCO3 40 gをMQに溶かす。
[飽和硫安溶液(飽和AMS)の調製]
−1.5リットルのMQを熱する。
−(NH4)2SO4を1200 g加えて充分に溶かす。
−室温まで下がったら低温室に放置する。
−AMSが析出するまで低温室に数日放置。
−微小な結晶が混ざらないよう注意して、上清を回収する。
−Tris(あるいはTrizmabase)で、pHを7.0にする。
−最終濃度が1mMになるように、粉末EDTAを溶かす。
−pHを確認し、7.0に合わせる。
−低温室に保存。
[手 順]
ウサギ屠殺前日に、ナイロンゴースの煮沸、ガーゼ、缶、液体廃液用の容器、テフ ロン棒2本、冷MQを用意しておく。
|
4% NaHCO3 溶液を室温に戻し、以下のものは低温室に移動。
遠心チューブ、ローター、G.S. solution(-2°C)、ミンサー、テフロン棒 |
ウサギ屠殺(早稲田大学のガイドラインに従って処置)
|
内臓を除去したウサギを氷中で15分放置 |
低温室で赤筋は取り除き、背筋、大腿筋などの白筋のみを集めて素早くミンチにす る = 1 vol.(weight)
|
-2°C に調製したG.S.を3vol.(weight)加える(イオン強度を上げる)
|
氷漬けの浴槽で2本のテフロン棒を使ってゆっくり撹拌 |
遠心 5K rpm × 10 min. at 0°C
18
遠心開始まで10分
2004年度 博士論文 鈴木 団
ガーゼで濾過
ペレット:アクチン、nTm調製用 上清:ミオシン調製用 |
ペレットの体積を見積もる
V(ペレット) = V(遠心前) – V(上清) 4 vol.の冷MQ
テフロン棒で手早く撹拌し、
ゴースで素早く濾過 5リットルの室温0.4% NaHCO3
|
撹拌し、室温で10分放置
ビーカーで浮いてきた脂肪を適宜取り除く。
|
ゴースで濾過 5リットルの室温0.4% NaHCO3(2回)
撹拌し、室温で10分放置
ビーカーで浮いてきた脂肪を適宜取り除く。
| 冷MQ5リットル(3回)
濾過 肉がふやけるまで。
素早く撹拌・濾過 |
肉がふやけたら冷MQで2リットルに合わせる。
|
氷中で数時間〜一晩放置(制御系を外すため)
|
遠心 5K rpm × 30 min. at 0°C
上清:nTm調製用 ペレット:アクチン調製用 粘性がある。
|
4重ガーゼで濾過し体積を量る。
体積は約1000∼2000 ml。粘性が強い場合は3重でもOK。
|
3リットルビーカーに移す。 100 mlあたりAMSを25 gだけ加える(∼40%)
19
2004年度 博士論文 鈴木 団
泡立てないようそっと。
氷中の3リットルビーカーでゆっくり10分撹拌。
白くにごってくる。
|
遠心 7K rpm × 33 min. at 0°C |
上清 |
4重ガーゼで濾過し体積を量る。
|
3リットルビーカーに移す。 100 mlあたりAMSを12.5 gだけ加える(〜60%) 泡立てないようそっと。
氷中の3リットルビーカーでゆっくり10分撹拌 白くにごってくる。
|
遠心 7K rpm × 65 min. at 0°C |
ペレット
非常に柔らかいので上清を捨てるとき注意。
|
ペレットを70 mlの冷MQで溶かす。
|
飽和AMSから調製した60% AMSを加え、210 mlにする(40% AMS)。 |
遠心 40K rpm × 100 min.又は30K rpm × 180 min. at 8°C(アクチン重合)with RP42 |
上清 |
体積を量る。 AMS 12.5 g / 100 ml
以下のものを低温室に準備。
- 500 ml メスシリンダー - 遠心チューブ (RP-42) - ホールピペット
以下のものを調製
- 飽和AMSから60% AMS sat. AMS 120 ml MQ 80 ml - 冷MQ
氷中で30分攪拌。
|
20
2004年度 博士論文 鈴木 団
遠心 7K rpm 46 min. at 0°C |
ペレット |
ペレットを1 mM NaHCO3(bc,透析外液を利用)で溶かす(約30∼40 ml / ウサギ)。 |
1 mM bcで透析。だいたい8回は外液を交換。
|
ゴミ落し遠心 30K rpm × 60 min. at 0°C |
上清 |
濃度測定。吸光係数:0.38 cm2 / mg at 280 nm |
-80°Cで保管。
2-2-3) プラズマゲルゾリン調製法
[Washing Buffer] 冷MQで調製。 2リットル×必要本数を適宜 50% (NH4)2SO4 25 mM Tris-HCl pH8.0
0.5 mM NaN3 1 mM DTT
[×10 BufferA] 2リットル×必要本数を適宜
0.45 mM NaCl 0.25 M Tris-HCl pH8.0 10 mM EGTA pH8.0 0.3% NaN3
10 mM DTT
[BufferB] 1リットル
30 mM NaCl 25 mM Tris-HCl pH8.0 0.1 mM EGTA pH8.0 0.03% NaN3 1 mM DTT
[BufferC] 1リットル
21
2004年度 博士論文 鈴木 団
30 mM NaCl 25 mM Tris-HCl pH8.0 2 mM CaCl2 0.03% NaN3
1 mM DTT
[その他]
ウシプラズマ:フナコシ株式会社 受託・特注品業務 #K20 保存条件4°C カラム樹脂:TOSOH TSK gel DEAE-TOYOPEARL 650S
A-PMSF : (p-Amidinophenyl) methanesulfonyl Fluoride Hydrochloride Wako#014-10391
DFP:Diisopropyl Fluorophosphate Wako#049-18921
[カラムの準備]
−樹脂を3リットルビーカーに入れ、2倍量の×10 BufferAを入れ攪拌。
−樹脂が沈むのを待つ(∼2.5 hrs)
−樹脂がしっかり沈殿したら上澄みを捨てる。
−×10 BufferAを入れ攪拌。
3回繰り返す
−樹脂が沈むのを待つ(∼2.5 hrs)
−樹脂をBufferAと薬さじで良く攪拌
−フローアダプターで樹脂を詰め、低温室に置く。
[手順(硫安分画)]
1週間前に、フナコシにウシプラズマを注文しておく。
|
前日晩に冷凍遠心機の電源を入れて冷やしておく。また低温室にビーカー+スター ラー、2リットルメスシリンダー+4重ガーゼを用意。
22
2004年度 博士論文 鈴木 団
|
プラズマが届いたら、プラズマ2リットル当たり、2.53 mg/mlに調製したA-PMSF を1 mlとDFPの原液を0.02 mlだけ即座に加える。
|
4重ガーゼを使って濾過し、2リットルメスシリンダーで計量 = A ml |
ビーカーへ泡立てないように傾けて入れる
final 50 mM Tris-HCl(pH8.0)=1 Mストックを A × 50/1000 [ml]
final 35% AMS = A × 0.209 [g]
Stir 30 to 60 min.
5K rpm × 35 min. at 2°C
x’ [ml] ガーゼを使って濾過し、計量。
final 50% AMS = x’ × 0.094 [g]
Stir 30 to 60 min.
6K rpm × 68 min. at 2°C
23
2004年度 博士論文 鈴木 団
Wash Bufferで洗う。
上澄み液の赤色が薄くなるまで(4本 × 3∼4回)。
回転数、時間は上と同じだが、時間は沈み具合で調節。
ペレットをよく砕いて遠心。ピペットを使う。
Wash Bufferは水でバランスできる程度にたっぷり入れる。
必要最小限の量のBuffer Aでペレットを溶かす。
これで最終的なタンパク濃度が決まるので薄め過ぎないこと。
しかし、完全に溶かす。
BufferAで透析。AMS抜き。
ゴミ落とし 35K rpm × 120 min. at 2°C 満タンなら 40K
用意しておいたビーカーへ移す(#)。
[手順(カラム)]
Buffer Aでカラムを洗う。カラム容量が150 [ml]として150 × 2 = 300 [ml]くらい。
流速3∼4 [ml/min]。ストップウォッチを使い流速を調べておく。
|
カラムにタンパク(#)をロード。
|
タンパクがなくなるまで目で確認。
|
24
2004年度 博士論文 鈴木 団
Buffer Aをロード。
3~4時間かそれ以上。何も出なくなるまで(ピークが下がって平坦に)。 |
Buffer Bをカラム容量×2だけロード。EGTA濃度を下げる。
|
Buffer Cでゲルゾリンを溶出。1本当り3~4 mlでチューブに取っていく。
|
濃度測定1.5 cm2 / mg at 280 nm |
-80°Cで保管。
2-2-4) G1-G3調製法 DAY1
Scrape top of frozen FX-45 gelsolin clone (E.coli K12) with a sterilized inoculation loop and plate on an LB plate with 100 µg/ml ampicillin and 25 µg/ml chloramphenicol. To plate, make initial streak across and incubate at 37°C overnight.
DAY2
Pick up a single colony into a 500 ml erlenmyer flask containing 100 ml of autoclaved LB media to which 200 µl of 50 mg/ml stock filter-sterilized ampicillin and 50 µl of stock 50 mg/ml chloramphenicol has been added. Shake at 37°C overnight.
DAY3
Add 1/2 of each flask (50 ml) of E.coli culture to a 1 l Erlenmyer flask containing 500 ml of autoclaved LB media with 1ml of 50 mg/ml filter-sterilized ampicillin stock
25
2004年度 博士論文 鈴木 団
and 0.25 ml of chloramphenicol 50 mg/ml stock. Prepare a total of 4 flasks in this manner.
Incubate in a rotary shaker at 200 rpm and 37°C. Remove aliquots every 1/2 hour with a sterilized Pasteur pipette and measure O.D.550.
Once the O.D.550 is approximately 0.6-0.8, induce expression by adding 1.1 ml of 0.2 M filter-sterilized IPTG to each 550 ml of culture. (To make stock IPTG, dissolve 0.476 g IPTG in 8 ml of MQ. Bring final volume to 10 ml. Divide into 1 ml aliquots, quick freeze with liquid nitrogen and store at –70°C).
Shake at 200 rpm and 37°C for approximately 3-4 more hrs, taking aliquots every 1/2 hour and measuring the O.D.550. Save aliquots to run on gel.
When O.D.550 is approximately 2.0, remove culture from shaker and spin in 500 ml centrifuge container at 5000 rpm for 10 min at 4°C.
Resuspend pellet in 20 ml of lysis buffer. Save a 20 µl aliquot to run on a gel.
[Lysis Buffer 1X Solution]
50 mM Tris-HCl 1 mM EGTA 1 µg/ml leupeptin
Add all of the above to 80 ml MQ and adjust pH to 8.0 with HCl. Bring the final volume to 99 ml and add 15 mg DTT and 1 ml of 100 mM PMSF directly before use.
[2 M Stock Tris-HCl pH9.0]
Add 120 g Tris to 400 ml MQ. Adjust to pH 9.0 with HCl. Bring the final volume to 500 ml.
[0.5 M EGTA Stock pH 8.0]
Add 19.02 g EGTA to 50 ml MQ. Adjust to pH 8.0 with 6 M KOH. Bring the final volume to 100 ml.
[6 M KOH]
Add 16.83 g KOH to 40 ml MQ. Stir until KOH is dissolved. Bring the final
26
2004年度 博士論文 鈴木 団
volume to 50 ml. KOH should be made on the day of use.
[100 mM PMSF Stock]
Add 174 mg PMSF to 10 ml isopropanol. Divide into 1 ml aliquots and store at –20°C.
[20 mg/ml Leupeptin Stock]
Add 200 mg leupeptin to 10 ml MQ. Divide into 1 ml aliquots, quick freeze in liquid nitrogen and store at –70°C. Thaw aliquots at 37°C for 1.5 min.
Pass the E.coli pellet suspended in lysis buffer through a French cell press 3X at 100 psi. (This step can be replaced by ultra sonication.)
Spin lysate at 14K rpm for 20 min.
Resuspend pellet with 5 ml of 20 mM Tris-HCl, pH 9.0.
Add 153.75 µl of 1 N NaOH to 5 ml of pellet suspended in 20 mM Tris to increase pH to 12.0. Vortex while adding NaOH.
After 20 sec, add 625 µl of 2 M Tris-HCl, pH 9.0 while vortexing.
Spin at 14K for 20 min. at 4°C.
Dialyze 5 ml of supernatant against 2x1 l of 20 mM Tris-HCl, 1 mM EGTA, 1 mM PMSF, 1 mM DTT, and 5 µg/ml leupeptin, pH 9.0 overnight at 4°C. Rinse dialysis tubing with MQ before use. (pH of Tris is adjusted at 4°C, not room temperature.
This means that the pH standard to calibrate the probe should also be at a temperature of 4°C.)
DAY4
Remove supernatant from dialysis tubing and spin at 14K rpm for 20 min at 4°C.
Dissolve the pellet in 5 ml 20 mM Tris and run on gel. Save 20 µl aliquot of dialyzed supernatant to run on gel.
27
2004年度 博士論文 鈴木 団
Load on a 50 ml DE-52 column that has been equilibrated with 20 mM Tris-HCl, pH 8.5 at 22°C, 1mM EGTA, 1 mM DTT, 1 mM PMSF and 5 µg/ml leupeptin. Run the sample at 1 ml/min. Add PMSF and DTT directly before use.
Wash the column with a total of 2 volumes of 20 mM Tris-HCl buffer and collect in a beaker. When A280 baseline is approximately 0, start collecting fractions at approximately 2 ml/fraction. At this time, apply a linear NaCl gradient with a total volume of 150 ml (75 ml of 20 mM Tris, 0 mM NaCl and 75 ml of 500 mM NaCl in DE-52 running buffer).
(For 500 mM NaCl, add 2.9 g NaCl to 100 ml of DE-52 running buffer.)
Run samples from protein peaks on Laemmli 5% stack and 12% resolving gels.
Pool Gelsolin fragment peaks. Measure volume and O.D.260, 280 and 280/260.
Pooled fractions were then dialyzed against buffer D, the pH of which was decreased from 9.0 to 7.0 in steps of 0.5 pH units. 10% glycerol was included during the first 4 steps.
[Buffer D]
10 mM MOPS 1.0 mM EGTA 1.0 mM Mg-acet.4H2O 20 mM K-propionate 1 mM DTT 5 µg/ml leupeptin 10 % glycerol
1. Make 2 l of 2X stock containing 10 mM MOPS, 1.0 mM EGTA, 1.0 mM Mg-acetate and 20 mM K-propionate. Bring the final volume to 2 l with MQ and cool to 4°C.
2. Remove a 400 ml aliquot from the above. Add 80 ml glycerol and 100 ml MQ. Bring to pH 9.0 at 4°C with KOH and add DTT stock and leupeptin stock at 20 mg/ml. Bring final volume to 800 ml with 4°C MQ. Check pH of final solution at 4°C. Repeat for pH steps of 8.5, 8.0 and 7.5. For pH step of 7.0, replace glycerol with MQ and tighten clamps on dialysis tubing as much as
28
2004年度 博士論文 鈴木 団
possible. Dialysis tubing with pooled fractions should be dialyzed against pH 9.0 and pH 7.0 buffer for 2 hrs. All other dialysis steps should be performed for approximately 45 min.
Upon completion of dialysis, spin gelsolin at 20K rpm for 10 min to remove any precipitated protein. Measure volume and O.D.280, 260 & 280/260. Divide into 1 ml aliquots, quick freeze and store at –70°C.
2-3) 顕微解析装置について
第3章と第5章で述べる研究では、以下に示すような、赤外(IR)レーザーによる 光ピンセット光学系を基本とした顕微解析装置を利用した。
2-3-1) ミクロ熱励起・ミクロ温度計測法で用いた装置について
一部の実験(3-4-3)及び図3-2、3-3)は、励起/モノクロメーターシステム(T.I.L.L Photonics, Germany)を用いて行なった。その際、FITCダイクロイックミラー、515 nmの蛍光フィルター、12bitのA/Dコンバーターにつなげたペルチェ冷却SensiCam カ メ ラ (640×480 ピ ク セ ル )、100 倍 の 対 物 レ ン ズ 及 び 、IGOR プ ロ グ ラ ム
(Wavemetrics, USA)で作成した、T.I.L.L.画像解析プログラムを利用した。以上の 装置は、0.05 mM Eu-TTA/DMSOの励起スペクトル(図3-2参照)及びEu-TTAを 閉じ込めたマイクロピペットの温度感受性を調べるとき(図3-3参照)に利用した。
その他の実験(図 3-4〜3-6)は、退色の影響をできるだけ少なくするために 50 mMという高濃度のEu-TTA/DMSOを用いて行なった。またCCDカメラはCCD-300
(DAGE-MTI, Michigan City, IN, USA)を利用し、励起・蛍光の光学系は上と同様
365 nm励起/FITCダイクロイックミラー/515 nmLPフィルターを利用した。赤
外レーザーはNd-YAG(1064 nm)を使った(T10-V-106C, 2.5W, Spectra-Physics Lasers Inc., Mountain View, CA, USA)。光学系は顕微鏡(IX70, Olympus, Tokyo,
29
2004年度 博士論文 鈴木 団
Japan)を基礎にしている。対物レンズは PlanApo100×/1.40/oil を使った。以上の 光学系を、図2-2にまとめた。
実験画像はデジタルテープレコーダー(DSR-11, Sony, Tokyo, Japan)を使って 30 Hzで録画した。その後、画像の取得と解析は画像ボード(LG3, Scion Corporation, Frederick, MD, USA)とコンピューター(Apple Japan, Tokyo, Japan)で行なった。
2-3-2) ナノ筋収縮系で用いた装置について
顕微鏡装置と画像解析の方法は、以前に報告のあったもの(Nishizaka et al., 2000)
を次のように改良して利用した。模式図を図2-3に示す。まず位相差像の取得はCCD カメラ(CCD-300, DAGE-MTI, Michigan City, IN, USA)を利用し、画像ボード(LG3, Scion Corporation, Frederick, MD, USA)を通してコンピューターに取り込んだ。ビ ーズの位置(画像の輝度重心)は30 Hzのビデオレート画像をデジタル化して計算 した。蛍光像はICCDカメラ(ICCD-350F; Video Scope International, Washington D.C., USA)で観察した。光ピンセットのバネ定数(0.10-0.25 pN/nm)は、西坂ら の方法(Nishizaka et al., 1995)に従って計測した。一方ビーズの変位はビデオレー トで記録されるため、それより速い時間周波数を持つ動きは、測定されない。例え ばブラウン運動の100 Hz成分が、30 Hz成分より激しくても30 Hzの計測では無視 され、それゆえ見かけ上の空間分解能は上がることになる。そこで、30 Hz の時間 分解能による計測中の空間分解能は、別に見積もった(Nishizaka et al., 2000)。装 置系のドリフトはここに含まれる。0.12 pN/nmのバネ定数で40秒間捕捉したビー ズの変位の標準偏差(SD)はx方向が1.43 nm、y方向が1.46 nmだった(n = 9)。 一方、ガラス面に40秒間吸着したビーズのSDは、x方向が2.15 nm、y方向が1.71 nmだった(n = 9)。
30
2004年度 博士論文 鈴木 団 2004年度 博士論文 鈴木 団
31 31
2004年度 博士論文 鈴木 団
図2-1 アセトンパウダーからのG-アクチン調製法
32
2004年度 博士論文 鈴木 団
温度計 ピペット 細胞
Nd-YAG レーザー
1064nm
CCDカメラ
角度が2軸で 調節可能なモーター
駆動のミラー
ND ND
水銀ランプ
フィルター LP 515
フィルター BP365 ダイクロイックミラー(BA515IF)
対物レンズ ヒーター
ピペット
シャッター レンズ1
レンズ2 レンズ3
ダイクロイックミラー(Infrared)
ミラー
ND
図 2-2 ミクロ熱励起・ミクロ温度計測法のための光学系概略図 点線と破線はそ れぞれIRレーザー及び蛍光イメージングのための光路を表す。Nd-YAGレーザーか らの光をレンズ1 で広げ、モーター駆動のミラーで反射する。続いて光量を調節す るための ND フィルターと、光束の太さを調節するためのレンズ 2,3 を通過して、
ヒーターピペット先端で焦点を結ぶように対物レンズに入射する。Eu-TTA励起用の 光は水銀ランプを用い(励起フィルターBP365)、ダイクロイックミラー(BA515IF)
で反射して、対物レンズに入れる。615 nmにピークを持つEu-TTAからの蛍光はフ ィルターLP515を通ってCCDカメラに投影される。
33
2004年度 博士論文 鈴木 団
図 2-3 ナノ筋収縮系で用いた顕微鏡光学系の概略図 赤線は光ピンセット用の IR
レーザー光路、緑線は蛍光イメージング用の光路、青線は位相差像観察用の光路を 表す。光ピンセットで捕捉したビーズの位相差像を、CCDカメラを用いてビデオレ ートで測定し、蛍光ラベルしたアクチンフィラメントを ICCD カメラで確認する。
これら蛍光像と位相差像は、画像合成機により同時録画・観察する。
34
2004年度 博士論文 鈴木 団
第3章 新しいミクロ熱励起・ミクロ温度計測法の開発について
3-1) 要約
µm サイズの微小領域を対象に、顕微鏡下の任意の箇所で室温から水の沸点までに 渡る広範な温度上昇と下降を行うことができる新しい技術を開発した(Zeeb et al., 2004)。
局所熱励起は、任意の領域へ移動できるようマイクロマニピュレーターにより操 作されるマイクロピペット先端(φ = 1 ∼ 2 µm)に固着した金属微粒子に、開口数の大 きな対物レンズ(N.A.1.3 ∼ 1.4)を通して集光した赤外レーザー(λ = 1064 nm)を照射 して行う。最高温度は入射レーザー光の強度を制御することで任意に変えられる。
温度上昇・下降の速さは固着した金属の熱容量に依存するが、約10 msであった。
ミクロ温度センサーには、熱以外の環境変化に応答しないよう水溶液から隔離さ れた微小領域に閉じ込めた蛍光色素、Europium-TTA の蛍光強度変化を利用する。
これまでにマイクロピペット先端(φ = 1 ∼ 2 µm)に閉じ込めることに成功している。
これにより、0.1°C の精度で局所的な温度変化を検出できる。また温度変化による 蛍光色素の蛍光強度変化は充分速く、時間分解能は30フレーム/秒である。
この技術は標準的な電気生理学、蛍光イメージングの技術と互換性が良いため実 験装置の融合が容易であり、これまでにない全く新しい細胞生物学研究へ即座に応 用可能である。
3-2) 研究の背景
一般に細胞機能の基盤は、細胞骨格や受容体などのタンパク質のみならず、様々 なイオン、細胞内外の電位差(膜電位)、温度といった多くのパラメータの時空間 ダイナミクスにある。現在、生きた単一細胞の膜電位や細胞内Ca2+濃度について
35
2004年度 博士論文 鈴木 団
は、ミリ秒オーダーで検出・制御することができる。しかし、正確な制御や局所 的な測定の難しい「温度」の場合は事情が異なる。というのも通常、加熱・冷却 や温度測定は、実験装置全体の温度を制御しつつ試料の近くに置いた「小さな」
温度計で行われるため、どうしても時間・空間分解能には限界があるからである。
温度制御の場合には、例えば実験試料にレーザーといった強力な光源からの照 射光をパルス的に当てることで、急速な温度上昇を実現できる。しかし、暖めら れた試料の体積にもよるが、冷却速度はずっと遅い。この問題は、温度パルスを 任意のµm領域に制限しつつ、実験試料周辺、実験装置を温度一定にしておけば解 決できる。急速な温度上昇と下降を局所的に起こすため、カバーガラスに固着し た金属粉や金属薄膜に波長約 1 µm の赤外レーザーを照射する方法が本研究室で 開発された。この方法を用いると、温度パルスを単一細胞程度の局所的な範囲に 限定して起こすことができる。
一方、温度を単一細胞と同程度の体積で測定することは簡単ではない。熱電対、
NMR、FTIR、ラマン分光、放射分析法、焦電気膜、などといった方法はどれも温 度検出に対して非常に感受性が高い。しかし、単一細胞の温度測定に用いるには技 術的に様々な問題がある。そこで、温度感受性の蛍光色素を細胞内に流し込む方法 が提案され、単一細胞における熱発生、局所的温度勾配の発生が報告されてきてい る(Zohar et al., 1998)。しかしこの魅力的な方法は、色素の環境変化(pHなど)
に対する感受性が高すぎるゆえに、正確な温度測定を行うには大きな問題があった。
さらに細胞膜上で色素が不均一に分布してしまうことや退色が速いことは、温度と 蛍光強度とのキャリブレーションを非常に困難にしていた。
こ れ ら の 問 題 点 を 克 服 す る た め 、100% DMSO に 溶 か し た 蛍 光 色 素
Europium-TTA(Eu-TTA)を、先を閉じたパッチクランプ用のピペットに封入する
ことにした。励起波長365 nmのEu-TTAは、非常に温度感受性のある蛍光を615 nm
36
2004年度 博士論文 鈴木 団
に持ち(Bhaumik, 1964; Zohar et al., 1998)、また集積回路における温度を計測す るために用いられたこともあった(Kolodner and Tyson, 1982)。本研究においては、
0.1°C の 分 解 能 の あ る 局 所 的 な 温 度 計 測 を 、µm サ イ ズ の ピ ペ ッ ト 先 端 に
Eu-TTA/DMSOを満たすことにより行なった。これにより、細胞を色素で直接染め
るときに生じる上記の問題点を克服でき、温度と蛍光強度とを正確に較正できるよ うになった。そして、細胞膜とぴったり接触したピペット先端部は閉じた系となっ ているために(エネルギーのやり取りは可能だが、物質のやり取りはできない)、
温度だけを唯一の変数とすることができた。
3-3) 材料と方法 3-3-1) 試薬
Europium(III) thenoyltrifluoroacetonate trihydrate (Eu-TTA)は、Acros Organics
(Pittsburgh, PA, USA)から購入した。DMSOはSigma-Aldrich(St. Louis, MO, USA)
から購入した。直径0.1 µmのアルミ粉はNilaco(Tokyo, Japan)から購入した。蛍 光ビーズ(FluoSpheres carboxylate-modified microspheres, 1.0 µm, red-orange fluorescent (565/580) F-8822)は、Molecular Probes Inc. (Eugene, OR, USA)か ら購入した。
ヒーターマイクロピペット及び温度計ピペットは、直径 1.2 mm のガラス管
(Drummond Sci. Co., Broomall, PA, USA)を引き伸ばして作成する(図3-1)。ガ ラス管の引き延ばしにはピペットプラー(PB-7, Narishige, Tokyo, Japan)を用いた。
ミクロピペット先端部をマイクロフォージ(MF-900, Narishige)で熱し、溶かして 閉じる。
37
2004年度 博士論文 鈴木 団
3-4) 実験結果
3-4-1) 温度計ピペットの作成
ピペットの作成方法の模式図を、図3-1に描いた。ガラス管は2段階で引き伸ばし、
引き伸ばしたガラス管先端部の直径を約1-2 µmにする。ピペットの先端はマイクロ フォージの高温になったフィラメントに注意深く触って、溶かして閉じる。こうし て作った、閉じたピペットの内部を約1 µlの、100% DMSOに溶かした0.05 mM、
あるいは50 mMのEu-TTAで満たす。先端部に残った気泡は、ピペットを指先では
じくことによって容易に取り除ける。ピペットの反対側については特に閉じるよう な操作をしないので、液面は開放されていることになる。そのためこうして作られ たミクロ温度計ピペットは繰り返し使用することができる。温度と蛍光強度との較 正は実験毎に、実験直後に行なう。まず 45°C の熱湯を実験容器に入れ、溶液の温 度が室温まで自然に下がることを利用して、ピペット先端部の同一箇所の蛍光強度 を2°C毎に測定する。温度と蛍光強度との関係は、50 mM Eu-TTAの場合は線形で 非常に良く近似できた。ただし一つだけ問題があった。温度を変化させたときに、
時々ベースラインがわずかに変わってしまうことである。これはおそらく、ピペッ トの位置がガラスの熱膨張によりドリフトするためだと思われる。この問題は、ピ ペットの形状を変えたり、あるいは溶液に浸けるピペットの長さをできるだけ短く したりすることで解消できる。
3-4-2) ヒーターピペットの作成
ミクロ温度計と同じ大きさ(先端の直径が約1 ∼ 2 µm)のマイクロピペットを準備 する(図3-1)。水に懸濁したアルミ粉(0.1 µm;50 mg/ml)に先端を3秒間浸けて 持ち上げ、ピペット先端の水分を自然に蒸発させる。すると10 ∼ 15秒で、ピペッ
38
2004年度 博士論文 鈴木 団
トの最先端部にアルミの粉の凝集体ができ上がる。次にこのピペット先端を加熱し たマイクロフォージのフィラメントへ近付けることで、凝集体はガラスに固定され る。この方法はパッチクランプ用のピペットを作成する際に行なわれる標準的なや り方と同じで良い。
3-4-3) 温度の測定
蛍光強度の変化を利用した温度測定の方法は、図3-2と3にまとめた。図3-2 に示 したEu-TTAの励起スペクトルから分かるように、Eu-TTAの蛍光強度は温度に敏感 に反応する。溶液を還流(3 ml/min)するためのシリコンチューブの一部を熱湯に 短時間(約 2 秒)だけ浸すことで、約1°Cの温度変化を実験容器(2.5 ml)に与え る。この温度変化は、容器内に置かれた熱電対によって計測される(図 3-3A)。熱
電対は、0.05 mM Eu-TTA/DMSOを封入したピペット先端から3 mm離れた場所に置
いてある。Eu-TTAの励起と、温度計ピペット先端の 25 µm2の領域における蛍光強 度の読み取りは同期を取っており、露光時間100 msを3 Hzで繰り返し行った(図 3-3E)。蛍光強度は、熱及び退色両方の影響を受ける。退色の過程は単一指数関数で 表せるので、計測される蛍光強度変化(温度変化を与える前)から除算して補正し た(図3-3C)。
3-4-4) レーザー焦点位置の調整
IRレーザーを任意の位置に持ってくるには、光ピンセットの光学系を利用する(図 2-2)。集光したレーザーは焦点で約0.8 µmまで絞られているので、モーターでコン トロールされるミラーによりµm の精度で、焦点位置をヒーターピペット先端の金 属粉凝集体に合わせることができる。周期的に金属粉凝集体を励起する目的で、こ こでは非常に単純な、振り子型のシャッターを用いた。金属粉凝集体の周辺部にお
39
2004年度 博士論文 鈴木 団
ける温度分布は、約10 msで定常状態になる(Kato et al., 1999)。本研究ではこの 緩和時間は約50 msだったが、その理由は振り子型シャッターの性能に依存してい る。
3-4-5) 局所熱励起・温度計測
普通、ある細胞に着目してそれが外部環境から熱的に隔離されているという定義に は、外来的あるいは内在的な熱発生過程は細胞内部の各部分に検出できるような温 度勾配を作るほど強力ではない、という前提がある。実際、水の熱伝導率(k = 0.597
W/m/K)は非常に低く(Weast et al., 1986)、断熱材に近いと言える。この水の性質
は、安定した急な温度勾配を熱源の周りに作るのに好都合である。実際、波長が約 1 µmのIRレーザーで水中に置かれたサブミクロンサイズの金属粒子や(Kato et al.,
1999)、カバーガラス上に円形(直径 10 µm)に蒸着したアルミニウムの薄膜を熱
すると(Kawaguchi and Ishiwata, 2001)、その周りにできる温度勾配は2 °C/µmで ある。さらにレーザー照射を開始してから約10 msで温度勾配が定常状態になる。
以上の性質を利用して、パッチクランプ用のピペット先端部に金属粉凝集体を固 定しピペットを任意の位置へ動かすことで、局所的に温度を制御するシステムを構 築した(図 2-2)。もう一本の先を閉じたパッチクランプ用ピペットは DMSO に溶
かした Eu-TTA で満たし、局所的温度を検出するのに用いる。また温度の測定は、
熱せられる微小体積内に置かれたピペット先端部から、蛍光強度を読み取って行な う。本研究で利用したIRレーザー強度の範囲では、金属粉凝集体を直接励起しない 限り、検出可能な温度勾配が水中にできることは無かった(図3-4)。一方で、金属 粉凝集体をレーザー焦点に持ってくると、金属のすぐ近くで目視できるほどの水の 沸騰が起こった。また沸騰は、NDフィルターでIRレーザーを弱めることで解消で
きた(図 3-5)。アルミ金属粉凝集体を励起したときに生じる安定した温度勾配は、
40
2004年度 博士論文 鈴木 団
5 ∼ 20 µmの範囲にわたって約10 °C/15µmという非常に急なものになる。もし最高
温度を変えたければ、光路中に置いてある ND フィルターを交換すれば良い。それ だけで、µmサイズの微小体積を任意の温度に約10 msの速さで変化させることが できる(図3-6)。
3-5) 考察
本研究で述べた手法の利点の一つは、ピペットが充分小さいので単一細胞と同程度 の領域を熱的に励起し、かつ同時にそこで温度を測定できることである。しかもヒ ーターピペット及び温度計ピペットは共にマニピュレーターで操作されるので、任 意の場所に温度刺激を与え、また任意の箇所の温度分布を測定することができる。
色素で細胞を直接染めたり(Zohar et al., 1998)タンパク質を蛍光染色したり
(Kato et al., 1999; Kawaguchi and Ishiwata, 2001)して温度測定をする方法と異な り、本手法における色素の蛍光強度の変数は温度のみである。なぜなら、マイクロ ピペットに閉じ込められた蛍光色素は外部環境との直接的な接触を免れているから である。さらにガラス製のピペットは、水と比べると熱伝導率は高く熱容量は小さ い(Weast et al., 1986)。それゆえ温度変化を捉えるには充分に「透明」だと言える。
以上の特徴によって、膜やタンパク質を染めるといった方法で細胞に直接導入さ れた蛍光色素が不均一に広がる、あるいは温度以外の環境変化に応答するという典 型的な人為的要因を、排除することができる。ガラスの熱膨張によるピペットのわ ずかな、しかし計測できる程度の機械的なズレは、今分かっている唯一の誤差原因 である。図 3-4や 3-6 に見られるベースラインのわずかな変化は、おそらくここに 起因する。ヒーターマイクロピペット周辺にできる温度勾配により、水に対流が発 生することが考えられる。しかし本研究では温度計ピペットとヒーターマイクロピ
41
2004年度 博士論文 鈴木 団
ペットとは充分離れていたためか、対流に伴うと思われる温度計ピペットの動きは 見られなかった。
µmサイズの微小領域に高速で可逆な温度変化を与え、かつその領域の温度を測 定する手法は、細胞内部器官における"in vivo"での熱力学を、単一細胞を対象に適用 しようとする多くの研究にとって有効だろう。温度というパラメーターが非常にゆ っくりと変化する非局所的な変数として捉えられてきたのと対照的に、高速かつ局 所的に簡単に制御できることが本研究により示された。
線形非平衡熱力学に従えば(熱流による力と、媒質の何らかの物理量の流れとが 線形関係にある)、熱流だけでなく温度勾配によっても、電気の流れ以外に物質の輸 送が引き起こされる(Kondepudi and Prigogine, 1998)。即ち、局所的温度変化は原 理的に、単一細胞レベルの局所的な膜横断的イオン流などの流れへと、変換されう る。明らかなのは、もし温度が細胞の局所的なパラメーターとして捉え得るように なれば、単一細胞を対象とした多くの興味深い問題を扱えるようになるということ である。微小領域に人工的に温度勾配を作ることが比較的簡単に達成できたという ことは、生きた細胞内でも温度変化が人工的に実現できるのではないかということ を意味する。そこで、プロトンやカルシウムの濃度勾配は、それと共役した熱発生 機構と共に強力な熱源として考えることができるのではないか、というアイディア を検証するための一つの手法を、我々は手にしたことになる。
42
2004年度 博士論文 鈴木 団
1~2µm
Eu-TTA/DMSO Eu-TTA/DMSO
アルミ金属粉
(0.1µm, 50mg/ml) 3秒
アルミ金属粉
(0.1µm, 50mg/ml) 3秒
10~15秒 10~15秒
マイクロフォージ
(ヒーター)
1.2mm
図 3-1 温度計ピペット(上、写真右)及びヒーターピペット(下、写真左)の作 成法模式図 先端部の直径が約1-2 µmとなるよう引き伸ばしたガラス管を、マイク ロフォージのフィラメントに軽く触れ、溶かして閉じる。そこへEu-TTA/DMSOを 注入し、温度計ピペットとする。これと同様に引き伸ばした別のガラス管を、水に 混濁したアルミ粉に約3 秒漬けて持ち上げ、自然乾燥させる。すると、先端部にア ルミ粉の凝集体ができる。これをマイクロフォージのフィラメントへ近付け凝集体 をガラスに固定し、ヒーターピペットとする。
43
2004年度 博士論文 鈴木 団
1400
1200
1000
800
600
400
200
0
蛍光強度 (a.u.)
420 400 380 360 340
励起波長 (nm)
420 400 380 360 340
B A
20°C
1400
1200
1000
800
600
400
200
0 15°C
25°C 30°C
35°C
15°C
25°C
35°C
蛍光強度 (a.u.)
励起波長 (nm)
1400
1200
1000
800
600
400
200
0
蛍光強度 (a.u.)
420 400 380 360
340 360 380 400 420 340
励起波長 (nm)
420 400 380 360
340 360 380 400 420 340
B A
20°C
1400
1200
1000
800
600
400
200
0 15°C
25°C 30°C
35°C
15°C
25°C
35°C
蛍光強度 (a.u.)
励起波長 (nm)
図 3-2 マイクロピペット先端の Eu-TTA の励起スペクトル マイクロピペット先 端部に閉じ込めた0.05 mM Eu-TTA/DMSOからの蛍光をLP515 nmのフィルターを 通して測定した。 (A) 15°C〜35°Cまで熱したとき。 (B) 35°C〜15°Cまで冷や したとき。キセノンランプからの光をモノクロメーターを用いて分光し、320 nmか ら420 nmまで5 nmごとに露光時間100 msで励起した。
44