博士
博士論文 論文
ルイス酸活性点として 14族原子を 含む遷移金属錯体の創成
Syn thes is of trans i t ion-me ta l comp lexes
con ta in ing a group 14 a tom as a Lew is ac id s i te
谷脇 旦
2016年 3月
謝辞
本研究の遂行および論文の執筆にあたり、御指導および多大なお力添えを賜りま した弘前大学大学院理工学研究科岡崎雅明教授に心から感謝致します。研究に際し、
有益な御助言および学術雑誌の執筆にお力添えいただきました弘前大学大学院理工学 研究科太田俊助教に感謝致します。
元素分析を測定していただきました弘前大学機器分析センターの氏家夏樹氏、京 都大学化学研究所元素分析室の平野敏子氏、NMR スペクトルの測定の際に有益な 御助言を頂きました弘前大学理工学部教育研究支援室の荒木宏孝氏に厚く御礼申し 上げます。硝子器具の製作および修繕をしていただきました弘前大学硝子工作室の 堀井智美氏、中村硝子製作所の中村俊一氏に感謝致します。
最後に、私の私生活を支えて頂き、常に温かく見守って下さいました家族の方々 に心から感謝致します。
略語表
本文中では以下の略語を用いる。
Me:メチル基 Et:エチル基
iPr:イソプロピル基
tBu:tert-ブチル基
Ph:フェニル基
NBS:N-ブロモコハク酸イミド THF:テトラヒドロフラン
PF6:ヘキサフルオロリン酸イオン BPh4:テトラフェニルホウ酸イオン DMAP:4-ジメチルアミノピリジン
TFPB:テトラキス[3,5-ビス(トリフルオロメチル)フェニル]
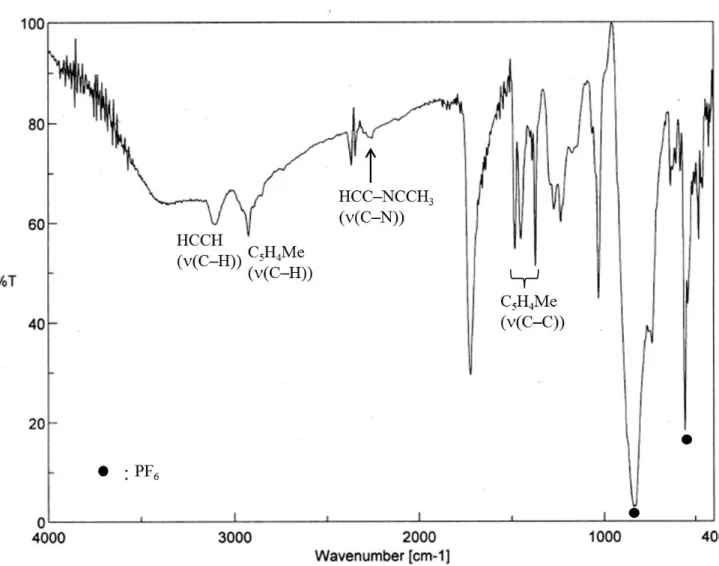
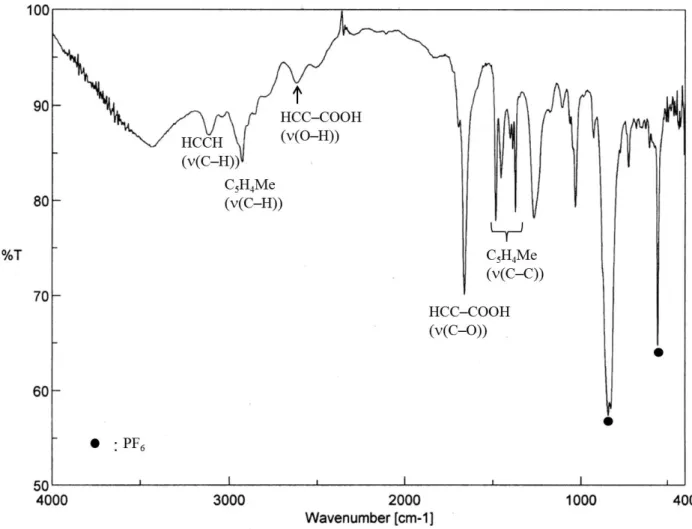
Ts:p-トルエンスルホニル基 [(η5-C5H4Me)4Fe4(HCCH)2](PF6) (1)
[(η5-C5H4Me)4Fe4(HCCH)(HCC–Br)](PF6) (2) [(η5-C5H4Me)4Fe4(HCCH)(HCC–NCCH3)](PF6)2(3) [(η5-C5H4Me)4Fe4(3–CO)(3–CH)(HCCH)](PF6) (4) [(η5-C5H4Me)4Fe4(HCCH)(HCC–OH)](PF6) (5) [(η5-C5H4Me)4Fe4(HCCH)(HCC–OMe)](PF6) (6)
[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–F)](PF
6) (7)
[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–NC
4H
4N)](PF
6)
2(8) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCCNC
5H
5)](PF
6)
2(9)
[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–(1,2–N
2C
4H
4)](PF
6)
2(10) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–COOH)](PF
6) (11) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–Cl)](PF
6) (12) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CH
2CN)](PF
6) (13) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CH
2CN)](BPh
4) (14)
[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–NC(Me)(NC
4H
2O
2))](PF
6) (15) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CN)](PF
6) (16)
[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CN)] (17)
[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CNC
6H
11)](PF
6)
2(18) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CNCH
2Ph)](PF
6)
2(19) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–C(O)NHCH
2Ph)](PF
6) (20) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CNC
6H
3(CH
3)
2)](PF
6)
2(21) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–CNC
6H
2(
tBu)
3)](PF
6)
2(22) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–C(O)NHC
6H
3(CH
3)
2)](PF
6) (23) [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–NC
4H
4N)](TFPB)
2(24)
[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–NC
4H
4N)](OTf)
2(25)
[(η
5-C
5H
4Me)
4Fe
4(
3–CO)(
3–CH)(HCCH)](OTf) (26) [(η
5-C
5H
4Me)
4Fe
4(HCCH)
2](OTf) (27)
[Ir(PMe
3)
3(H)(η
2-SSiMe
2)] (28)
[Ir(PMe
3)
3(H)(SH)(Si(OH)Me
2)] (29)
[Ir(PMe
3)
3(H)(SH)(Si(OMe)Me
2)] (30)
[Ir(PMe
3)
3(H){
2(S,Si)-SC(O)OSiMe
2}] (31)
目次
第一章 緒言 1
第二章 四鉄骨格に架橋配位したエチニルカチオンの発生と性質 32
第三章 四鉄上で発生したエチニルカチオンによるアセトニトリル 94 の活性化
第四章 四鉄上で発生したエチニルカチオンによるイソシアニドの 122 活性化
第五章 四鉄上で発生したエチニルカチオンによる有機ケイ素化合物 204 の活性化
第六章 遷移金属に配位したシランチオン錯体の合成と反応性 248
第七章 結語 295
1
第一章
緒言
この章のみ、化合物番号は別個に割り振った。
2
ルイス酸触媒は基質の求電子性を向上させることで反応を促進させる重要な触 媒である。これまでに、炭素と炭素をつなぐ多くの反応がルイス酸触媒により達成 されており、様々な有機合成反応が展開されてきた。図
1-1
にルイス酸触媒による 基質活性化の機構を示す。図
1-1.
ルイス酸触媒による基質活性化ルイス酸により捕捉された基質
(Lewis adduct)
では、ルイス酸の最低空軌道(LUMO)
と基質の最高被占軌道(HOMO)
との電子的相互作用により、結合が大きく分極するため、基質への求核攻撃が容易になる。このため、求核剤の添加により基 質へ官能基が導入され、生成物の脱離とともにルイス酸触媒が再生する。
これまでにルイス酸活性中心としてはホウ素、アルミニウム、鉄、チタン、スカ ンジウムなどの原子が主に用いられており、これらの原子を用いたルイス酸触媒と しては、
B(C
6F
5)
3、AlCl
3、FeCl
3、TiCl
4、Sc(OTf)
3などがあり、様々な有機合成に使 われている。1) 代表的な例としてFriedel-Crafts
反応があり、この反応は1877
年にCharles Friedel
とJames Mason Crafts
によって初めて報告された芳香族化合物への 求電子置換反応である。1b,c)例えば、塩化アルミニウムをルイス酸触媒として用いた
Friedel-Crafts
アルキル化 では、芳香族化合物とアルキルクロリドを用いることで、芳香環へアルキル基の導 入が可能である(
スキーム1-1)
。この反応では、ルイス酸である塩化アルミニウム の空の軌道にアルキルクロリドの塩素上の非共有電子対が供与されることにより、分極した炭素-塩素結合が開裂することで、カルボカチオンが発生する。発生した カルボカチオンは芳香環に求電子置換したのち、塩化物イオンによってプロトンが 引き抜かれることで、塩化水素の発生を伴いながらアルキル化芳香族生成物および 触媒が再生する。
3
スキーム
1-1. Friedel-Crafts
アルキル化反応このように古典的ルイス酸触媒により有機分子の基本骨格を形成する炭素-炭 素結合の生成反応が見出されることで様々な炭素骨格の合成が可能とされた。近年 では適切な化学修飾を施したルイス酸反応場が多数設計されており、古典的ルイス 酸触媒では達成しえなかった反応性および選択性の制御に成功している。例えば、
アルミニウムに
2,6-
ジフェニルフェノキシドを導入したルイス酸(1)
を用いること で、, -
不飽和アルデヒド(2)
とtBuMgCl
との反応では選択的に1,4-
付加体(3a)
が生 成する(
スキーム1-2)
。1
は、ルイス酸活性中心であるアルミニウム上に嵩高い置換 基を有するため、1
によりアルデヒドのカルボニル部位が覆われ、求核剤が選択的 に位を攻撃することで、1,4-
付加体を得ている。2)スキーム
1-2.
ルイス酸1
による立体選択的な求核付加反応4
また、光学活性ルイス酸触媒についても設計されており、医薬品や農薬に使われ る有機分子の合成に必須とされる不斉合成をも可能としている。その例としては、
スキーム
1-3
に示すように、キラルなボラン4
をルイス酸触媒として用いることで 化合物5
のエナンチオ選択的な環化付加反応が起こり、生成物6a
が高い選択性を もって得られることが報告されている。3)スキーム
1-3.
キラルなルイス酸触媒による不斉合成現在もルイス酸触媒となる反応場は活性、選択性、安定性、触媒回転数などが考 慮され、よりポテンシャルの高い触媒系の開発が行われており、炭素-炭素結合を 形成する実用的な触媒系が展開されている。従来のルイス酸触媒では、その活性中 心としては主に典型金属あるいは遷移金属が用いられた例がほとんどであり、典型 元素がルイス酸活性中心となる実用的な触媒系は数少ない。
本研究では、
14
族典型原子である炭素およびケイ素がルイス酸活性中心として 作用する新しいタイプの反応場を構築し、これまでのルイス酸触媒では達成し得な かった新反応の開拓に取り組んだ。以下、ルイス酸として作用しうる炭素陽イオン 種、ケイ素陽イオン種およびケイ素不飽和化合物について概観し、これらを用いた ルイス酸活性種の合成戦略を述べる。5
1-1 カルボカチオンの性質
正電荷を持つ炭素陽イオン種はカルボカチオンと総称されており、これは、有機 反応において、重要な反応である求核置換反応の中間体としてその発生が仮定され ている。
S
N1
反応では、置換基L
が脱離することで中間体としてカルボカチオンが 発生し、求核剤が陽イオン炭素上を攻撃することで、求核置換された立体が異なる 生成物を1:1の比で与える(
図1-2)
。図
1-2.
求核置換反応(S
N1
反応)
カルボカチオンは正電荷を持つ炭素原子に隣接する炭素原子上の
C – H
結合お よびC – C
結合との超共役によって安定化される。つまり、そのような結合の数が 最も多い第三級が最も安定で生成しやすく、逆に第一級カルボカチオンは不安定で 生成しにくい(
図1-3)
。図
1-3.
カルボカチオンの安定性このため、メチルカチオンのようなアルキル基を持たないカルボカチオンは非常 に不安定であると考えられているが、言い換えると非常に高い活性を有すると考え ることができ、このようなフラグメントの発生とその反応性については興味深い。
これまでに、電子不足な陽イオン性炭素種の発生は様々な手法で取り組まれている。
6
1-2 カルボカチオンの発生と単離
一般的な有機反応によってカルボカチオンを発生させるための様々な手法がこ れまでに確立されており、中にはカルボカチオンを安定な形で単離した例も報告さ れている。カルボカチオンの発生方法を以下に示す。
(a)
ソルボリシス反応炭素原子上に優れた脱離基をもつ化合物はソルボリシス反応によって、イオン化 される。例えば、ハロゲン化アルキルあるいは、スルホン酸エステルを有するアル キル化合物はソルボリシス反応によって、カルボカチオンとアニオン化学種を発生 する
(
スキーム1-4)
。スキーム
1-4.
ソルボリシス反応(b)
官能基を優れた脱離基に変換する方法官能基を脱離基に変換し、カルボカチオンを発生させることが可能な特定の反応 がある。アルコールにプロトンを付加させ、オキソニウムイオンとすることで、カ ルボカチオンと水を生成する方法である。もう1つは、第一級アルキルアミンと亜 硝酸との反応により、アミンをジアゾニウムに変換させ、カルボカチオンと窒素を 発生させる方法である
(
スキーム1-5)
。スキーム
1-5.
官能基の脱離基への変換7
(c)
アルケンまたはアルキンに正電荷を帯びた化学種を導入する方法プロトンなどの正電荷を帯びた化学種をアルケンまたはアルキンへ付加させる ことで、カルボカチオンを発生させることができる
(
スキーム1-6)
。スキーム
1-6.
アルケン、アルキンへの陽イオン性化学種の付加(d) C=X
結合に正電荷を帯びた化学種を導入する方法(X = O, S, N)
炭素とヘテロ原子との間に二重結合を有する化学種に、正電荷を帯びた化学種を 付加させることで、共鳴安定化されたカルボカチオンを発生する
(
スキーム1-7)
。スキーム
1-7. C=X
結合への正電荷を帯びた化学種の導入8
(e)
超酸中でのカルボカチオンの発生フルオロ硫酸と五フッ化アンチモンの混合溶液は超酸と呼ばれ、非常に強力な酸 である。超酸存在下では、アルキル化合物から、ヒドリドが引き抜かれることで、
カルボカチオンが発生する
(
スキーム1-8)
。4)スキーム
1-8.
超酸によるヒドリドの引き抜き五フッ化アンチモンはフッ化アルキルからフッ素を引き抜くことができる。この 性質を利用してカルボカチオンを発生させた例も存在する
(
スキーム1-9)
。5)スキーム
1-9.
フッ化アルキルと五フッ化アンチモンとの反応また、カルボカチオンには安定な形で単離されているものがある。
Reed
らはジ クロロメタン中で、n -ブタンとメチルカルボラン CH
3(CHB
11Me
5Cl
6)
との反応を–30 ºC
で行うことでtert -ブチルカチオンの安定な塩 [Me
3C][CHB
11Me
5Cl
6]
が得られるこ とを報告している(
スキーム1-10)
。6)スキーム
1-10. n -ブタンと CH
3(CHB
11Me
5Cl
6)
との反応[Me
3C][CHB
11Me
5Cl
6]
の構造は単結晶X
線構造解析によって明らかにされている(
図1-4)
。表1-1
より、tert -ブチルカチオンの 3
つの結合角度(C – C
+– C)
の合計は360 °
となっていることから、
tert -ブチルカチオンの中心炭素は sp
2混成に特徴的な平面 構造をとっていることが明らかにされている。9
図
1-4. Molecular structure of [Me
3C][CHB
11Me
5Cl
6]
・CH
2Cl
2with 50% thermal ellipsoids. The disordered solvent molecule is omitted for clarity.
表
1-1. Selected bond lengths (Å) and bond angles (°) of [Me
3C][CHB
11Me
5Cl
6]
・CH
2Cl
2. C(1)-C(2) 1.429(4) C(2)-C(1)-C(3) 120.3(3)
C(1)-C(3) 1.438(4) C(2)-C(1)-C(4) 120.0(3)
C(1)-C(4) 1.459(4) C(3)-C(1)-C(4) 119.7(3)
10
1-3 カルボカチオンをルイス酸として用いた例
カルボカチオンの中心炭素は
sp
2混成軌道からなる3
つの結合と上下に均等に張 り出した空のp
軌道を有する。空の軌道に電子を受け入れることができるため、カ ルボカチオンはこれまでにルイス酸として様々な反応に用いられてきた。7) 近年で は、工業的に重要なプロセスとして、メタノールをオレフィンに変換する反応(MTO)
において、1,3-
ジメチルシクロペンタジエニルカチオンや1,1,2,4,6-
ペンタメチルベンゼニウムカチオン
(
図1-5)
が重要な中間体としてNMR
スペクトルによっ て観測されている。8)図
1-5
1-3-1 第三級カルボカチオンをルイス酸触媒として用いた例
第三級カルボカチオンのように安定なカルボカチオンはルイス酸触媒として利 用されている。向山らはトリチル塩をルイス酸触媒として用いて、マイケル付加反 応やアルドール反応に適用している。9) 例えば、シリルエノールエーテルと
, -
不 飽和ケトンとの反応をトリチル塩の存在下で行うことで、1,5-
ジカルボニル化合物 が得られるマイケル付加反応を報告しており、この反応では中間体としてシリルエ ノールエーテルが単離されている。(
スキーム1-11)
。9c)11
スキーム
1-11.
トリチル塩を触媒として用いたマイケル付加反応アルドール反応の一例としては、トリチル塩を触媒量用いてベンズアルデヒドと シリルエノールエーテルとの反応から、アルドール生成物を得る反応が報告されて いる
(
スキーム1-12)
。9e)スキーム
1-12.
トリチル塩を触媒として用いたアルドール反応この反応では、ジアステレオマーの関係にあるアルドール生成物が得られており、
エリトロ型とトレオ型の生成比はトリチルの対陰イオンによって大きく異なるこ とがわかっている。また、
Chen
らはトリアリールカルベニウムを用いて向山アル ドール反応をエナンチオ選択的に達成している。10) 最近では、Franzén
らによって、トリチルカチオンを触媒量用いて様々な有機分子変換反応が展開されている。11) 例えば、ディールス・アルダー反応では
, -
不飽和アルデヒドがトリチルカチオン に活性化されることで、中間体A
とジエンが反応して中間体B
を形成し、ディー ルス・アルダー付加物の生成を伴い、触媒が復活する(
スキーム1-13)
。11a)12
スキーム
1-13.
トリチルカチオンを触媒として用いたディールス・アルダー反応しかしながら、触媒として用いられるカルボカチオンはトリチルカチオンがほと んどである。トリチルカチオンよりも活性の高いカルボカチオンはその不安定性か ら、発生してもすぐに分解してしまうため、触媒として利用することが難しいと考 えられる。
1-3-2 第二級および第一級カルボカチオンの発生と反応性
第三級カルボカチオンよりも、不安定な第二級および第一級カルボカチオンでは、
一般的には触媒としては利用されていないが、発生させること自体は可能であり、
第二級カルボカチオンについては反応性についても研究されている。アルコキシド 塩と四塩化チタンあるいは塩化鉄
(III)
との反応により、第二級カルボカチオンを発 生させることができる。12) 例えば、リチウムアルコキシド化合物と四塩化チタン との反応では、第二級カルボカチオンが発生し、末端アルキンと反応してビニルカ チオンを生成し、ビニルカチオンはクロリドで捕捉された形で最終生成物として得 られる(
スキーム1-14)
。12a)13
スキーム
1-14.
四塩化チタンを用いた第二級カルボカチオンの発生また、第二級カルボカチオンを発生させる方法としては、スキーム
1-15
に示す ようなカチオンプール法が採用されている。この方法は低温下で電気化学的な酸化 を行うことで、炭素-水素結合の切断を促進させる方法であり、ジアリールメタン から第二級カルボカチオンを発生させ、溶液中に蓄積させることが可能である。13)スキーム
1-15.
カチオンプール法による第二級カルボカチオンの発生このようなカチオンプール法で発生させた第二級カルボカチオンは基質との反応 性が検討されている
(
スキーム1-16)
。第二級カルボカチオンと芳香族化合物との反 応では求電子置換生成物が得られている。ケテンシリルアセタールとの反応では不 飽和結合部位への求電子攻撃を可能とする。また、アリルシランとの反応ではアリ ル化生成物が得られる。14
スキーム
1-16.
第二級カルボカチオンの反応性同様の電気化学的な酸化による方法で第一級カルボカチオンを発生させることも 可能である。
DMSO
中でアニソールの電気化学的酸化を行うことで、ベンジルカ チオンが発生し、DMSO
による捕捉を受けた形で生成が確認されている(
スキーム1-17)
。14)スキーム
1-17.
電気化学的酸化による第一級カルボカチオンの発生この反応系で発生するベンジルカチオンはそのままの状態では安定に存在せず、
溶媒である
DMSO
が配位した形をとる。このように、カルボカチオンの安定性が 低下することで高い反応性を示すことが期待されるが、発生および保存が難しくな り、触媒としての利用は困難である。15 1-4 アルキニルカチオンの発生
カルボカチオンの中でも、
sp
混成をとるエチニルカチオンは特に不安定であり、その発生は困難であると考えられる。図
1-6
にメチルカチオンを基準としたカルボ カチオンの安定性を示す。図
1-6.
メチルカチオンを基準としたカルボカチオンの安定性15)エチニルカチオンは、メチルカチオンよりも約
55 kcal/mol
不安定であるといえ、これはより電気陰性である
sp
混成軌道上に正電荷が存在するためと考えられる。Helwig
らは、アルキニルジアゾニウム塩[PhCCN
2](SbCl
5Ts)
を用いても、アルキ ニルカチオンは発生しないことを報告している。16) このように、優れた脱離能を 持つジアゾニオ基(N
2+)
を用いても、通常の条件下では、アルキニルカチオンは発 生しない。Hanack
らは、アルキニル基の水素をトリチウムに変換した化学種を用いて、崩
壊によるアルキニルカチオンの発生に成功している
(
スキーム1-18)
。17)スキーム
1-18.
アルキニルカチオンの発生この反応では、トリチウムから、ベータ粒子と反電子ニュートリノが放出される ことによって23
He
が発生している。炭素とヘリウムの結合は非常に不安定である16
ため、瞬時に開裂することでエチニルカチオンが生成し、ベンゼンとの反応によっ て求電子置換生成物へと変換される。アルキニルカチオンはこれまで、質量スペク トルによってのみ観測されているが18)、このように反応系中での発生例は極めて 珍しく、その反応性研究は皆無といえる。
一般的に、電子不足な有機フラグメントは、金属へ配位することで逆供与を受 けて、電子的に安定化する。電子不足な陽イオン性化学種は、金属骨格からの 逆 供与を利用することで、安定化されることが期待できる。
このような効果に期待すると、エチニルカチオンも多金属骨格へ架橋配位させる ことで発生させることが可能ではないかと考えられる。アルケンおよびアルキンは 金属から
逆供与を受ける(
図1-7)
。図
1-7.
金属とアルケンまたはアルキンとの相互作用図
1-7
に示すように、金属に配位したアルキンは逆供与を受けることで安定化 されており、これまでにアルキンが金属に架橋配位している錯体あるいはクラスタ ーの合成例は、多数報告されている。17
1-5 金属と結合したアルキンクラスター
アルキンあるいはアルキン由来の配位子は金属と様々な相互作用をすることが 報告されている (図 1-8)。19)
図 1-8. 金属とアルキンの結合様式
図 1-8に示すように、アルキンの金属への結合様式は様々なものが存在しており、
それはアルキンの置換基や金属の数によって異なり、アルキンの C–C結合長や、
金属とアルキン炭素の M–C結合長は幅広い範囲をとる。異なる数の金属と相互作 用したアルキンは、それぞれ性質が異なり、これまでに多種多様な遷移金属アルキ ン錯体が合成されている。しかしながら、エチニルカチオンあるいはアルキニルカ チオンの前駆体となりうる、良い脱離基を置換基としてもつアルキンが配位した遷 移金属錯体およびクラスターの例は極めて少ない。
これまでに報告されているハロアルキンが遷移金属に架橋配位した錯体として
は 、 AgBF4の 存 在 下 、 [Mo2(CO)6(η–C5H5)2]と PhCCBr と の 反 応 に よ り 、 [Mo(η2–PhCCBr)2(CO)(η–C5H5)](BF4)が得られることが Beevorらによって報告され
ている(スキーム 1-19)。20)
18
スキーム 1-19. [Mo(η2–PhCCBr)2(CO)(η–C5H5)](BF4)の合成
また、Langらは[Co2(CO)8]と BrCCR (R =nPr,nBu, SiMe3, Ph)との反応により、
[Co2(η2–BrCCR)(CO)6]が得られることを報告している(スキーム 1-20)。21)
スキーム 1-20. [Co2(η2–BrCCR)(CO)6]の合成
ハロアルキンが遷移金属に架橋配位したクラスターの報告例は1例のみであり、
[Fe3(CO)9(3–CF)2]と [Cp*Co(CO)2] (Cp* = C5(CH3)5)と の 反 応 に よ っ て 、 [Cp*CoFe3(CO)9(4–η2–FCCF)]が得られることが Lentzらによって報告されている (スキーム 1-21)。22)
スキーム 1-21. [Cp*CoFe3(CO)9(4–η2–FCCF)]の合成
このような、ハロアルキンが遷移金属に配位した錯体の合成例は極めて少なく、
クラスターの合成例は1例のみであり、アルキニルカチオンに相当する [RCC]+フ ラグメントの発生を試みた例はない。
19
1-6 ケイ素の陽イオン種および不飽和化学種について 1-6-1 シリルカチオンの発生と反応性
14族元素で炭素の下に位置するケイ素は電気陰性度が低いため、+性が炭素よ り大きい。したがって、より電気陰性度の高い原子との結合が強くなり、実際に結 合開裂エネルギーを炭素と比較するとケイ素-水素結合は炭素-水素結合よりも 弱くなるが、電気陰性度の高いハロゲンとの結合ではいずれもケイ素の方が強い結 合をつくる(図 1-9)。
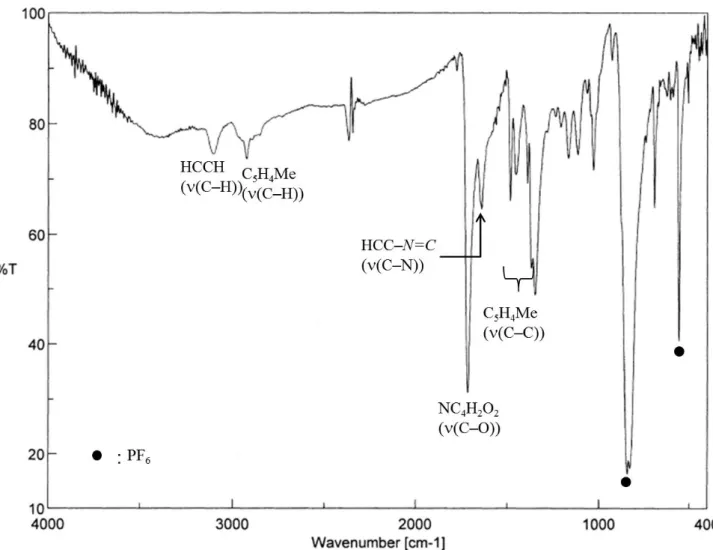
図 1-9. 結合開裂エネルギー(kJ/mol)23)
このため、ケイ素陽イオン種であるシリルカチオンの発生は、トリチルカチオン を用いた方法で行われており、ヒドロシランとトリチル塩との反応により、シリル カチオンを発生させることができる(スキーム 1-22)。24)
スキーム 1-22. トリチルカチオンによるシリルカチオンの発生
このようなヒドロシランとトリチルカチオンとの反応ではシランのケイ素上の 水素がトリチルカチオンに引き抜かれ、トリフェニルメタンの生成を伴いながら、
シリルカチオンが発生する。この反応では、弱いケイ素-水素結合が切れて、より 強い炭素-水素結合を形成することを駆動力に進行していると考えられ、同様の手 法により、シリルカチオンを発生させた例が多数報告されている。また、シリルカ チオンは、図 1-7からも明らかなとおり、ケイ素-フッ素結合が熱力学的に安定で
20
あるため、容易にフッ化物イオンを引き抜くことが可能である。例えば、カルボラ ンを対陰イオンとしてもつトリエチルシリルカチオンをフルオロベンゼン中で加 熱することにより、アリールカチオンがカルボランに捕捉された生成物が得られて いる
(
スキーム1-23)
。25)スキーム
1-23.
シリルカチオンによる脱フッ素化反応水素との結合とは逆に、フッ素との結合ではケイ素の方が炭素よりも強く安定であ るため、フルオロベンゼンとの反応ではシリルカチオンがフッ化物イオンを引き抜 き、フルオロシランの生成とアリールカチオンの発生が起こる。このように、シリ ルカチオンはフッ素の引き抜きに適しており、脱フッ素化反応における触媒として も利用されている。
Ozerov
らは、トリエチルシランとトリチルカチオンとの反応 から、トリエチルシリルカチオンを発生させ、これを触媒として用いることで、C(sp
3)–F
結合の切断とC(sp
3)–H
結合の形成反応に適用している(
スキーム1-24)
。26)スキーム
1-24.
シリルカチオンを触媒として用いた脱フッ素化反応これは、シリルカチオンを用いてトリフルオロメチルベンゼンのフッ素をすべて 水素に交換する反応であり、この反応では、まずヒドロシランの水素がトリチルに 引き抜かれることで、シリルカチオンが発生し、これがメチル炭素上のフッ素を引 き抜くとカルボカチオンが発生し、カルボカチオンが再度、ヒドロシランから水素
21
を引き抜くことで、再びシリルカチオンが発生するといった過程を繰り返すことで、
炭素フッ素結合がすべて炭素水素結合へと変換される
(
スキーム1-25)
。スキーム
1-25.
シリルカチオンを触媒とする脱フッ素水素化反応の機構以上のように、シリルカチオンはフッ素の引き抜きに適しており、主に炭素-フ ッ素結合開裂を引き起こし、ケイ素-フッ素結合の形成させる反応が主に報告され ている。
22
1-6-2 不飽和結合を有するケイ素化合物
元素周期表の第
2
周期にある炭素、窒素、酸素などは、二重結合や三重結合など の不飽和結合が安定に形成される。例えば、炭素-酸素二重結合を有するアセトン は安定な化合物であり、工業的にも学術的にも幅広く利用されている。一方、周期 表で炭素の下に位置するケイ素では不飽和結合が極めて不安定である。ケイ素は炭 素に比べて原子が大きいため、p
軌道の重なりが小さく、
結合のエネルギー(C=C 65 kcal/mol, Si=Si 25 kcal/mol)
が小さいためである。27) 例えば、二酸化炭素(O=C=O)
では安定な気体分子として存在するのに対して、ケイ素の酸化物ではポリマー状(Si-O-Si-O-Si…)
となり、O=Si=O
のような二重結合を含む形では存在しない。この不安定性ゆえに、
1980
年代までは二重結合則(double bond rule)
28)という「第3
周期 元素以降の元素について、同一元素同士または他の元素との間で、二重結合や三重 結合を形成できない」といった法則が多くの教科書に記載されていた。1981
年に安定な炭素-ケイ素二重結合を有するシレン化合物がBrook
らにより 初めて合成された(
スキーム1-26)
。29)スキーム
1-26. Brook
らによるシレンの合成同じく、
1981
年にWest
らによって、ケイ素-ケイ素二重結合を有するジシレン が合成され(
スキーム1-27)
30)、その翌年に正宗らは別な合成方法でジシレンを得て いる(
スキーム1-28)
。31)23
スキーム 1-27. Westらによるジシレンの合成
スキーム 1-28. 正宗らによるジシレンの合成
このように、シレンおよびジシレンはいずれも嵩高い置換基を導入した立体保護 により、二重結合を安定化させており、ポリマー化を防いでいる。しかしながら、
これらの二重結合部位は極めて不安定であり反応性が高く、スキーム 1-29に示す とおりジシレンは水や酸素と容易に反応することがわかっている。32)
スキーム 1-29. ジシレンと水および酸素との反応性
24
このような嵩高い置換基によって安定化されたジシレンの合成については現在、
多数報告されている。一方、ケイ素と酸素や硫黄などの
16
族元素との二重結合化 合物が安定な形で単離された例は少ない。その理由としては、ケイ素上にしか置換 基を導入できないため、安定化に適した置換基が見つけにくかったためである。これまでにケイ素-酸素二重結合をもつ化合物は塩基によって安定化された形 で単離された例が多数報告されているが、33)ケイ素中心が四配位で四面体型構造で あり、二重結合性は失われていると考えられる。最近、別な方法によって、安定化 したシラノンの合成方法が報告された
(
スキーム1-30)
。スキーム
1-30.
遷移金属シラノン錯体の合成この反応では、フルオロベンゼン中、ブロモシリリデン錯体と
Na[B(Ar
F)
4] (Ar
F= C
6H
3(CF
3)
2)
との反応によって、ブロミドが引き抜かれて、クロミウムシリリジン錯 体[(
5-C
5Me
5)(CO)2Cr≡Si(SIdipp)][B(Ar
F)
4] (SIdipp = 1,3-
ビス(2,6-
ジイソプロピルフ ェニル)
イミダゾリジン-2-
イリデン)
が得られており、ここにCO
を加えることで、メタロシリレン錯体が生成する。これと
N
2O
との反応において、窒素の脱離を伴 い、メタロシラノン錯体が得られている。34)この錯体ではシラノンが遷移金属であ るクロムに末端配位しており、ケイ素中心は平面三角系の構造である。このように、ケイ素-酸素二重結合が保持されている例が
2014
年に初めて報告された。その翌 年には、タングステンにSi=O
が配位したシラアルデヒド錯体が合成されており、ケイ素および酸素原子がタングステンに2
-
型で配位した形が初めて得られている(
スキーム1-31)
。35)この錯体は、THF
中でアニオン性のシリレン錯体にピリジン-N-
オキシドを作用させることで、室温で速やかに得られる。
25
スキーム
1-31.
遷移金属シラアルデヒド錯体の合成次に、シラノンの酸素原子を同族の重原子である硫黄に置き換えたシランチオン について述べる。シランチオンのケイ素-硫黄二重結合はシラノンのケイ素-酸素 二重結合と比較して、熱力学的にも速度論的にも安定であることが理論計算によっ て明らかとされている。36) しかしながら、シランチオンの合成例は少なく、その 反応性についてはほとんど知られていないといえる。
1989
年にCorriu
らは5
配位 のシランと硫黄あるいは二硫化炭素とを反応させることによって、熱力学的に安定 化したシランチオンの合成に成功している(
スキーム1-32)
。37)スキーム
1-32.
熱力学的に安定化されたシランチオンの合成26
この方法によって得られたシランチオンのケイ素中心は窒素上の孤立電子対が 供与された四配位四面体型構造であり、ケイ素-硫黄結合は真の二重結合とはいえ ない。また、速度論的に安定化されたシランチオンはこれまでに時任および岡崎ら によって報告されている。立体保護基として非常に嵩高い置換基である Tbt (2,4,6- トリス{ビス(トリメチルシリル)メチル}フェニル)および Tip (2,4,6-トリイソプロピ ルフェニル)をケイ素上に導入したジブロモシランとリチウムナフタレニドを作用 させた後、硫黄を加えることでテトラチオシロランを得ている。これに脱硫試剤で あるトリフェニルホスフィンを作用させることで、シランチオンが単離されている (スキーム 1-33)。38)
スキーム 1-33. 熱力学的に安定化されたシランチオンの合成
また、この合成では、ケイ素上の置換基に Tbt基と Mes 基(Mes = 2,4,6-トリメチ ルフェニル)を用いた系では立体保護が不十分であることがわかっており、発生し たシランチオンが二量化することが明らかとなっている(スキーム 1-34)。
スキーム 1-34. シランチオンの二量化
シランチオンはこれまでに、非常に嵩高い置換基をケイ素上に導入することでの み、単離されている。一方で、遷移金属に安定化されたシランチオンはこれまでに 報告例が無く、メチル基などの一般的な置換基を有するシランチオンの合成は行わ れていない。
27
1-7 本研究の目的
これまでに当研究室では、四面体型構造をとる四鉄上に
4
つカルボニル配位子が 架橋した四鉄キュバン型テトラ(
カルボニル)
クラスター[(η
5–C
5H
4Me)
4Fe
4(CO)
4]
の 合成に成功している。[(η
5–C
5H
4Me)
4Fe
4(CO)
4]
にLiAlH
4を作用させることで、カル ボニル配位子の還元的カップリング反応により、四鉄の上下にアセチレン配位子が 架橋した四鉄ビス(
アセチレン)
クラスター[(η
5-C
5H
4Me)
4Fe
4(HCCH)
2](PF
6) (1)
へ変換 されることが報告されている(
スキーム1-35)
。39)スキーム
1-35.
四鉄ビス(
アセチレン)
クラスターの合成さらに、
1
とNBS
との反応により、アセチレン水素の選択的な臭素化に成功し ており、ブロモアセチレンクラスター[(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–Br)](PF
6) (2)
を 定量的に得ることに成功している(
スキーム1-36)
。40)スキーム
1-36.
ブロモアセチレンクラスターの合成得られた
2
は各種求核剤との反応により置換されたアセチレンクラスターへと 変換されることが報告されている。2
の[HCC–Br]
フラグメントは、四鉄に配位する ことで逆供与を受けて、電子的に安定化していると考えられる。2
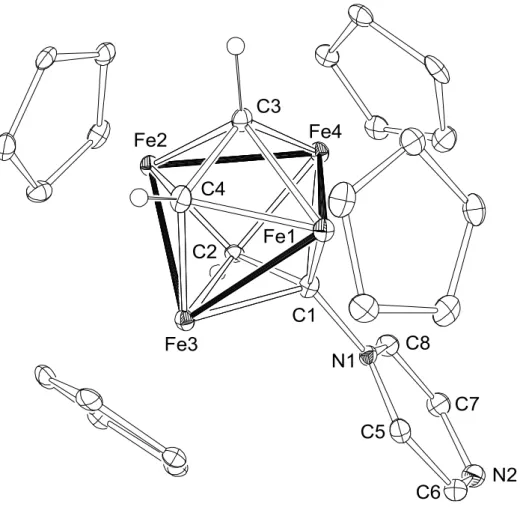
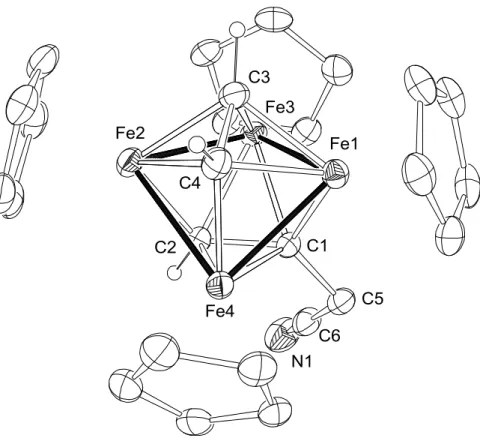
から臭化物イ オンが解離すると、図1-10
に示すような四鉄上でのエチニルカチオンの発生が期 待できる。28
図
1-10.
四鉄上でのエチニルカチオンの発生本研究では、図
1-10
に示した四鉄上において、エチニルカチオンを発生させ、この点をルイス酸反応場として用いた基質活性化に取り組んだ。様々な基質との反 応を行い、得られた知見に基づき、そのルイス酸性を評価した。
一方、周期表で炭素の下に位置するケイ素では、不飽和結合部位を活性中心とし て用いる。ケイ素は炭素に比べて原子半径が大きく、
p
軌道を十分重ねあわせるこ とができないため、エネルギー準位の低い空の*軌道をもち、ルイス酸性が高いと 考えられる。中でも、ケイ素と16
族元素との二重結合化合物は、ケイ素上にしか 置換基を導入できないため、合成難易度が高く、報告例が少ない。特に、ケイ素と 硫黄との間に二重結合をもつシランチオンはこれまでに非常に嵩高い置換基を用 いた化合物に限られている。本研究では、シランチオンを立体保護基により安定化 するのではなく、遷移金属に配位させ、電子的に安定化した形で合成および単離を 目指した。本研究では、スキーム
1-37
に示した方法により、シランチオンの二重結合部位 の
電子がイリジウムに供与された錯体の合成および単結晶X
線構造解析に成功し た。合成したシランチオン錯体は電気陽性なケイ素部位を有しており、この点をル イス酸反応場としてとらえ、その反応性についても調査した。スキーム
1-37.
イリジウムに配位したシランチオン錯体の合成29
参考文献
1) a) K. Ishihara, H. Yamamoto, Eur. J. Org. Chem. 1999, 527. b) C. Friedel, J. M. Crafts, C. R. Hebd, Seances Acad. Sci. 1877, 84, 1392. c) C. Friedel, J. M. Crafts, C. R. Hebd, Seances Acad. Sci. 1877, 84, 1450. d) J. Kischel, I. Jovel, K. Mertins, A. Zapf, M.
Beller, Org. Lett. 2006, 8, 19. e) L. Ackermann, Organometallics 2003, 22, 4367. f) L.
Ackermann, L. T. Kaspar, J. Org. Chem. 2007, 72, 6149. g) K. Ishihara, M. Kubota, H.
Kurihara, H. Yamamoto, J. Am. Chem. Soc. 1995, 117, 4413.
2) K. Maruoka, H. Imoto, S. Saito, H. Yamamoto, J. Am. Chem. Soc. 1994, 116, 4131.
3) H. Guo, E. Herdtweck, T. Bach, Angew. Chem. Int. Ed. 2010, 49, 7782.
4) G. A. Olah, J. Lukas, J. Am. Chem. Soc. 1967, 89, 4739.
5) a) G. A. Olah, E. B. Baker, J. C. Evans, W. S. Tolgyesi, J. S. McIntyre, I. J. Bastien, J.
Am. Chem. Soc. 1964, 86, 1360. b) D. M. Brouwer, E. L. Mackor, Proc. Chem. Soc.
1964, 147. c) G. M. Kramer, J. Am. Chem. Soc. 1969, 91, 4819.
6) T. Kato, C. A. Reed, Angew. Chem. Int. Ed. 2004, 43, 2908.
7) R. R. Naredla, D. A. Klumpp, Chem. Rev. 2013, 113, 6905.
8) D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, V. V.
Speybroeck, M. Waroquier, J. F. Haw, Angew. Chem. Int. Ed. 2008, 47, 5179.
9) a) T. Mukaiyama, S. Kobayashi, M. Murakami, Chem. Lett. 1984, 1759. b) T.
Mukaiyama, S. Kobayashi, M. Murakami, Chem. Lett. 1985, 447. c) S. Kobayashi, M.
Murakami, T. Mukaiyama, Chem. Lett. 1985, 953. d) T. Mukaiyama, H. Nagaoka, M.
Murakami, M. Ohshima, Chem. Lett. 1985, 977. e) S. Kobayashi, M. Murakami, T.
Mukaiyama, Chem. Lett. 1985, 1535. f) S. Kobayashi, S. Matsui, T. Mukaiyama, Chem.
Lett. 1988, 1491. g) T. Mukaiyama, H. Akamatsu, J. S. Han, Chem. Lett. 1990, 889.
10) a) C.-T. Chen, S.-D. Chao, K.-C. Yen, C.-H. Chen, I.-C. Chou, S.-W. Hon, J. Am. Chem.
Soc. 1997, 119, 11341. b) C.-T. Chen, S.-D. Chao, K.-C. Yen, Synlett 1998, 924.
11) a) J. Bah, J. Franzén, Chem. Eur. J. 2014, 20, 1066. b) J. Bah, V. R. Naidu, J. Teske, J.
Franzén, Adv. Synth. Catal. 2015, 357, 148. c) V. R. Naidu, J. Bah, J. Franzén, Eur. J.
Org. Chem. 2015, 1834.
12) a) M.-L. Yao, T. R. Quick, Z. Wu, M. P. Quinn, G. W. Kabalka, Org. Lett. 2009, 11, 2647. b) G. W. Kabalka, M.-L. Yao, S. Borella, L. K. Goins, Organometallics 2007, 26, 4112.
13) M. Okajima, K. Soga, T. Nokami, S. Suga, J. Yoshida, Org. Lett. 2006, 8, 5005.
14) Y. Ashikari, T. Nokami, J. Yoshida, Org. Lett. 2012, 14, 938.
30
15) F. A. Carey, R. J. Sundberg, In Advanced Organic Chemistry Part A: Stracture and Mechanism Fifth Edition, Springer, New York, 2007.
16) a) R. Helwig, M. Hanack, Chem. Ber. 1985, 118, 1008. b) R. Glaser, J. Am. Chem. Soc.
1987, 109, 4237.
17) M. Hanack, J. Vermehren, M. Speranza, J. Am. Chem. Soc. 1988, 110, 1298.
18) a) P. C. Burgers, J. L. Holmes, A. A. Mommers, J. E. Szulejko, J. Am. Chem. Soc. 1984, 106, 521. b) S. J. Huang, F. Di Battista, D. Picker, G. Wilson, J. Org. Chem. 1975, 40, 124.
19) E. Sappa, A. Tiripicchio, P. Braunstein, Chem. Rev. 1983, 83, 203.
20) R. G. Beevor, M. Green, A. G. Orpen, I. D. Williams, J. Chem. Soc., Dalton Trans.
1987, 1319.
21) H. Lang, G. Rheinwald, U. Lay, L. Zsolnai, G. Huttner, J. Organomet. Chem., 2001, 634, 74.
22) D. Lentz, H. Michael, Angew. Chem. Int. Ed. Engl. 1988, 27, 845.
23) Speight, J. G. Lange’s Handbook of Chemistry Sixteenth Edition, McGraw-Hill.
24) J. B. Lambert, S. Zhang, J. Chem. Soc., Chem. Commun. 1993, 383.
25) S. Duttwyler, C. Douvris, N. L. P. Fackler, F. S. Tham, C. A. Reed, K. K. Baldridge, J.
S. Siegel, Angew. Chem. Int. Ed. 2010, 49, 7519.
26) V. J. Scott, R. Ҫelenligil-Ҫetin, O. V. Ozerov, J. Am. Chem. Soc. 2005, 127, 2852.
27) a) M. W. Schmidt, P. N. Truong, M. S. Gordon, J. Am. Chem. Soc. 1987, 109, 5217. b) P. v. R. Schleyer, D. Kost, J. Am. Chem. Soc. 1988, 110, 2105.
28) K. S. Pitzer, J. Am. Chem. Soc. 1948, 70, 2140.
29) A. G. Brook, F, Abdesaken, B. Gutekunst, G. Gutekunst, R. K. Kallury, Chem.
Commun. 1981, 191.
30) R. West, M. J. Fink, J. Michl, Science 1981, 214, 1343.
31) S.Masamune, Y. Hanzawa, S. Murakami, T. Bally, J. F. Blount, J. Am. Chem. Soc. 1982, 104, 1150.
32) a) A. J. Millevolte, D. R. Powell, S. G. Johnson, R. West, Organometallics 1992, 11, 1091. b) D. N. Roark, G. J. D. Peddle, J. Am. Chem. Soc. 1972, 94, 5837.
33) a) Y. Xiong, S. Yao, M. Driess, J. Am. Chem. Soc. 2009, 131, 7562. b) Y. Xiong, S. Yao, R. Müller, M. Kaupp, M. Driess, J.Am. Chem. Soc. 2010, 132, 6912. c) Y. Xiong, S.
Yao, R. Müller, M. Kaupp, M. Driess, Nature Chem. 2010, 2, 577. d) R. S. Ghadwal, R.
Azhakar, H. W. Roesky, K. Pröpper, B. Dittrich, S. Klein, G. Frenking, J. Am. Chem.
31
Soc. 2011, 133, 17552. e) T. Muraoka, K. Abe, Y. Haga, T. Nakamura, K. Ueno, J. Am.
Chem. Soc. 2011, 133, 15365. f) Y. Xiong, S. Yao, M. Driess, Angew. Chem. 2013, 125, 4398; Angew. Chem. Int. Ed. 2013, 52, 4302. g) R. Rodriguez, T. Troadec, D. Gau, N.
Saffon-Merceron, D. Hashizume, K. Miqueu, J.-M. Sotiropoulos, A. Baceiredo, T. Kato, Angew. Chem. 2013, 125, 4522; Angew. Chem. Int. Ed. 2013, 52, 4426. h) R.
Rodriguez, D. Gau, T. Troadec, N. Saffon-Merceron, V. Brandchadell, A. Baceiredo, T.
Kato, Angew. Chem. 2013, 125, 9150; Angew. Chem. Int. Ed. 2013, 52, 8980.
34) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev, G. Schnakenburg, Angew. Chem.
Int. Ed. 2014, 53, 565.
35) T. Fukuda, H. Hashimoto, S. Sakaki, H. Tobita, Angew. Chem. Int. Ed. 2016, 55, 188.
36) T. Kudo, S. Nagase, Organometallics 1986, 5, 1207.
37) P. Arya, J. Boyer, F. Carré, R. Corriu, G. Lanneau, J. Lapasset, M. Perrot, C. Priou, Angew. Chem. Int. Ed. Engl. 1989, 28, 1016.
38) a) H. Suzuki, N. Tokitoh, S. Nagase, R. Okazaki, J. Am. Chem. Soc. 1994, 116, 11578.
b) H. Suzuki, N. Tokitoh, R. Okazaki, S. Nagase, M. Goto, J. Am. Chem. Soc. 1998, 120, 11096.
39) a) M. Okazaki, T. Ohtani, S. Inomata, N. Tagaki, H. Ogino, J. Am. Chem. Soc. 1998, 120, 9135. b) M. Okazaki, T. Ohtani, M. Takano, H. Ogino, Inorg. Chem. 2002, 41, 6726. c) M. Okazaki, T. Ohtani, M. Takano, H. Ogino, Organometallics 2004, 23, 4055.
d)
大谷健夫, 修士論文(
東北大学) ,
平成11
年。40) a) M. Takano, M. Okazaki, H. Tobita, J. Am. Chem. Soc. 2004, 126, 9190. b) 高野正
人, 修士論文(
東北大学) ,
平成14
年。32
第二章
四鉄骨格に架橋配位したエチニルカチオンの発生と性質
33
2-1 序
アセチレンから水素がヒドリドとして外れた化学種であるエチニルカチオンは、
非常に優れた脱離基であるジアゾニオ基を用いても発生しない ( スキーム 2-1) 。
スキーム 2-1. ジアゾニオ基を用いたエチニルカチオンの発生
一般的に、電子不足な有機フラグメントは、金属へ配位することで 逆供与を受け て、安定化する。この効果を期待して、四鉄上でエチニルカチオンの発生を試みた。
第 一 章 で 述 べ た よ う に 、 四 鉄 ビ ス ( ア セ チ レ ン ) ク ラ ス タ ー [(η
5-C
5H
4Me)
4Fe
4(HCCH)
2](PF
6) (1) は NBS との反応により、アセチレン水素を選択的 に臭素化することができ、 [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–Br)](PF
6) (2) を定量的に合 成することが可能である
1)。 2 のように脱離基としてブロモ基がアセチレンに導入さ れた化学種では、四鉄によって安定化されたエチニルカチオンの発生が期待される
(スキーム 2-2 )。
スキーム 2-2. 四鉄上でのエチニルカチオンの発生
本章では、 2 から銀塩によって臭化物イオンを引き抜き、エチニルカチオンの発
生を検討した。さらに、発生したエチニルカチオンに塩基を配位させることによっ
てシュレンクテクニックで取り扱い可能な化学種へと変換し、これをエチニルカチ
オンの合成等価体としてとらえ、反応性を検討した。
34
2-2 実験
操作
特に断らない限り、実験はすべて充分に乾燥した器具を用いて行い、窒素雰囲気 下あるいはアルゴン雰囲気下のグローブボックス中で行った。
試薬
[(η
5-C
5H
4Me)
4Fe
4(HCCH)
2](PF
6) (1)
2)、 [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCCBr)](PF
6) (2)
1)は文献記載の方法で合成した。ピリジンは水酸化ナトリウムで乾燥した後、蒸留し、
モレキュラーシーブス 4A 存在下で保存したものを用いた。 tert- ブチルブロミドはモ レキュラーシーブス 4A で脱水したものを用いた。他の試薬は市販のものを用い、
特に精製せずに使用した。
溶媒
アセトニトリルは水素化カルシウムから使用直前に窒素雰囲気下で蒸留して用い た。 NMR スペクトル測定に使用したアセトニトリル -d
3は水素化カルシウムから使 用直前に窒素雰囲気下で蒸留したのち、モレキュラーシーブス 4A で乾燥して用い た。他の溶媒は市販の脱水溶媒を用いた。
測定機器
1
H NMR 、
13C NMR スペクトルは日本電子製 JNM–ECA 500 を用いて測定した。質
量スペクトルは島津高速液体クロマトグラフ質量分析計 LCMS–2010 EV を用いて測
定した。赤外吸収スペクトルは日本分光製 FT/IR–4100 を用いて測定した。単結晶 X
線構造解析は(株)リガク製 R–AXIS RAPIDII を用いて行なった。
35
2-2-1 [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–NCCH
3)](PF
6)
2(3) の合成
グローブボックス中で、ナスフラスコに [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCCBr)](PF
6) (2) (0.184 g, 0.226 mmol) 、 AgPF
6(68 mg, 0.27 mmol) を加えたのち、アセトニトリル (10 mL) 溶液とし、反応溶液を室温で 30 分攪拌した。溶液をろ過したのち、ろ液から減 圧下揮発成分を留去した。残渣をジクロロメタンで洗浄し減圧下乾燥することで、
暗緑色の固体として 3 を得た。 収量 0.160 g (0.174 mmol) 。 収率 77% 、 分子量 921.91 。
3 の各種スペクトルデータと元素分析の結果を表 2-1 に示す。
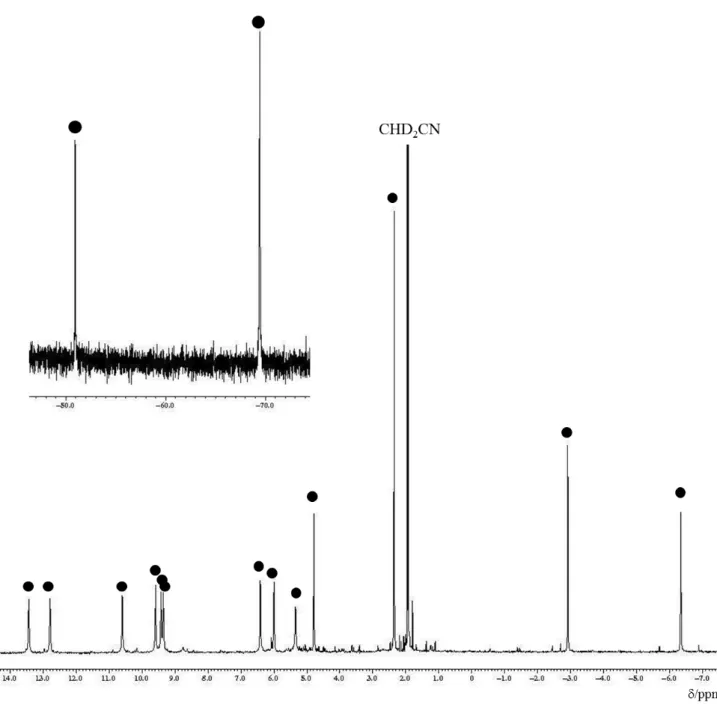
36
表 2-1. [(η
5-C
5H
4Me)
4Fe
4(HCCH)(HCC–NCCH
3)](PF
6)
2(3) の各種スペクトルの測 定値と元素分析の結果
元素分析
組成式 C
30H
34NFe
4P
2F
12測定値 / % ( 計算値 / %)
C : 38.57 (39.08) H : 4.053 (3.72).
1
H NMR スペクトル (500 MHz, CD
3CN)
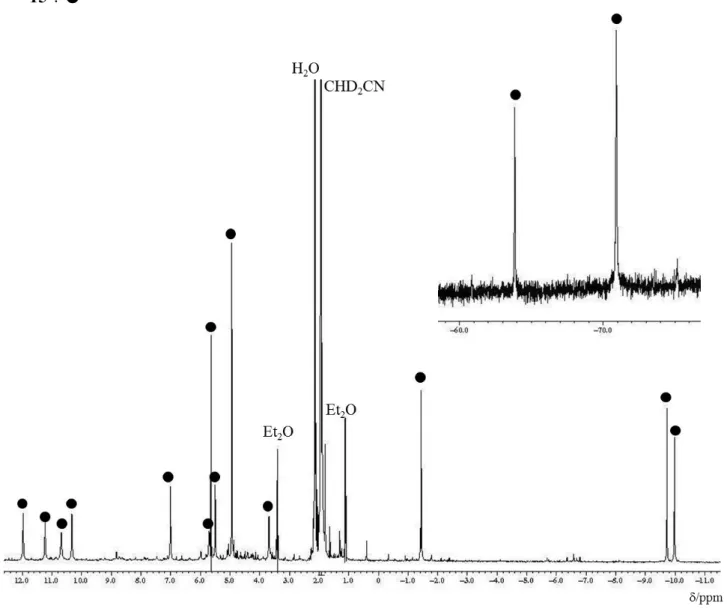
3)δ / ppm –73.4 (s, 2H, HCCH)
–54.0 (s, 1H, HCC–NCCH
3) –4.5, –4.0 (s, 3H×2, 2C
5H
4Me) 1.3 (s, 6H, 2C
5H
4Me)
5.0, 6.7, 8.0, 8.9, 9.5, 10.9, 11.5, 11.5 (s, 2H×8, 4C
5H
4Me).
2
H NMR スペクトル (77 MHz, CD
3CN) δ / ppm –4.8 (s, 3D, HCC–NCCD
3).
13
![図 1-4. Molecular structure of [Me 3 C][CHB 11 Me 5 Cl 6 ] ・ CH 2 Cl 2 with 50% thermal ellipsoids](https://thumb-ap.123doks.com/thumbv2/123deta/5923654.2055534/15.892.261.614.118.729/図-molecular-structure-chb-cl-ch-thermal-ellipsoids.webp)