医療機関における病院薬剤師の 医療安全への貢献に関する研究
日本大学医学部附属板橋病院・薬剤部
早坂 正敏
目 次
序章...1
基礎となる原著論文...7
第
1章 医薬品添付文書情報に基づく薬物間相互作用情報の構築とその評価...9
第
1節 諸言...9
第
2節 処方入力時における薬物間相互作用自動チェックシステムの構築...10
第
1項 目的...10
第
2項 方法...10
第
3項 結果...11
第
4項 考察...12
第
3節 薬物間相互作用チェックシステムによる中毒患者の服用薬剤の検討...13
第
1項 目的... ...13
第
2項 方法... ...14
第
3項 結果...14
第
4項 考察... ....15
第
4節 本章のまとめ
...16第
2章 医療現場に則した医薬品安全性情報の補完に関する検討...17
第
1節 諸言...17
第
2節 がん化学療法後の好中球減少症に対する
G-CSF製剤使用法に対する情報の 検討...18
第
1項 目的...18
第
2項 方法...19
第
3項 結果...20
第
4項 考察...24
第
3節 注射用抗がん剤希釈濃度の違いによる副作用情報の検討...25
第
1項 目的...25
第
2項 方法...27
第
3項 結果...30
第
4項 考察...36
第
4節 副作用情報を補完するための患者背景因子の検討...38
第
1項 目的...38
第
2項 方法...39
第
3項 結果...40
第
4項 考察...42
第
5節 本章のまとめ
...43第
3章 医療機関における未知の医薬品安全性情報の収集に関する検討
...45第
1節 諸言...45
第
2節 日本大学医学部データウェアハウスの医療情報を使用した副作用に対する薬 剤疫学的検討...46
第
1項 目的...46
第
2項 方法...47
第
3項 結果...51
第
4項 考察...54
第
3節 臨床現場における新規薬物間相互作用情報の探索...55
第
1項 目的...55
第
2項 方法...56
第
3項 結果...57
第
4項 考察...61
第
4節 本章のまとめ
... ...64総括... ...65
謝辞...6 7
引用文献... ...68
1
序 章
病院薬剤師は, 「医薬品の適正使用への貢献」, 「患者サービスの向上」, 「チーム医療の 充実」を目的とし,薬剤管理指導業務を実践している。薬剤管理指導業務の実践には,
医薬品や薬物療法に関するエビデンスに基づいた情報を適確に入手する必要がある。ま た,これらの情報の正確性や妥当性を評価したうえで,臨床現場に提供できるかどうか の判断を求められることが多い。さらに,臨床現場において患者,医師および看護師か ら医薬品や薬物療法に関する様々な情報提供を求められている。
病院における医薬品情報の根源は,薬事法第
52条で定められた医療用医薬品添付文 書(医薬品添付文書)である。医薬品添付文書は唯一の法的根拠に基づく文書であり,
1995
年に施行された製造物責任法(
PL法)により指示および警告義務が極めて重要視 され,医薬品の適正使用のためには必要不可欠な情報源と位置付けされている。医薬品 添付文書は,
1993年に起きた薬物相互作用による薬害であるソリブジン事件を契機に
1 ),
1997年 に 記載 様式 が改 訂 され
2 ~ 4 ), 現 在では 独 立行 政法 人医 薬品医 療 機器 総合 機構
(PMDA;Pharmaceuticals and Medical Devices Agency)からインターネットで公開さ れている。さらに,医薬品添付文書よりも詳しい情報源としては日本病院薬剤師会が記 載要領を定めたインタビューフォームがある。
医薬品添付文書の有効性と安全性情報および効能・効果に関しては治験データを検証 した結果で承認されたものであり,副作用に関しては製造販売に至るまでの動物実験や 治験の結果をもとに収集されたものであるが,ほとんどが羅列された表記となっている
5 )
。医薬品添付文書における副作用情報は,製造販売後調査などから随時追加・修正さ
れてはいるが,主として新たな副作用項目の追加記載である。このように,医薬品添付
文書の情報は治験および製造販売後調査での画一的な情報であり,臨床の現場が求める
情報であるかは疑問が生じるところではある。しかし,薬物療法における最大のリスク
2
である副作用については,医薬品添付文書に様々な注意喚起を行うことで,その症状を 軽減し,さらには予防することが可能であり,中でも重篤な副作用は,医療従事者が医 薬品添付文書の内容を把握して使用し,患者が決められた用法・用量を守ることで
3~7割は防ぐことができるといわれている
6 )。これらのことから医薬品を適正に使用する上 で医薬品添付文書は最も重要な情報であることが明白である。その情報を医師による処 方作成時に提供することは大きな意義があると考える。特に,製造販売後に薬物療法を 行う場合,複数の医薬品が併用されることから,医薬品を有効かつ安全に使用するため には,薬物間相互作用のチェックが重要である。
医薬品添付文書の主たる情報源である治験(第Ⅰ相~第Ⅲ相)では治験薬の有効性と 安全性を適正に評価する方法が採用されている。しかしながら,医薬品の安全性を評価 するには,①発現頻度が著しく低い副作用があること,②大方の副作用が出そろうまで に長い期間の観察が必要であること,③副作用の発現には病態や合併症および併用薬と の相互作用が複雑に関与していることなどの理由により,治験段階でその医薬品の安全 性を全般的に把握するには限界がある。これを端的に表す言葉として ,Rogers は
5つ の
too (Five toos)を挙げている
7 )。①Too few : 米国の治験では総被験者数は
2,000人 未満である。仮に
10,000人に
1件の副作用を
95%の確率で検出するには,30,000人の 被験者が必要となる
8 )。このような稀な副作用を製造販売前に検出することは困難であ る。②Too simple : 治験段階では薬効評価のために,複雑な病状の患者や薬物療法を受 けている患者は除外される。③Too median-aged : 極端な若年や高齢者は,治験ではめ ったに組み込まれない。④Too narrow : 治験では被験者の適応は明確に求められている。
製 造 販 売 後 は リ ス ク
/ベ ネ フ ィ ッ ト 比 の ま っ た く 異 な る 病 態 に も 用 い ら れ る 。 ⑤
Toobrief :
数年にわたって使用した後に出現する副作用や,1 回用いた後,長期間を要して
出る副作用(例:ジエチルスチルベストロールによる膣がん)を製造販売前に知ること
は困難である。
3
次に,製造販売後の医薬品使用に関する重要な課題の一つは安全性つまり副作用情報 の管理である。製造販売後では治験段階で予測しなかった様々な環境下で医薬品が患者 に使用される。医薬品の製造販売後では製薬企業による製造販売後調査,医療関係者か らの自発報告,データベースを使用した薬剤疫学調査などを通して
Five toosを補完し ていく必要があると考える。すなわち,①Too few を補完するには,使用成績調査等で 症例集積を検討すること,②Too simple を補完するには,肝・腎障害等の合併症を有す る患者や妊婦・授乳婦に実際に投与された情報や病態および他の薬物との相互作用を検 討すること,③Too median-aged を補完するには,特定使用成績調査等で小児または高 齢者に使用された情報を検討すること,④Too narrow を補完するには,適応疾患の周 辺領域や適応外疾患に実際に投与された情報を検討すること,⑤Too brief においては,
治験薬が使用されていた期間より長い期間投与される薬が多く存在し,治験の時には確 認できなかった副作用が初めて発現する可能性が高いことから,製造販売後の安全性に 関する情報について継続して評価することが必要である
9 ~ 1 2 )。さらに,医薬品の有用 性を確保しつつ,副作用をできるだけ減らすための工夫が必要であり新しいバイオマー カーや患者背景因子の検討が必要と考える。
医薬品安全性情報の基本は個別症例を念頭に置いた副作用報告であり,その情報源に
は医学雑誌で報告される副作用症例(case report)と行政報告対象の副作用自発報告の
2種類に大別され,中でも自発報告に大きく依存してきた。自発報告は,臨床現場での
使用実態を反映し,報告方法が比較的容易で,未知・重篤な副作用を収集するために最
も効果的な手段であるが,常に過少報告の懸念がつきまとい,詳細な情報や追加情報が
得られにくく,使用患者数等の分母情報がないために発生頻度が示せないという 欠点が
ある。医薬品の副作用対策(予防も含む)を直接的および間接的に向上させるためには ,
薬剤師がエビデンス・データや蓄積情報を収集して評価し,自発報告における事象と医
薬品との因果関係を自ら明確にすることが重要と考える。
4
以下に,医薬品の安全対策に結びついた代表的な自発報告について
2件紹介する。
1.カプトプリルによる空咳出現の報告
アンジオテンシン変換酵素(ACE)阻害薬による空咳は現在では周知された副作用で あるが,日本でカプトプリルが発売されてから
2年経過した
1985年に瀬底らにより世 界で初めて報告された
13 )。67 歳女性の高血圧症患者に
ACE阻害薬であるカプトプリ ルを投与して
3ヵ月後に空咳を訴え始め,3 ヵ月間持続した。そのためカプトプリルか らメチルドパに変更し,他の併用薬もすべて投与中止した結果,空咳は
2,3日で消失 した。
3週間後メチルドパからカプトプリルに変えて再投与すると
2,3日以内に咳が出 現し,再度中止して咳が消失した。さらに,カプトプリルの投与を再開すると,咳が再 発し,カプトプリルによる咳であることが確認された。この経過から,瀬底らは
ACE阻害薬と咳嗽の関連が考えられると報告している。
2.納豆によるワルファリンカリウム作用減弱の報告
ワルファリンカリウム服用患者に納豆を摂取しないよう指導するのは現在では周知さ れているが,日本で
1962年にワルファリンカリウムが発売されてから
16年経過した
1978年に工藤らにより初めて納豆によるワルファリンカリウムの作用減弱が報告され
た
1 4 )。ワルファリンカリウムによる抗凝血療法中でトロンボテスト値(TT 値)が安定
していた患者
3例のうち,
2例は週に
2~3回,
1例は週に
4~5回納豆を食べた結果
TT値が上昇した,その後納豆の摂取を止めると
TT値が低下した。さらに,健康成人によ
る納豆摂取試験においても同様に
TT値が上昇することを確認した。ビタミンKはワル
ファリンカリウムの作用を減弱する。納豆の原料である大豆自体のビタミンK含有量は
ホウレン草やキャベツなどの緑黄色野菜のビタミンK含有量に比べ少ない。しかし,納
豆菌はビタミンK合成能が高い
Bacillus subtilisに属しており,納豆の摂取により腸
内で多量のビタミンKを合成し,その結果ワルファリンカリウムの作用を減弱させるこ
とが確認された。
5
これら
2つの事例のように,自発報告からでも未知の副作用の特定や相互作用の検出 が可能であることから,安全性情報への貢献の基本は,副作用個別症例報告にあると考 えられる。
薬剤師は,数ある医薬品情報を十分に熟知したうえで,薬物治療における資質の向上 や患者の治療認識度の改善に寄与することで,チーム医療の一員として貢献することが 求められている。薬剤管理指導業務を通して,薬剤師は副作用を自発報告できるような クリニカルクエスチョンを見出し,薬学的な観点から副作用情報を蓄積したうえで個々 の事象を検証する必要があると考える。医療機関における医薬品安全対策と安全性情報 収集への薬剤師の関与についてはいずれにおいても検討すべき課題は多いが,以上を踏 まえ,本研究の第
1章では医薬品添付文書を情報源とした薬物間相互作用情報のシステ ム構築とその評価を,各診療科における①処方作成時と②薬物中毒により救命救急病棟 に搬送された時点において検討する。第
2章では③医薬品添付文書情報を逸脱した医師 の裁量による顆粒球コロニー形成刺激因子製剤(G-CSF 製剤)の使用法のあり方,④治 験時の溶解液量の設定を臨床現場に持ち込んだことによる副作用の発生頻度,⑤副作用 を事前に回避するための情報作成についてそれぞれ検討し,医療現場に則した医薬品安 全性情報の補完に関して分析する。第
3章では⑥日本大学医学部データウェアハウスを 使用した副作用に対する薬剤疫学的評価および⑦臨床現場で発覚した新規薬物間相互作 用情報の評価を通じて,臨床現場における自発報告を検討する。
本稿で検討する上記④および⑦に関する研究については,倫理審査委員会の承認を受 け実施され,③および④に関する研究については,入院時に主治医が「医療・介護関係 事業者における個人情報の適正な取扱いのためのガイドライン」,「症例報告を含む医学 論文及び学会研究会発表における患者プライバシー保護に関する指針」に基づき,患者 情報の取り扱いに十分留意するとともに個人の臨床情報や治療結果を学術目的で発表,
公表することを説明し,書面による同意が得られた症例を対象とした。また,①,②お
6
よび⑥に関する研究について取得したデータは既に匿名化されていて個人を特定するこ とができないものであったので,①,②および⑥のいずれの研究もわが国の「疫学研究 に関する倫理指針」に従い倫理審査委員会による承認を必要としなかった。
本稿で検討する上記すべての研究については,利益相反はなかった。
7
基礎となる原著論文
(1)処方(注射薬を含む)入力時における薬物相互作用自動チェックシステムの開発.
早坂正敏,青柳京子,木村高久,牧原剛,牧村瑞惠.
医療薬学 2001; 27: 380-385.
(2)救命救急センターの初療時における薬剤師
24時間対応の必要性.
早坂正敏,藏内恭子,葉山達也,吉田善一,丹正勝久.
医療薬学 2012; 38: 313-321.
(3)
G-CSF製剤の投与開始時期が化学療法による骨髄抑制および投与スケジュールに
与える影響-進行大腸がんに対する一次療法
FOLFOX4療法における検討-.
平井利幸,今井徹,早坂正敏,関利一.
医療薬学 2011; 37: 127-131.
(4)Docetaxel 希釈濃度による過敏症発現率の相違に及ぼす検討.
葉山達也,早坂正敏,小沼芽生,中山敏光,天野定雄,吉田善一.
医療薬学 2012; 38: 547-558.
(5)FOLFOX4 療法における末梢神経障害発症に関与する臨床的因子の検討.
今井徹,早坂正敏,中馬真幸,濃沼政美,葉山達也,中山敏光,吉田善一,丹正 勝久.
医療薬学 2010; 36: 347-351.
8
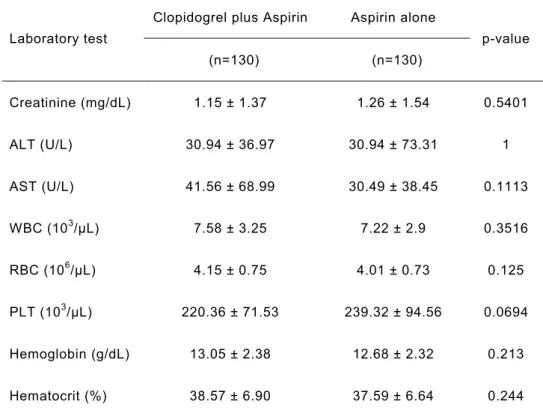
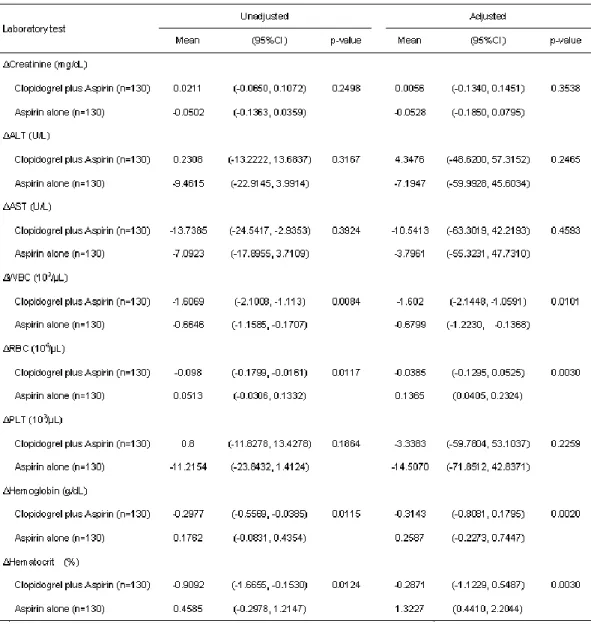
(6)Comparative effect of clopidogrel plus aspirin and aspirin monotherapy on
hematological parameters using propensity score matching.Masatoshi Hayasaka, Yasuo Takahashi, Yayoi Nishida, Yoshikazu Yoshida, Shinji Hidaka, Satoshi Asai.
Vascular Health and Risk Management. 2013; 9: 65-70.
( 7 )
Proton pump inhibitors may increase the risk of delayed bleeding complications after open heart surgery if used concomitantly with warfarin.Mitsumasa Hata, Masatoshi Hayasaka, Akira Sezai, Tetsuya Niino, Masataka Yoda, Satoshi Unosawa, Makoto Taoka, Syunji Osaka, Nobuyuki Furukawa, Haruka Kimura, Kazutomo Minami.
Thorac Cardiov Surg 2008; 56: 274-277.
9
第
1章 医薬品添付文書情報に基づく薬物間相互作用情報の構築とその評価
第
1節 諸言
医療用医薬品添付文書(医薬品添付文書)は,薬事法第
52条で定められた文書であ り,唯一の法的根拠に基づく文書である。医薬品添付文書は,
1995年には製造物責任法
(PL 法)が施行され,医薬品添付文書による指示および警告が極めて重要視され,医 薬品の適正使用のためには必要不可欠な情報源と位置づけされている。
1993年に発生し た薬物間相互作用による薬害であるソリブジン事件を契機に
1 ),
1997年に記載様式が改
訂され
2 ~ 4 ),現在では独立行政法人医薬品医療機器総合機構(PMDA)からインターネ
ットで公開されている。また,コンピュータシステムの進歩により,電子カルテシステ ム,オーダリングシステムおよびインターネットを通して医薬品添付文書情報が病院内 で閲覧できるよう運用されている施設が多い。しかし,コンピュータシステムにおいて は,医薬品添付文書情報が単に閲覧できるだけでは医薬品情報システム機能としては乏 しいことから,保険適応マッチング,薬物間相互作用チェック,疾病禁忌チェックなど の機能が導入されている。特に臨床の現場で薬物療法を行う場合,複数の医薬品が併用 されることから,医薬品を有効かつ安全に使用するためには,医師および薬剤師が薬物 間相互作用をチェックすることが重要である。しかし,現在においてもコンピュータ自 身によるレスポンス低下,コンピュータシステム上の登録医薬品の限定や各施設の薬剤 部が作成した標準化されていない相互作用データの使用などの問題点が報告されている
1 5 ~ 2 0 )
。
そこで本研究では薬剤師が医薬品添付文書情報を基に作成した薬物間相互作用監視用
データの有用性を検討した。
10
第
2節 処方入力時における薬物間相互作用自動チェックシステムの構築
第
1項 目的
薬剤師が医薬品添付文書情報を基に作成した薬物間相互作用監視用データを使用し,
医師が処方を入力する時点において薬物間相互作用チェックシステムを稼働することが 有用であるかを検討する。
第
2項 方法
2-1
薬物間相互作用監視用データ
一般財団法人 医療情報システム開発センター(MEDIS-DC)が開発した医薬品情報 データベース【医薬品添付文書】の相互作用の記載がある項目を使用して医薬品相互作 用データベース「相互作用情報データ(
DRIS-DB)」を構築した。DRISは相互作用の データのうち,併用禁忌のみではなく併用注意に関してもデータベース化した。DRIS の相互作用情報は,①薬剤情報データ,②相互作用知識データ,③相互作用辞書データ の3つで構成した(Fig. 1)。
Fig.1
相互作用情報データ
DRIS-DBのしくみ
11
2-2
薬物間相互作用の監視機能
入院や外来の同一診療科および他診療科を問わず,医師が注射薬を含む処方を入力す る時点で,その処方薬の投与期間中に重複している医薬品すべてに対して相互作用の照 合が自動的に行われるように設定した(Fig. 2)。
Fig.2
相互作用監視システムのフローチャート
第
3項 結果
医薬品添付文書から情報を構築しデータベースを作成し,それを処方・注射オーダリ
ングシステム上で運用する相互作用チェックシステムを開発した。
1999年から稼働を開
始し現在に至っている。併用禁忌を処方した件数は,システム導入前の
1998年
2月で
は
13件,3 月
14件,4 月
12件,5 月
14件,6 月
12件,7 月
13件,8 月
12件および
9月
14件であったが,導入後の
1999年
2月で
4件,3 月
5件,4 月
0件,5 月
5件,6
月
3件,7 月
4件,8 月
0件および
9月
4件と減少した。
12
第
4項 考察
医薬品添付文書の問題点として,医薬品添付文書中の相互作用情報をそのまま用いた 場合,推察にすぎない相互作用誘発医薬品の組み合わせなど,必要としない情報も多く あると報告されている
2 1 )。さらに,ドイツの一般医のアンケート調査結果では,医薬 品添付文書情報の重大さ,作用機序,用量調節について半数以上が不満足 であると報告 されている
2 2 )。これらは医薬品添付文書の情報の必要性の高さを示しているものと考 えられると同時に情報源の問題点としては,類薬の報告を引用した曖昧な表現が記載さ れていること,定量的数値に関して十分な記載がされていないことなどが考えられる
2 3~ 2 4 )
。実際に医薬品添付文書の情報は各製薬会社で記載方法が異なるまたは多様なため,
機序コメントと処置コメントは統一化し代表語を設定する必要がある。さらに,情報作 成で最も重要な部分は医薬品添付文書に記載されている相手薬剤の特定方法である。医 薬品添付文書の相手薬剤の記載の方法は個別薬剤名を記載している場合もあるが,ほ と んどが薬理・薬効群名の記載が多い。従って,薬理・薬効群名に所属する一般名を慎重 に選択する必要がある
20 ~ 2 5 )。これら
2点が相互作用自動チェックシステム導入にあ たり最も注意し苦労したことである。
1998
年にはテルフェナジンとアステミゾールに代表されるような併用注意から併用 禁忌へ変更されたもの,利尿剤やフェノチアジン系医薬品に代表されるような慎重投与 から禁忌へ変更されたものなど,医薬品添付文書の改訂が相次いだ。そのような背景を 踏まえ,突然の医薬品添付文書改訂が生じた場合でも薬剤師が
DRIS-DBを容易に変更 できる仕様としたため対応が迅速にでき,適切に情報提供できたと考える。また,シス テム導入したことで患者一人当たりの処方が複数科からオーダ発行されても適確に確認 でき,そのため併用禁忌の処方が減ったことは医師への支援を果たしていると考える。
さらに,院外処方せんに相互作用を確認したことを印字できるシステム としたため,保
険薬局からは「調剤業務の支援になる」(70%),「服薬指導業務の支援になる」(61%)
13
と回答があることからも相互作用自動チェックシステムを導入したことはメリットがあ ったと考える。
第
3節 薬物間相互作用チェックシステムによる中毒患者の服用薬剤の検討
第
1項 目的
救命救急センターの初療では患者背景が不明であることが多く,既往歴や薬歴を把握 できないまま救命を目的とした侵襲を伴う検査や治療手技が施行され,気道,呼吸,循 環,意識状態に直接作用する医薬品が医師の口頭指示により使用される。薬剤師は医師 が指示した医薬品の薬品名・投与量・投与速度・投与経路などを迅速に確認した後に調 製した医薬品を医師に手渡している。また,バイタルサインや血液ガス分析結果をモニ ターしたうえで必要な薬剤を判断し,医師からの指示を受ける前に準備をするほか,必 要時には薬剤投与について医師に助言をすることもある。特に心肺停止の患者に対して はアドレナリンの一定時間毎の投与が必要であり薬剤師はタイムキーパーの役割を担う。
さらに,中毒患者が搬送された場合には,薬剤師は直ちに中毒の原因物質の同定を行い,
有毒物質の有無,過量摂取時の副作用,相互作用および対処法,解毒剤・拮抗薬の有無 などの情報提供を行う。中毒原因物質の特定ができない場合には薬剤師が尿を用いた迅 速検査によるスクリーニングを行っている。これらの業務以外に患者のベッド移動,患 者の衣服の切断・脱衣,ルート確保後の固定,滴数指示医薬品の調製および輸液ポンプ の設定,心臓マッサージなど薬剤師の枠を超えてスキルミックスを実践している。 これ らの実務の中,特に中毒患者の情報の作成と提供が薬の専門家である薬剤師に求められ ている。
そこで中毒患者が持参した推定服用薬毒物の種類と服用数,薬物間相互作用監視用デ
ータを使用した薬物間相互作用チェックとその情報提供の有用性を検討した。
14
第
2項 方法
2-1
対象
2009
年
8月
1日から
2009年
12月
31日までの
5ヵ月間に当センターへ搬送された 患者を勤務シフトによる搬送時間帯別(9 時から
17時[9-17 群],
17時から
1時[17-1 群],
1
時から
9時[1-9 群]の
3群)に分類し連続
135名を対象とした。
2-2
統計的処理
情報提供実績と割合には m×n カイ二乗検定で有意差があった場合,各群間において
Bonferroni
の補正(有意水準×1/3)を行った(多重比較)。
第
3項 結果
初療時における情報提供の実績
初療時における情報提供は中毒情報が
73件(54%)と半数以上を占めた。その他,投与 量および効果に関する情報提供が多かった(Fig. 3a)。時間帯別情報提供の割合は[9-17 群]と[1-9 群]では有意差はなかったが,[17-1 群]においては両群と比較し有意に低かっ た(Fig. 3b)。
中毒患者に対する推定服用薬毒物調査の時間帯別実施割合は
3群とも
70%程度の患者に対して行われた。また,
3群における服用薬剤の種類および服用数の平均は
7.1品目,
115.7
剤であった(
Fig. 4)。相互作用の検索から医師への伝達文書作成までは患者一人当たり
5.4±1.2分であった。
15
中毒 54%
投与量 15%
投与方法 6%
効果 10%
副作用 4%
持参薬 1%
その他 10%
a. b.
Fig.3 初療における情報提供実績と割合
a.
初療における情報提供の実績(n=135) b. 初療における時間帯別情報提供の割合
Fig.4 中毒患者に対する服用薬剤調査時間帯別実施割合
第
4項 考察
初療時の情報提供に関して,中毒情報の提供が最も多く,これは当センターに搬送さ
れた中毒患者に対して中毒物質に関する情報収集および情報提供が薬剤師に一任されて
16
いることによるものと考えられる。中毒患者の搬送時には救急隊が推定服用薬毒物を持 参してくる。薬剤師が直ちに薬剤の空包などを確認し,医薬品名,含有量および空包か ら服用量を推定し,その中毒量,薬物動態,大量服用時の中毒作用,相互作用,治療法 などを情報提供する。推定服用薬毒物調査が
3群間で同程度実施され,その服用薬剤の 種類や平均服用錠数などに変化はなかった。相互作用の検索は,本検索システムを使用 することで
5分程度に短縮できるが,本検索システムがなければ患者一人当たり
30分 以上の時間を必要とした。薬剤師が作成した相互作用検索システムは薬物中毒患者の対 応においても大きく貢献できることが明らかとなった。しかし,薬物の過量投与時の薬 物吸収速度,吸収量,初回通過効果の飽和による
bioavailability,蛋白結合部位の飽和による血漿蛋白結合率などのそれぞれの変化および代謝酵素の飽和による代謝速度の飽
和現象
2 6 )などの体内動態情報は医薬品添付文書には記載が少ない。さらに,過量投与
時の相互作用の十分な情報がないことから医師,薬剤師および看護師における評価は決 して高くはなく,現在のままでは中毒患者に使用するには堪えない医薬品情報である こ とが示唆された。
第
4節 本章のまとめ
薬剤師が医薬品添付文書情報を基に作成した薬物間相互作用監視用データを使用する ことは,医師が処方を入力する時点および薬物中毒患者の相互作用検索だけでなく保険 薬局での調剤業務においても有用である。一方,情報源である医薬品添付文書情報には,
類薬での結果を引用した曖昧な表現が用いられていること,定量的数値に関して十分な
情報が記載されていないことなど現時点では多くの問題を抱えることが確認された。
17
第
2章 医療現場に則した医薬品安全性情報の補完に関する検討
第
1節 諸言
医薬品添付文書の効能・効果および効能・効果に関連する使用上の注意は,治験デー タを検証した結果に基づいて承認されたものである
5 )。保険診療を実施する上では,こ の医薬品添付文書の適応情報が重要であるということはいうまでもない。しかし ,治験 時には,肝・腎障害等の合併症を有する患者や妊婦・授乳婦には実際に投与されず,ま た病態および他の薬物との相互作用を回避しており,さらに,適応疾患の周辺領域の病 態に投与されることは少ない。臨床現場における医薬品の使用に関しては,実際に患者 に対して当該医薬品を使用できるかどうかの情報が少ないことから,患者の利益になる と考えた医師の裁量で使用されることが多い。先述した
Too few,Too simple および
Too narrow
を補完するには,臨床で投与された患者において,症例集積を検討すること,
肝・腎障害等の合併症を有する患者や妊婦・授乳婦に実際に投与された情報や病態およ び他の薬物との相互作用を検討すること,適応疾患の周辺領域や適応外疾患に実際に投 与された情報を検討することが必要である。
そこで,薬剤師は薬のプロフェッショナルという立場から抗がん剤や支持療法を含め
たレジメンのマネジメント,投与時投与後の副作用モニタリングとそのマネジメントお
よび患者指導など多くの場面で薬学的な視点での対応が求められること,さらに,抗が
ん剤は症例数が少ない情報で製造販売承認されることから,Too few を補完する材料と
して取り上げた。本研究ではがん化学療法後の好中球減少症に対する 顆粒球コロニー形
成刺激因子製剤(G-CSF 製剤)の使用方法と抗がん剤の希釈濃度の違いによる過敏症の
発現頻度に関して検討する。次いで,医薬品添付文書の副作用の項に関しては製造販売
に至るまでの動物実験や治験の結果をもとに収集されたものであるが,ほとんどが羅列
された表記となっている
5 )。医薬品の有用性を確保しつつ,副作用をできるだけ減らす
18
ためには患者背景因子の検討が必要である。これらの情報を医薬品添付文書の副作用の 項に記載することで安全性を担保できる可能性があることから ,本研究では
FOLFOX(オキサリプラチン(L-OHP),フルオロウラシル(5-FU)/レボリナートカルシウム
(l-LV)の持続点滴)療法における副作用発症に関する因子を検討する。
第
2節 がん化学療法後の好中球減少症に対する
G-CSF製剤使用法に対する情報の検 討
第
1項 目的
G-CSF
製剤をがん化学療法施行
24時間以内に投与することは,分裂を促進させた骨
髄細胞ががん化学療法により障害を受けることで,白血球や好中球の減少をさらに悪化 させるとの報告がある
27 )。それに基づき本邦における
G-CSF製剤の医薬品添付文書上 の適応は好中球数の減少が観察された時点の投与であり,がん化学療法前後
24時間以 内の投与は避けることとされている
2 8 ~ 2 9 )。一方,G-CSF 製剤の同時併用の有用性に
ついて,
Imatinib療法において重篤な感染を抑制し高用量での継続投与が可能であった
とする報告や
cyclophosphamide,mitoxantrone,etoposideおよび
prednisoloneを用 いた化学療法(CMEP 療法)において無顆粒球状態の期間や顆粒球の完全回復期間の有 熱期間を短縮したという報告もある
3 0 ~ 3 1 )。これらの報告のように,G-CSF 製剤をが ん化学療法
24時間以内に投与することについて,確立した見解が得られていないのが 現状である。臨床現場では
FOLFOX4療法の次クールの投与予定日と白血球数および好 中球数の最低値時期とが重なってしまい,投与予定日に投与基準を満たせないことが患 者に不利益であると判断された場合,医師の裁量で化学療法施行
24時間以内に
G-CSF製剤が投与されることがある。
そこで,本研究では
FOLFOX4療法施行患者に対する
G-CSF製剤の投与開始時間に
19
よる骨髄抑制に対する影響について適応基準との比較検討を行った。
第
2項 方法
2-1対象患者
2006
年
1月
1日から
2008年
12月
31日までに日本大学医学部附属板橋病院(以下,
当院)で進行大腸がんに
FOLFOX4療法を実施した患者に対し,治療期間中に
G-CSF製剤を投与した連続
21症例を対象とした。その中で,FOLFOX4 療法開始
24時間前か ら投与終了後
24時間以内に
G-CSF製剤を投与した患者
12人を
24時間以内群,
FOLFOX4
療法後
24時間を経過してから投与した患者
9人をその他群とした。また,
各個人で
G-CSF製剤の投与開始時期が両群におよぶ患者は今回の調査から除外した。
各群における
G-CSF製剤の投与開始基準は好中球数
1,000個/mm
3未満かつ
38℃以上 の発熱の観察,あるいは好中球数
500個/mm
3未満が観察された時点より医師の判断で 投与され,
2,000個/mm
3以上の好中球数に到達した時点で投与中止とした。なお,すべ ての患者においてフィルグラスチム
150μgを皮下投与した。
2-2
調査項目
調査は
24時間以内群とその他群のそれぞれにおいて,FOLFOX4 療法施行後の白血 球数と好中球数の減少を
Common Terminology Criteria for Adverse Events, Version3.0,日本語訳JCOG/JSCO
版(CTCAE ver. 3.0)の有害事象共通用語基準に基づいて評
価を行った。また,発熱の有無,1クールあたりの
G-CSF製剤の投与回数,14 日間隔 での投与遵守率,FOLFOX4 療法投与総クール数を
retrospectiveに調査した。
2-3
統計的処理
データは中央値(範囲)で表した。
2群間における変数の比較には
Mann-Whitney’ s20
U test,カテゴリー変数にはFisher’s exact probability test
を用いた。各検定におけ る有意水準はいずれも危険率
5%未満とした。2-3 relative dose intensity (RDI)
の算出
FOLFOX4
療法各薬剤における
RDIの割合(%)は下記の方法で算出した。
RDI(%)=〔実投与量(mg/m2/week)/標準的投与量(mg/m2/week)〕×100
第
3項 結果
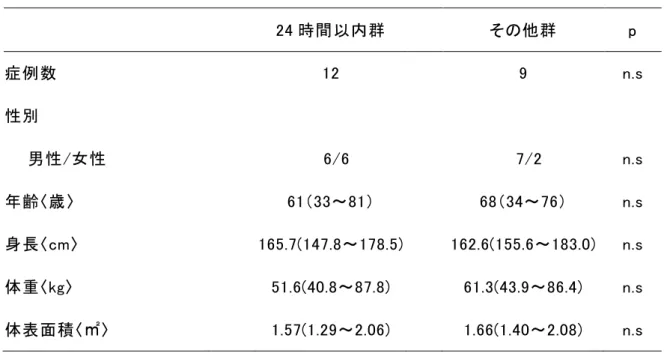
3-1 患者背景調査対象患者は
21人であり,
24時間以内群の患者は
12人で男性
6人,女性
6人,
年齢
61(33~81)歳,身長 165.7(147.8~178.5)cm,体重51.6(40.8~87.8)kg,体表面積
1.57(1.29~2.06)㎡であった。一方,その他群の患者は 9人で男性
7人,女
性
2人,年齢
68(34~76)歳,身長
162.6(155.6~183.0)
cm,体重 61.3(43.9~86.4)
kg,体表面積1.66
(1.40~2.08)㎡であり,両群に有意な差は認められなかった(Table
1)。FOLFOX4
療法の各薬剤における
RDI(%)および総投与クール数は,24時間以
内 群 で は
L-OHP 60.7(42.0~
99.0)%,
l-LV 72.0(42.0~
111.0)%,
5-FU急 速 静 注
64.0(34.5~100.0)%,5-FU持続静注 64.0(36.5~103.0)%,FOLFOX4 療法総施行数は
76クールであり,その他群では
L-OHP 50.0(35.0~100.0)%,l-LV 52.0(44.5~107.0)%,5-FU
急速静注
49.8(37.5~102.0)%,5-FU持続静注 50.0(44.5~102.0)%,FOLFOX4 療法総施行数は
62クールであり,両群に有意な差は認められなかった(
Table 2)。なお,対象患者は
FOLFOX4療法の投与条件を満たしており,群間において全身状態,肝機能
および腎機能に有意な違いは認められなかった。
21
Table 1.
患者背景
24 時間以内群 その他群 p
症例数 12 9 n.s
性別
男性/女性 6/6 7/2 n.s
年齢〈歳〉 61(33~81) 68(34~76) n.s
身長〈cm〉 165.7(147.8~178.5) 162.6(155.6~183.0) n.s
体重〈kg〉 51.6(40.8~87.8) 61.3(43.9~86.4) n.s
体表面積〈㎡〉 1.57(1.29~2.06) 1.66(1.40~2.08) n.s
n.s:有意差なしTable 2. FOLFOX4
療法薬剤の
RDIおよび総投与クール数
24 時間以内群 その他群 p
L-OHP〈%〉 60.7(42.0~99.0) 50.0(35.0~100.0) n.s l-LV 〈%〉 72.0(41.5~111.0) 52.0(44.5~107.0) n.s 5-FU 急速静注〈%〉 64.0(34.5~100.0) 49.8(37.5~102.0) n.s 5-FU 持続静注〈%〉 64.0(36.5~103.0) 50.0(44.5~102.0) n.s
FOLFOX4 実施総クール数 76 62
G-CSF 製剤投与開始クール 2(1~24) 3(1~11) n.s
G-CSF 製剤投与開始投与日 0(0~3) 19(7~22) p=0.00001
n.s:有意差なし22
3-2
骨髄抑制に対する影響
FOLFOX4
療法後の
CTCAE v3.0による
Grade3以上の白血球数および好中球数の減
少の出現率について,白血球数の減少は
24時間以内群で
14症例中
5症例の
35.7%,その他群は
12症例中
2症例の
16.7%であった。また,好中球数の減少は 24時間以内群が
10
症例中
5症例の
50.0%,その他群は7症例中
2症例の
28.6%に認められた(Fig. 5)。1クールでの
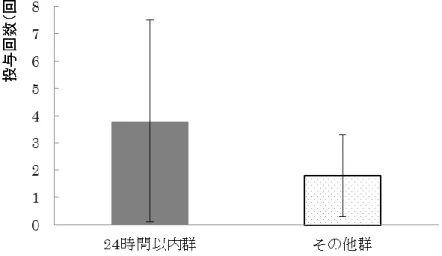
G-CSF製剤の使用回数については,
24時間以内群では1クールあたり
3.8±3.7 回,その他群では1クールあたり
1.8±1.5回であった(
Fig. 6)。FOLFOX4療法 後に発熱した割合は
24時間以内群では
14症例中
4症例の
28.6%,その他群では 12症 例中
1症例もなかった(
Fig. 7)。Fig. 5 Grade3
以上の白血球減少および好中球減少の出現率
減少および好中球減少の出現率
23
Fig. 6 1
クールあたりの
G-CSF製剤投与回数の比較
エラーバーは標準偏差を表す
Fig. 7
発熱の出現率の比較
24
3-3
投与スケジュールに対する影響
24
時間以内群とその他群の治療期間での
FOLFOX4療法の
14日間隔での投与遵守率 については投与間隔を遵守出来たのは
24時間以内群で
76クール中
27クールの
35.5%, その他群で
63クール中
13クールの
20.6%であった(Fig. 8)。また,総投与クール数については
24時間以内群で
6.9±7.09クール,その他群で
6.9±4.46クールであった。
第
4項 考察
がん化学療法は休薬せず施行することが予後に良好な影響を与えること
3 2 ),G-CSF 製剤を使用すれば
FOLFOX4療法を定期的に継続できること
3 3 )から,本来の適正な投 与開始時期ではない
FOLFOX4療法施行
24時間以内にやむを得ず
G-CSF製剤を使用し,
がん化学療法を実施する症例が存在する。そこで,本研究では大腸がん,ステージⅣ,
一次療法として
FOLFOX4療法の投与条件を満たした患者を
2群に分けて
G-CSFの投
Fig. 8 FOLFOX4療法投与遵守率の比較
25
与に関して解析を行った。エルプラット
®注射用
100 mg使用成績調査の最終集計結果で は,FOLFOX4 療法による
Grade3以上の白血球数の減少は
17.9%,好中球数の減少は27.5%と報告されているが3 4 )
,24 時間以内群は使用成績調査と比べて白血球減少や好
中球減少の出現率が高いことが認められた
3 2 )。その結果,FOLFOX4 療法施行
24時間
以内に
G-CSF製剤を投与することは,一時的に白血球や好中球を増殖させ,
FOLFOX4療 法 の 投 与 遵 守 率 を 向 上 さ せ る が , 総 投 与 ク ー ル 数 の 向 上 に は 影 響 し な い こ と や
Grade3
以上の重篤な白血球数や好中球数の減少を出現させ,発熱のリスクを増大させ
ることが示唆された。したがって,FOLFOX4 療法において
24時間以内の
G-CSF製剤 投与は避けることが望ましく,白血球や好中球が減少している場合には,
G-CSF製剤を 本来の適正な開始時期に投与することで,発熱性好中球減少症のリスクを増大させるこ となく,治療を継続できる可能性があると考える。
本研究から,FOLFOX 療法においては,FOLFOX4 療法施行
24時間以内に
G-CSF製剤を投与しないことという医薬品添付文書の記載内容と同様の結果が得られた。症例 数のさらなる蓄積と検討が望まれるが,本研究から医薬品添付文書情報を補完すること ができ,医薬品の適正使用の観点から医師等に対して
G-CSF製剤の使用法について助 言することができた。
第
3節 注射用抗がん剤希釈濃度の違いによる副作用情報の検討
第
1項 目的
ドセタキセル(以下,Doc)の主たる重大な副作用としては骨髄抑制,浮腫,末梢神
経障害などが列挙されるが,その多くは適切な支持療法や減量・休薬基準の遵守により
忍容できるものである。しかし,安全性情報として医薬品添付文書から入手できる過敏
症は,発現頻度が
8.1% (重篤例 0.7%)であるものの3 5 ),実際にはその対策に難渋し薬
26
剤変更を余儀なくされるケースが少なくない。過敏症は,発赤,紅斑,嘔吐といった軽 度なものから血圧低下,呼吸困難,全身性の蕁麻疹,血管浮腫のような致死的な症状を
呈する
3 6 )。これらの症状はⅠ型アレルギー様反応ではあるが,過去の曝露や感作の経
緯がなく出現する事から,肥満細胞の脱顆粒に伴う
IgE非依存型反応(Ⅰ型アレルギー 様反応)としても推察されている
3 6 ~ 3 7 )。国内では
Docによる過敏症は比較的軽度と され
Docの前投薬は必須とされていないが,タキサン系薬剤投与患者の
42 %で過敏症が観察され,その内
2 %が重篤な症状を呈し3 8 ),発現時期として
95 %が初回または 2回目投与時に出現するとの報告がある
3 9 )。
Docによる過敏症に対した前投薬において,
有効性が示されている薬剤として
dexamethasone,diphenhydramineまたは H
2受容 体拮抗薬があげられる
40 )。その他,脱感作法および
Docと
cyclophosphamideの投与 順 位 を 逆 に す る 事 の 有 効 性 を 示 し た 報 告 も あ る
4 1 ~ 4 2 )。 一 方 で
dexamethasone,
diphenhydramine
または H
2受容体拮抗薬の前投薬を行ったにもかかわらず対象患者
の
41 %で過敏症が出現した報告もあり4 3 ),完全な予防法の確立まで至っていない。
当院の外来化学療法室では,乳癌に対する標準療法となった
Docと
cyclophosphamideの併用療法(q3wTC レジメン)が導入された時期から過敏症が高頻度に出現している傾 向があった。他の
Doc含有レジメンと比較した際,明らかに異なる背景として
q3wTCレジメンにおける
Doc希釈液が 250 mL であり,その他大多数の
Doc含有レジメンは
Doc
希釈液が
500 mLであった。以上より,Doc 希釈濃度が過敏症発現に関連する 1 つ
の要因であると推察した。医薬品添付文書による調製法としては「250 又は
500 mLの 生理食塩液又は
5%ブドウ糖液に混和する」と記載されていることから ,当院における登録レジメンにおいても希釈液量は統一化されていない。また過去において,Doc 希釈
液の
250 mLにおける安全性を担保した報告はない。したがって医薬品添付文書に記載
がある希釈液量を問題とし,希釈濃度と過敏症発現の関連性が明確にできるか検討した。
本研究は当院での外来化学療法において
Doc投与を受けた患者を対象とし,
Doc希釈
27
濃度に起因した過敏症発現率の相違を
retorospectiveに調査した。副次的に,過敏症発 現時期,重篤度,要因因子および過敏症の発現を抑止できる可能な濃度設定の探索を行 った。
第
2項 方法
2-1対象患者
選択基準は
2006年
1月から
2011年
5月の期間に,当院での外来化学療法室において 乳がん,胃がん,非小細胞肺がん,前立腺がんおよび皮膚がんに対する
Docを含む化学 療法を導入した患者連続
471名を対象とした。
除外基準は登録レジメンに逸脱した前投薬 (メチルプレドニゾロンコハク酸エステル ナトリウム,クロルフェニラミンマレイン酸塩,ヒドロキシジン塩酸塩等)を施行した症 例とした。対象の
Doc希釈液は
250 mLまたは
500 mLの
5%ブドウ糖液または生理食塩液であった。
2-2 対象レジメン
q3wTC:術前,術後および進行・再発乳がんに対する初期治療
3wDoc:術前,術後および進行・再発乳がんに対する初期治療および進行・再発非小細
胞肺がんに対する二次治療
TS-1+Doc:腹膜転移を伴う進行・再発胃がんに対する初期治療
3wDoc+ZDA:ホルモン抵抗性かつ骨転移を有する前立腺がんに対する初期治療 Weekly Doc:ホルモン抵抗性前立腺がんおよび皮膚がんに対する初期治療 DG:プラチナ製剤不耐性進行・再発非小細胞肺がんに対する初期治療 M-Doc+CBDCA:非小細胞肺がんに対する術後補助化学療法
臓器別レジメンにおける各薬剤の投与量,投与時間および投与期間は
Table 3,前投薬処方は
Table 4に示す。
28
Table 3. Docetaxel regimen
Table 4.
前投薬処方
29 2-3
調査方法
外来化学療法室における独自の患者データ管理システム,薬剤管理指導記録,診療録 または看護指導記録に基づき
retrospectiveに調査した。過敏症の重篤度は
Common Terminology Criteria for Adverse Events, Version 4.0,日本語訳JCOG版(CTCAE ver.
4.0)の有害事象共通用語基準に基づいて評価を行った。
2-4
評価項目 <主要評価項目>
希釈液
250 mL群と
500 mL群の
2群間における過敏症発現率の相違。
<副次的評価項目>
1) 2
群間における過敏症発現時期。
2) 2
群間における過敏症発現症例に対する投与中止率および重篤度。
3) 2
群 間 に お け る 過 敏 症 発 現 の 要 因 因 子 と し て の 希 釈 濃 度
(w/v%), 実 投 与 量 (mg),relative dose intensity (RDI) (%), 投 与 時 間 (min) お よ び 流速 濃 度
(w/v%/min)。尚,RDI (%)
は下記の方法で算出した。
RDI (%) = {実投与量 (mg/m2/week)/標準的投与量 (mg/m2/week)}×100
4) 過敏症が抑止可能とする希釈濃度 (カットオフ値)。
過敏症発現症例の希釈濃度中央値を境界濃度の基準とし,段階的に境界濃度 を設定する。全症例において各々の境界濃度以上を
above群,未満を
below群 と割付する。両群における過敏症発現率および中止率に有意差が認められなく なった時点の濃度を抑止可能希釈濃度
(カットオフ値)と定義する。
なお,全症例における各希釈濃度は以下の方法で算出した。
希釈濃度 (w/v%) = 実投与量 (g) / 希釈液 (250 mL または 500 mL)
30 2-5
統計解析
2
群間における過敏症の発現率,過敏症発現による投与中止率,重篤度および基準希 釈濃度における過敏症発現率の比較にはイエーツ補正χ二乗検定を用いた。また,発現 時期,希釈濃度,実投与量,投与時間,
RDIおよび流速濃度の比較には Mann-Whitney
U-testを用いた。
第
3項 結果
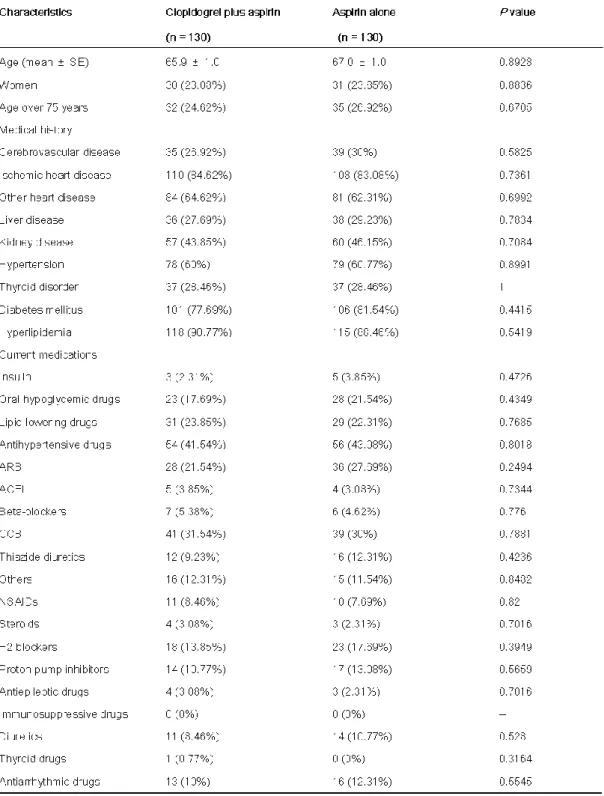
3-1 患者背景対象患者となった 471 名に対して,docetaxel 希釈液が 250 mL 群と 500 mL 群 の患者背景を比較した (Table 5)。
250 mL群の患者は
90名であり,男女比は男性 2 名,
女性 88 名,年齢中央値は 56 歳 (95% 信頼区間 (CI):53 - 58),体表面積は 1.50±
0.14 m2 (Mean±SD, 95% CI:1.47 - 1.53) に対し,500 mL
群の患者は 381 名であ り男女比は男性 207 名,女性 174 名,年齢中央値は
67歳 (95% CI:65 - 67),体表 面積は 1.52±0.15 m
2 (Mean±SD, 95% CI:1.51 - 1.54)であった。また,原発巣の割 合は両群で比較すると,250 mL 群において乳がん患者がほぼ全例を占める割合であっ た {250 mL 群; 85 例(乳がん),0 例, 0 例,0 例,5 例 (皮膚がん), 500 mL 群;
118
例 (乳がん),122 例 (胃がん),103 例 (非小細胞肺がん),38 例 (前立腺がん),0
例 (皮膚がん
)}。この結果に連動するように導入レジメンは 250 mL群で q3wTC が
90%以上施行された結果であった。
31 Table 5.
患者背景
3-2
過敏症発現の頻度と発現時期
250 mL
群と 500 mL 群における過敏症発現率は 250 mL 群
14.4%,500 mL群
2.4%で あ り ,250 mL 群 で 有 意 に 過 敏 症 発 現 率 が 高 い 事 が 示 さ れ た
(p<0.01) (Fig.9a)
。また,発現時期を比較したところ 250 mL 群では 1.69±0.75 クールに対し,
500 mL群 で は 2.22 ±1.71 ク ー ル で あ り 両 群 に 発 現 時 期 の 有 意 な 差 は 認 め ら れ な か っ た
(p=0.76)(Fig. 9b)。32
Fig.9 2
群間における過敏症発現率と発現時期の比較
3-3
過敏症発現による投与中止と重篤度
過敏症が発現した 22 名に対し過敏症発 現に よって投与中止とな っ た割合は両群で 有意な差は認められなかった (p=0.87)(Fig. 10a)。また,過敏症の重篤度を比較したと ころ 250 mL 群 (n=13)では
Grade 1 (G1)が
61.5% (n=8),Grade 2 (G2)が 30.8%
(n=4),Grade 3 (G3) / Grade 4 (G4)
が
7.7% (n=1),500 mL群 (n=9)では
G1が 77.8%
(n=7),G2
が
11.1% (n=1),G3/G4が
11.1% (n=1)であり,両群で重篤度に有意な差は認められなかった (p=0.86)(Fig. 10b)。
33
Fig.10 2
群間における過敏症による投与中止率と重篤度の比較
3-4
過敏症発現の要因
過敏症発現の原因因子として 希釈濃度 (w/v%),実投与量 (mg),RDI (%),投与時間 および流速濃度 (w/v%/min) を
250 mL群と 500 mL 群の
2群間で比較したところ希 釈 濃 度 に 関 し て
250 mL群 は
0.0384±
0.0066w/v% (95% CI:
0.0370w/v% - 0.0398w/v%),500 mL群は 0.0151±0.0050w/v% (95% CI :
0.0146w/v% - 0.0155w/v%)であり 250mL 群が有意に高かった
(p<0.01) (Fig. 11a)。また,実投与量に関して
250 mL群は 92.54±1.1 mg (95% CI:90.41 mg - 94.67 mg),500 mL 群は 71.2±0.56 mg
(95% CI:70.10 mg - 72.30 mg)であり 250 mL 群が有意に高かった
(p<0.01)(Fig.11b)
。しかし,
RDIに関して 250 mL 群は 90.1±9.3% (95% CI:88.1% - 92.0%),
500 mL群は 87.3±18.1% (95% CI:85.4% - 89.1%) であり,投与時間では
250 mL群は
61.7±6.9 min (95% CI:60.2 min - 63.1 min),500 mL群は 64.3±13.9 min (95% CI:
62.8 min - 65.6 min)
と
RDI, 投 与 時 間 で は 両 群 で 有 意 な 差 は 認 め ら れ な か っ た (p=0.051, p=0.329) (Fig. 11c, 11d)。さらに,流速希釈濃度に関して
250 mL群 6.3×
34
10-4±1.2×10-4 w/v%/min (95%Cl:6.1×10-4 w/v%/min – 6.6×10-4 w/v%/min),500 mL
群は 2.4×10
-4±0.9×10-4 w/v%/min (95%Cl:2.3×10
-4 w/v%/min – 2.5×10-4 w/v%/min)であり
250 mL群が有意に高かった
(p<0.01)(Fig. 11e)。Fig.11 2
群間における過敏症発現要因の探索
3-5
過敏症発現の抑止可能な希釈濃度 (カットオフ値) の探索
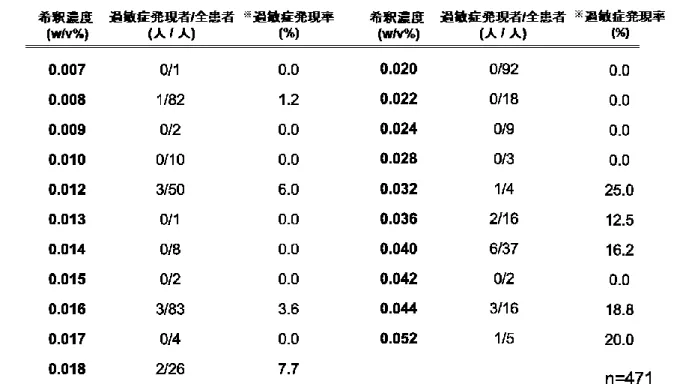
全症例における希釈濃度および過敏症発現者の分布を
Table 6に示す。過敏症発現症例
22名の希釈濃度中央値 0.034w/v% を境界濃度の基準とし,段階的に境界濃度を設定 した。全症例に対して各々の境界濃度以上を
above群,未満を below 群と新たに割付 け,両群における過敏症発現率の違いを各々の境界濃度において比較した。その結果,
境界濃度 0.021w/v%~0.034w/v% では above 群において過敏症発現率が有意に高か
った (p<0.01)。しかし,境界濃度が 0.02 w/v% まで低下させた時点において両群間に
おける過敏症発現率に有意な差が認められなくなった (p=0.137) (Table 7)。尚,境界濃
度 0.034 w/v%以上の全濃度において
above群で過敏症発現率に有意な差が認められ,
35
逆に境界濃度
0.02w/v%以下では両群間で過敏症発現率に有意な差は認められなかった
(data not shown)。同様に中止率に関して探索した結果,0.024w/v%
まで低下させた時
点において両群間における有意な差が認められなかった (p=0.065)。
Table 6. 全対象患者における希釈濃度および過敏症発現者の分布
36
Table 7. 境界希釈濃度毎における過敏症発現率・中止率の比較
第
4項 考察
過敏症発現リスクには性別,年齢および個体のウイルス感染等が要因との報告
4 4 ~ 45 )
があるが断定的な見解は得られていない。国内前・後期第Ⅱ相臨床試験
(評価対象例数 100 例以上) およ び 第Ⅲ相臨床試験にお け る過敏症発現率の男 女 比 と年齢差
4 6 ~ 50 )
との比較,国内外の疫学的検討
5 1 ~ 5 3 )との比較から本研究において性差および年齢 差が過敏症発現リスクの要因に寄与する可能性は希薄と考える。また,前投薬における 過敏症抑止への有用性
54),投与時間の緩徐や投与間隔の延長による過敏症への有用性
55 ~ 5 6)
の報告があるが,対称的に
Eisenhauerらによると
paclitaxelにおいて
3時間投
与と
24時間投与では過敏症発現率に差がなかったとの報告がされている
5 7)。その他,
37
過敏症発現リスク因子として「薬剤自身の持つ特性や投与方法」と「個体側の要因」に 大別した際,希釈濃度は前者に該当すると考える。宇野の報告では
5 8),薬物濃度とア レルギーの関係性に対し,局所で生体のセットポイン以上に薬物濃度が上昇した場合に 抗原として認識され,アレルギー反応が誘発されると示唆している。一方,後者は生体 の抗原認識の閾値を低下させる原因とされ,その主な因子としてアレルギー体質,感染,
環境因子,性ホルモンおよび遺伝的因子などが列挙され複雑に関与していると推察され る。後者の詳細なメカニズムとして,Itoh らの報告
5 9 ~ 6 2)では同じタキサン系薬剤で あるパクリタキセルで
in vivoにおける検討がなされており,知覚神経ペプチド(サブ スタンスP,ニューロキニンA,カルシトニン遺伝子関連ペプチド)が過剰遊離するこ とで肺における血管透過性が亢進すると結論付けている。以上の報告より,過敏症発現 には様々な要因が関与していることが推察できる。
本研究において,Doc の希釈濃度が異なる 2 群間 (250 mL vs 500 mL) の過敏症発
現率は,
500 mL群に対し
250 mL群で有意に高い発現率を示した。各群間の発現率は,
500 mL
群では
2.4 %と500 mL希釈液を使用した国内前・後期第Ⅱ相臨床試験
3 5 )の発
現率
7.4 %と比較し低かったが,250 mL群では
14.4 %とより高頻度に発現する傾向を示した。また,段階的に境界濃度以上,未満における過敏症発現率の相違を検討した結 果,0.02w/v% を境界濃度と設定した時から過敏症発現率に有意な差が認められなくな った。つまり
Doc 100 mg (q3wTC平均投与量 101.5±12.1 mg) とした時,希釈液を
500 mLと す れ ば 過 敏 症 発 現 率 を 低 下 さ せ る 事 を 示 唆 し て お り , 本 研 究 で 対 象 と な っ た
qw3TC
レジメンにおいて希釈液が
250 mLであった事が過敏症発現率を有意に高めた
大 き な 要 因 と 推 察 で き る 。 一 方 ,
Doc過 敏 症 に 対 す る 主 な 支 持 療 法 と し て
dexamethasone,diphenhydramineまたは H
2受容体拮抗薬の前投薬があげられる。し
かし,前投薬を施行したにも関わらず過敏症が発現するケースが見られる点,発現メカ
ニズムが不明瞭な点を含め前投薬のみでは完全な抑止までは至っていないのが現状と考
38
える。従って,過敏症発現を完全に抑止するためには,多側面的にアプローチをする事 が重要と考える。すなわち,エビデンスに基づいた前投薬の実施に加え,本研究で見出 された希釈濃度を 0.02w/v% 以下に設定する事でより的確に過敏症発現を 抑止するこ とが期待できる。
本研究により,調製法として「250 又は
500 mLの生理食塩液又は
5%ブドウ糖液に混和する」とのみ記載されている医薬品添付文書の情報では不足であることが明確にな り,安全かつ有効な化学療法を実施するためには,本研究で示した
Docの希釈濃度に関 する情報を医薬品添付文書に補完する必要性があると考える。
第
4節 副作用情報を補完するための患者背景因子の検討
第
1項 目的
大腸がん治療ガイドラインには切除不能転移・再発大腸がんに対して
FOLFOX療法 が標準治療として推奨されている
6 3 )。FOLFOX 療法の普及により切除不能転移・再発 大腸がん患者の生存期間は大幅に延長されているが,FOLFOX 療法に使用されている
L-OHP
の特徴的副作用に末梢神経障害があり,用量規制因子の一つとなっている
6 4 )。
末梢神経障害は治療のスケジュールや患者の
Quality of Lifeに対し,多大な影響を及ぼ す重大な副作用であり,
L-OHPの末梢神経障害は投与後すぐに出現する急性末梢神経障 害と,投与を繰り返すことによる慢性末梢神経障害に分けられる。急性末梢神経障害は
L-OHP