リコモジュリン点滴静注用 12800
トロンボモデュリン アルファ(遺伝子組換え)
第 2 部 CTD の概要
2.5 臨床に関する概括評価
2.5 項 略号一覧(1/2)
略号 省略していない表現
AL-P alkaline phosphatase(アルカリホスファターゼ) AML acute myeloid leukemia(急性骨髄性白血病) AML(M3)又は
APL
acute promyelocytic leukemia(急性前骨髄球性白血病)
AML(M3 以外) acute promyelocytic leukemia(急性前骨髄球性白血病)以外の AML APC activated protein C(活性化プロテイン C)
APTT activated partial thromboplastin time(活性化部分トロンボプラスチン時間) ATIII antithrombin III(アンチトロンビン III)a)
ATL adult T-cell leukemia(成人 T 細胞白血病)
ATRA all-trans retinoic acid(オールトランスレチノイン酸)
AUC area under the plasma concentration-time curve(血漿中濃度時間曲線下面積) AUCinf area under the plasma concentration-time curve from zero to infinite(無限大時間まで
の血漿中濃度時間曲線下面積) C0 initial concentration(投与初期濃度)
C5min plasma concentration at 5 min after administration(投与 5 分後の血漿中濃度) Cmax maximum plasma concentration(最高血漿中濃度)
ChE cholinesterase(コリンエステラーゼ)
Cl クロール
CLtot total clearance{血漿クリアランス(全身クリアランス)} CV coefficient of variation(変動係数)
CYP cytochrome P450(チトクロム P450)
DIC disseminated intravascular coagulation(汎発性血管内血液凝固症)b) ELISA enzyme-linked immunosorbent assay(酵素固定化免疫測定) FAS full analysis set
FDP fiblin/fibrinogen degradation products(フィブリン・フィブリノゲン分解産物) GCP good clinical practice
GOT glutamic-oxaloacetic transaminase GPT glutamic-pyruvic transaminase IC50 50% inhibitory concentration(50%阻害濃度) iv intravenous(静脈内) K カリウム LDH lactate dehydrogenase(乳酸デヒドロゲナーゼ) a) アンチトロンビン III の正式名称としては、アンチトロンビン(AT)が推奨されているが、本 申請資料では、試験報告書で使用される用語が ATIII であるため、混同を避ける目的でアン チトロンビン III(ATIII)の用語を用いた。 b) 一般に、DIC を示す疾患名としては「播種性血管内血液凝固症」の用語が用いられることが 多いが、同効薬の添付文書において「汎発性血管内血液凝固症」が用いられているため、本
2.5 項 略号一覧(2/2)
略号 省略していない表現
MDS myelodysplastic syndrome(骨髄異形成症候群) MOF multiple organ failure(多臓器不全)
Na ナトリウム
PAI-1 plasminogen activator inhibitor-1(プラスミノゲンアクチベーターインヒビター-1) PIC plasmin-α2 plasmin inhibitor complex(プラスミン・α2プラスミンインヒビター複
合体)
PK Pharmacokinetic(薬物動態)
PPK population pharmacokinetics(母集団薬物動態) PPS per protocol set
PT prothrombin time(プロトロンビン時間) QOL Quality of Life
sc subcutaneous(皮下)
S-Cr serum creatinine(血清クレアチニン) T1/2 elimination half-life(消失半減期)
T1/2α α-phase elimination half-life(α相の消失半減期) T1/2β β-phase elimination half-life(β相の消失半減期)
TAT thrombin-antithrombin III complex(トロンビン・アンチトロンビン III 複合体) TF tissue factor(組織因子)
TM thrombomodulin(トロンボモジュリン)
t-PA tissue plasminogen activator(組織プラスミノゲンアクチベーター) U unit(単位) α2PI α2-plasmin inhibitor(α2プラスミンインヒビター) 厚生省 DIC 診断 基準 厚生省特定疾患血液凝固異常症調査研究班による DIC 診断基準(1988 年改訂) c) 本剤、本薬 2.5 項において、トロンボモデュリン アルファ注射製剤を示す場合「本剤」、ト ロンボモデュリン アルファ薬剤原薬を示す場合「本薬」と略した。 低用量群、中用 量群、高用量群 低用量群、中用量群、高用量群は、それぞれ 38 U/kg 群(0.006 mg/kg 群)、130 U/kg 群(0.02 mg/kg 群)、380 U/kg 群(0.06 mg/kg 群)を示す。 c) 次頁に厚生省 DIC 診断基準を添付した。
I.基礎疾患 得点 あり 〔1〕 なし 〔0〕 II.臨床症状 1) 出血症状(注 1) あり 〔1〕 なし 〔0〕 2) 臓器症状 あり 〔1〕 なし 〔0〕 III.検 査 成 績 1) 血清 FDP 値(μg/mL) 40≦ 〔3〕 20≦ <40 〔2〕 10≦ <20 〔1〕 10> 〔0〕 2) 血小板数(×103/μL)(注 1) 50≧ 〔3〕 80≧ >50 〔2〕 120≧ >80 〔1〕 120< 〔0〕 3) 血漿フィブリノゲン濃度(mg/dL) 100≧ 〔2〕 150≧ >100 〔1〕 150< 〔0〕 4) プロトロンビン時間 時間比(正常対照値で割った値) 1.67≦ 〔2〕 1.25≦ < 1.67 〔1〕 1.25> 〔0〕 IV.判 定(注 2) 1) 7点以上 DIC 6点 DIC の疑い(注 3) 5点以下 DIC の可能性少ない 2) 白血病その他注 1 に該当する疾患 4点以上 DIC 3点 DIC の疑い(注 3) 2点以下 DIC の可能性少ない V.診断のための補助的検査所見 1) 可溶性フィブリンモノマー陽性 2) D-Dダイマーの高値 3) トロンビン・アンチトロンビン III 複合体の高値 4) プラスミン・α2プラスミンインヒビター複合体の 高値 5) 病態の進展に伴う得点の増加傾向の出現。特に数 日内での血小板数あるいはフィブリノゲンの急激 な減少傾向ないし FDP の急激な増加傾向の出現 6) 抗凝固療法による改善 VI.注 1:白血病および類縁疾患、再生不良性貧血、抗 腫瘍剤投与後などの骨髄巨核球減少が顕著 で、高度の血小板減少をみる場合は血小板数 および出血症状の項は 0 点とし、判定は IV-2) に従う*1。 注 2:基礎疾患が肝疾患の場合は以下の通りとする。 a. 肝硬変および肝硬変に近い病態の慢性肝炎 (組織上小葉改築傾向を認める慢性肝炎)の 場合には、総得点から 3 点減点した上で IV-1) の判定基準に従う。 b. 劇症肝炎および上記を除く肝疾患の場合は、 本診断基準をそのまま適用する。 注 3:DIC の疑われる患者でV.診断のための補助 的検査成績、所見のうち 2 項目以上満たせば DICと判定する。 VII.除外規定 1) 本診断基準は新生児、産科領域の DIC の診断には 適用しない。 2) 本診断基準は劇症肝炎の DIC の診断には適用しな い。 厚生省 DIC 診断基準(厚生省特定疾患血液凝固異常症調査研究班、1988 年改訂) *1 本資料中では、「VI.注 1」の「白血病および類縁疾患、再生不良性貧血、抗腫瘍剤投与後な どの骨髄巨核球減少が顕著で、高度の血小板減少をみる」に該当し、判定を「IV-2)」で行った 症例を DIC の疾患群分類として「白血病群」と称した。上記に該当せず、判定を「IV-1)」で行っ た症例を DIC の疾患群分類として「非白血病群」と称した。
目次
[2.5-頁] 2.5 臨床に関する概括評価... 1 2.5.1 製品開発の根拠... 1 2.5.1.1 目標適応症の臨床的/病態生理学的側面... 1 2.5.1.2 目標適応症に対する試験実施の科学的根拠... 7 2.5.1.3 関連するガイドライン... 8 2.5.1.4 臨床開発計画の概略... 9 2.5.2 生物薬剤学に関する概括評価... 21 2.5.3 臨床薬理に関する概括評価... 23 2.5.3.1 薬物動態試験... 24 2.5.3.2 生体反応と血漿中濃度の関係... 34 2.5.3.3 臨床薬理試験のまとめ... 37 2.5.4 有効性の概括評価... 39 2.5.4.1 試験対象集団の特性... 39 2.5.4.2 試験方法 ... 40 2.5.4.3 有効性評価の主要な結果... 44 2.5.4.4 部分集団における結果の類似性、相違について... 55 2.5.4.5 推奨される用法・用量... 56 2.5.4.6 観察された効果の臨床的意義及びその限界... 59 2.5.4.7 まとめ ... 66 2.5.5 安全性の概括評価... 67 2.5.5.1 安全性情報の収集と評価の方法... 67 2.5.5.2 曝露状況と人口統計学的特性... 67 2.5.5.3 動物における毒性学的情報及び製品品質に関する情報... 71 2.5.5.4 安全性に関する結果の分析... 71 2.5.5.5 有害事象の予防、軽減、管理方法... 81 2.5.5.6 本剤と他の抗凝固薬との相互作用... 86 2.5.5.7 過量投与に対する反応... 86 2.5.5.8 依存性、反跳現象、乱用を誘発する可能性... 87 2.5.5.9 海外での使用経験(世界における市販後使用経験)... 87 2.5.6 ベネフィットとリスクに関する結論... 89 2.5.6.1 ベネフィット... 89 2.5.6.2 リスク ... 90 2.5.6.3 用量とベネフィット・リスクに関する考察... 94 2.5.6.4 効能・効果とベネフィット・リスクに関する考察... 95 2.5.6.5 ベネフィットとリスクのまとめ... 96 2.5.7 参考文献 ... 992.5 臨床に関する概括評価 2.5.1 製品開発の根拠 トロンボモデュリン アルファは、ヒトトロンボモジュリンの活性部位を含む細胞外ドメインの みを可溶型分子として遺伝子工学的に動物細胞で産生させた新規物質である。本剤は、既存薬に ない新しい血液凝固調節作用メカニズムを有する。 本剤の目標適応症は、「汎発性血管内血液凝固症(DIC)」である。 2.5.1.1 目標適応症の臨床的/病態生理学的側面 2.5.1.1.1 DIC の患者数 DICは、造血器悪性腫瘍・重症感染症・固形癌などの基礎疾患の存在下に発症する重篤な合併 症である。基礎疾患に DIC を合併することにより、患者の予後は悪化する場合が多い1)。本邦で は、厚生省特定疾患血液凝固異常症調査研究班により 1997 年に実施された全国的アンケート調査 の結果2)、年間の DIC 患者数は、73,000 人と推定されている。同アンケート調査によれば、DIC を合併した患者の生命予後は極めて悪く、6 診療科 243 施設(内科・外科・小児科・産科婦人科・ 集中治療部・救急部)2,193 例における転帰は、死亡が 56.0%、生存が 39.8%であった(不明 4.2%)。 死亡原因の内訳は、基礎疾患によるものが 54%、DIC によるものが 24%、合併症によるものが 20% であった。同調査によれば、上記 6 診療科における DIC の基礎疾患の内訳は表 2.5.1-1 のとおり であり、患者数は、感染症、造血器悪性腫瘍、固形癌の順に多く、その他ではショック、肝硬変 などが多かった。 表 2.5.1-1 DIC の各基礎疾患別の患者数 基礎疾患分類 基礎疾患 患者数a) 敗血症 303 呼吸器感染症 144 胆道感染症 55 感染症 成人呼吸促迫症候群 53 555 急性骨髄性白血病 104 急性リンパ性白血病 76 急性前骨髄球性白血病 73 造血器悪性腫瘍 非ホジキン悪性リンパ腫 161 414 肝細胞癌 142 肺癌 99 胃癌 93 固形癌 結腸癌 65 399 ショック 222 肝硬変 123 その他 大動脈瘤 69 414 a) 厚生省特定疾患血液凝固異常症調査研究班による調査の結果2)、DIC の 基礎疾患について回答のあった 6 診療科 243 施設における患者数
2.5.1.1.2 DIC の発症機序と病態 DICの発症機序を図 2.5.1-1 に示した。DIC は、造血器悪性腫瘍、感染症、固形癌などの重篤な 基礎疾患に合併し、血液凝固系の過度な活性化により全身の微小血管内に血栓を生じ、重症化す ると微小循環障害により臓器障害を合併する、あるいは線溶系活性化及び消費性凝固障害により 出血症状が生じる病態である。 図 2.5.1-1 DIC の発症機序3) 図 2.5.1-1 に示すとおり DIC の発症機序は基礎疾患により異なる。すなわち、造血器悪性腫瘍・ 固形癌といった悪性腫瘍を基礎疾患とする DIC においては、腫瘍細胞に発現している組織因子が 血液と接触することにより、血液凝固系が過度に活性化される4)。重症感染症を基礎疾患とする DICにおいては、エンドトキシンなどによる炎症性サイトカインネットワークの活性化を介して 凝固系の過度の活性化が起こる4)。 このように、各基礎疾患により凝固系活性化の機序は異なるものの、各基礎疾患に共通するの は、トロンビンの過剰生成が起こることである4)。 トロンビンの過剰生成の結果、全身の微小血管内に血栓が多発すると、その結果として虚血性 の臓器障害、すなわち DIC の臓器症状が生じる5)。脳、肺、肝臓、腎臓などの主要臓器での血栓 形成は特に問題であり、症状が高度な場合は直接生命に関わる。血栓形成の程度が致死的でない 場合でも、これら臓器の機能不全が全身状態を悪化させる場合が多い。特に、臓器障害が複数の 臓器で起こり、多臓器不全(以下、MOF)の状態に陥ると、患者の予後は極めて不良となる6)。 一方、血液凝固系の過度な活性化による全身性微小血栓の多発に対して二次線溶が過度に活性 化すると、止血血栓の溶解が必要以上に起こるため、出血が生じる。また、過剰に生成したトロ ンビンにより血小板や凝固因子が消費され、血液中での血小板数や凝固因子濃度が低下すると、 止血血栓が十分に形成されないため、やはり出血が生じる。すなわち、DIC における出血の原因 は、トロンビンの過剰生成により引き起こされる、線溶系の過度な活性化及び消費性凝固障害で ある5)。出血症状に関しては、出現部位が脳、肺、消化管などの重要臓器の場合、出血そのもの 組織障害 単球・マクロ ファージ活性化 組織因子 血管内皮 細胞障害 トロンビン生成 微小血栓 MOF 出血 血小板・凝固因子の消費性低下 凝固系活性化 造血器悪性腫瘍、固形癌 感染症(エンドトキシン、サイトカイン) 二次線溶 外傷 MOF:多臓器不全 組織障害 単球・マクロ ファージ活性化 組織因子 血管内皮 細胞障害 トロンビン生成 微小血栓 MOF 出血 血小板・凝固因子の消費性低下 凝固系活性化 造血器悪性腫瘍、固形癌 感染症(エンドトキシン、サイトカイン) 二次線溶 外傷 MOF:多臓器不全
が死因となる場合がある7)。DIC における出血症状は、さまざまな部位に起こることが知られて おり8)、これらの出血症状が持続した場合には、患者の QOL が著しく損なわれる9)。 以上のように、DIC の基礎疾患は様々でありその症状の発現状況も多様であるが、DIC の本態 がトロンビンの過剰生成であることは、基礎疾患あるいは発現する症状の種類によらず共通であ る。 2.5.1.1.3 DIC の診断 DICの診断は、出血症状、臓器症状の観察に加え複数の凝血学的検査を組み合わせて行うのが 一般的である。 凝固線溶系のバランスが破綻した DIC においては、凝固系の活性化と線溶系の活性化が同時に 起こり一見相反する複雑な病態を呈する。病態が凝固系の活性化、線溶系の活性化のどちらに傾 いているかによって、臓器症状及び出血症状の発現程度が異なる。したがって、個々の DIC 患者 の病態把握には、凝固系検査、線溶系検査を複数組み合わせて評価することが必要である10)。 また DIC では、心筋梗塞や脳血栓症などといった局所性の血栓症と異なり、全身性に微小血栓 が多発するため、血栓の存在を画像検査などの方法で直接捉えることができない。したがって、 DICの病態把握のためには、複数の凝血学的検査により間接的に微小血栓の存在を把握する必要 がある。本邦で長年使用されてきた厚生省 DIC 診断基準においても、出血症状、臓器症状、凝血 学的検査を組み合わせて DIC の診断を行っており、凝血学的検査値に得点が重点的に配点されて いる11)。 本邦においては、1980 年に厚生省特定疾患血液凝固異常症調査研究班により DIC 診断基準が作 成され12)、さらに 1988 年に改訂された11)。この厚生省 DIC 診断基準は臨床現場で広く使用され ている。これまでに本邦で実施されてきた DIC 治療薬の臨床試験のほとんどが厚生省 DIC 診断基 準を選択基準として用いている。また、2004 年には日本血栓止血学会・日本救急医学会により救 急領域の DIC 診断基準が作成され13)、さらに改訂されたものが急性期 DIC 診断基準として 2005 年に公表された14,15)。 欧米においては、DIC は治療すべき疾患として認識されながらも、長らく統一的な DIC の診断 基準がなかったため、臨床試験実施の妨げとなってきた16,17)。このため、欧米においても DIC の 診断基準の必要性が指摘され、2001 年になり国際血栓止血学会により、厚生省 DIC 診断基準を踏 襲、一部変更する形で overt-DIC(顕性化した DIC)の診断基準が作成され、使用され始めている 16,18,19)。 2.5.1.1.4 DIC 治療の現状 2.5.1.1.4.1 DIC の治療意義
DICの治療意義は、「DIC による患者の死亡を防ぐこと」及び「DIC により患者の全身状態が 悪化するのを防ぎ基礎疾患に対する積極的な治療機会を増やすこと」である。したがって DIC の 治療においては、以下の 2 点が重要であると指摘されている5)。 • DIC の基礎疾患を治療し DIC の原因を除去すること • 出血症状や臓器症状といった DIC の症状をコントロールすること DICの治療においては、DIC の原因となっている基礎疾患を治療し取り除くことが最優先され る。しかしながら、DIC の基礎疾患はいずれも重篤な疾患であるため、基礎疾患自体に対する治
療が奏効しない場合あるいは奏効するまでに時間を要する場合が多い。また、感染症に合併した DICの場合、たとえ抗菌剤による治療が奏効していたとしても、それだけでは不十分であり、DIC そのものに対する治療が必要である17,20)。基礎疾患の病態に加えて DIC による出血症状や臓器症 状が重なると患者の全身状態は悪化するので、DIC に対する治療を適切に行って出血症状や臓器 症状をコントロールし、DIC による患者の状態悪化を軽減することが求められる。 2.5.1.1.4.2 DIC 治療の実際 基礎疾患の治療と並行して実施される DIC に対する治療は、以下のとおりに大別される。 (1) 抗凝固薬による抗凝固療法 (2) 濃厚血小板・新鮮凍結血漿による補充療法 (3) 抗線溶薬による抗線溶療法 (1)の抗凝固療法の位置付けについては次項に述べる。 (2)の濃厚血小板・新鮮凍結血漿による補充療法は、DIC により消費された血小板や凝固因子を 体外から補う療法であり、血小板数あるいは血液凝固因子が高度に低下している DIC 患者に実施 される21)。 (3)の抗線溶療法は、線溶系の活性化が高度で出血傾向が顕著な一部の患者に適用となり、本邦 ではトラネキサム酸などが使用されている。しかしながら、線溶系を阻害することは、使用方法 を誤ると血栓による臓器障害を悪化させるため、抗線溶療法は一般的には実施されていないのが 現状である22)。 2.5.1.1.4.3 DIC 治療における抗凝固療法の位置付け DICの本態は血液凝固系の過度な活性化であるため、それを抑制する抗凝固療法が基礎疾患に 対する治療と並行して広く行われている。 抗凝固療法が、出血症状や臓器症状のコントロール、転帰の改善に有用であることは以下のと おり多くの基礎的研究、臨床研究により明らかにされている。 (1) 出血症状 DIC患者では、出血症状が高頻度に認められ、その原因は上述したように凝固系の過度 な活性化により引き起こされる線溶系活性化と消費性凝固障害である5)。抗凝固薬による 凝固系の抑制は、線溶系活性化及び消費性凝固障害の是正につながるため、DIC 患者の出 血症状を改善させることができる。DIC 患者に抗凝固療法を施すことにより、出血死を減 少させることが白血病に合併した DIC 患者を対象とした試験により示されている23)。 (2) 臓器症状 DIC患者では、微小血栓の多発による臓器障害が起こり、しばしば MOF の状態に陥る。 抗凝固薬による凝固系の抑制は、微小血栓の形成を阻害することができるため、臓器症状 の軽減につながる。動物を用いた基礎研究により、抗凝固薬の投与により敗血症の DIC モ デルにおいて肝臓などの臓器不全の改善、死亡率の改善などの効果が得られることが明ら かにされている24,25)。 (3) 転帰(生命予後)
感染症に合併する DIC に対して抗凝固療法を施すことが、生命予後の改善に結び付くこ とは、主に欧米で実施された臨床研究より明らかにされている。そのような臨床研究が行 われるようになった経緯も含めて臨床試験結果の概略を以下に記載した。 欧米においては、「2.5.1.1.3 項」で記載したとおり、2001 年に overt-DIC(顕性化した DIC)の診断基準が作成されるまで統一的な診断基準がなかったため、選択基準として 「DIC」が明示された抗凝固薬の比較臨床試験はほとんど実施されてこなかった。しかしな がら、医療現場では DIC 患者への抗凝固薬投与として、凝固系活性化に対する予防的な低 用量ヘパリン投与が、急性前骨髄球性白血病や重症敗血症の治療に用いられてきた26,27)。 また近年になって、DIC 合併例を多く含む重症敗血症患者を対象とした抗凝固療法の大 規模臨床試験が行われるようになり、遺伝子組換え型 APC による抗凝固療法が、重症敗血 症患者の死亡率を低下させることが証明された28)。 さらに、overt-DIC の診断基準が作成されたことを受けて、敗血症患者に対する臨床試験 の対象患者から overt-DIC の患者を抽出する事後的サブグループ解析が行われるように なった。この結果、上記の遺伝子組換え型 APC の試験にエントリーされた症例のうち、 overt-DICと診断された症例での検討から遺伝子組換え型 APC による抗凝固療法で DIC 患 者の死亡率が有意に低下することが示された29)。さらにこの治験データから、DIC におけ る微小血栓の形成が患者の予後悪化に寄与することが指摘されている30)。同様な結果が、 高用量の ATIII 製剤を重症敗血症患者に投与した試験のサブグループ解析からも得られて いる31)。すなわち、DIC 患者に対して抗凝固療法を施すことが、患者の生命予後を改善さ せる上で有用であることが複数のサブグループ解析から示された。 古くから DIC の治療に用いられてきたヘパリンに関しても以下のような知見が最近得ら れている。重症敗血症患者を対象とした遺伝子組換え型 APC、高用量の ATIII 製剤、遺伝 子組換え型 tissue factor pathway inhibitor の大規模臨床試験においては、血栓形成防止目的で 低用量ヘパリン(低分子量ヘパリン含む)が約 7 割の患者に投与されていた。ヘパリン投 与に対して無作為化されていないため結果の解釈には留意が必要であるが、これらの試験 の事後的サブグループ解析結果から、ヘパリンを投与した患者は非投与と比較して 28 日目 の生存率が有意に高いことが報告されている32,33)。
2.5.1.1.4.4 DIC における抗凝固療法のハードエンドポイント
DICにおける抗凝固療法による治療意義は、基礎疾患の悪化に伴い発症する DIC を抑制し、DIC による直接的な死亡を防ぐことや、DIC の悪化による基礎疾患の治療機会の逸失を防ぎ、結果的 に患者の生命予後を改善することにあると考えられる。すなわち、DIC における抗凝固療法のハー ドエンドポイントは、患者の生命予後改善であると考えられる。しかしながら、その評価時期は 以下に述べるとおり、基礎疾患により異なると考えられる。 感染症を基礎疾患とする DIC においては、海外で行われた重症敗血症における大規模臨床試験 で投与開始後 28 日目の死亡率が主要評価項目として設定されている28,34,35)ことから、抗凝固薬投 与開始後およそ 1 ヶ月の生命予後が抗凝固療法のハードエンドポイントとして適切であると考え られる。 造血器悪性腫瘍あるいは固形癌を基礎疾患とする DIC においては、感染症を基礎疾患とする DICの場合と異なり、しばしば再発・寛解を繰り返すこともあり、基礎疾患の再発により再び DIC が発症することも少なくない。すなわち、造血器悪性腫瘍あるいは固形癌の長期間にわたる治療
経過においては、患者が死亡のリスクにたびたびさらされることになるため、これらの累積する 死亡リスクを低下させることが臨床的に重要である。したがって、造血器悪性腫瘍あるいは固形 癌を基礎疾患とする DIC の抗凝固薬による治療においては、長期間の治療経過における生命予後 がハードエンドポイントであると考えられる。 本剤の第 3 相臨床試験で主要評価項目とした DIC 離脱率とハードエンドポイントとの関係につ いては、「2.5.4.6.2 項」に記載した。 2.5.1.1.4.5 DIC 治療における抗凝固療法の問題点 現在本邦で DIC 治療に用いられている抗凝固薬は、ヘパリン、低分子量ヘパリン、低分子ヘパ リノイド、蛋白分解酵素阻害剤、及び ATIII 製剤である。
これらの中でヘパリンは、DIC におけるプラセボ対照の controlled study に基づいたエビデンス はないものの、古くから DIC 治療に広く使用されてきた36,37)。したがって、ヘパリンは DIC に対 する抗凝固療法の中では実質的な標準薬として位置付けられている。このことは、上記の既存薬 のほとんどがヘパリンを対照薬として比較試験を行ってきたことからも裏付けられる。 DIC患者にヘパリンを使用する目的は、凝固系の活性化を抑制することである38)。DIC 患者に 対しては 1 日量 7,200~12,000 U のヘパリンを持続点滴するのが一般的である4)。 DIC以外の一般的な血栓症の治療においてヘパリンで十分な抗凝固作用を得るには、1 日用量 で 30,000~40,000 U 程度が必要である39)。このような用量では、ヘパリンは、その強力な抗凝固 作用によって止血系の反応も阻害する場合がある。特に DIC 患者においては、血小板や凝固因子 の低下、線溶系活性化が原因で出血が生じやすい状況に陥っているため、20,000 U 以上のヘパリ ン投与では出血症状の発現・増悪が問題となる26,40)。欧米では当初、一般の血栓症の患者と同様 に DIC 患者に対する 30,000~40,000 U 程度のヘパリン投与がなされたが、出血発現例が多発し、 ヘパリン療法の評価が比較的低くなる原因となった41)。今日においては、DIC 患者における抗凝 固効果と出血に対する影響のバランスから、欧米、本邦共に DIC における最適化された標準的な ヘパリンの用量は、一般の血栓症に対する治療量よりも低用量の 1 日 7,200~12,000 U である4,41,42)。 しかしながら、この低用量ヘパリン療法は、一般の血栓症に対する用量よりも低い用量に抑えら れていることから明らかなように、効果の面では必ずしも十分な抗凝固作用が発現する治療では ない38)。ヘパリン以外の抗凝固薬においても、基本的にはヘパリンと同様な問題点を有しており、 例えば一部の蛋白分解酵素阻害剤においては臨床的に抗凝固効果が十分に発揮されていないと指 摘する報告がある43)。 すなわち、ヘパリンを初めとする従来の抗凝固薬の DIC 治療における問題点は、「全身的に高 度な凝固系の活性化状態にある DIC に対し抗凝固療法を施す必要がある一方で、DIC は出血が起 こりやすい状態にあるため、十分な抗凝固療法ができない」という点である。 DIC治療において抗凝固薬投与により、出血を悪化させずに十分な抗凝固効果を得ることがで きれば、出血症状・臓器症状の改善を通じた患者の全身状態の改善、さらに生命予後の改善が期 待できる。同時に、十分な抗凝固療法により患者を DIC 状態から離脱させることができれば、濃 厚血小板や新鮮凍結血漿など供給量に限界のある血液製剤の使用頻度を減少させられることが期 待できる。DIC は致死的疾患であり、臨床現場においては、出血症状の悪化が生じない範囲で、 十分な抗凝固効果を得ることができる抗凝固薬の出現が切望されている。
2.5.1.2 目標適応症に対する試験実施の科学的根拠 トロンビンは、単独で存在するとその基質特異性がフィブリノゲンや血小板上のレセプターに 向いており、血液を凝固させるように作用する。ところが、図 2.5.1-2 に示したように、トロンビ ンは一旦 TM と結合すると、その基質特異性は変化しプロテイン C に向き、プロテイン C を限定 分解し APC へと活性化させる。生じた APC は、プロテイン S を補酵素とし血液凝固系の活性化 第 VIII 因子、活性化第 V 因子を失活させるため、TM は結果的に血液凝固系の活性化を阻害する。 すなわち、TM はトロンビンの基質特異性を変化させることで、血液凝固系の活性化にネガティ ブフィードバックをかける。トロンボモデュリン アルファは、この天然型 TM の血液凝固に対す るネガティブフィードバック作用を保持するヒト型の可溶型分子である。したがって、生体内で 血液凝固系が過度に活性化されるため発症する血栓症に本薬は効果を発揮すると考えられた。 PL,Ca2+ FⅧa FⅨ FⅨa FⅩ PL,Ca2+ FⅩa FⅤa プロトロンビン トロンビン フィブリノゲン フィブリンモノマー プロテインC プロテインS 組織因子 FVIIa 活性化 血栓 活性化プロテインC (APC) 不 活 化 不 活 化 太実線:トロンビン生成阻害に関る反応 太点線:トロンビン直接阻害に関る反応 PL,Ca2+ FⅩa FⅤa プロトロンビナーゼ複合体 TMあるいは トロンボモデュリン アルファ PL,Ca2+ FⅧa FⅨ FⅨa FⅩ PL,Ca2+ FⅩa FⅤa プロトロンビン トロンビン フィブリノゲン フィブリンモノマー プロテインC プロテインS 組織因子 FVIIa 活性化 血栓 活性化プロテインC (APC) 不 活 化 不 活 化 太実線:トロンビン生成阻害に関る反応 太点線:トロンビン直接阻害に関る反応 PL,Ca2+ FⅩa FⅤa プロトロンビナーゼ複合体 TMあるいは トロンボモデュリン アルファ PL,Ca2+ FⅧa FⅨ FⅨa FⅩ PL,Ca2+ FⅩa FⅤa プロトロンビン トロンビン フィブリノゲン フィブリンモノマー プロテインC プロテインS 組織因子 FVIIa 活性化 活性化 血栓 活性化プロテインC (APC) 不 活 化 不 活 化 不 活 化 不 活 化 太実線:トロンビン生成阻害に関る反応 太点線:トロンビン直接阻害に関る反応 PL,Ca2+ FⅩa FⅤa プロトロンビナーゼ複合体 TMあるいは トロンボモデュリン アルファ 図 2.5.1-2 血液凝固カスケードと TM あるいはトロンボモデュリン アルファの 抗凝固作用機序
FVIIa:活性化第 VII 因子、FIX:第 IX 因子、FIXa:活性化第 IX 因子、FX:第 X 因子、 FXa:活性化第 X 因子、FVIIIa:活性化第 VIII 因子、FVa:活性化第 V 因子、PL:リン脂質、 Ca2+:カルシウムイオン 非臨床試験の結果、本薬は、in vitro におけるヒト血漿への組織因子添加によって惹起されるト ロンビンの生成を、プロテイン C 活性化促進作用を介して阻害した。さらに、本薬のこの作用は、 トロンビンの凝固活性を直接的に阻害する作用発現濃度の 1/110 の濃度域で発現することが明ら かとなった(2.4.2.1.3 項)。トロンビンは血栓形成のみでなく止血反応に必須な因子であること から、トロンビンを直接的に阻害しその凝固活性を過度に抑制することは出血につながる。トロ ンビンの凝固活性を直接的に阻害しない濃度で、トロンビンの生成そのものを抑制することがで きる本薬は、ヘパリンを初めとする従来の抗凝固薬で困難であった「出血が発現あるいは増悪す るリスクを増大させない濃度範囲で強力な抗凝固活性を示すこと」が可能になる初めての抗凝固 薬であることが期待された。 さらにサルの DIC モデルを用いて本薬の静脈内投与による DIC 発症抑制効果を調べた結果、本
ビンの凝固活性に対する直接阻害発現濃度域(in vitro)の 1/44 の濃度域で発現したことより、in vitro の試験で想定した本薬の特徴が in vivo においても認められることが示唆された(2.4.2.1.5.1 項)。 以上、非臨床試験の結果より本薬は「全身的に高度な凝固系の活性化状態にあるため DIC に対 して抗凝固療法を施す必要がある一方で、DIC は出血が起こりやすい状態にあるため、十分な抗 凝固療法ができない」という DIC 治療の根本的な問題を解決し得る薬剤である可能性が示唆され た。 これらの知見をもとに DIC を目標適応症とした本剤の臨床試験を 19 年に開始した。 2.5.1.3 関連するガイドライン DIC治療に関するガイドラインは、本邦及び他地域においていずれも作成されていないが、現 在、日本血栓止血学会において作成が検討されている。 DIC治療薬の臨床試験に関するガイドラインも、本邦及び他地域においていずれも作成されて いない。 本剤の第 1 相臨床試験開始時から第 3 相臨床試験開始時までの期間において、一般的に広く認 知・使用されていた DIC の診断基準は、厚生省 DIC 診断基準のみであったので、本剤の臨床試験 においては一貫して厚生省 DIC 診断基準を用いた。「2.5.1.1.3 項」に記載したように、本剤の第 3相臨床試験開始後、2001 年に国際血栓止血学会の overt-DIC 診断基準が作成されたが19)、厚生 省 DIC 診断基準を改変したもので、大幅な変更はなされていない18)。また、本邦においても 2004 年に日本血栓止血学会・日本救急医学会により救急領域の DIC 診断基準が作成され13)、さらに改 訂されたものが急性期 DIC 診断基準として 2005 年に公表された14,15)。
2.5.1.4 臨床開発計画の概略 本剤の臨床開発計画の概略は、表 2.5.1-2 のとおりで、臨床試験は全て国内で実施した。 では、海外(米国等)において を対象と して臨床試験が 20 年 月より開始された。 また、北米において本剤の「 を対象とした を適応症と する第 2 相臨床試験(皮下投与)」が実施された。適応症、投与経路、剤型共に異なるため、本 試験結果は申請データパッケージには含めなかったが、公表論文を参考資料として添付した{添 付資料番号:5.3.5.4-2(参考)}。 上記の北米での第 2 相臨床試験に先立ち、米国において健康成人を対象とした第 1 相臨床試験 が実施された。本試験結果は、申請データパッケージには含めなかったが、公表論文を参考資料 として添付した{添付資料番号:5.3.5.4-1(参考)}。 表 2.5.1-2 臨床開発計画の概略 試験名 試験時期 添付 資料 番号 第 1 相臨床試験 (静脈内持続投与) 19 年 月~ 19 年 月 5.3.3.1-1 第 1 相臨床試験 (静脈内急速投与) 19 年 月 5.3.3.1-2 前期第 2 相臨床試験 19 年 月~ 19 年 月 5.3.5.2-1 後期第 2 相臨床試験 19 年 月~ 19 年 月 5.3.5.1-1 第 3 相臨床試験 20 年 月~ 20 年 月 5.3.5.1-2 治験相談 第 2 相終了後相談 19 年 月 日 治験相談 個別相談 19 年 月 日 医薬品申請前相談 20 年 月 日
2.5.1.4.1 第 1 相臨床試験(静脈内持続投与) 試験の概略を表 2.5.1-3 に示した。本試験の計画における主な設定根拠は以下のとおりである。 • 用法: DICの治療には緊急性を要する場合が多いため、本剤の投与法としては、短時間で有効 血漿中濃度に到達させる静脈内投与が望ましいと考えられた。 本剤の血漿中濃度半減期は、6~7 時間と長いことが非臨床試験で明らかとなり、ヒトに おいても半減期は長いと予想された。一般的に半減期の長い薬剤を 24 時間持続静脈内投与 すると血漿中濃度の上昇に時間を要する。本剤において血漿中濃度の上昇に時間がかかる 24時間持続静脈内投与は DIC 治療には適切ではなく、短時間での静脈内投与の用法が適切 と考えられた。さらに、安全性の観点から被験者の状態を観察しながらの投与が可能で、 万一投与中に不都合が生じた際、即座に投与を中止できる「2 時間静脈内持続投与」を用 法として選択した。 • 用量: 本剤は、トロンビンと結合し凝固系にネガティブフィードバックをかけトロンビン生成 阻害効果を発揮する。健康成人においては、DIC 患者と異なりトロンビンが血液中にほと んど存在しないため、本剤の作用は DIC 患者と比較して健康成人では発現し難いと考えら れた。本剤は、健康成人に投与する試験では忍容性の確認が困難と考え、DIC 患者を対象 とした試験で安全性を確認しながら慎重に用量を上げていくべき薬剤であると判断した。 したがって、第 1 相臨床試験では健康成人において作用が発現するまで増量することは避 け、薬物動態の把握が可能な用量までを投与することとし、190 U/人(0.03 mg/人)~1,900 U/人(0.3 mg/人)に設定した。 • 皮膚テスト: 本剤は蛋白製剤であるため、治験薬投与に先立ち皮膚テストを実施し、陰性が確認され た被験者のみに治験薬を投与した(以下、全ての臨床試験において同様である)。 表 2.5.1-3 第 1 相臨床試験(静脈内持続投与)の概略 目的 健康成人を対象に本剤を単回静脈内投与し、その安全性及び体内薬物動態について 安全性に十分留意しながら検討する。単回投与試験の成績を考慮して、健康成人を 対象に本剤を 3 日間反復静脈内投与し、同様の検討を行う。 単回投与試験 反復投与試験 対象 健康成人男性 健康成人男性 投与量 a)及 び投与方法 190 U(0.03 mg)、640 U(0.1 mg)、あるいは 1,900 U(0.3 mg)を、2 時間で静脈内持続投 与した。 1,300 U(0.2 mg)を、2 時間で静脈内持続投 与した。これを 3 日間繰り返した。 ステップ及 び被験者数 単回投与試験 ステップ 1:190 U/人(0.03 mg/人)2 時間静脈内持続投与(4 名) ステップ 2:640 U/人(0.1 mg/人)2 時間静脈内持続投与(4 名) ステップ 3:1,900 U/人(0.3 mg/人)2 時間静脈内持続投与(4 名) 反復投与試験 :1,300 U/人(0.2 mg/人)2 時間静脈内持続投与 3 日間(4 名) ガイドライ ンなど 遵守:旧 GCP a) 体重当たりの投与量で示すと、体重 50kg として、190 U/人(0.03 mg/人)~1,900 U/人(0.3 mg/人)は、3.8 U/kg(0.0006 mg/kg)~38 U/kg(0.006mg/kg)に相当する。
2.5.1.4.2 第 1 相臨床試験(静脈内急速投与) 試験の概略を表 2.5.1-4 に示した。 • 本試験の背景について: 2時間静脈内持続投与試験の結果を踏まえ、臨床現場における用法の選択のひとつとし て静脈内急速投与も可能と考えられたため、前期第 2 相臨床試験の開始後ではあるが、そ の安全性を検討することとした。 また、本試験では、本剤の体内における活性を把握するため、トロンボモジュリン活性、 すなわちプロトロンビナーゼに対する阻害活性を ex vivo で測定した。 なお、静脈内急速投与の用法については、申請用法としなかった。その理由は、万一投 与中に不都合が生じた際、即座に投与を中止できる静脈内持続投与の安全性と静脈内急速 投与の利便性のバランスを考慮し、安全性を優先させるべきと考えたためである。 表 2.5.1-4 第 1 相臨床試験(静脈内急速投与)の概略 目的 健康成人を対象に本剤を単回静脈内急速投与し、その安全性及び体内薬 物動態について安全性に十分留意しながら検討する。 対象 健康成人男性 投与量a)及び投与方法 1,900 U/人(0.3 mg/人)・静脈内急速投与 被験者数 4名 ガイドラインなど 遵守:旧 GCP a) 体重当たりの投与量で示すと、体重 50kg として、1,900 U/人(0.3 mg/人)は、38 U/kg(0.006mg/kg)に相当する。 2.5.1.4.3 前期第 2 相臨床試験 試験の概略を表 2.5.1-5 に示した。本試験の計画における主な設定根拠は以下のとおりである。 • 対象患者: 厚生省 DIC 診断基準を用いて選択し「DIC」又は「DIC の疑い」と診断された患者とし、 基礎疾患は限定しなかった。 • 用法: 30分間静脈内持続投与とした。第 1 相臨床試験では 2 時間の静脈内持続投与を検討した が、2 時間の静脈内持続投与と 30 分の静脈内持続投与の推定最高血漿中濃度はほとんど同 等であり(2.7.2.2.1.1.2 項)、安全性に問題ないと考えられた。このため、前期第 2 相臨床 試験以降は、DIC 治療における緊急性及び臨床現場における利便性を考慮し 30 分間静脈内 持続投与を選択した。 • 用量: 有効性と安全性を確認しつつ低用量からステップ毎に上げていくこととした。投与量を 上げる次のステップへの移行に関しては、その時点までに得られた試験成績を十分検討し、 治験総括医師の判断により決定した。また同一患者内においては、3 日間投与して効果が 不十分な場合には残りの 3 日間の投与に関しては増量可とした。投与量の設定については、 臨床現場における利便性を考慮し、ヒト当たりの投与量とした。サルの DIC モデル試験、 サルにおける 1 ヶ月反復投与毒性試験、及び第 1 相臨床試験の結果をもとに、最低用量を 1,900 U/人(0.3 mg/人)、最高用量を 19,000 U/人(3 mg/人)に設定した。
各ステップの用量に関しては、開始用量を 1,900 U/人(0.3 mg/人)、3,800 U/人(0.6 mg/ 人)、7,700 U/人(1.2 mg/人)、13,000 U/人(2.0 mg/人)の 4 ステップとし、それぞれ増量 後の用量を 3,800 U/人(0.6 mg/人)、7,700 U/人(1.2 mg/人)、13,000 U/人(2.0 mg/人)、 19,000 U/人(3.0 mg/人)と設定した。 • 投与期間と評価時期: 投与期間に関しては、医療現場での抗凝固薬の投与期間及び類薬の臨床試験での投与期 間を考慮し、以下のとおりとした。 白血病を基礎疾患とする DIC における抗凝固薬の投与期間に関しては以下のような報告 があった。すなわち、急性白血病に対する寛解導入療法時のヘパリンの投与期間は、凝血 学的検査値が回復するまで、あるいは白血病細胞が消失するまでの期間である 5~10 日間 とすべきであり44)、実治療においてはヘパリンの投与期間の中央値が 9 日間であった40)と 報告されていた。感染症を基礎疾患とする DIC に関しては、敗血症性ショックを基礎疾患 とする DIC 患者を対象とした ATIII 製剤の治療効果を検討した臨床試験で、ATIII 製剤の投 与期間に相当する 4 日目には 71%の患者が DIC 状態を脱しており、10 日目には生存例の全 例が DIC 状態を脱していたと報告されていた45)。これらの報告から、DIC における抗凝固 薬の適切な投与期間は、4~10 日程度と考えた。また、当時における最新の DIC の臨床試 験では、投与期間は 5 日間とされていた46)。以上のことから投与期間は 6 日間に設定した。 評価時期に関しては、DIC は致死的疾患であり、可能な限り早期に治療効果が発揮する ことが求められることから、本剤の治療効果は投与開始後 1 週間程度で判定すべきと考え た。本剤の血漿中濃度半減期が約 20 時間と長いこともあわせて考慮し、評価時期は投与終 了日の翌日(7 日目)とした。 表 2.5.1-5 前期第 2 相臨床試験の概略 目的 本剤の DIC に対する有効性並びに安全性を検討する。 対象 「DIC」又は「DIC の疑い」と診断a)された患者 1,900 U/人(0.3 mg/人)より開始し、投与期間は 6 日間投与とした。ただし、3 日間 投与しても効果が十分でない場合には、下記の計画に従って増量した。次のステッ プへの移行については、各施設から寄せられた症例の内容を十分に検討した上で、 治験総括医師の指示に従って行った。
ステップ 0 1,900 U(0.3 mg) 3,800 U(0.6 mg) ×3 日間(増量)×3 日間 1,900 U(0.3 mg)×3 日間 ステップ 1 3,800 U(0.6 mg) 7,700 U(1.2 mg)×3 日間(増量)×3 日間 3,800 U(0.6 mg)×3 日間 ステップ 2 7,700 U(1.2 mg) 13,000 U(2.0 mg)×3 日間(増量)×3 日間 7,700 U(1.2 mg)×3 日間 投与量b)
ステップ 3 13,000 U(2.0 mg) 19,000 U(3.0 mg)×3 日間(増量)×3 日間 13,000 U(2.0 mg)×3 日間 投与方法 1日 1 回 30 分間の静脈内持続投与を 6 日間繰り返した。 評価時期 治験薬投与開始 4 日目及び 7 日目(又は治験薬投与中止時) 評価項目 臨床症状改善度・凝血学的検査値改善度・DIC スコア改善度・全般改善度・安全度・ 有用度 目標症例数 各ステップ 10~15 例 ガイドライ ンなど 遵守:旧 GCP
b) 体重当たりの投与量で示すと、体重 50kg として、1,900 U/人(0.3 mg/人)~19,000 U/人(3.0 mg/人)は、38 U/kg(0.006 mg/kg)~380 U/kg(0.06mg/kg)に相当する。 2.5.1.4.4 後期第 2 相臨床試験 試験の概略を表 2.5.1-6 に示した。本試験の計画における主な設定根拠は以下のとおりである。 • 対象患者: 前期第 2 相臨床試験と同様に厚生省 DIC 診断基準を用いて選択し「DIC」又は「DIC の 疑い」と診断された患者とし、基礎疾患は限定しなかった。 • 主要評価項目: 客観性を高めるため、凝血学的検査値をスコア化して判定する凝血学的検査値改善度と 出血症状・臓器症状改善度とを予め設定したマトリックスを用いて組み合わせて評価する 全般改善度とした。 • 試験デザイン: 並行群間比較試験法を採用し、有効性評価に大きく影響をおよぼすと考えられる背景因 子について用量群間のバランスを保つため、「DIC の基礎疾患」「投与前 FDP 値」「年齢」 を層化要因とする非盲検動的割り付け法とした。 • 用法・用量: 用法については前期第 2 相臨床試験と同様であるが、用量については以下のとおりであ る。 最低用量に関しては、DIC が致死的な疾患であるため、明らかな無効量を選択すること は避けるべきであると考えた。第 1 相臨床試験(静脈内急速投与)において、1,900 U/人(0.3 mg/人)の用量で、プロトロンビナーゼ活性の有意な低下が認められたこと、また前期第 2 相臨床試験において、開始用量が 1,900 U/人(0.3 mg/人)に該当するステップ 0 で 3 例中 2 例が有効であったことから、1,900 U/人(0.3 mg/人)は DIC に対して無効量ではないと判 断した。以上のことより、最低用量は 1,900 U/人(0.3 mg/人)に設定した。 最高用量に関しては、前期第 2 相臨床試験のステップ 3{13,000 U/人(2.0 mg/人)~19,000 U/人(3.0 mg/人)}までの投与成績があり、そのときの全般改善度の「中等度改善」以上 率は 83.3 %(10/12 例)で、この改善率は DIC における他の抗凝固薬の成績と比較して同 等以上に高く十分な有効性を示しているものと判断した。また、この用量での安全性に特 に問題は認められなかった。 一方、サルでの反復投与毒性試験において、出血の毒性発現が 3,800 U/kg(0.6 mg/kg)で認 められた。この用量を薬物動態の差を考慮してヒトに外挿すると、1,300 U/kg(0.2 mg/kg)、 すなわち 64,000 U/人(10 mg/人)となる。前期第 2 相臨床試験の最高用量 19,000 U/人(3.0 mg/人)は、その約 1/3 量に相当するので、個人間の血漿中濃度のばらつきを考慮すると、 安全性の観点からこれ以上の高い用量は設定すべきでなく、19,000 U/人(3.0 mg/人)が後 期第 2 相臨床試験の最高用量として適切と考えられた。 以上より、最低用量を 1,900 U/人(0.3 mg/人)、最高用量を 19,000 U/人(3.0 mg/人)と し、公比 3 で、中用量を 6,400 U/人(1.0 mg/人)とした。 後期第 2 相臨床試験は、投与量と効果の相関性を検証することが主な目的であり、より 正確に有効性の用量相関性をみることができるように血漿中濃度のばらつきの要因はでき る限り少なくすることが望ましいと考えた。そこで用量は、前期第 2 相臨床試験までで採
用したヒト当たりの設定に代えて、体重当たりの設定を採用することとした。したがって、 体重を 50 kgとして換算し、低用量を 38 U/kg (0.006 mg/kg)、中用量を 130 U/kg (0.02 mg/kg)、 高用量を 380 U/kg (0.06 mg/kg)と設定した。本試験以降は、この体重当たりの設定を採用す ることとした。 • 投与期間と評価時期: 前期第 2 相臨床試験の結果より、DIC スコアは投与期間の 6 日間を通じて経時的に低下 していたことから、投与期間として 6 日間は必要と考えた。一方、評価時期については、 投与開始 7 日目におけるステップ 3 の改善率は高く十分な有効性を示していると考えられ たことから、6 日間投与で 7 日目に評価することが妥当と判断した。 表 2.5.1-6 後期第 2 相臨床試験の概略 目的 DIC に対する本剤の用量反応関係を非盲検動的割り付け群間比較試験により検討す る。 対象 「DIC」又は「DIC の疑い」と診断a)された患者 試験デザイ ン 非盲検動的割り付け法による群間比較試験(電話登録法)
投与量 38 U/kg(0.006 mg/kg)、130 U/kg(0.02 mg/kg)、380 U/kg(0.06 mg/kg)のうち、いずれか の投与量を登録時の情報に従い割り付けた。 投与方法 1日 1 回 30 分で静脈内持続投与し、これを 6 日間繰り返した。 評価時期 治験薬投与開始 7 日目(又は治験薬投与中止時) 評価項目 全般改善度・凝血学的検査値改善度・出血症状改善度・臓器症状改善度・安全度・ 有用度 目標症例数 各群 40 例(計 120 例) ガイドライ ンなど 遵守:旧 GCP a) 厚生省 DIC 診断基準による 2.5.1.4.5 治験相談 (1) 第 2 相終了後相談 第 3 相臨床試験開始に先立ち、本試験計画の妥当性に関しては、19 年 月 日に医 薬品副作用被害救済・研究振興調査機構(現 独立行政法人医薬品医療機器総合機構、以 下「医薬品機構」)と治験相談(第 2 相終了後相談)を実施し、以下の助言を得た(議事 録:1.13 項-1)。
(2) 個別相談 第 3 相臨床試験開始に先立ち、(1)の第 2相終了後相談で検討できなかった事項に関して、 19 年 月 日に医薬品機構と治験相談(個別相談)を実施し、以下の助言を得た(議事 録:1.13 項-2)。 2.5.1.4.6 第 3 相臨床試験 上記した 2 回の治験相談の助言に基づき、本剤の有効性(DIC 離脱率を主要評価項目とした非 劣性検証)及び安全性を検討するために第 3 相臨床試験を実施した。試験の概略を表 2.5.1-7 に示 した。本試験の計画における主な設定根拠は以下のとおりである。 • 対象患者: 後期第 2 相臨床試験までは、厚生省 DIC 診断基準に基づき「DIC」又は「DIC の疑い」 と診断された患者としていたが、本試験では同診断基準に基づき「DIC」と診断された患者 とした。 DICの基礎疾患に関する規定はこれまで設けていなかったが、本試験においては、直接 誘因基礎疾患が「固形癌」「その他」の患者は対象とせず、「造血器悪性腫瘍」あるいは 「感染症」の患者のみを対象とした。このようにした根拠を本項の「DIC の基礎疾患の選 択理由」に詳細に記載した。造血器悪性腫瘍あるいは感染症を基礎疾患とする DIC 患者は、 薬効評価をそれぞれの基礎疾患で行えるように独立に動的割り付けし、両基礎疾患共に最 低 100 例ずつ集積することとした。 • 試験デザイン: ヘパリンナトリウムを対照薬としたダブルダミー法による二重盲検無作為化並行群間比 較法を採用した。各薬剤への割り付けは、有効性評価に大きく影響をおよぼすと考えられ る背景因子について薬剤群間のバランスを保つため、「開始時 DIC スコア」「出血症状の 有無」を層化要因とする動的割り付け法により行った。 • 用法・用量: 本剤は、後期第 2 相臨床試験で用いた最大用量 380 U/kg(0.06 mg/kg)の 30 分間静脈内持続 投与が、その有効性・安全性から用法・用量として妥当であると考えられた。 ヘパリンナトリウムは、DIC 治療で標準的に使用されている用法・用量(8 U/kg/hr を 24 時間静脈内持続投与)を採用した。 • 主要評価項目: 後期第 2 相臨床試験までは全般改善度であったが、本試験では DIC の治療目標のひとつ である DIC からの離脱率とした。検証方法は、対照薬ヘパリンに対する非劣性検証(非劣
性限界値 5%)としたが、両薬剤群間の DIC 離脱率の差の下側信頼限界値が 0%を上回った ときは、優越と判定することとし治験実施計画書に取り決めた。なお、本治験実施計画作 成後ではあるが、2000 年に欧州の CPMP(Committee For Proprietary Medical Products)より 「Points to consider on switching between superiority and non-inferiority」のガイドラインが発行 された47)。このガイドラインに照らしても上記判定方法は妥当であると考えられた。 • 薬剤の特徴を評価するための副次的評価項目: 有効性と安全性の両側面を反映する指標である「出血症状の経過」を設定した。DIC に おける出血症状は、さまざまな部位に起こることが知られており8)、これらの出血傾向が 持続した場合には、患者の QOL が著しく損なわれ9)、また全身状態の悪化を招き、基礎疾 患に対する積極的な治療機会が失われることもある。さらに、頭蓋内出血、肺・気管出血、 消化管出血といった重要臓器での出血は、致死的な状況につながる可能性も高い7)。一方、 DICの治療においては、「全身的に高度な凝固系の活性化状態にある DIC に対し抗凝固療 法を施す必要がある一方で、DIC は出血が起こりやすい状態にあるため、十分な抗凝固療 法ができない」という問題点があり、出血を発現あるいは悪化させずに十分な抗凝固効果 を発揮できる薬剤が医療現場で求められている。本剤は、作用機序及び後期第 2 相臨床試 験までの結果から、出血を発現あるいは悪化させる可能性は少ないと考えられた。さらに、 凝固系の活性化を強力に抑制することで、血小板、凝固因子の消費性低下や二次線溶の活 性化を抑制し、DIC に伴う出血症状を改善することが期待された。以上のことから、「出 血症状の経過」は本剤の特徴を評価する項目として適切と考えられた。 • DIC の基礎疾患の選択理由: 造血器悪性腫瘍及び感染症が、本剤の第 3 相臨床試験(検証試験)の対象として妥当と 考えられた理由は以下のとおりである。 - 造血器悪性腫瘍においては抗癌剤による治療、骨髄移植による治療などで基礎疾患その ものの治療が可能である場合が多い。同様に、感染症においても抗菌剤による治療、外 科的処置などで基礎疾患そのものの治療が可能である場合が多い。したがって、造血器 悪性腫瘍及び感染症では、合併する DIC の病勢を抑制できれば、DIC が直接的あるいは 間接的に関与する患者の死亡を防ぐことができると考えられ、抗凝固薬の投与の臨床的 意義が大きい。 - 造血器悪性腫瘍あるいは感染症を基礎疾患とする DIC は、急性型が多くその時間経過が 比較的類似していると考えられた。 - 本剤及び対照薬であるヘパリンの至適用量が造血器悪性腫瘍あるいは感染症を基礎疾 患とする DIC で同様と考えられた。 「固形癌」及び「その他の基礎疾患」が第 3 相臨床試験(検証試験)の対象として妥当 でないと考えられた理由は以下のとおりである。 固形癌を直接誘因基礎疾患とする DIC(以下、固形癌 DIC)の特徴として次のことが挙 げられる。すなわち、転移が進行した末期癌にしばしば DIC が合併するため、固形癌 DIC 患者においては、基礎疾患に対する治療が一般的に造血器悪性腫瘍や感染症より困難で DIC治療の効果も低い8)。したがって、末期癌に合併した DIC においては、基礎疾患の治 療が困難な患者が多く、抗凝固薬による DIC 治療が生命予後の改善に結び付き難いと考え られた。固形癌 DIC の中でも、固形癌自体の治癒が可能な患者に関しては抗凝固薬による
治療の意義が大きいが、そのような患者を予め予測し臨床試験に組み入れることは困難で あると考えられた。 「2.5.1.1.4 項」に記載したとおり、DIC の治療の成否には基礎疾患の治療や経過が影響す る。したがって、検証試験においては基礎疾患の治療や経過が類似した患者を対象とする ことが精度の高い有効性・安全性の評価のために必要である。上述したとおり、固形癌 DIC は、固形癌の治療が困難で治療に時間を要するため DIC も慢性の経過をたどることが多く、 基礎疾患の治療や経過が造血器悪性腫瘍あるいは感染症を基礎疾患とする DIC とは異なる と考えられた。 以上の理由から、第 3 相臨床試験で固形癌 DIC を造血器悪性腫瘍あるいは感染症を基礎 疾患とする DIC と同じ治験実施計画で評価することは、不適切であると判断した。 「その他の疾患」を直接誘因基礎疾患とする DIC を対象としなかった理由は以下のとお りである。すなわち、「その他の基礎疾患」としては、大動脈瘤、産科的疾患、外傷など 様々な疾患が挙げられる。これら「その他の基礎疾患」の DIC 患者では、基礎疾患の治療 や経過が多様性に富むため、本剤の第 3 相臨床試験(検証試験)の対象とすることは精度 の高い薬効評価を行う上で不適切と判断した。
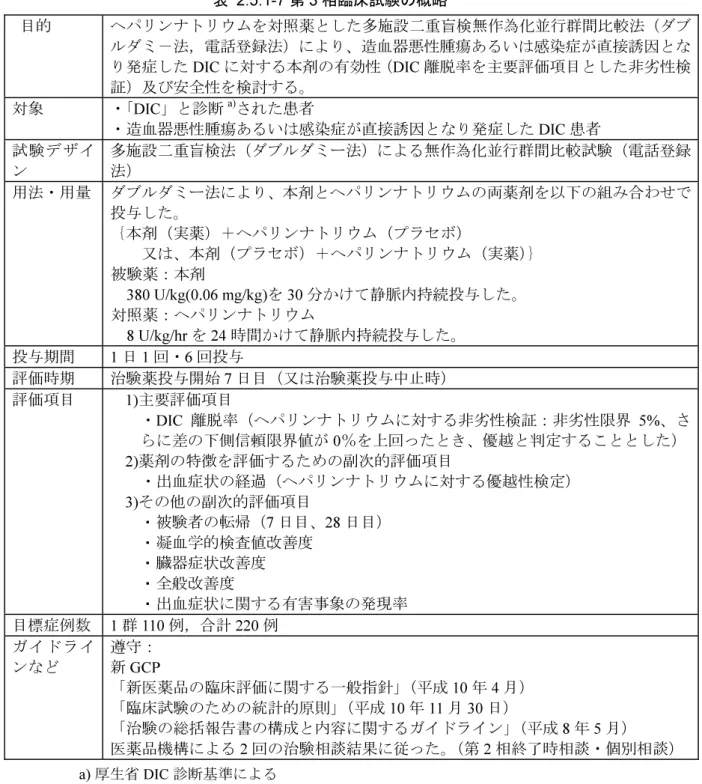
表 2.5.1-7 第 3 相臨床試験の概略 目的 ヘパリンナトリウムを対照薬とした多施設二重盲検無作為化並行群間比較法(ダブ ルダミ-法,電話登録法)により、造血器悪性腫瘍あるいは感染症が直接誘因とな り発症した DIC に対する本剤の有効性(DIC 離脱率を主要評価項目とした非劣性検 証)及び安全性を検討する。 対象 ・「DIC」と診断a)された患者 ・造血器悪性腫瘍あるいは感染症が直接誘因となり発症した DIC 患者 試験デザイ ン 多施設二重盲検法(ダブルダミー法)による無作為化並行群間比較試験(電話登録法) 用法・用量 ダブルダミー法により、本剤とヘパリンナトリウムの両薬剤を以下の組み合わせで 投与した。 {本剤(実薬)+ヘパリンナトリウム(プラセボ) 又は、本剤(プラセボ)+ヘパリンナトリウム(実薬)} 被験薬:本剤 380 U/kg(0.06 mg/kg)を 30 分かけて静脈内持続投与した。 対照薬:ヘパリンナトリウム 8 U/kg/hrを 24 時間かけて静脈内持続投与した。 投与期間 1日 1 回・6 回投与 評価時期 治験薬投与開始 7 日目(又は治験薬投与中止時) 評価項目 1)主要評価項目 ・DIC 離脱率(ヘパリンナトリウムに対する非劣性検証:非劣性限界 5%、さ らに差の下側信頼限界値が 0%を上回ったとき、優越と判定することとした) 2)薬剤の特徴を評価するための副次的評価項目 ・出血症状の経過(ヘパリンナトリウムに対する優越性検定) 3)その他の副次的評価項目 ・ 被験者の転帰(7 日目、28 日目) ・ 凝血学的検査値改善度 ・ 臓器症状改善度 ・ 全般改善度 ・ 出血症状に関する有害事象の発現率 目標症例数 1 群 110 例,合計 220 例 ガイドライ ンなど 遵守: 新 GCP 「新医薬品の臨床評価に関する一般指針」(平成 10 年 4 月) 「臨床試験のための統計的原則」(平成 10 年 11 月 30 日) 「治験の総括報告書の構成と内容に関するガイドライン」(平成 8 年 5 月) 医薬品機構による 2 回の治験相談結果に従った。(第 2 相終了時相談・個別相談) a) 厚生省 DIC 診断基準による 2.5.1.4.7 医薬品申請前相談 本申請に先立ち、 に関して、20 年 月 日に独立行政 法人医薬品医療機器総合機構と医薬品申請前相談を実施し、以下の助言を得た(議事録:1.13 項
当該相談の助言を参考に、CTD 資料を作成した。
以上のように、各臨床試験は適切に計画、実施されると共に計画した症例数でそれぞれの試験 目的が達成された結果、本剤は DIC 治療に有用な薬剤であることが明らかとなったため、今回製 造販売承認申請するに至った。
2.5.2 生物薬剤学に関する概括評価 市販予定製剤と治験用製剤の概観を表 2.5.2-1 に示した。なお本剤は静脈内投与製剤であること から生物学的同等性試験を実施する必要はないと判断した。 表 2.5.2-1 市販予定製剤と治験用製剤の概観 治験用製剤(第 1 相臨床試験) 治験用製剤(前期第 2 相臨床試験) 原体 トロンボモデュリン アルファ トロンボモデュリン アルファ 製剤 トロンボモデュリン アルファとし て 0.1 mg を含有する凍結乾燥注射剤 トロンボモデュリン アルファとし て 1.0 mg を含有する凍結乾燥注射剤 用法・用量 設定投与量にあわせて必要量を、予 めトロンボモデュリン アルファ乾 燥製剤を適量の電解質液で溶解した 溶液から 10 mL の生理食塩液に希釈 し、約 2 時間かけて 1 日 1 回で単回 あるいは反復点滴静注を行う。 用量:単回 190、640、1,900 U/人 (0.03、0.1、0.3 mg/人) 反復 1,300 U/人 (0.2 mg/人) 反復投与:3 日間 設定投与量にあわせて必要量を、予 めトロンボモデュリン アルファ乾 燥製剤を 5 mL の生理食塩液で溶解 した溶液から 100 mL に希釈し、約 30 分かけて 1 日 1 回の点滴静注を行 う。 用量:1,900~19,000 U/人 (0.3~3.0 mg/人) 治験薬剤の投与期間:6 日間 使用製剤数 0.1 mg バイアル×1~3 本 1.0 mg バイアル×1~6 本 0.1 mg、1.0 mg はそれぞれ 640 U、6,400 U に相当する。 治験用製剤(後期第 2 相臨床試験) 治験用製剤(第 3 相臨床試験) 原体 トロンボモデュリン アルファ トロンボモデュリン アルファ 製剤 トロンボモデュリン アルファとし て 1.0 mg を含有する凍結乾燥注射剤 トロンボモデュリン アルファとし て 2.0 mg を含有する凍結乾燥注射剤 用法・用量 通常、体重にあわせて必要量を、予 めトロンボモデュリン アルファ乾 燥製剤を 2 mL の生理食塩液で溶解 した溶液から 100 mL の生理食塩液 に希釈し、約 30 分かけて 1 日 1 回の 点滴静注を行う。 用量:38、130、380 U/kg (0.006、0.02、0.06 mg/kg) 治験薬剤の投与期間:6 日間 通常、体重にあわせて必要量を、予 めトロンボモデュリン アルファ乾 燥製剤を 4 mL の生理食塩水で溶解 した溶液から 100 mL の生理食塩水 に希釈し、約 30 分かけて 1 日 1 回の 点滴静注を行う。 用量:380 U/kg (0.06 mg/kg) 治験薬剤の投与期間:6 日間 使用製剤数 1.0 mg バイアル×1~6 本 2.0 mg バイアル×3 本 1.0 mg、2.0 mg はそれぞれ 6,400 U、13,000 U に相当する。
市販予定製剤 原体 トロンボモデュリン アルファ 製剤 トロンボモデュリン アルファとし て 12,800 U を含有する凍結乾燥注射 剤 用法・用量 1 バイアル(12,800U)当り 2mL の日局 生理食塩液で溶解し、この溶液から 患者の体重にあわせて必要量をとり 日局生理食塩液 100mL に希釈し、点 滴静注する。 用量:380 U/kg 使用製剤数 12,800 U バイアル×3 本