九州大学学術情報リポジトリ
Kyushu University Institutional Repository
酸化還元酵素を範としたエネルギー変換反応の構築
徳永, 泰介
https://doi.org/10.15017/1931871
出版情報:Kyushu University, 2017, 博士(工学), 課程博士 バージョン:
権利関係:
九州大学大学院工学府 物質創造工学専攻
博士論文
酸化還元酵素を範とした エネルギー変換反応の構築
徳永泰介
目次
第1章 緒言 1-1. 序
1-2. FHLとそのモデル研究:ギ酸からの水素発生
1-3. 光合成系IIとそのモデル研究:光阻害および自己修復
1-4. 本論文の構成 1-5. 参考文献
第2章 ギ酸水素リアーゼの機能を模倣した二核Ru錯体によるギ酸からの水素発生 のメカニズム
2-1. 序 2-2. 実験
2-2-1. 試薬および測定機器
2-2-2. [RuI2(CO)4(µ-HCOO)2(DMSO)2] (1) の合成 2-2-3. 1を触媒とするHCOOHからのH2発生
2-2-4. 同位体標識による1を触媒としたH2、HD及びD2発生 2-2-5. 1の濃度に対する触媒的H2発生の初速度
2-2-6. HCOOHのプロトンに対するヒドリド種2の反応性
2-2-7. 2のヒドリド配位子のH+/D+交換 2-2-8. 1のX線結晶解析
2-3. 結果および考察
2-3-1. 錯体の合成および同定 2-3-2. 1から2の生成
2-3-3. 2におけるヒドリド配位子の性質 2-3-4. 1を触媒とするHCOOHからのH2発生
2-3-5. 同位体標識による1を触媒としたH2、HD及びD2発生 2-4. 結論
2-5. 参考文献
1 1 4 7 9
12 13 14 14 15 15 15 16 16 16 16 18 18 21 23 23 25 29 29
第3章 酸素発生複合体における光阻害種のMnIモデル 3-1. 序
3-2. 実験
3-2-1. 試薬および測定機器
3-2-2. [MnI(cyclam)(CO)2](Br or BPh4) {[3](Br or BPh4)} の合成 3-2-3. [3](Br) から生じた [Mn2III,IV
(cyclam)2(µ-O)2](Br)3 {[5](Br)3} の単離
3-2-4. 3の酸化反応における同位体標識実験
3-2-5. 4から3への反応に伴い発生するCOの定量分析 3-2-6. 4から5への反応に伴い発生するCOの定量分析 3-2-7. 3から5への反応に伴い発生するCOの定量分析 3-2-8. 3および4の電気化学分析
3-2-9. 3および4のX線結晶解析 3-3. 結果および考察
3-3-1. 錯体の合成および同定
3-3-2. 3および4のO2及びH2Oとの反応性 3-3-3. 3とO2との反応
3-4. 結論 3-5. 参考文献
第4章 結言
発表論文目録
謝辞
32 33 34 34 35 35 36 36 36 36 36 36 38 38 43 44 48 49
53
56
57
1 第1章 緒言
1-1. 序
自然界の酸化還元酵素によって触媒される多くのエネルギー変換反応は、全てが人 類にとって有用な化学反応である。ギ酸脱水素酵素 (Formate hydrogen lyase, FHL) は、
ギ酸から水素を作り出す酸化還元酵素の複合体であり 1)、光化学系 II (Photosystem II,
PSII) は、水から電子を取り出す酸化酵素である 2)。これらの酸化還元酵素は脱炭素社
会の鍵となりうる反応を触媒するが、未だにその反応メカニズムの解明には至ってい ない。本論文では、モデル研究によりFHLとPSIIの反応メカニズムを検討した。
1-2. FHLとそのモデル研究:ギ酸からの水素発生
水素は、燃焼して排出される副生成物が水のみであることから、近年注目されてい る重要な代替エネルギー源である 3)。また、現在の水素はおもに石油の水蒸気改質お よび水性ガスシフト反応により合成されているが、バイオマスからも同様に水素を製 造することができ、水の電気分解を行うことによっても得られるため、水素は再生可 能なエネルギー源でもある 3b)。水素は常温常圧で気体であり、可燃性で爆発限界が広 いため、実用・安全面を踏まえた運搬・貯蔵方法が求められる。ギ酸を水素と二酸化 炭素に分解する反応およびその逆反応は、気体である水素を液体のギ酸として貯蔵・
運搬することを可能にするため有用であり、現在活発に研究されている 4)。また、ギ 酸は他の水素キャリアであるメチルシクロヘキサンやメタノールと比較して引火点が 高く (69 °C)、二酸化炭素を水素キャリアとして利用することができる。
FHL はギ酸を可逆的に水素と二酸化炭素に変換する酵素複合体であり、MoSe 錯体 を活性中心とするギ酸デヒドロゲナーゼ (Formate dehydrogenase, FDH) ユニット1c,d)、 NiFe錯体を活性中心とするヒドロゲナーゼ ([NiFe]H2ase) ユニット1e)及び電子伝達を行 う複数のユニットからなる (図 1-1)。 即ち、FDH は HCOOH から電子を取り出して CO2と2H+に変換し、[NiFe]H2aseはH+に電子を与えてH2に変換する。
2
図1-1. 提唱されている生体内 (Escherichia coli) のギ酸水素リアーゼ (FHL) の構造の概 略図. FdhF: ギ酸デヒドロゲナーゼ、HycE: ヒドロゲナーゼ、HycB、HycC、HycD、 HycF、HycG: 電子伝達系、黄色四角: [4Fe-4S]クラスター.
ギ酸を水素と二酸化炭素に変換する反応は、他の水素キャリアと比較して熱力学的 に有利であるため (式 1)、少ない投入エネルギーで水素を得られる 4a)。逆反応にあた る二酸化炭素をギ酸に変換する反応は、水溶液中で行うことにより若干熱力学的に有 利に進行する (式 2)。また、アンモニアなどの塩基を加えることによっても熱力学的 に有利となる (式3)。
HCOOH(l) CO2(g) + H2(g) ΔGº = –32.9 kJmol–1 (1)
CO2(aq) + H2(aq) HCOOH(l) ΔGº = –4 kJmol–1 (2)
CO2(g) + H2(g) + NH3(aq)
HCOO–(aq) + NH4+(aq) ΔGº = –9.5 kJmol–1 (3)
3
FHL そのもののモデル錯体の研究は 1 例のみであるが、1-2 の冒頭で述べたように ギ酸を水素と二酸化炭素に変換する反応およびその逆反応を触媒する錯体の研究は盛 んに行われている 5)。ギ酸を水素と二酸化炭素に変換する反応においては、燃料電池 の触媒毒となり得る一酸化炭素の発生量が不均一系触媒に対して少ない均一系の触媒 がアドバンテージを得ている。触媒にはIr、Ru、Au、Agおよび Pd などの重金属が用 いられている一方で、Fe や Co などの第一遷移金属を用いた触媒系も報告されている
5b)。単核錯体が多く、多核錯体の報告例は少ない。図 1-2 に代表的な錯体触媒を示す。
多くの研究において提唱されている反応機構にヒドリド錯体中間体の存在が示唆され ている。しかし、ヒドリド錯体におけるヒドリド配位子の性質、即ちプロトン的であ るかヒドリド的であるかを注意深く検証した例はない。中には、提唱している反応機 構においてヒドリド配位子がプロトンとヒドリドの両方の性質を有すると論じる報告
もある4a,5c,d)。
FHLのモデル錯体については Nguyen らにより二核NiRu錯体が報告されている (図
1-2e)6)。しかし、提唱している反応機構を詳細に解析しておらず、中間体として生じ
る低原子価種へのプロトン化や生じるヒドリド錯体中間体におけるヒドリド配位子の 反応性について十分な検討がなされていない。また、ギ酸デヒドロゲナーゼにおいて はギ酸からの脱炭酸の過程で、ギ酸と多点相互作用することが提唱されている。それ
に対し、Nguyen らの錯体はギ酸配位子が末端配位しており、モデル研究としては不十
分であった。
4
図1-2. 従来の触媒. (a) Himedaらの触媒、(b) Fukuzumiらの触媒、(c) Laurenczyらの触 媒、(d) Bellerらの触媒、(e) Nguyenらの触媒.
1-3. 光化学系IIとそのモデル研究:光阻害および自己修復
水が酸化されることにより酸素を生じる反応は熱力学的に不利である。ゆえに、反 応を進行させるには外部からのエネルギーが必要である。高等植物やシアノバクテリ アなどの生物による酸素発生型光合成では光エネルギーを用いて水を酸化、即ち電子 を取り出し酸素に変換することにより、変換したエネルギー源 (ATPやNADPH) を炭 素固定に利用する 7)。酸素発生型光合成生物のように水を電子源、即ち電気エネルギ ー源として活用できることは無尽蔵の太陽光エネルギーを電気エネルギーに変換でき ることを意味している。ゆえに、エネルギー利用としての光合成研究はグリーンケミ ストリーの重要な研究分野である。
光合成は高等植物が持つ葉緑体やシアノバクテリアが有するチラコイド膜内に存在 す る 複 数 の タ ン パ ク 質 複 合 体 が 行 な っ て お り 、 そ の 複 合 体 の 一 つ に 光 化 学 系 II (Photosystem II, PSII)2) がある。PSII は主に光エネルギーの吸収、電子伝達および水の 酸化を行なっている。即ち、PSII は光エネルギーを利用して水から電子を取り出して
NiII RuII H S S N
N
2+
O O H
N N
N N
N N N
N
RuII IrIII Cp*
OH2 4+
N
N IrIII
Cp*
OH2 HO
HO
2+
in situ [Ru(H2O)6](tos)2 + in situ [RuCl2(benzene)]2 + SO3Na
P
SO3Na
NaO3S P
P
(a) (b)
(c) (d) (e)
5
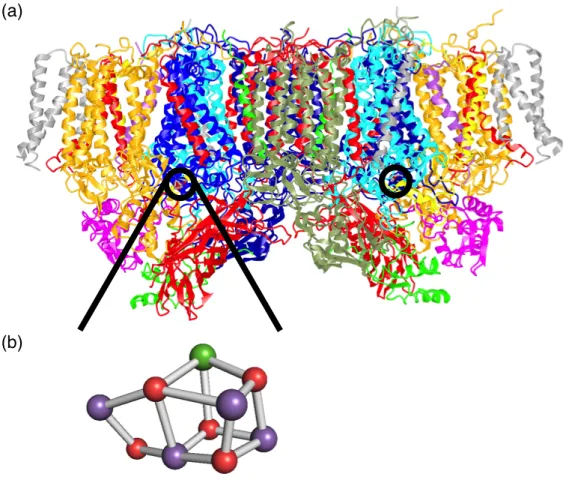
酸素に変換する光合成の反応中心である。PSII は活性中心に酸素発生複合体 (Oxygen evolving complex, OEC) を有し、Mn4CaO5で構成されるクラスターが存在することがX 線結晶構造解析により明らかにされている (図1-3) 8)。
図1-3. 酸素発生型光合成生物 (Thermosynecochoccus vulcanus由来8a)) の (a) PSII内部に おけるOEC の位置 (赤丸で示した場所) および (b) OECにおけるMn4CaO5クラスター の構造 (Mn: 紫色、Ca: 緑色、O: 赤色). 簡略化のためMn4CaO5クラスターとタンパク 質以外は除外している.
生物化学的に PSII の構造を明らかにし、機能解析を行ったり、錯体化学的に OEC のモデル錯体を構築し反応性を調べたりするなど、分野横断的に光合成研究が進めら れている。PSII は通常、光をエネルギー源として有効に利用しているが、同時に OEC が光により損傷してしまうことが知らており、光阻害と呼ばれている。一方で、光阻
6
害された OEC が自己修復することも知られているが、光阻害された OEC の分子構造 およびその自己修復のメカニズムは明らかにされていない。OEC の光阻害および自己 修復は水の酸化と複合的に行われているため、OEC における光阻害種の自己修復シス テムを明らかにすることは地上にほぼ無限に降り注ぐ太陽の光エネルギーを有効利用 する上で重要な知見となるはずである。これまでの光阻害および光修復の研究におい て、従来は過剰の光が照射されることによって生成した活性酸素種により、PSII を構 成するタンパク質が破壊されてしまうことで光阻害が起こると考えられてきた。しか し近年では、活性酸素種は光阻害を受けたOECの修復を阻害することが明らかとなっ ており、光阻害の最初の過程は光に通常の光によるOECの分解であり、脱離したMnII イオンが検出されている9)。そして検出された MnIIがOECの光阻害種ではないかとさ れているが、詳細な知見はなく、また自己修復のメカニズムも依然として不明である。
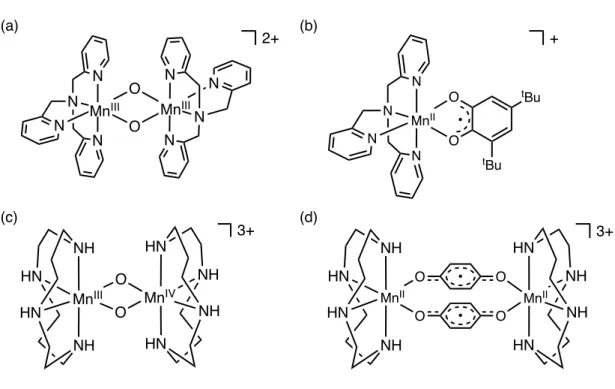
近年報告されている、錯体化学的にモデル錯体を構築した例について説明する 10)。 2014 年に Yatabe らは二核 Mn2III,IIIビス(µ-オキソ)錯体をモデル錯体として (図 1-4a)、
光阻害による分解と酸素による修復過程を提案している (図 1-4b)10a)。また、同年に Nakamoriらは二核Mn2III,IVビス(µ-オキソ)錯体を用いて (図1-4c) 同様の修復過程を提案 している (図1-4d)10b)。これら二つの系ではいずれも光阻害種としてMnII錯体が生成し、
酸素によって元の二核Mnビス(µ-オキソ)錯体を再生している。しかし、これら二つの 例は PSII の光阻害種から ESR 測定によって検出された MnIIイオンがそのまま光阻害 種の構成要素に含まれているという仮定に基づいている。即ち、OEC 内部の真の光阻 害種における Mn の電子状態は確実な証拠がない。異なる酸化状態の Mn 錯体を光阻 害種のモデルとし、先行研究と同様に酸素分子によるOEC修復システムを実現したら、
光阻害状態のOECにおけるMnの酸化状態についてより理解が深まるのではないかと 考えている。
7
図1-4. これまでに合成されたOEC光阻害種のモデル錯体. (a) YatabeらのMn2III,IIIビス (µ-オキソ)錯体および (b) その光阻害種モデル、(c) Nakamori らのMn2III,IVビス(µ-オキ ソ)錯体および (d) その光阻害種モデル.
1-4. 本論文の構成
本論文では、FHL とPSII の反応メカニズムの検討をテーマに図 1-5のような構成で 議論する。第 2 章では、水素の利用方法としてギ酸からの水素発生に着目し、FHL の モデルとして二核 Ru 錯体を用いたギ酸からの水素発生について反応機構を、特に中 間体であるヒドリド種およびジヒドリド種のプロトン性について議論する。第 3 章で は水を電子源として利用する光合成における光阻害および修復過程に着目し、MnI カ ルボニル錯体をOECにおける光阻害種のモデルとし、酸素によって自己修復されるメ カニズムを提唱する。また、カルボニル配位子の数の違いによる反応性の違いについ て議論する。これらの知見を第 4 章で総括し、本研究の意義および今後の展望につい て述べる。
3+
MnIII O O
MnIV NH
HN HN
NH
HN
NH NH HN
O O N
N N
N
N
N N
N
2+
MnIII MnIII
MnII O O
MnII O O
•
•
3+
NH HN
HN
NH
HN
NH NH HN
MnII O O N
N N
N
tBu
tBu
•
(a) (b) +
(c) (d)
8
図1-5. 本論文の構成.
MnI CO CO NH HN HN
NH
+
3+
MnIII O O
MnIV NH
HN HN
NH
HN NH NH HN MnI 2
CO CO HN
HN HN
NH
+ CO
2
CO2 CO2
RuI RuI H
O H O
RuI RuI O H O
O O
H
OC CO
S S
CO O OC
O 0
0
HCOO–
H2 H+
O2 あるいは H2O 2CO 光照射
第2章 第3章
9 1-5. 参考文献
1 (a) C. Pinske, R. G. Sawers, Anaerobic formate and Hydrogen Metabolism, EcoSal Plus, 2016, 7. (b) J. S. McDowall, B. J. Murphy, M. Haumann, T. Palmer, F. A. Armstrong, F.
Sargent, Bacterial formate hydrogenlyase complex, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3948–E3956. (c) H. C. A. Raaijmakers, M. J. Romão, Formate-reduced E. coli formate dehydrogenase H: the reinterpretation of the crystal structure suggests a new reaction mechanism, J. Biol. Inorg. Chem., 2006, 11, 849–854. (d) J. C. Boyington, V. N.
Gladyshev, S. V. Khangulov, T. C. Stadtman, P. D. Sun, Crystal Structure of Formate Dehydrogenase H: Catalysis Involving Mo, Molybdopterin, Selenocysteine, and an Fe4S4
Cluster, Science, 1997, 275, 1305–1308. (e) W. Lubitz, H. Ogata, O. Rüdiger, E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148.
2 (a) J. P. McEvoy, G. W. Brudvig, Water-Splitting Chemistry of Photosystem II, Chem.
Rev., 2006, 106, 4455–4483. (b) D. G. Nocera, The Artificial Leaf, Acc. Chem. Res., 2012, 45, 767–776.
3 (a) T. C. Johnson, D. J. Morris, M. Wills, Hydrogen generation from formic acid and alcohols using homogeneous catalysts, Chem. Soc. Rev., 2010, 39, 81–88. (b) R. M.
Navarro, M. A. Peña, J. L. G. Fierro, Hydrogen Production Reactions from Carbon Feedstocks: Fossil Fuels and Biomass, Chem. Rev., 2007, 107, 3952–3991.
4 (a) D. Mellmann, P. Sponholz, H. Junge, M. Beller, Formic acid as a hydrogen storage material – development of homogeneous catalysts for selective hydrogen release, Chem.
Soc. Rev., 2016, 45, 3954–3988. (b) J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N.
Lo, T. J. Williams, A prolific catalyst for dehydrogenation of neat formic acid, Nat.
Commun., 2016, 7, 11308. (c) T. Reda, C. M. Plugge, N. J. Abram, J. Hirst, Reversible interconversion of carbon dioxide and formate by an electroactive enzyme, Proc. Natl.
Acad. Sci. U. S. A., 2008, 105, 10654–10658.
5 (a) E. Fujita, J. T. Muckerman, Y. Himeda, Interconversion of CO2 and formic acid by bio-inspired Ir complexes with pendent bases, Biochim. Biophys. Acta, 2013, 1827, 1031–
10
1038. (b) W. H. Bernskoetter, N. Hazari, Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts, Acc. Chem.
Res. 2017, 50, 1049–1058. (c) W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita, T.
Hirose, Y. Himeda, Highly Efficiet D2 Generation by Dehydrogenation of Formic Acid in D2O through H+/D+ Exchange on an Iridium Catalyst: Application to the Synthesis of Deuterated Compounds by Transfer Deuterogenation, Chem. Eur. J. 2012, 18, 9397–9404.
(d) S. Fukuzumi, T. Kobayashi, T. Suenobu, Unusually Large Tunneling Effect on Highly Efficient Generation of Hydrogen and Hydrogen Isotopes in pH-Selective Decomposition of Formic Acid Catalyzed by a Heterodinuclear Iridium–Ruthenium Complex in Water, J.
Am. Chem. Soc., 2010, 132, 1496–1497.
6 N. T. Nguyen, Y. Mori, T. Matsumoto, T. Yatabe, R. Kabe, H. Nakai, K.-S. Yoon, S. Ogo, A [NiFe]hydrogenase model that catalyses the release of hydrogen from formic acid, Chem. Commun., 2014, 50, 13385–13387.
7 (a) V. Balzani, A. Credi, M. Venturi, Photochemical Conversion of Solar Energy, ChemSusChem, 2008, 1, 26–58. (b) G. D. Scholes, G. R. Fleming, A. Olaya-Castro, R. van Grondelle, Lessons from nature about solar light harvesting, Nat. Chem., 2011, 3, 763–774.
8 (a) Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å, Nature, 2011, 473, 55–60. (b) M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago, J.-R. Shen, Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses, Nature, 2015, 517, 99–103.
9 (a) Y. Nishiyama, S. I. Allakhverdiev, N. Murata, A new paradigm for the action of reactive oxygen species in the photoinhibition of photosystem II, Biochim. Biophys. Acta, 2006, 1757, 742. (b) X. Hou, H. J. M. Hou, Roles of manganese in photosystem II dynamics to irradiations and temperatures, Front. Biol., 2013, 8, 312–322.
10 (a) T. Yatabe, M. Kikkawa, T. Matsumoto, H. Nakai, K. Kaneko, S. Ogo, A model for the water-oxidation and recovery systems of the oxygen-evolving complex, Dalton Trans.,
11
2014, 43, 3063–3071. (b) H. Nakamori, T. Matsumoto, T. Yatabe, K.-S. Yoon, H. Nakai, S. Ogo, Synthesis and crystal structure of a dinuclear, monomeric MnII p-semiquinonato complex, Chem. Commun., 2014, 50, 13059–13061.
12
第 2 章 FHL の機能を模倣した二核 Ru 錯体によるギ酸からの水素発生のメカ ニズム
概要
本研究では、ギ酸水素リアーゼのモデルとしてギ酸架橋二核 Ru 錯体を用いて水中 におけるギ酸からの触媒的水素発生の反応機構について検討した。反応基質および溶 媒の同位体標識を行うことにより、Ru 錯体触媒の中間体であるヒドリド種がプロトン 的な性質を有することを明らかにした。
Tokunaga, T.; Yatabe, T.; Matsumoto, T.; Ando, T.; Yoon, K.-S.; Ogo, S.
Sci. Technol. Adv. Mater. 2017, 18, 870–876.
1
Ru0 Ru0 B
0 –H+
H2 RuI RuI
H
H
C A
CO2
0 HCOO–
2 RuI RuI
H
O H O
CO2
RuI RuI
H +
HCOOH pKa = 3.75
H+
RuI RuI O H O
O O
H
OC CO
S S
CO O OC
O 0
+H+ 0
1 RuI RuI
O H O
O O
H 0
=
13 2-1. 序
大腸菌 (Escherichia coli) などの数種の微生物は発酵により水素ガスを産生すること が知られている。嫌気的条件下でグルコースが解糖系により代謝される過程でピルビ ン酸からギ酸が生じ、発生したギ酸がギ酸水素リアーゼ (formate hydrogen lyase, FHL) によりCO2とH2に変換する(式 1)1)。この酵素複合体はMoを含むギ酸デヒドロゲナー ゼ (formate dehydrogenase, FDH)2)と[NiFe]ヒ ド ロ ゲ ナ ー ゼ ([NiFe] hydrogenase, [NiFe]H2ase)3)から構成されており、触媒的なギ酸の酸化 (式2) と水素発生 (式3) をそ れぞれ担っている。二つの酵素間で授受される電子は、電子伝達を担う複数のタンパ ク質を経て移動している。
第 1 章で述べたように、ギ酸を水素と二酸化炭素に変換する触媒は近年盛んに報告 さ れ て い る 。 当 研 究 室 で 報 告 さ れ た NiIIRuII モ デ ル 錯 体 [NiII(X)(HCOO)(µ- H)RuII(C6Me6)] (X = N,N’-dimethyl-3,7-diazanonane-1,9-dithiolato) はギ酸からの水素およ び二酸化炭素発生を素早く触媒する [触媒回転数 {TOF = (発生した水素の物質量/触媒
の物質量) 毎時} = 857 h–1]4)。しかし、この極めて速い反応速度のために水素発生のメ
カニズムを解明することは困難であった。即ち、錯体触媒において中間体として生じ るヒドリド錯体がH+として (プロトン的に) 振る舞うのか、あるいはH–として (ヒドリ ド的に) 振る舞うのか明らかにされていない。ヒドリド配位子の性質はプロトンとの反 応により明らかにすることができる。即ちヒドリド配位子がプロトン交換する場合は ヒドリド配位子がプロトン的な性質を有するのに対し、プロトン化されて水素を発生 する場合はヒドリド配位子がヒドリド的な性質を有すると決定できる。
HCOOH CO2 + 2H+ + 2e– [NiFe]H2ase
H2 2H+ + 2e–
(2)
(3) FDH
HCOOH FHL CO2 + H2 (1)
14
ギ酸からの水素及び二酸化炭素の発生メカニズムを解明するにあたり、今回我々は、
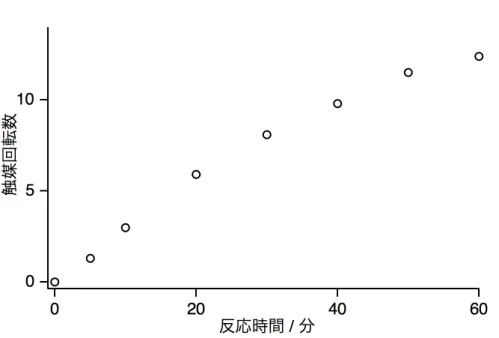
上記の反応をとても遅い反応速度で触媒する (TOF = 13.1 h–1) 二核ルテニウム錯体 [RuI2(CO)4(µ-HCOO)2(DMSO)2] (1) を報告する。基質および溶媒の同位体標識を行い、
発生する水素の同位体比を測定することにより、中間体として生じるヒドリド錯体の 性質を調べた。
2-2. 実験
2-2-1. 試薬および測定機器
全ての実験をシュレンク (Schlenk) 技術またはグローブボックスを用いることにより 窒素又はアルゴン雰囲気下で行なった。テトラヒドロフラン (THF) は使用前に窒素雰 囲 気 下 で Na/ベ ン ゾ フ ェ ノ ン で 脱 水 し 蒸 留 後 使 用 し た 。HCOOH、DCOOD、40%
NaOD/D2O およびジメチルスルホキシド (DMSO) は和光純薬工業株式会社から購入し
た。HCOOD および DCOOH は東京化成から購入した。これらの試薬は精製を行わず に使用した。
プロトン核磁気共鳴 (1H NMR) スペクトルの測定に JEOL JNM-AL300 spectrometerを 用い、測定温度を25 °Cとして、クロロホルム-d1中のテトラメチルシラン (TMS) また
は DMSO-d6中の DMSO の化学シフトを内部標準とした。UV-vis 吸収スペクトルの測
定に JASCO V-670 UV-Visible-NIR分光光度計 (光路長 1.0 cm) を用いた。25 °Cにおけ る赤外 (IR) スペクトルの測定に Thermo Nicolet NEXUS 870 フーリエ変換赤外 (FTIR) 装置を用いた。H2、HD及びD2の定量ガスクロマトグラフィー (GC) 分析は Shimadzu GC–8A (ヘリウムキャリア) にMnCl2–アルミナカラム (model: Shinwa OGO–SP) を用い て測定し、測定温度を–196 °C (液体窒素) とした。そしてH2、HD、D2及びCO2の定量
分析はShimadzu GC-2014 (アルゴンキャリア) に活性炭カラムを用いて測定し、測定温
度を100 °Cとした。元素分析にはPerkinElmer 2400II series CHNS/O analyzerを用い、
アルゴンをキャリアガスとした。動的光散乱測定にMalvern Zetasizer Nanoを用いた。
Xバンド電子スピン共鳴 (ESR) スペクトルの測定にJEOL JES-FA200 spectrometerを用 い、測定温度を–150 °Cとした。
15
溶液の pH を HCOOH 及び NaOH/H2O (1.0–7.0) を用いて調整した。溶液の pD を HCOOH、DCOOH、HCOOD、DCOOD及び40% NaOD/D2O (1.0–7.0) を用いて調整し、
D2O 中における D+濃度と比較して HCOOH 及び DCOOH の H+濃度は微量であるとし た。pH (又は pD) 1.0–7.0 の領域において、溶液の pH (又は pD) 値を pH 複合電極 (model: TOA GST-5725C、東亜ディーケーケー株式会社) を付けた pH メータ (model:
TOA HM20J、東亜ディーケーケー株式会社) を用いて決定した。pD値は測定した値に
0.4を加算することで校正した (pD = pHメータの値 + 0.4)5)。
2-2-2. [RuI2(CO)4(µ-HCOO)2(DMSO)2] (1) の合成
[Ru03(CO)12] (1.00 g、1.56 mmol) 及びHCOOH (12 mL、0.32 mol) のTHF溶液を3.5時 間還流撹拌した。溶液を室温に冷却し、DMSO (335 µL、4.72 mmol) を加えて30分撹 拌した。溶媒を減圧留去し真空乾燥して 1 の黄色粉末を得た {加えた[Ru03(CO)12]に基 づく収率: 85%}。1H NMR (300 MHz、クロロホルム-d1、TMS 基準): d 8.23 (s, 2H, HCOO), 3.06 (s, 12H, (CH3)2SO)。元素分析においては1 (C10H14O10Ru2S2) の計算値: C, 21.43; H, 2.52% に対し、測定値: C, 21.45; H, 2.22% であった。
2-2-3. 1を触媒とするHCOOHからのH2発生
セプタム付き3.0 mLサンプル管に1 (1.25 µmol)、HCOOH (2.60 mmol) 及びH2O (1.0 mL) を加え、溶液のpHを1.0–7.0に調節し80 °Cに加熱し1時間反応させた。サンプ ル管中の気相をガスタイトシリンジ (500 µL) に取り、H2及びCO2をGCで分析した。
COは検出されなかった。動的光散乱測定により微粒子の生成は確認されなかった。
2-2-4. 同位体標識による1を触媒としたH2、HD及びD2発生
セプタム付き3.0 mLサンプル管に1 (1.25 µmol) を加え、同位体標識された基質の水 溶液DCOOH (2.60 mmol) とH2O (1.0 mL)、HCOOD (2.60 mmol) とD2O (1.0 mL) 又は DCOOD (2.60 mmol) とD2O (1.0 mL) を加えた。NaOHまたはNaODを加えることで溶 液中のpH 又はpDを 3.5 に調節し、80 °Cに 1時間加熱した。サンプル管中の気相を
16
ガスタイトシリンジ (500 µL) に取り、H2、HD、D2及びCO2をGCで分析した。COは 検出されなかった。動的光散乱測定により微粒子の生成は確認されなかった。
2-2-5. 1の濃度に対する触媒的H2発生の初速度
セプタム付き3.0 mLサンプル管に1 (0.63、1.25、2.5または5.0 mM) 及びHCOOH (2.60 M) の水溶液1 mLを入れた。溶液のpHを3.5に調節し80 °Cで300秒反応させ た。サンプル管中の気相をガスタイトシリンジ (500µL) に取り、H2を GCで分析した。
触媒反応速度は触媒濃度に対して一次に比例していた。
2-2-6. HCOOHのプロトンに対するヒドリド種2の反応性
セプタム付き3.0 mLサンプル管に1 (14 mg、25 µmol) 及びHCOONa (1.7 mg、25 µmol) のDMSO (2.0 mL) 溶液を入れ80 °Cで1時間反応させた。生成した2に対し10
等量のHCOOH (9.4 µL、250 µmol) を加えた。GC分析によりH2が発生しなかったこ
とを確認した。同様の反応について、DMSOの代わりにDMSO-d6 (450 µL) を溶媒とし
て行い、1H NMRスペクトルで反応を追跡した。2のヒドリドに由来するピークの減少
は見られなかった。
2-2-7. 2のヒドリド配位子のH+/D+交換
NMRサンプル管に1 (14 mg、25 µmol) 及びHCOONa (1.7 mg、25 µmol) のDMSO-d6 (400 µL) 溶液を入れ、80 °Cで1時間反応させて2を生成させた。1H NMRにより2の 生成を確認後、窒素雰囲気下でDMSO-d6溶液にD2O (50 µL) を加えた。CH2Br2を内部 標準としヒドリド及びギ酸配位子に由来するピークの強度を 1H NMR により追跡する ことにより、2のヒドリド配位子のH+/D+交換が確認された。
2-2-8. 1のX線結晶解析
1のTHF溶液にジエチルエーテルを拡散させることでX線解析に適した1の単結晶 を得た。Rigaku/MSC Saturn CCD回折計を用いて共焦点単色Mo-Ka線 (λ = 0.7107 Å) を
17
線源として測定した。CrystalClear program を用いてデータの収集および処理を行なっ た 。 最 適 化 計 算 に SHELXL-97 を 用 い 、 そ れ 以 外 の 計 算 は 全 て CrystalStructure
crystallographic ソフトウェアパッケージを用いて行なった。1の結晶学的データはケン
ブリッジ結晶学データセンターに登録されている (登録番号: CCDC1556459)。
18 2-3. 結果及び考察
2-3-1. 錯体の合成および同定
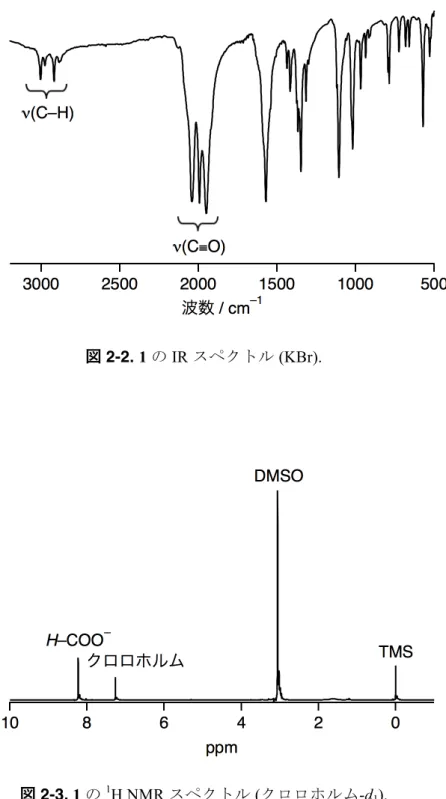
窒素雰囲気下、THF 溶液中で [Ru03(CO)12]、HCOOH および DMSO を反応させるこ とによりビス (µ-ギ酸) RuI2錯体である [RuI2(CO)4(µ-HCOO)2(DMSO)2] (1) を合成し、X 線解析 (表2-1、図2-1)、IR (図2-2)、1H NMR (図2-3) 及びUV-visスペクトル (図2-4) により確認した。二つの Ru 原子はどちらもそれぞれ二つの C(CO)、一つの S(DMSO)、
二つのO(µ-ギ酸) 及び一つの隣接するRu原子が配位している歪んだ八面体型の立体構 造を有する (図2-1)。二つのRu原子はRu–Ru単結合と二つのギ酸配位子により繋げら れている。Ru–Ru間の結合距離 {2.6654(3) Å} は他のギ酸架橋二核ルテニウム錯体の値 (2.679および2.720 Å) と類似している6)。各々のHCOO–配位子におけるC–O間の結合 距離 {C1–O1 = 1.258(2)、C1–O2 = 1.262(2)、C2–O3 = 1.263(2)、C2–O4 = 1.259(2) Å} は ほぼ同一であり、二つの C–O 結合において二重結合が非局在化していることを示して いる。四つのCO配位子におけるC–O間結合距離 {1.142(2)–1.148(2) Å} は遊離CO分 子のそれ (1.128 Å) よりも少し長いことから、低原子価 RuIの t2g軌道からCOの π*反 結合性軌道へ電子密度の逆供与が生じていることを示唆している。
表2-1. 1のX線結晶データ 1
Empirical Formula C10H14O10Ru2S2
Formula Weight 560.47
Crystal System monoclinic
Space Group P21/c
a (Å) 16.020(2)
b (Å) 7.9505(10)
c (Å) 14.045(2)
α (deg) 90.000
β (deg) 92.996(3)
γ (deg) 90.000
Volume (Å3) 1786.4(4)
Z 4
Dcalc (g/cm3) 2.084
Number of reflection 4089
R1 (I>2σ(I)) 0.0159
wR2 (all data) 0.0512
GOF 1.359
19
図2-1. 1のORTEP図. DMSOの水素原子は省略されている. 原子間距離 (l/Å) 及び角度 (φ/º): Ru1–Ru2 = 26654(3)、Ru1–O2 = 2.1152(13)、Ru1–O3 = 2.1290(13)、Ru1–S1 = 2.4107(5)、Ru1–C3 = 1.855(2)、Ru1–C5 = 1.853(2)、Ru2–O2 = 2.1374(13)、Ru2–O4 = 2.1221(13)、Ru2–S2 = 2.4134(5)、Ru2–C4 = 1.855(2)、Ru2–C6 = 1.849(2)、C1–O1 = 1.258(2)、C1–O2 1.262(2)、C2–O3 = 1.263(2)、C2–O4 = 1.259(2)、C3–O5 = 1.145(2)、
C4–O6 = 1.142(2)、C5–O7 = 1.148(2)、C6–O8 = 1.146(2)、O1–C1–O2 = 126.67(18)、O3–
C2–O4 126.76(17).
1の IRスペクトルにおいて 1950、1994及び2041 cm–1に RuIに配位している COの 伸縮振動由来のピークが見られ (図 2-2)、遊離 CO 分子の伸縮振動由来の波数 (2143 cm–1) よりも低い。この結果もまた低原子価 RuIから CO配位子への逆供与によるもの
である。1H NMRスペクトルのシグナルが反磁性領域に現れることから、二つのRuI中
心の金属–金属結合を介した反強磁性交換相互作用により 1が反磁性となることを示し ている (図2-3)。H2O中における1の紫外-可視吸収スペクトルにおいて、280 nmに鋭 い吸収帯 (8480 M–1cm–1)、420 nmに幅の広い吸収帯 (550 M–1cm–1) が見られる (図2-4)。
C2
O2 O1 C1
O3 O4
H2
H1
C5 O7 C6
O8
O5 C3
O6 C4 Ru1
Ru2
S2 C10
O10
C9 S1
C8 C7
O9
20
図2-2. 1のIRスペクトル (KBr).
図2-3. 1の1H NMRスペクトル (クロロホルム-d1).
21
図2-4. 1のUV-vis吸収スペクトル (0.060 mM水溶液).
2-3-2. 1から2の生成
ビス (µ-ギ酸) RuI2錯体1はDMSO 中において1等量の HCOONa存在下で80 °Cに 加熱することによりCO2を放出すると同時に (ヒドリド)(ギ酸) 種 2を生じる。二つの 金属中心により β-水素脱離が促進され、CO2 の放出とヒドリド配位子の形成が進行す ると考えられる7)。DMSO-d6中における 2の 1H NMRスペクトルからヒドリド及びギ 酸由来のシグナルがそれぞれ–12.4 及び 7.76 ppm に同じ強度で検出される (図 2-5a)。 ヒドリド由来のピークは既存のヒドリド架橋Ru錯体に見られるピークと同様である8)。 2 が二量体構造を有し、二つの RuI中心間が反強磁性交換相互作用していることは 1H NMR スペクトルの反磁性領域にシグナルが観測されること及び ESR が観測されない ことから支持される。
22
図2-5. 2の1H NMR. (a) D2O添加前および (b) D2O添加後. 2は1のDMSO-d6溶液を1
等量のHCOONa存在下80 °Cで1時間加熱することで得られた。内部標準: CH2Br2 の
ピーク.
23
2-3-3. 2におけるヒドリド配位子の性質
我々は 2 のヒドリド配位子がヒドリド的でなくプロトン的な性質を有することを以 下の実験により明らかにした。2 は HCOOH のプロトンに対して反応不活性であり、
即ちヒドリド配位子をプロトン化して水素ガスを発生しないことが 1H NMR スペクト ルおよびGC分析により確認された。そして、2のヒドリド配位子が H+/D+交換するこ
とを1H NMRスペクトルにより確認した (図 2-5)。DMSO-d6中で2 のヒドリド配位子
のピーク強度はD2Oを添加することによってのみ減少し (図 2-5b)、ヒドリド配位子が D+とH+/D+交換することを示している。
2-3-4. 1を触媒とするHCOOHからのH2発生
錯体1はpH 1–7の水中においてHCOOH からH2及びCO2への変換触媒となり、反
応性は pHに依存する (図 2-6–10及び表 2-2)。GC を用いてH2及びCO2を検出した。
動的光散乱測定において、触媒反応におけるナノ粒子の生成は見られなかった。図2-6 には水中で80 °Cにおける1と過剰量のHCOOHとの反応により生じたH2の触媒回転 数 (Turnover number、TON、発生した H2の物質量/1の物質量) の経時変化を表したも のである。TON は pH に依存し、pH 3.5 で最大となった (図 2-7)。1 の濃度 (0.63–5.0 mM) に対する H2発生の初速度の依存性を調べた。触媒反応が触媒濃度に対して一次 に比例することを明らかに示している線形な相関が得られた (図2-8)。
24
図2-6. 1 (1.25 mM) を触媒とするH2O中80 °CにおけるHCOOH (2.60 M) からのH2発 生の時間に対する触媒回転数.
図2-7. 1 (1.25 mM) を触媒とするH2O中80 °CにおけるHCOOH (2.60 M) からのH2発 生のpH依存性. 触媒回転数の最大値はpH 3.5において13.1である.
25
図2-8. 1の濃度 (0.63–5.0 mM) に対するH2O中pH 3.5、80 °CにおけるHCOOH (2.60 M) からのH2発生初速度のプロット. 300秒後のH2発生量を測定.
2-3-5. 同位体標識による1を触媒としたH2、HD及びD2発生
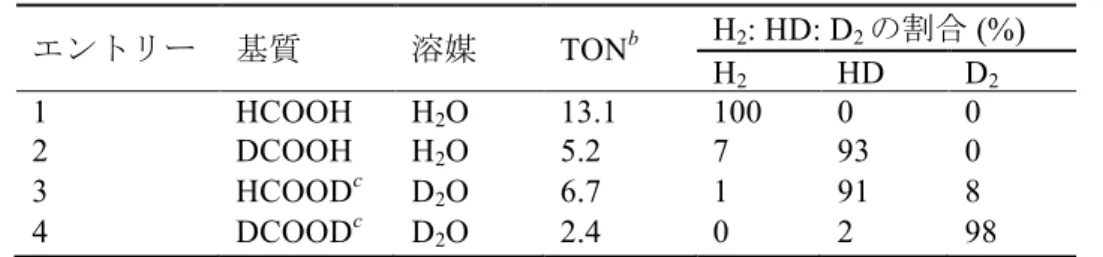
HCOOH の水素脱離の反応機構を調べるため、表 2-2 に示すように重水素同位体標
識実験を行なった。エントリー2 (DCOOH / H2O、pH 3.5) 及び3 (HCOOD / D2O、pD 3.5) ではTONがそれぞれ5.2及び6.7であり、エントリー1 (HCOOH / H2O、pH 3.5) に おける 13.1 よりも低い値である。これらの結果は HCOOH における C–H 結合及び H2OにおけるO–H結合の解離が反応律速段階に関与していることを示唆している。エ ントリー4 (DCOOD / D2O、pD 3.5) におけるTONは、エントリー2及び3から予想さ れるように、2.4 であり最も低い。エントリー1–4 の結果に基づいて図 2-9 に反応機構 を示す。ビス (µ-ギ酸) 錯体1はCO2の放出を伴って (ヒドリド)(ギ酸) 種2に変化する。
続く2 の脱炭酸によりジヒドリド種 Aを生じることが、報告されている類似した構造 からも予想される9)。そして H2の還元的脱離により低原子価種 Bを与える。Bのプロ トン化によりモノヒドリド種Cが生成し、HCOO–が配位することにより2に戻る。提 案した反応機構には B のプロトン化する段階、及び共役酸の pKa 3.75 の HCOO–が C に結合する段階が含まれており、そのために TON の最大値が pH 3.5 付近に生じる。
26
酸性条件ではプロトン化する段階が促進され、塩基性条件では HCOOH を脱プロトン 化することによりCにHCOO–が結合する段階が促進されるためである。
表 2-2. H2O または D2O 中における 1 を前駆体とする HCOOH、DCOOH、HCOOD ま
たはDCOODの脱水素化a
エントリー 基質 溶媒 TONb H2: HD: D2の割合 (%)
H2 HD D2
1 HCOOH H2O 13.1 100 0 0
2 DCOOH H2O 5.2 7 93 0
3 HCOODc D2O 6.7 1 91 8
4 DCOODc D2O 2.4 0 2 98
a 反応条件: 1 (1.25 mM) およびHCOOH (2.60 M)、DCOOH (2.60 M)、HCOOD (2.60 M) またはDCOOD (2.60 M)をpHまたはpD 3.5のH2OまたはD2O中80 °Cで1時間撹拌.
b 触媒回転数: 発生した水素ガス (H2、HDまたはD2) の物質量/1の物質量. c HCOODお
よびDCOODには2%のH原子が含まれている.
27
図2-9. H2O中1を触媒とするHCOOHからH2及びCO2への変換の反応機構.
表 2-2 に示すように発生した水素ガスを観測することにより水素の同位体交換反応 について検討した。エントリー2ではH2O中における1とDCOOH との反応によりH2
(7%) 及び HD (93%) を生じた。この結果は発生した水素ガス (H2及び HD) の原子が
H2OのHと DCOOHの Dに由来していることを明確に示している (図 2-10a)。発生し
たH2はH2OのH+のみがH2を与えることを示すため、反応機構の解明に重要な生成物 である。この生成物により、ヒドリド配位子のプロトン化により水素ガスが発生する 可能性を除外することができる。エントリー3においても同様である (図2-10b)。プロ トン性ヒドリド配位子の H+/D+交換を伴い、水素ガスの還元的脱離を起こす水素同位 体交換の反応機構を図2-10に提案する10)。
1
Ru0 Ru0 B
0 –H+
H2 RuI RuI
H
H
C A
CO2
0 HCOO–
2 RuI RuI
H
O H O
CO2
RuI RuI
H +
HCOOH pKa = 3.75
H+
RuI RuI O H O
O O
H
OC CO
S S
OC CO O
O 0
+H+ 0
1 RuI RuI
O H O
O O
H 0
=
28
図2-10. 表2-2の (a) エントリー2および (b) エントリー3における二核Ru錯体を触媒 とする水素発生における同位体交換反応.
2 RuI RuI
H
O H O
0 RuI RuI
D
O H O
0
2-d1
RuI RuI H
H
0
A RuI RuI
D
H
0
A-d1 RuI RuI
D
D
0
A-d2
Ru0 Ru0 B
0 C-d1 RuI RuI
D +
CO2
D+
H+ H+ D+
D+ H+
CO2
D2 8%
HD 91% H2 1%
D+
HCOO–
2-d2 RuI RuI
D
O D O
0 RuI RuI
H
O D O
0
2 (C-d1)
RuI RuI D
D
0
A-d2 RuI RuI
D
H
0
A-d1 RuI RuI
H
H
0
A
Ru0 Ru0 B
0 C
RuI RuI
H +
CO2
H+
D+ D+ H+
H+ D+
CO2
H2 7%
HD 93% D2 0%
H+
DCOO–
(b) 表2-2、エントリー 3、基質: HCOOD、溶媒: D2O (a) 表2-2、エントリー 2、基質: DCOOH、溶媒: H2O
29 2-4. 結論
合成した二核ルテニウムビス(µ-ギ酸)錯体1を用いた溶媒および基質の同位体標識実 験により、H2O/D2O中の1によるギ酸 (HCOOH又は DCOOH) からの水素発生はプロ トン性ヒドリド配位子の H+/D+交換とプロトン性ジヒドリド配位子の還元的脱離を経 ることが示された。中間体であるヒドリド錯体のヒドリド配位子について H+/D+交換 が見られた一方、プロトン化による水素発生は見られなかったことが 1H NMR より明 らかとなった。溶媒と基質のどちらか一方を重水素で同位体標識した場合、HD ガス が主生成物であった。FHLの機能モデル錯体1はFHLにおけるギ酸からの水素発生と 同様に、先にギ酸からの脱炭酸が進行して 2 そしてジヒドリド中間体 A を生成し、プ ロトン性のジヒドリドから水素を発生した。
2-5. 参考文献
1 (a) C. Pinske, R. G. Sawers, Anaerobic Formate and Hydrogen Metabolism, EcoSal Plus, 2016, 7. (b) C. Pinske, M. Jaroschinsky, R. G. Sawers, Levels of control exerted by the Isc iron-sulfur cluster system on biosynthesis of the formate hydrogenlyase complex,
Microbiology, 2013, 159, 1179–1189. (c) L. Forzi, R. G. Sawers, Maturation of [NiFe]- hydrogenases in Escherichia coli, Biometals, 2007, 20, 565–578. (d) K, Bagramyan, A.
Trchounian, Structural and Functional Features of Formate Hydrogen Lyase, an Enzyme of Mixed-Acid Fermentation from Escherichia coli, Biochemistry (Moscow), 2003, 68, 1159–
1170.
2 (a) R. Hille, J. Hall, P. Basu, The Mononuclear Molybdenum Enzymes, Chem. Rev., 2014, 114, 3963–4038. (b) P. J. Gonzalez, M. G. Rivas, C. S. Mota, C. D. Brondino, I. Moura, J.
J. G. Moura, Periplasmic nitrate reductases and formate dehydrogenases: Biological control of the chemical properties of Mo and W for fine tuning of reactivity, substrate specificity and metabolic role, Coord. Chem. Rev., 2013, 257, 315–331.
3 (a) Special issue on Hydrogenases Eur. J. Inorg. Chem., 2011, 2011, 915–1171. (b) Special issue on Renewable energy Chem. Soc. Rev., 2009, 38, 1–300. (c) C. Tard, C. J.
30
Pickett, Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases, Chem. Rev., 2009, 109, 2245–2274. (d) Special issue on Hydrogen Chem. Rev. 2007, 107, 3900–4435.
4 N. T. Nguyen, Y. Mori, T. Matsumoto, T. Yatabe, R. Kabe, H. Nakai, K.-S. Yoon, S. Ogo, A [NiFe]hydrogenase model that catalyses the release of hydrogen from formic acid, Chem.
Commun. 2014, 50, 13385–13387.
5 (a) P. K. Glasoe, F. A. Long, Use of glass electrodes to measure acidities in deuterium oxide, J. Phys. Chem. 1960, 64, 188–190. (b) K. Mikkelsen, S. O. Nielsen, Acidity measurements with the glass electrode in H2O-D2O mixtures, J. Phys. Chem. 1960, 64, 632–637.
6 (a) D. J. Morris, G. J. Clarkson, M. Wills, Insights into Hydrogen Generation from Formic Acid Using Ruthenium Complexes, Organometallics, 2009, 28, 4133–4140. (b) M. Czaun, A. Goeppert, J. Kothandaraman, R. B. May, R. Haiges, G. K. S. Prakash, G. A. Olah, Formic Acid As a Hydrogen Storage Medium: Ruthenium-Catalyzed Generation of Hydrogen from Formic Acid in Emulsions, ACS Catal., 2014, 4, 311–320.
7 A. Roberson, T. Matsumoto, S. Ogo, The development of aqueous transfer hydrogenation catalysts, Dalton Trans., 2011, 40, 10304–10310.
8 (a) M. R. Churchill, T. S. Janik, T. P. Duggan, J. B. Keister, Synthesis, Characterization, and Crystal Structure of (µ-H)2Ru3(µ3-η2-CHC(O)OCH3)(CO)9, a Stabilized Intermediate in the Reductive Elimination of Hydrocarbons from Trimetallic Clusters, Organometallics, 1987, 6, 799–805. (b) Y. Gao, J. K. Kuncheria, H. A. Jenkins, R. J. Puddephatt, G. P. A.
Yap, The interconversion of formic acid and hydrogen/carbon dioxide using a binuclear ruthenium complex catalyst, J. Chem. Soc., Dalton Trans., 2000, 3212–3217. (c) J. B.
Pelayo-Vázquez, F. J. González, M. A. Leyva, M. Campos, L. A. Torres, M. J. Rosales- Hoz, A ruthenium carbonyl cluster containing a hydroquinone ligand: A layered structure with a polymetallic species. Structure and electrochemical characterization, J. Organomet.
Chem., 2012, 716, 289–293. (d) Md. J. Hossain, S. Rajbangshi, Md. M. M. Khan, S. Ghosh,
31
G. Hogarth, E. Rosenberg, K. I. Hardcastle, M. G. Richmond, S. E. Kabir, Experimental and computational studies on the reaction of silanes with the diphosphine-bridged triruthenium clusters Ru3(CO)10(µ-dppf), Ru3(CO)10(µ-dppm) and Ru3(CO)9{µ3- PPhCH2PPh(C6H4)}, J. Organomet. Chem., 2014, 767, 185–195.
9 (a) T. Mayer, H.-C. Böttcher, Protonation of metal-metal bonds in coordinatively unsaturated diruthenium cores, Polyhedron, 2013, 50, 507–511. (b) A. R. Siedle, R. A.
Newmark, L. H. Pignolet, Organometallic Chemistry of Fluorocarbon Acids. Synthesis, Structure, and Solvolysis of a Sulfinate-Bridged Diruthenium Dihidride Cluster,
[(Ph3P)4Ru2(µ-H)2(µ-CF3SO2)(CO)2]HC(SO2CF3)2, Inorg. Chem., 1986, 25, 1345–1351.
(c) K. Geetharani, S. K. Bose, S. Sahoo, B. Varghese, S. M. Mobin, S. Ghosh, Cluster Expansion Reactions of Group 6 and 8 Metallaboranes Using Transition Metal Carbonyl Compounds of Groups 7–9, Inorg. Chem. 2011, 50, 5824–5832. (d) H. Nagashima, T.
Fukahori, K. Aoki, K. Itoh, Partial Hydrogenation of Acenaphthylene on the Face of a Triruthenium Cluster: Formation of (µ2:η1:η5-C12H10)Ru3H2(CO)7 from (µ3:η2:η3:η5- C12H8)Ru3(CO)7, J. Am. Chem. Soc., 1993, 115, 10430–10431. (e) J. van Buijtenen, J.
Meuldijk, J. A. J. M. Vekemans, L. A. Hulshof, H. Kooijman, A. L. Spek, Dinuclear Ruthenium Complexes Bearing Dicarboxylate and Phosphine Ligands. Acceptorless Catalytic Dehydrogenation of 1-Phenylethanol, Organometallics, 2006, 25, 873–881.
10 S. Ogo, Electrons from hydrogen, Chem. Commun., 2009, 3317–3325.
32
第3章 酸素発生複合体における光阻害種のMnIモデル
概要
本研究では、酸素発生複合体における光阻害種のモデルとして、MnI(cyclam)錯体 (cyclam: 1,4,8,11-tetraazacyclotetradecane) を用いてO2およびH2Oとの反応性について検 討 し た 。 反 応 性 は CO 配 位 子 の 数 に よ っ て 変 化 し た 。MnI ジ カ ル ボ ニ ル 錯 体 、 [MnI(cyclam)(CO)2]+は H2O とは反応せず O2と反応してビス(µ-オキソ)Mn2III,IV錯体を生 じた。一方でMnIトリカルボニル錯体、[MnI(cyclam)(CO)3]+は O2および H2O のどちら にも反応性を示さなかった。新規に合成したMnI(cyclam)ジカルボニル錯体を ESI質量
分析、UV-vis吸収スペクトル、IRスペクトル及びX線解析により特定した。
Yatabe, T.; Tokunaga, T.; Matsumoto, T.; Kikkawa, M.; Yoon, K.-S.; Ogo, S.
Chem. Lett. 2018, 47, 34–36.
C2 O2
C1 O1 Mn1
N1
N2
N4
N3
33 3-1. 序論
自然界の生物には自己修復システムが備わっている。光化学系 II (Photosystem II、
PSII) における活性中心である酸素発生複合体 (Oxygen-evolving complex、OEC) にも自 己修復システムが備わっている1)。OEC の Mn4CaO5クラスターは光阻害により損傷す るとMnII種を生じ 2)、自己修復過程により元の Mn4CaO5クラスターを回復すると提唱 されてきた (図3-1)1)。
しかしながら今日まで、光阻害を受けた Mn 種は完全には特定されておらず、修復 機構は明らかにされていない。即ち、どのようなMn種がH2OまたはO2と反応して架 橋オキソ配位子を回復するか明らかにされていない。
図3-1. PSIIにおける活性中心、OECの光阻害及び修復システム.
我々は二種類の MnII種、MnII p-セミキノナト錯体および MnII(ClO4)2·6H2O について H2O ではなく O2と反応して、従来の OEC モデルとされるビス(µ-オキソ)Mn2III,IV錯体、
即ち[Mn2III,IV(cyclam)2(µ-O)2]3+ (5、cyclam = 1,4,8,11-tetraazacyclotetradecane) を生じるこ とを報告した 3-5)。その研究に続き、我々は異なる酸化数を有する Mn 種を光阻害モデ ルとして研究し、O2およびH2Oとの反応性について検討してきた。
本 研 究 で は 二 つ ま た は 三 つ の CO 配 位 子 を 有 す る MnI(cyclam)錯 体 、 [MnI(cyclam)(CO)2]+ (3) および[MnI(cyclam)(CO)3]+ (4)6) について、合成および特性評価
光阻害 光照射 修復
Mn4CaO5 クラスター Mn光阻害種
34
をおこなった。加えて、O2および H2O との反応性についてビス(µ-オキソ)Mn2III,IV錯体 5を生じるかどうか検討した (図3-2)。
図 3-2. ビス(µ-オキソ)Mn2III,IV 錯体、[Mn2III,IV(cyclam)2(µ-18O)2]3+ (5-18O2) を生成する MnI(cyclam)ジ カ ル ボ ニ ル 錯 体 [MnI(cyclam)(CO)2]+ (3) お よ び ト リ カ ル ボ ニ ル 錯 体 [MnI(cyclam)(CO)3]+ (4) の18O2又はH218Oとの反応性.
3-2. 実験
3-2-1. 試薬および測定機器
全ての実験をシュレンク (Schlenk) 技術またはグローブボックスを用いることにより 窒素又はアルゴン雰囲気下で行なった。アセトニトリル (CH3CN) 及びメタノール (CH3OH) は使用前に窒素雰囲気下で CaH2または Mg/I2 で脱水して蒸留後使用した。
[MnI(Br)(CO)5]および1,4,8,11-テトラアザシクロテトラデカン (cyclam) はアルドリッチ 社から購入した。18O2 (98 atom%) は昭光通商株式会社から購入した。これらの試薬は 精製を行わずに用いた。[MnI(cyclam)(CO)3](Br) {[4](Br)} は文献に従って合成した6)。
MnI CO CO NH HN HN
NH
+
3+
MnIII
18O
18O
MnIV NH
HN HN
NH
HN NH NH HN
18O2
5-18O2 3
4CO + e– MnI 2
CO CO HN
HN HN
NH
+ CO
4
18O2
H218O
H218O 2
2CO 光照射
![図 1-1. 提唱されている生体内 (Escherichia coli) のギ酸水素リアーゼ (FHL) の構造の概 略図 . FdhF: ギ酸デヒドロゲナーゼ、 HycE: ヒドロゲナーゼ、 HycB 、 HycC 、 HycD 、 HycF、HycG: 電子伝達系、黄色四角: [4Fe-4S]クラスター](https://thumb-ap.123doks.com/thumbv2/123deta/9914627.1917705/6.892.160.741.153.475/提唱いるリアーゼ酸デヒドロゲナーゼヒドロゲナーゼクラスター.webp)


![図 3-2. ビス (µ- オキソ )Mn 2 III,IV 錯体、 [Mn 2 III,IV (cyclam) 2 (µ- 18 O) 2 ] 3+ (5- 18 O 2 ) を生成する Mn I (cyclam)ジ カ ル ボ ニ ル 錯 体 [Mn I (cyclam)(CO) 2 ] + (3) お よ び ト リ カ ル ボ ニ ル 錯 体 [Mn I (cyclam)(CO) 3 ] + (4) の 18 O 2 又は H 2 18 O との反応性](https://thumb-ap.123doks.com/thumbv2/123deta/9914627.1917705/38.892.220.680.262.683/ビスµオキソIIIIV錯体IIIIVµO+O生成するcyclamジカルボ.webp)
