九州大学学術情報リポジトリ
Kyushu University Institutional Repository
糖鎖付加を伴う無細胞蛋白質合成系の構築
樽井, 寛
九州大学農学研究科遺伝子資源工学専攻
https://doi.org/10.11501/3180746
出版情報:Kyushu University, 2000, 博士(農学), 課程博士 バージョン:
権利関係:
Construction of a Novel Cell-Free Translation/Giycosylation System
Hiroshi Tarui
2000
CONTENTS
INTRODUCTION 1
CHAPTER
1. Insect cell extract preparation by the nitroge n de com pression method for cell-free translation
1-1 Introduction 8
1-2 Materials and Methods 10
1-2-1 Cell culture conditions 10
1-2-2 Detection of cell disruption efficiency 10 1-2-3 Preparation of insect cell extract 11
1-2-4 Cell-free protein synthesis 12
1-2-5 Detection of synthesized protein 12
1-3 Results 13
1-3-1 Disruption efficiency with Mini-Bomb cell disruption
chamber 13
1-3-2 Cell-free translation of BMV mRNA 16 1-3-3 Optimal disruption conditions with the Mini-Bomb cell
disruption chamber 16
1-3-4 Translation efficiency and stability of synthesized
proteins 21
1-3-5 Comparison of translational ability among various
cell-free systems 23
1-4 Discussion 23
1-5 Summary 28
2. Glycoprotei n synthesis i n the cell-free translation/glycosylation system derive d from insect cells
2-1 Introduction
2-2 Materials and Methods
2-2-1 Cells and culture conditions 2-2-2 Subcloning the gp120 gene
30 31 31 31
2-2-3 Preparation of gp120 mRNA 32 2-2-4 Preparation of insect cell extract 33
2-2-5 Cell-free protein synthesis 34
2-2-6 Detection of gp 120 34
2-2-7 Endo H treatment 35
2-2-8 Protease protection assay 35
2-2-9 F ACS analysis 35
2-3 Results 36
2-3-1 Construction of plasmids cloned gp120 gene
for template mRNA 36
2-3-2 Cell-free translation of gp120 with insect cell extract 36 2-3-3 Glycosylation of synthesized gp120 41
2-3-4 Protease protection assay 41
2-3-5 Binding activity of gp120 to CD4 41
2-4 Discussion 44
2-5 Summary 48
3 Establishment and characterization of cell-free translation/
glycosylation in insect cell extract
3-1 3-2
3-3
Introduction 50
Materials and Methods 51
3-2-1 Cell culture conditions 51
3-2-2 Preparation of insect cell extract 52
3-2-3 RNA preparation 52
3-2-4 Cell-free protein synthesis in insect cell extract 53 3-2-5 Analysis of biotinylated proteins synthesized in insect
cell extract 53
3-2-6 Deglycosylation 53
3-2-7 Western blot analysis 53
Results 54
3-3-1 Glycosylated gp120 synthesized in the cell-free
translation/glycosylation system 54
3-3-2 Optimization of glycoprotein synthesis in the cell-free
translation/glycosylation system 56
3-3-3 Influence of disruption conditions with Mini-Bomb cell
disruption chamber on glycosylation 58 3-3-4 Comparison of the Mini-Bomb cell disruption chamber
and the Potter-Elvehjem homogenizer 59 3-3-5 Productivity of the insect cell-free system 61 3-4 Discussion
3-5 Summary
64 68
4 Characterization of oligosaccharides linked to gp120 synthesized in the insect cell-free translation/glycosylation system
4-1 Introduction 69
4-2 Materials and Methods 70
4-2-1 Cells and culture conditions 70 4-2-2 Construction of transfer vectors and generation of
recomb in ant b acul oviruses 70
4-2-3 Expression of gp120 in Sf21 cells and silkworm
larvae 71
4-2-4 Preparation of gp120 mRNA 72
4-2-5 Preparation of insect cell extract 72
4-2-6 Cell-free protein synthesis 72
4-2-7 Detection of gp120 72
4-2-8 Deglycosylation 73
4-3 Results 73
4-3-1 Western blot analysis of g-gp120 synthesized in the insect cell-free system and gp120 expressed in
baculovirus-infected Sf21 cells 73
4-3-2 Deglycosylation of synthesized gp120 75 4-3-3 Characterization of oligosaccharides linked to gp120s
synthesized in vitro and in vivo 77
4-4 Discussion 77
4-5 Summary 81
5 Construction of cell-free protein synthesis systems derived from various cell lines
5-1 Introduction 82
5-2 Materials and Methods 83
5-2-1 Cells and culture conditions 83 5-2-2 Construction of template plasmids encoding hiL-6 84
5-2-3 Preparation of hiL-6 mRNA 86
5-2-4 Preparation of cell extracts 86
5-2-5 Cell-free protein synthesis 86
5-2-6 Detection of biotinylated protein 87
5-2-7 Western blot analysis 87
5-2-8 Deglycosylation 87
5-2-9 Subcellular fractionation 87
5-2-10 Characterization of subcellular fractions 88
5-2-11 Protease protection assay 88
5-3 Results 89
5-3-1 Translational ability of various extracts 89 5-3-2 Glycoprotein production in insect cell extracts 89 5-3-3 Sucrose density gradient of extracts 91
5-3-4 Proteinase protection assay 94
5-4 Discussion 94
5-5 Summary 98
CONCLUSION 99
ACKNOWLEDGMENT 105
REFERENCES 107
INTRODUCTION
In the last few years, large-scale sequencing projects throughout the world have yeilded new sequences at an ever-increasing rate. Of the roughly 140,000 genes in the human genome sequence will be completed within a few years. Although a large quantity of sequences data have been accumulated, the function of an estimated two thirds is likely to be unknown.
Now that the sequences of genomes from several species have been, or are about to be, completed, researchers and their funding bodies are looking towards the next step: the understanding of gene function. Consequently, there is a growing need for tools to analyze and organize these data.
A technology of expression of genes in cell culture has already led to the successful production of a number of proteins, however, there are some serious limitations: it needs long time to obtain desired protein; the synthesis of proteins, which are toxic for the host cells, is not possible; aggregation or degradation of expressed proteins sometimes occur in host cells.
With the completion of an increasing number of genomic sequences, attention is currently focused on how the data contained in sequence databases might be interpreted in terms of the structure, function, and control of biological systems. Approaches for global profiling of gene expression at the mRNA level as a function of the cellular state have been developed and are widely used to identify clusters of genes for which the expression is idiotypic for a specific state. These methods, though exquisitely sensitive, do not indicate changes in protein expression.
Moreover, the correlation between mRNA and protein levels is poor, because the rates of degradation of individual mRNAs and proteins differ, and because many proteins are modified after they have been translated, so that one mRNA can give rise to more than one protein. In comparison to
gene expression analysis at the mRNA level, proteome analysis, the global analysis of protein expression, provides more accurate information about biological systems and pathways because the measurement directly focuses on the actual biological effecter molecules. However, it is difficult to correspond the informations from this proteosome analysis method to the set of mRNAs existed in the cells or genomic sequence.
The cell-free protein synthesis systems, containing translation machinery, amino acids, energy source and energy regeneration system, can translate exogenous mRNA. These systems have potential to
synthesize desired proteins including not only native proteins, but also proteins toxic to living cells and the artificially modified proteins as we wishes. In addition, it is possible to synthesize proteins incorporated modified and unnatural amino acids (Noren et al., 1989). Many kinds of cell-free translation systems derived from wheat germ (Erickson and Blobel, 1983), rabbit reticulocyte (Jackson and Hunt, 1983), Escherichia.
coli (Chen and Zubay, 1983), yeast (Hussain and Leibowitz, 1986), Chinese hamster ovary (CHO) cells (Moldave and Fischer, 1983), Ehrlich ascites tumor cell (Henshaw and Panniers, 1983), yeast (Chanda and Kung, 1983) and many other sources. Of these systems, the traditional systems, i.e., wheat germ extract, rabbit reticulocyte lysate and E. coli extract systems, are widely used in order to synthesize endogenous and artificial proteins for characterization of the protein or to elucidate the translational mechanisms.
However, these systems were relatively inefficient production of foreign proteins comparing with living cells because of their instability (Roberts and Paterson, 1973). Recently, great deals of efforts have put into improvement of the protein productivity in the cell-free protein synthesis systems. Spirin et al. (1988) proposed a continuous flow cell-free
translation system, in which a solution containing amino acids and energy
sources is continuously supplied to the reaction and simultaneously extrude the products and wastes through a ultrafiltration membrane. As a result, protein synthesis is maintained at a constant rate for more than
20
h so that more than100
�g synthesized proteins is obtained. On the other hands, the translation efficiency was increased by condensation of the extract with ultrafiltration membrane(
Nakano et al., 1994) or with a polyethylene glycol(
Nakano et al., 1996; Kim et al., 1996; Kigawa et al., 1999), or by modification of the reaction condition(
Nakano et al., 1993; Kawarasaki et al., 1994; Kawarasaki et al., 1995; Kim et al., 1996; Kim and Swartz, 1999;Kigawa et al., 1999; Shen et al.,
2000),.
Recently, Madin et al.(2000)
eliminated a suicide facters, which is partly derived from ribosome
inactivating proteins, from the wheat germ cell-free system, resulting successfully to prepare a robust cell-free protein synthesis system. Of the system, the reaction was prolonged for 4 h in batch system and for above 60 h with continuously supply of substrates and removal of wastes. These systems achieved to synthesize 1-10 mg of active proteins in 1 ml of reaction mixture. Now, the development of cell-free protein synthesis system has been evolved to next stage to produce active proteins in preparative scale.
The wheat germ extract and rabbit reticulocyte lysate systems ,however, cannot compare the phenomenon of translation in vivo with in vitro using the same sources. While existing system are attempted to improve their productivity, the development of a new system derived from another source is also required.
On the other hands, many proteins require posttranslational modification to become functional. Especially, oligosaccharides of glycoproteins were utmost influence to the biological activity of the glycoproteins. In order to synthesize glycosylated protein in vitro, the cell-free assay systems for proteins with posttranslational modification have
3
been constructed to combine a eukaryotic cell-free translation system and rough microsomes
(
Blobel and Bobberstein,1975,
Walter and Blobel,1983,
Rothblatt and Meyer,
1986).
These reconstituted assay systems for coupled protein translational and posttranslational processing in higher eukaryotes has allowed to characterize the translocational machinery(
Walter and Blobel,1981,
Gorlich et al.,1992),
to define the topology of membrane proteins(
Popov et al.,1997),
and to reveal the regulation of N-linked core glycosylation(
Kasturi et al.,1997).
However, these reconstituted assay systems may lack some components for the posttranslation process during preparation of micrisomal membrane fraction. Furthermore, heterologous collection of components may complicate the results(
Rothblatt et al.,1986;
Walter et al.,1981;
Meyer etal.,
1985;
Prehn et al.,1987).
Cell-free glycoprotein synthesis systems have been also constructed from yeast(
Rothblatt and Meyer,1986),
Xenopus eggs
(
Matthews and Colman,1991)
and Trypanosoma brucei(
Duszenko et al.,1999).
The yeast system is homologous but reconstituted system supplemented with microsomal fraction prepared from yeast.Xenopus eggs system is required supplementation of
S-100
fractionprepared from rabbit reticulocyte lysate to increase its translation efficiency, and Trypanosoma brucei system does not allow initiation of protein synthesis.
Hence, this study aimed to construct coupled cell-free
translation/glycosylation system with homological extract that was prepared from a single source, with a single-step extraction, and without
supplementation of fractions separately prepared. Among several cells for expression of recombinant proteins, as a source for extract, the author selected lepidopteran cells which have ability to produce many recombinant proteins with high yield.
The baculovirus-insect cell system is now one of the most widely used to express a variety of proteins in large qualities under the control of a strong polyhedrin promoter of Autographa californica nuclear
polyhedrosis virus (AcMNPV) (Summers and Smith, 1985, and 1987).
One of the major advantages of this invertebrate virus expression vector over bacterial, yeast and mammalian expression is very abundant
expression of recombinant proteins, which are in many cases, antigenicall y, immunogenically and functionally similar to their authentic counterparts (Luckow and Summers, 1988). Furthermore, many mammalian proteins which require posttranslational modification for acquisition of biological function have been successfully expressed in recombinant-baculovirus infected insect cells. For instance, recombinant proteins produced in insect cells can be secreted (Smith et al., 1983), targeted to the nucleus (Jeang et al., 1987), targeted to the cell surface (Matsuura et al., 1986);
assembled into oligomeric complexes (Estes et al., 1987) and disulfide
linked dimmers (Giese et a!., 1989); proteolytically cleaved (Smith et a!., 1983; Matsuura et al., 1986); phosphorylated (Jeang et al., 1987; O'Reilly and Miller, 1988; Overton et al., 1989); N-glycosylated (Smith et al., 1983;
Matsuura et al., 1986; Estes et al., 1987); 0-glycosylated (Greis et al., 1994); myristylated (Overton et al., 1989).
In this study, the purpose is to construct a coupled cell-free
translation/glycosylation system derived from Spodoptera frugiperda 21 (Sf21) cells. Moreover, the author attempted to construct cell-free
translation/glycosylation systems from many kinds of cells. The obtained knowledge was described in five parts as follows.
First, construction of exogenous mRNA dependent cell-free
translation system derived from insect cells was examined. In chapter 1, the optimal condition for cell disruption was decided toward preparation of
the insect cell extract retaining translation activity using the Brome mosaic virus
(
BMV)
mRNA as a template. In addition, the translatability of the insect cell-free system was compared with those of wheat germ system and rabbit reticulocyte system.Second, a coupled cell-free translation/glycosylation system derived from insect cells was constructed. In chapter 2, the human
immunodeficiency virus type-1 gp 120 was constructed as a reporter protein and it was revealed that the insect cell-free system have intact microsome and can synthesize glycosylated proteins that was folded into the mature conformation.
Third, the reaction condition of insect cell-free system was optimized for improvement of protein translation and glycosylation efficiency. In chapter 3, the concentration of low-molecular components of cations, nucleotide triphosphates, creatine kinase, creatine phosphate, and
spermidine, were systematically optimized. Moreover, cell disruption condition was re-optimized according to glycosylation. Then, the
glycoprotein productivity of this insect cell-free system was estimated by comparison with commercial available gp120.
Next, the gp120 synthesized in the insect cell-free system was
compared with the gp120 expressed in baculovirus-infected insect cells and the gp120 produced in hemolimph of baculovirus infected silkworm larvae.
In chapter 4, the amount and type of oligosaccharides bound to the
synthesized gp120 was compared with the gp120s derived from baculovirus infected insect cells and baculovirus infected silkworm larvae.
Finally, the universal applicability of this method for preparation of the cell-free translation/glycosylaion system was examined. In chapter 5, translation and glycosylation ability of several extracts are described, of which the extracts was prepared from several sources including fish,
mammalian, and insect cells.
CHAPTER 1
Insect cell extract preparation by the nitrogen decompression method for cell-free translation
1-1 INTRODUCTION
Several types of cell-free translation systems, e.g., wheat germ extract (Erickson and Blobel, 1983), rabbit reticulocyte lysate (Jackson and Hunt, 1983) and Escherichia. coli extract systems (Chen and Zubay, 1983), are widely used for synthesizing endogenous and artificial proteins for characterization of the protein or in order to elucidate the translational mechanisms. The productivities of these traditional systems have steadily increased (Madin et al., 2000; Kawarasaki et al., 1995; Nakano et al., 1994;
Kim et al., 1996), though it is still difficult to compare in vivo with in vitro phenomena using the same sources. Cell-free protein synthesis systems from cultured cells have been developed (Hussain and Leibowitz, 1986;
Moldave and Fischer, 1983; Henshaw and Panniers, 1983). However, the major disadvantage of these eukaryote cell-free systems is comparatively low protein productivity. With attempts to increase the productivity of existing systems, the development of a new system derived from another source has been anticipated.
The insect cell-baculovirus expression system has been used for high level expression of a wide variety of recombinant proteins (Lukow and Summers, 1988; Vlak et al., 1988). Many mammalian proteins which require post-translational modifications have been successfully expressed in insect cells infected by recombinant baculovirus (Lukow and Summers,
1988). A cell-free translation system derived from insect cells described by
Scheper et al. (1997) had the properties of the untranslated region (UTR) of the baculovirus p 10 mRNA. However, this system was prepared using a method of Drosophila cell-free system and the productivity and the
characteristics of the system were not described. Moreover, post
translational ability was not mentioned.
In order to prepare a highly efficient cell-free protein synthesis system, it is of the utmost importance to extract components that participate in protein synthesis with high efficiency. Furthermore, traditional cell-free protein synthesis systems such as the wheat germ and rabbit reticulocyte cell-free systems cannot synthesize glycosylated protein. To produce glycosylated protein, the addition of canine pancreatic microsomes to the traditional cell-free protein synthesis system is necessary (Walter and Blobel, 1983). Wheat germ is disrupted by grinding and reticulocytes burst in water, such that the microsomes in the wheat germ extract and reticulocyte lysate may also be broken or burst. Likewise, most extracts derived from cultured cells are swelled in hypotonic buffer and disrupted with the use of a tight-fitting Dounce homogenizer (Moldave and Fischer, 1983; Henshaw and Panniers, 1983), in which microsomes may also swell and undergo homogenization. The osmotic shock can lead to inefficient translation and failure of posttranslation.
The Mini-Bomb cell disruption chamber (Mini-Bomb) is a unique device designed to disrupt many biological materials while allowing high activity to be preserved. In the Mini-Bomb, compressed gas from a standard high-pressure tank is applied to the biological material at the desired pressure. Following equilibration, the sample is released through a needle valve. The sudden decompression causes controlled disruption of the biological material.
In this chapter, construction of cell-free translation system derived
from insect cells was described. The optimal conditions for cell disruption with the Mini-Bomb, aimed at preparation of an insect cell extract retaining translational activity, were determined. In addition, the author also found that the insect cell-free system had translatability different from that of the wheat germ extract and reticulocyte lysate systems.
1-2 MATERIALS AND METHODS
1-2-1 Cell culture conditions
Spodoptera frugiperda (
IPLB-Sf21-AEII) (
Vaughnet al.,
1977)
cell was routinely maintained as a spinner culture at 27°C at densities between 0.1 and 1.5 x 106 cells per milliliter in IPL-41 medium(
Gibco BRL-Life Technologies, Grand Island, NY, USA)
supplemented with 10%(
v/v)
heat-inactivated fetal bovine serum
(
Gibco BRL-Life Technologies)
.1-2-2 Detection of cell disruption efficiency
Sf21 cell
(
4.0 x 108 cells)
suspension in 4 ml of extraction buffer(
40 mM Hepes-KOH pH 7.95 at 25°C, 100 mM KOAc, 1 mM Mg(
OAc)
2, 2 mM CaC12, and 4 mM DTT)
was placed in a cold Mini-Bomb and thenequilibrated for 30 min under nitrogen gas pressure as indicated in Fig. 1- 2. The suspension was then released from the Mini-Bomb
(
Kontes Glass Company, Vineland, NJ, USA)
at a flow rate of approximately 4 ml/min.On the other hand, suspended cells were also disrupted by 5 strokes at 500 rpm with a motor-driven Potter-Elveh
j
em homogenizer. The disrupted cells were stained with trypan blue to visualize the nuclei and counted in a hemocytometer under a microscope. Incomplete disruption was indicated by persisting nuclei.1-2-3 Preparation of insect cell extract
Cells were harvested at a density of 1.0 x 106 cells /ml ( 4.0 x 108 cells in total), then washed three times with extraction buffer ( 40 mM Hepes
KOH pH 7.95 at 25°C, 100 mM KOAc, 1 mM Mg(OAc)2, 2 mM CaC12, and 4 mM DTT). The cell pellet was resuspended in a half volume of
extraction buffer (approximately 1.0 x 108 cells/ml). The cells in the extraction buffer were transferred to the cold Mini-Bomb. The cell suspension was pressurized and allowed to equilibrate for 30 min at nitrogen pressure of 10 kg/cm2• The cells were then disrupted to flow under atmospheric pressure at the flow rate of 4 ml/min. This preparation ("pressate") was kept on ice. The efficiency of cell lysis was checked by counting the cells in a lysate diluted with extraction buffer and stained with trypan blue in a hemocytometer under a microscope. For the Potter
Elvehjem homogenizer, a cell suspension at a density of 1.0 x 108 cells/ml was homogenized with 5 strokes of the motor-driven Potter-Elvehjem homogenizer at 500 rpm. The disrupted cells were centrifuged for 15 min at 14000 rpm at 4°C in a SW40Ti rotor (Beckman, Palo Alto, CA, USA).
The pellet was discarded and the supernatant (approximately 2 ml) was collected and chromatographed at 4 through a Sephadex G-25 fine column (16.0 x 1.0 em), previously equilibrated with column buffer ( 40 mM Hepes-KOH pH 7. 95 at 25°C, 100 mM KOAc, 5 mM Mg(OAc)2, and 4 mM DTT). Fractions of 0.5 ml were eluted with the same buffer. The two fractions with the highest RNNprotein concentration were pooled and aliquoted, immediately frozen in liquid nitrogen, and stored at -80°C.
Prior to using the extract for cell-free protein synthesis, endogenous mRNA was digested with micrococcal nuclease (Boehringer Mannheim, Indianapolis, IN, USA) (Pelham and Jackson, 1976) as follows: 1 �I of nuclease (1000 U/ml) and 2 �1 of 40 mM CaC12 were added to 20 �I of
extract diluted with the same volume of water and incubated for 5 min at 20°C. The digestion was stopped by addition of 2 �1 of 80 mM EGTA.
After the digestion reaction, 3.5 �1 of creatine phosphokinase (10 mg/ml dissolved in 10 mM Hepes-KOH pH 7.95) (Boehringer Mannheim) were added to the nuclease-treated extract.
1-2-4 Cell-free protein synthesis
The insect cell-free protein synthesis reaction (5 �1) contained 3.2 �1 of insect cell extract, 20 �g/ml of brome mosaic virus (BMV) mRNA, 20.6 mM Hepes-KOH (pH 7.95 at 25°C), 1.5 mM Mg(OAc)2, 100 mM KOAc, 2.5 mM DTT, 1.2 mM ATP, 0.25 mM GTP, 444 �g/ml creatine phosphokinase, 8.0 mM creatine phosphate, 0.25 mM spermidine, 0.2 �1 of biotinylated lysyl-tRNA (Amersham Pharmacia Biothech, Piscataway, NJ, USA), and 25
�M of 19 amino acids (minus lysine). Assays were carried out at 27°C for 90 min.
1-2-5 Detection of synthesized protein
Aliquots from the protein synthesis reactant were dissolved in sodium dodecyl sulfate -sample buffer and boiled for 5 min. The obtained samples were electrophoresed on 12% polyacrylamide gels together with molecular weight markers (Bio-Rad, Hercules, CA, USA). Following
electrophoresis, biotinylated polypeptides were transferred to PVDF membranes. Non-specific binding sites were blocked by incubation in 5%
skim milk dissolved in PBS for 1 h at room temperature. After washing the PVDF membranes three times with PBS containing 1% Tween-20, the membranes were incubated with 1/1000 diluted streptavidin conjugated horseradish peroxidase (Amersham Pharmacia Biotech) for 1h.
Following washing as described above, the transferred proteins were
incubated with ECL reagents (Amersham Pharmacia Biotech) for 1 min according to the supplier's instructions. The biotinylated bands were visualized by exposure to an X-ray film (Fuji photo film, Tokyo, Japan).
Quantitation of products was carried out using the computing densitometer ImageQuant (Molecular Dynamics, Sunnyvale, CA, USA).
1-3 RESULTS
1-3-1 Disruption efficiency with Mini-Bomb cell disruption chamber
With the aim of extracting intact translation machinery, the Mini
Bomb was used to break the cell membrane. The Mini-Bomb is a device designed to disrupt biological materials with nitrogen pressure. Figure 1-1 illustrates the Mini-Bomb. The cell suspension of biological materials in the Mini-Bomb was placed under pressure, from nitrogen gas derived by a pressure tank, and equilibrated for an adequate time. The needle valve was opened gently and slowly such that the entire suspension was extruded through the orifice. Alteration of nitrogen pressure and flow rate resulted in controlled disruption. Furthermore, nitrogen gas prevents the
biological materials from oxidating.
The pressure placed on the cell suspension might be the most remarkable factor contributing to the disruption of cells while allowing translational efficiency to be retained. Figure 1-2 shows the effect of nitrogen pressure on the degree of cell membrane disruption. When a nitrogen pressure of 5 kglcm2 was applied, the membranes of half of all cells placed in the Mini-Bomb were ruptured, though nearly 40% of the nuclei remained intact. More than 80% of cells placed in the Mini-Bomb were disrupted at a nitrogen pressure of 15 kglcm2 or higher. Even
t:J
Cell suspensionMini-Bomb
-- cell disruption chamber
Orifice
Disrupted cells
Fig. 1-1. Mini-Bomb cell disruption chamber.
14
4
.-. J!2 3
Q) () co 0 T"""
c. 2 L..
..c Q)
E
::J c 1 Q3 ()
0
2 5 10 15 30 H
Pressure (kg/cm2)
Fig. 1-2. Disruption efficiency of Sf21 cells using the Mini-Bomb cell disruption chamber and the Potter-Elvehjem homogenizer.
The number of cells disrupted completely or incompletely was indicated as solid bars or hatched bars, respectively. Open bars indicated undisrupted cells. The disruption efficiency achieved with the Potter-Elvehjem homogenizer is indicated by column H.
though the higher-pressure treatment resulted in more efficient disruption, nuclei and other organelles were more likely to be damaged, resulting in contamination of the pressate with these components. In contrast, lower pressure (2 kg!cm2) treatment caused inefficient disruption. Only about 30% of all cells were disrupted when the motor-driven Potter-Elvehjem homogenizer was used. Furthermore, half of the cells placed in the Potter-Elvehjem homogenizer were disrupted incompletely and cytosoles of the cells remained with partially damaged cell membranes.
1-3-2 Cell-free translation of BMV mRNA
The insect cell extract preparation and the conditions of cell-free translation were based on a method described by Erickson and Blobel (1983) for preparation of wheat germ extract, with minor modifications.
Cell-free translation in an insect cell extract was carried out with addition of BMV mRNA as an exogenous mRNA. The BMV has three genomic RNAs (RNAs 1-3) and one subgenomic RNA4 (Shin et al., 1972; 1976; Bastin et
al., 1976). RNA1-3 and subgenomic RNA4 code proteins 1a, 2a, and 3a and coat protein, respectively. When the BMV mRNAs were translated in the insect cell-free system, mainly two types of protein, i.e., protein 2a and coat protein, were synthesized (Fig. 1-3A). Hence, in this experiment, the protein productivity of the insect cell-free system was monitored according to the amounts of protein 2a and coat protein synthesized.
1-3-3 Optimal disruption conditions with the Mini-Bomb cell disruption chamber
Optimal disruption conditions for preparing a high productivity extract were systematically determined, in terms of nitrogen pressure, cell density upon placement in the Mini-Bomb, equilibration time and flow rate.
A B
2 3 4 5 6 II Protein 2a
kDa D Coat protein
Protein 2a_. -107
-- -- 100
74 �
� 80
">
49 u
::J 60
""0
Protein 3a_. 3 6 e 0..
Q) 40
29 :.;::::; >
en Q) 20
Coat protein__. 21 a:
0 5 10 15
Pressure (kg/em 2)
Fig. 1-3.
Effect of nitrogen gas pressure on protein productivity of the insect cell extract.
(A) Following preparation of the insect cell extracts from 4.0 x
108cells disrupted using the Mini-Bomb cell disruption chamber at nitrogen pressures of 5 (lanes 1, 2),
10(lanes
3,4) and 15 (lanes 5, 6) kg/cm2, translational reactions were carried out with (lanes 2, 4 and 6) or without (lanes
1, 3and 5) BMV mRNA. (B) Relative productivity was densitometorically estimated by calculating the amounts of protein 2a and coat protein synthesized. The amounts of protein 2a and coat protein synthesized, in the extract prepared with a nitrogen pressure of
10
kg/cm2, were each set at
100%.First, the effect of nitrogen pressure on protein productivity was examined (Fig. 1-3). Harvested cells ( 4 x 108 cells) were resuspended to 4 ml in extraction buffer and subjected to a pressure of 10 kglcm2 for 30 min, after which the cells were disrupted with decompression. The extracts derived from the pressate prepared at a nitrogen pressure of 10 kglcm2 had the highest productivity of both protein 2a and coat protein. Higher
pressure treatment disrupted insect cells efficiently, but reduced the protein productivity of the obtained extract. Higher-pressure treatment appears to be excessive for preparing the extract for cell-free translation, as the translation machinery may also be damaged with higher nitrogen pressure.
Secondly, the influence of suspension at various cell densities applied to the chamber, on the protein productivity of the obtained extract, was examined. Harvested cells were resuspended to 4 ml in extraction buffer at various cell densities and subjected to a pressure of 10 kglcm2 for 30 min, after which the cells were disrupted with decompression. Figure 1-4 shows the protein productivity of the obtained extract. The highest
productivity was observed in the extract prepared from cell suspensions of 1.0 and 1.5 x 108 cells/ml, which represent the suspension in the buffer of half of the packed cell volume and the cell pellet obtained with
centrifugation at 600 x g for 10 min, respectively. These results indicate that the Mini-Bomb has the ability to efficiently extract intact translation machinery from cells suspended at a high density.
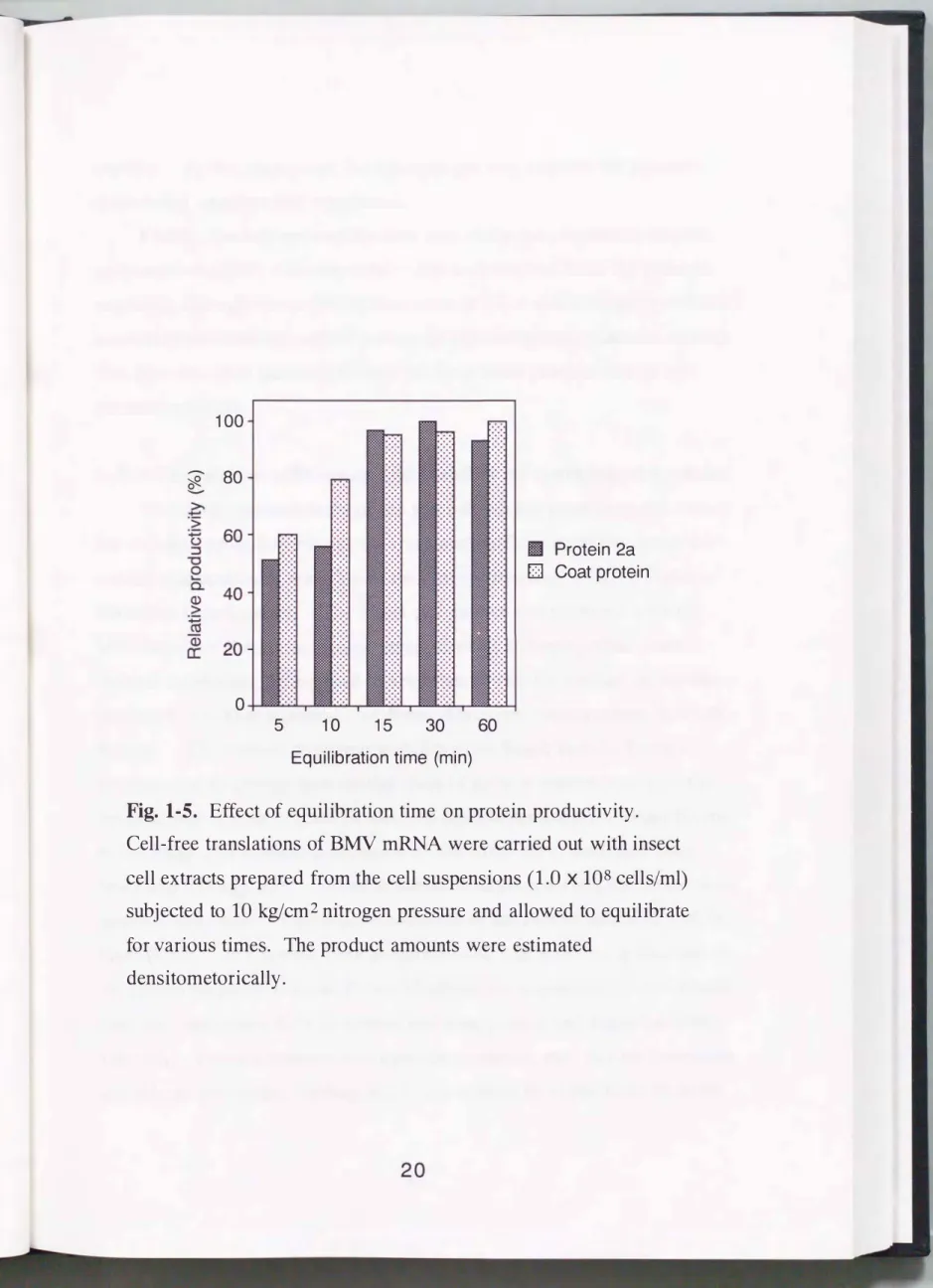
Next, the effect of equilibration time on the protein productivity of the extract prepared from the pressates obtained using various equilibration times was examined. After applying a pressure of 10 kglcm2 on
suspensions of 1.0 x 108 cells/ml, the suspensions were equilibrated for desired periods (Fig. 1-5). The results revealed that the cell suspension needed to equilibrate for at least 15 min to acquire efficient translation
100
?ft
80_..
>.
·s: +-'
� u 60
:::J
"'0 0 Ill
Protein 2a
"-
a. D
Coat protein
Q) 40
>
�
a: Q)
20
0.25 0.5 1 1.5
Cell number (x
1 oacells/ml)
Fig.
1-4. Effect of cell density on protein productivity of insect cell extract.Cell-free translations of BMV mRNA were carried out with insect cell extracts prepared from the cell suspensions at various densities as indicated under nitrogen pressure of 10 kg!cm2 using of the Mini-Bomb. The product amounts were estimated densitometorically.
100
�
80-
0
5 10 15 30
Equilibration time (min)
60
II
Protein 2a
D
Coat protein
Fig.
1-5. Effect of equilibration time on protein productivity.Cell-free translations of BMV mRNA were carried out with insect cell extracts prepared from the cell suspensions
(1.0
x108
cells/ml) subjected to10 kg!
em 2 nitrogen pressure and allowed to equilibrate for various times. The product amounts were estimateddensitometoricall y.
activity. At this time point, the nitrogen gas may prevent the pressate from being oxidized and inactivated.
Finally, the influence of the flow rate at the decompression step on protein productivity was examined. Extracts derived from the pressate extruding through the orifice at flow rates of 12,
4and 1 ml/min produced essentially the same amounts of protein 2a and coat protein (data not shown).
The flow rate thus had no influence on the protein productivity of the obtained extracts.
1-3-4 Translation efficiency and stability of synthesized proteins
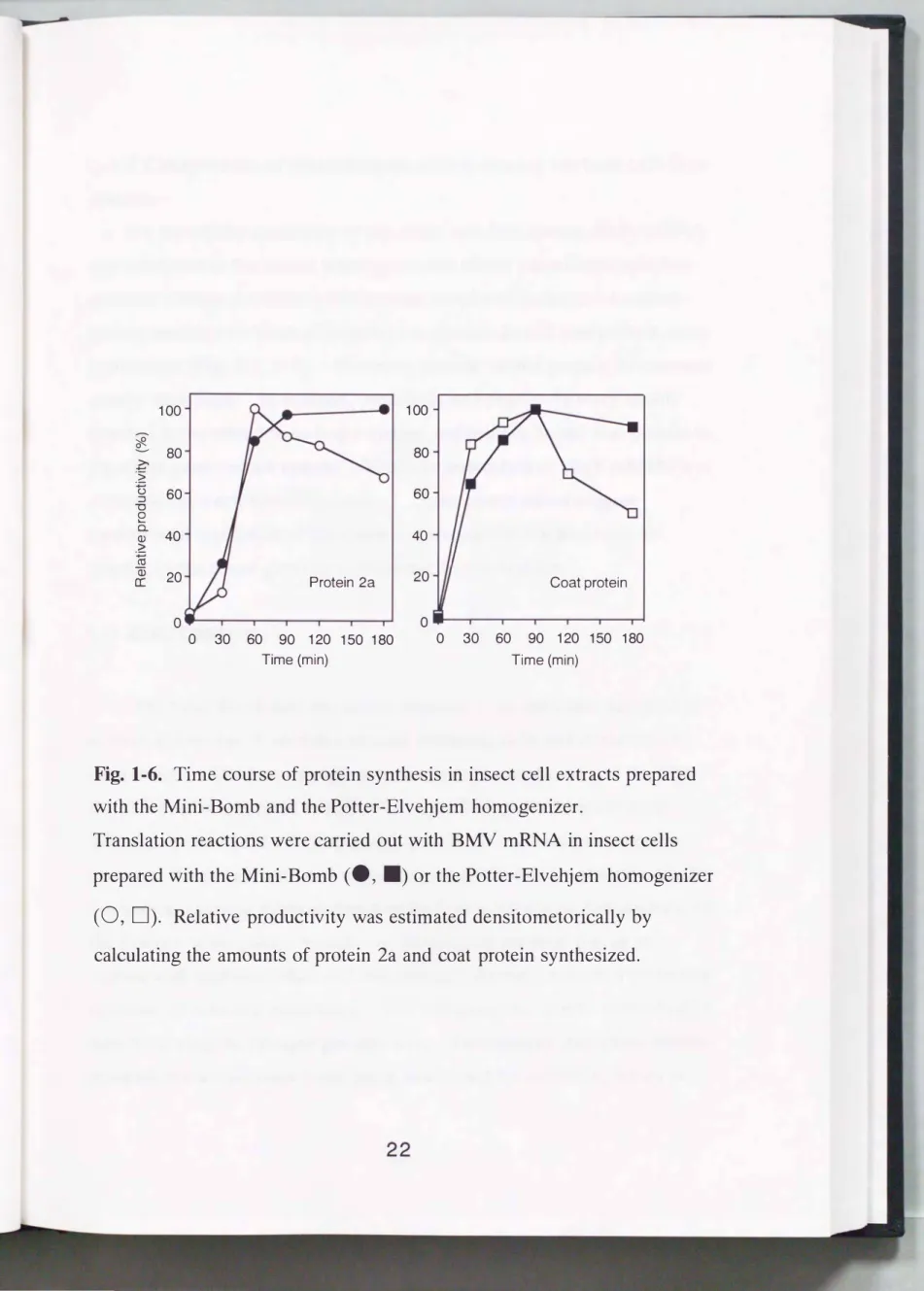
To clarify the usefulness of the Mini-Bomb for producing the extract for cell-free protein synthesis, the translation efficiency of the insect cell extract was compared with that of the extract prepared with the Potter
Elvehjem homogenizer. The insect cell extract was prepared with the Mini-Bomb or the motor-driven Potter-Elvehjem homogenizer under optimal conditions, determined in accordance with the amount of protein produced in a 90 min period (for Potter-Elvehjem homogenizer, data not shown). The extracts prepared with the Mini-Bomb and the Potter
Elvehjem homogenizer had similar rates of protein synthesis (Fig. 1-6).
Translations of both protein 2a and coat protein persisted for about 90 min in the insect cell extracts prepared with the Mini-Bomb and the Potter
Elvehjem homogenizer. A fter termination of the reaction, the synthesized proteins were stable in the reaction mixture of the extract prepared with the Mini-Bomb. In contrast, both protein 2a and coat protein, synthesized in the extract prepared with the Potter-Elvehjem homogenizer, were degraded over time and about 60% of synthesized coat protein had degraded within
180 min. Potter-Elvehjem homogenizer treatment may disrupt lysosomes and release proteinase, leading to the degradation of synthesized proteins.
2 1
100 100
�
80 80c .>
� () :::::l 60 60
-o e
0.. 40 40
<D
>
� en
Q5 20 20
cr: Protein 2a Coat protein
0 0
0 30 60 90 120 150 180 0 30 60 90 120 150 180
Time (min) Time (min)
Fig. 1-6. Time course of protein synthesis in insect cell extracts prepared with the Mini-Bomb and the Potter-Elvehjem homogenizer.
Translation reactions were carried out with BMV mRNA in insect cells prepared with the Mini-Bomb
(e, .)
or the Potter-Elvehjem homogenizer(0, D).
Relative productivity was estimated densitometorically by calculating the amounts of protein 2a and coat protein synthesized.1-3-5 Comparison of translational ability among various cell-free systems
To clarify the specificity of the insect cell-free system, BMV mRNA was translated in the insect, wheat germ and rabbit reticulocyte cell-free systems. When the BMV mRNAs were translated in the insect extract system, mainly two kinds of protein, i. e. protein 2a and coat protein, were synthesized (Fig. 1-3, 1-7). However, protein 1a and protein 3a were not clearly visualized. In contrast, protein 2a and protein 3a were mainly detected in the reticulocyte lysate system, and protein 3a and coat protein in the wheat germ extract system. Thus, the translation of BMV mRNAs was controlled in a cell-specific manner. These observations suggest
translational regulation of the insect cell translation machinery to be specific to the wheat germ or rabbit reticulocyte systems.
1-4 DISCUSSION
The Mini-Bomb cell disruption chamber is an apparatus designed to achieve disruption of several materials including cells and intracellular components. Gas from a high-pressure tank is introduced into the Mini
Bomb where it is applied to individual cells during the pressure cycle.
When the pressurized cell suspension is suddenly exposed to atmospheric pressure, the gas expands and ruptures the cells forming a pressate. Unlike mechanical shearing methods based on the Potter-Elvehjem homogenizer or the Dounce homogenizer which rely on bringing external forces into contact with each individual cell, this nitrogen disruption method uniformly ruptures all cells in a suspension. The cell disruption can be controlled by simply varying the nitrogen gas pressure. The nitrogen disruption method prevents cytosol proteins from being inactivated by oxidation, which is
Insect Rabbit Wheat cell reticulocyte germ extract lysate extract BMV mRNA - + - + - + Protein 2a _.
Protein 3a _.
Coat protei�
kDa
-107
- 74 - 49
- 36 - 29
- 21
Fig. 1-7. Comparison of translational profile among various cell-free systems.
sometimes sustained by mechanical shearing methods operating in air.
Many chambers operate on the same principle and are used for a variety of purposes. For example, the Yeda press (Shneyour and Avaron, 1970) is widely used for several biological materials. Under low pressure, it is used to yield a preparation of Oass A chloroplasts (Belknap and Togasaki, 1981 ), intact mitochondria (Belknap and Togasaki, 1981 ), outer and inner envelope membranes from chloroplasts (Block et al., 1983) and to isolate inside-out thylakoid membrane vesicles from chloroplasts (Anderson et al.,
1980). The Yeda press is also used for complete disruption of the
organelle membrane, aimed at isolating biologically active proteins within the organelle membrane (Walker and Harwood, 1985), which are difficult to dislodge from the membrane by osmotic shock. The nitrogen
disruption method is suitable for breaking down biological materials such as cell and organelle membranes.
In this experiment, the optimal conditions for cell disruption using the Mini-Bomb were determined based on translational efficiency. The cell disruption efficiency increased with rising nitrogen pressure. The degree of disruption was found to be controlled by varying the nitrogen pressure.
Furthermore, compared to the Potter-Elvehjem homogenizer, the Mini
Bomb was capable of disrupt cells with high efficiency, while allowing the translation efficiency of the cells to be retained. The protein concentration of the extract prepared with the Mini-Bomb, at a nitrogen pressure of 10 kg/cm2, was approximately 3-fold that of the Potter-Elvehjem homogenizer (data not shown). More severe treatment with the Potter-Elvehjem
homogenizer reduced the translational efficiency (data not shown). The Dounce homogenizer, which is generally used to homogenize cultured cells, was unable to homogenize insect cells, as it was less efficient than the
Potter-Elvehjem homogenizer (data not shown).
A higher protein concentration, in the extract condensed with
ultrafiltration membranes, raises the translatjon efficiency (Nakano et al., 1994; Kim et al., 1996). Although the rate of protein synthesis was increased with condensed extract, microsomes in the insect cell extract are apparently damaged and a posttranslation factor, serving as a signal
recognition particle, might also be eluted through the ultrafiltration membrane during the condensation procedure.
To increase the protein concentration of the extract without
condensation, the cell density of the cell suspension needs to be increased.
Usually, when extracts for cell-free translation are prepared from various cultured cells, the cell pellets are resuspended in packed cell volumes of hypotonic buffer the same to three times volume, then allowed to swell for efficient homogenization. Conversely, the Mini-Bomb allows disruption of the cells resuspended in half the volume of packed cells or even the cell pellet obtained by centrifugation at 600 x g for 10 min. In this experiment, the extract derived from cells suspended in extraction buffer, instead of
hypotonic buffer, (at a density of 1.0 x 108 cells/ml) had more than double the protein productivity of the extract derived from cells suspended in 2 volumes of extraction buffer (at 0.5 x 108 cells/ml). The ability to disrupt the cell suspension at high density resulted in the insect extract having high protein productivity. These observations showed the Mini-Bomb, which has properties of controlled and efficient disruption, to be a suitable device for preparing the extract for a cell-free protein synthesis system.
Furthermore, time course analysis revealed synthesized protein to be stable in the reaction mixture derived from the extract prepared with the Mini
Bomb. In contrast, synthesized proteins in the reaction mixture derived from the extract prepared with the Potter-Elvehjem homogenizer were not stable. This observation suggested that the Mini-Bomb may only disrupt
the cell membrane, leaving organelles, i.e. lysosome, of the cells with intact.
That is in agreement with the finding that the extract prepared with the Mini-Bomb had intact microsomes and had the ability not only to synthesize protein, but also to glycosylate the protein in the microsomal lumen.
However, the extract prepared with the Potter-Elvehjem homogenizer failed to synthesize glycosylated protein, producing only the polypeptide backbone (Tarui et. al., 2000).
The insect cell extract, in particular, translated protein 2a and coat protein (Fig. 1-3, 1-6). The 3' UTRs of the four RNAs of BMV contain 3'-terminal nucleotides forming a tRNA-like structure which stimulates the translation of capped (Daniel and Kobayashi, 1994) or uncapped mRNA (Karpova eta!., 1995). However, the region of four RNAs might not affect cell-specific translation because these RNAs have high similarity in this region (Ahlquist eta!., 1981). The effect of 5' UTR of BMV mRNA on translation remains unclear. The intrinsic translational efficiency of the mRNA is dependent on cis-acting elements alone or a cis-acting element with the trans-acting factors. Several cis-acting elements exist on 5' UTR which enhance or inhibit the translational efficiency of eukaryotic mRNA (Yamaguchi et al., 1982; Imataka eta!., 1994; Roberts eta!., 1997). For instance, the 5' UTR of growth related protein p23, which affects cell
specific translation efficiency, inhibits translation efficiency in Ehrlich ascites tumor cells, reticulocyte lysate and Xenopus oocytes, whereas the 5' UTR does not affect translation efficiency in the wheat germ extract (B"hm eta!., 1991). The 5' UTRs of RNAs that contain cap structures at the terminus of each mRNA show weaker homology, except for RNAs 1 and 2 (Ahlquist eta!., 1984). The 5' UTR of BMV RNAs 1 and 2 share
substantial homology, there being only two mismatches between the first 42 bp, though the remaining 74 bp 5' UTR of RNA1 constitute a subset of the
103 bp 5' UTR of RNA2. Although some regions of the 5' UTR of RNA3 bear distinguishable homology to RNAl and 2, the remainder show no such homology. Cell-specific translation of RNA2 and RNA3 observed among these cell-free protein synthesis systems is probably regulated by the cis
acting element on the non-homological region of these 5' UTRs.
Conversely, the RNA4 of BMV has the 5' UTR of a short non-coding region of
9
nucleotides(
Ahlquist et al.,1984),
which might be too short toconstruct a secondary structure at this region to act as the cis-acting element.
The rate of RNA4 translation might be limited by affinity of the cap binding protein elF 4E to 5' cap structure.
The author found the novel Mini-Bomb device to be useful for cell breakage, aimed at preparing an extract for cell-free protein synthesis.
Moreover, insect cell extracts, prepared with this device, had the unique property of allowing proteins synthesis, a property lacking in traditional rabbit reticulocyte lysate and wheat germ extract systems.
Many proteins are routinely synthesized using either a rabbit reticulocyte cell-free system or a wheat germ cell-free system. An alternative system is used in the event of one system not being able to synthesize a particular protein efficiently. This insect cell-free system may serve as a novel assay system because of its unique enhancement of mRNA translation, as compared to the traditional systems.
1-5 SUMMARY
An insect cell extract for cell-free translation was prepared using a Mini-Bomb cell disruption chamber, a unique device which disrupts biological materials by altering the pressure on these materials. The highest potential extract for cell-free translation was prepared from the cell
suspension at a density of 1.0 x 108 cells/ml, then disrupted at a nitrogen pressure of 10 kg/cm2• Furthermore, synthesized proteins were stable in this extract, but were degraded by proteinase in an extract prepared with a Potter-Elvehjem homogenizer. The insect cell extract showed efficient activity for translation of protein 2a and coat protein of the brome mosaic virus, while inefficiently translating protein 1a and protein 3a. On the other hand, mainly protein 3a and coat protein were translated in a wheat germ system while mainly proteins 2a and 3a were translated in a rabbit reticulocyte system. The insect cell-free system appears to possess an unusual property for enhancing translation of exogeneous mRNA, making it different from the rabbit reticulocyte and wheat germ systems.
29
CHAPTER 2
Glycoprotein synthesis in the cell-free translation/ glycosylation system derived from insect cells
2-1 INTRODUCTION
Cell-free assay systems for proteins with posttranslational
modification have been constructed by combining a eukaryotic cell-free translation system and rough microsomes (Blobel and Dobberstein, 1975;
Walter and Blobel, 1983; Rothblatt and Meyer, 1986). These reconstituted assay systems for assessing protein translation and posttranslational
processing in higher eukaryotes have allowed characterization of the translocational machinery (Walter and Blobel, 1981; Gorlich et al., 1992), and are being actively used to define the topology of membrane proteins (Popov et al., 1997) and to elucidate the regulation of N-linked core glycosylation (Kasturi et al., 1997). However, they may lack certain components of the posttranslation process (Hegde and Lingappa, 1997), and heterology may complicate the results (Rothblatt and Meyer, 1986; Walter
et al., 1981; Meyer, 1985; Prehn et al., 1987).
The baculovirus expression system has been used to express various proteins in insect cells under the control of a strong polyhedrin promoter (Hara et al., 1993; Luckow and Summers, 1988). Most of the recombinant proteins produced in this system have been shown to be antigenicall y, immunogenically, and functionally similar to their authentic counterparts, because the insect cells are able to carry out many types of posttranslational modifications, such as addition of N- and 0-linked oligosaccharides
(Luckow and Summers, 1988).
In this chapter, construction of a cell-free translation/glycosylation system prepared from insect cells that can produce functional glycosylated proteins was described. In the present study, human immunodeficiency virus type-1
(
HIV-1)
envelope glycoprotein gp120 that is glycosylated and folded in a mature conformation that allows it to bind to CD4 \vas produced.This system was prepared from a single source and includes almost all translational and posttranslational components maintained intact by a single-step extraction. It is completely different from conventional cell
free assay systems, such as the rabbit reticulocyte lysate system
supplemented with canine pancreatic microsomes, which are mixtures of heterologous fractions. This insect cell-free system will be a useful tool for synthesizing functional glycoproteins and will serve as a model system for elucidating the mechanisms of posttranslational modification.
2-2 MATERIALS AND METHODS
2-2-1 Cells and culture conditions
Spodoptera frugiperda (
IPLB-Sf21-AEII)
cells(
Vaughnet al.,
1977)
were grown at 27°C in IPL41 medium
(
GIBCO BRL-Life Technologies, Grand Island, NY, USA)
supplemented with 10% heat-inactivated fetal bovine serum(
GIBCO BRL-Life Technologies)
, in either monolayer or spinner cultures.2-2-2 Subcloning the gp120 gene
The pUC19-subcloned EcoRI fragment containing the
env
region of HIV-1 sF162(
Cheng-Mayeret al.,
1990) (
GenBank accession numberM38428
)
was used as the source of the HIV-1sF162 gp120 gene. The nucleotide numbers are based from the sequence of this gene, and thenumbering of the amino acids begins at the start of the open reading frame.
A eDNA fragment encoding gp120 was amplified by PCR using oligonucleotide primers to delete the gp41 env coding sequence by
introducing an in-frame stop codon directly following R502. The PCR product was cloned into the Bam Ill and N otl sites of p VL1393 (Invitrogen, San Diego, CA, USA), and the resulting recombinant plasmid was
designated p VLgp120. The integrity of the insert was confirmed by dideoxynucleotide chain termination DNA sequencing (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
2-2-3 Preparation of gp120 mRNA
The template DNA for generating gp120wt, authentic gp120, mRNA was constructed by ligating three fragments into pUC18 as follows. The fragment encoding gp120 was amplified by PCR with pVLgp120 as the template. Two unique restriction sites were introduced using
oligonucleotides that altered codons G5-16-R7 and R494-R495 without altering their coding ability. Modification of GGG to GGA at G5, A TC to ATT at 16, and AGG to CGA at R7, and modification of AGA to CGT at R494 and AGA to CGA at R495 created EcoRI and Sall/Xhol sites,
respectively. The fragment encoding gp120 was cloned into the EcoRI and Sall sites of pUC18, producing pUCgp. The Xhoi-Pstl fragment of the 3' end of the gp120-coding sequence following the polyhedrin 3'
untranslated region (UTR) amplified by PCR with AcNPV DNA as the template was ligated into the Sall and Pstl sites of pUCgp, producing pUCgp3. The EcoRI-EcoRI fragment of the T7 RNA polymerase
promoter and polyhedrin 5' UTR following the 5' end of the gp120 coding sequence, amplified by PCR using a synthetic oligonucleotide as the
template, was ligated into the EcoRI site of pUCgp3. The resulting
32
recombinant plasmid was designated pgp120wt. The EcoRI-Kpnl
fragment, which lacked a signal coding sequence, was ligated into the EcoRI site of pUCgp3 and the Kpnl site of the gp120-coding region instead of the EcoRI-EcoRI fragment to produce pgp120�s. The integrities of the inserts and resulting mutations were confirmed by dideoxynucleotide chain termination DNA sequencing (Amersham Pharmacia Biotech).
The plasmids pgp120wt and pgp120�s were linearized with Pstl and transcribed with T7 RNA polymerase using a MEGAscript kit (Ambion, Austin, TX, USA) according to the manufacturer's instructions. The
transcripts were purified by phenol/chloroform extraction and isopropanol precipitation, and fractionated by formaldehyde agarose gel electrophoresis to check the size and the homogeneity. The samples were resuspended in diethylpyrocarbonate treated water and stored at -80°C.
2-2-4 Preparation of insect cell extract
After washing three times with extraction buffer ( 40 mM Hepes-KOH (pH 7.95), 100 mM KOAc, 1 mM Mg(OAc)2, 2 mM CaC12, and 4 mM DTT), the Sf21 cell pellet was resuspended in 0.5 volume of extraction buffer at approximately 1.0 x 108 cells/mi. The cells were disrupted using a Mini
Bomb cell disruption chamber (Kontes Glass Company, Vineland, NJ, USA), with the cell suspension maintained under nitrogen gas for 1 h. The homogenate obtained was centrifuged in a SW40 Ti rotor (Beckman, Palo Alto, CA, USA) for 15 min at 14,000 rpm at 4 °C, then passed through a Sephadex G-25 fine column (1.0 x 16.0 em) equilibrated with extraction buffer containing 5 mM Mg(OAc)2 but no CaC12• The extract was frozen in aliquots and stored in liquid nitrogen.
2-2-5 Cell-free protein synthesis
Before using the insect cell extracts for cell-free protein synthesis, their endogenous mRNA was digested by adding 50 �1 of a nuclease mixture, containing 20 U/�1 of micrococcal nuclease (Boehringer Mannheim,
Indianapolis, IN, USA), 1.8 mM CaC12 and 5 U/�1 placental ribonuclease inhibitor, to a 40-�1 aliquot of thawed extract and incubating the mixture for 5 min at 20°C. The reaction was terminated by the addition of 4 !-!1 of 80 mM EGT A and 7 �1 of 10 mg/ml creatine kinase (Boehringer
Mannheim).
A 5-�1 cell-free protein synthesis reaction mixture contained (final concentrations): 25 �M of each of the 20 amino acids, 30.6 mM Hepes-KOH (pH 7.95), 1.5 mM Mg(OAc)2, 100 mM KOAc, 2.5 mM DTT, 0.25 mM spermidine, 1.2 mM ATP, 0.25 mM GTP, 8.0 mM creatine phosphate, 0.2 mg/ml mRNA, and 3.2 �1 of nuclease-treated extract. The reaction was carried out at 27°C for 40 min and terminated by adding sodium dodecyl sulfate (SDS) gel loading buffer, and the mixture was then boiled for 5 min.
Translations of mRNA in the wheat germ extract (Promega, Madison, WI, USA) or in the rabbit reticulosyte lysate (Amersham Pharmacia Biotech) were carried out according to the manufacturer's instructions.
2-2-6 Detection of gp120
Cell-free protein synthesis reactions were carried out as described above. The reaction mixtures were separated by 8.5% SDS
polyacrylamide gel electrophoresis and proteins were transfered to a PVDF membrane. Non-specific binding sites were blocked by incubation in 5%
skim milk dissolved in PBS for 1 h at room temperature. The membrane was incubated with monoclonal antibody (International Enzymes, Fallbrook, CA, USA) at a 1:1000 dilution for 1 h. Following washing three times
with T-PBS, the membrane was incubated with HRP-rabbit anti-sheep IgG (Zymed Laboratories, San Francisco, CA, USA) as a second antibody at a 1:2000 dilution for 1 h. Synthesized gp120 and standard gp120 were visualized with the ECL system. The products were quantified using the computing densitometer ImageQuant (Molecular Dynamics, Sunnyvale, CA, USA).
2-2-7 Endo H treatment
After the cell-free protein synthesis reaction, the reaction mixture was boiled in the presence of 0.5% SDS and 2.5% 2-mecaptoethanol. Four volumes of endo H solution (25 mM citrate buffer, pH 5.5, 12.5 mM EDT A, pH 8.0, 12.5% Triton X-100, 0.05 U/ml endo-B-N-acetylglucosaminidase H) was added and samples were incubated at 37°C for 24 h.
2-2-8 Protease protection assay
Proteinase K digestion was carried out as described by Walter and Blobel (1983) and Rothblatt and Meyer (1986) with minor modifications.
After the cell-free protein synthesis reaction, 8 �1 of translation reactions were diluted to 20 �1 in 10 mM CaC12, 100 mM KOAc and 1.5 mM Mg(OAc)2, 0.38 mg/ml proteinase K (Ambion) with or without 0.5%
Triton X-100 at 0°C. After incubation for 60 min, proteinase K was inhibited with 7 mM phenylmethylsulfonyl fluoride, and then all samples were boiled with SDS gel loading buffer for 5 min.
2-2-9 FACS analysis
The surface-associated gp120 was analyzed as described previously (Tahara-Hanaoka et al., 2000). Briefly, quail QT6 cells stably expressing CD4 containing a cytoplasmic tail deletion were incubated with gp120 in the
reaction mixture of the insect cell-free system or culture medium of recombinant baculovirus-infected insect cells. After washing, the cells were reacted with anti-HIV human IgG purified from the serum of an HIV-infected person. After washing again, the cells were reacted with FITC-conjugated rabbit anti-human IgG (DACO, Carpenteria, CA, USA).
The amount of surface-associated gp120 was measured using a FACS Calibur (Becton-Dickinson, San Jose, CA, USA).
2-3 RESULTS
2-3-1 Construction of plasm ids cloned gp120 gene for template mRNA
HIV-15F162 envelope glycoprotein gp120 is a secretory protein having an amino-terminal extension of 29 amino acid residues described as a signal sequence that is involved in the first step of the secretory pathway that translocates proteins across the membrane of the endoplasmic reticulum in eukaryotic cells. The gp 120wt mRNA with the natural signal sequence and the gp 120L1s mRNA without the natural signal sequence were
constructed (Fig. 2-1 ). However, to increase the translational efficiency of the insect cell-free system, the 5' UTR of gp120 was replaced with a polyhedrin 5' UTR, and a polyhedrin 3' UTR was added. The gp120 mRNA and gp120L1s mRNA were synthesized with T7 RNA polymerase using pgp120 and pgp120L1s as templates, respectively. The gp120wt mRNA and the gp120L1s mRNA encoded proteins having a molecular mass of 56 kDa and 53 kDa, respectively.
2-3-2 Cell-free translation of gp120 with insect cell extract To prepare an insect cell extract, a Mini-Bomb cell disruption
36
8
gpl20P.stl
Synthesized DNA
1
PCREcoRl EcoRl
�- · · I
T7 RNA Polyhedrin polymerase 5 · UTR promoter
Polyhedrin 5'U1R T7RNA polymerase promoter EcoRl
gpl20
�
17wl
1
PCR P"lI sssssssss J
Polyhedrin 3'UTR
Polyhed:rin
Pstl
Synthesized DNA
1
PCREcoRI Kpni
�- · · I
T7 RNA Polyhedrin polymerase 5' UTR promoter
gpl20
Polyhedrin
3' UTR
5' Uffi
Kpn8,
Sa!IXhoi Polyhedrinpgpl20Lls 3'UTR Pstl
EcoRI Pstl
�
ln vitro t ranscription�
[n vitro t ranscription I.Polyhedrin 5'UTR
S\SSS\9
gpl20wt Polyhedrin Polyhedrin
3' UTR 5' l.JTR gp 120wt mRNA
gpl20
gpl20Lls mRNA
ssssss5J Polyhedrin
3' UTR
Fig. 2-1. Construction of the vectors for synthesizing gp 120wt mRNA and gp120L\s mRNA for cell-free protein synthesis and in vitro transcription.