Title
非真空プロセスで作製する太陽電池材料に関する研究( 本文
(Fulltext) )
Author(s)
堀, 茂雄
Report No.(Doctoral
Degree)
博士(工学) 甲第455号
Issue Date
2014-06-30
Type
博士論文
Version
ETD
URL
http://hdl.handle.net/20.500.12099/49116
※この資料の著作権は、各資料の著者・学協会・出版社等に帰属します。博士論文
非真空プロセスで作製する太陽電池材料に関する研究
平成
26 年度
岐阜大学大学院 工学研究科
博士後期課程 環境エネルギーシステム専攻
1103815002 堀 茂雄
本論文の要旨 本論文は、非真空プロセスで作製可能な太陽電池の材料について、合成と解析を行ったも のである。特に化合物薄膜太陽電池の光吸収材料、透明電極材料について研究した。化合物 薄膜太陽電池は安価な材料やプロセスを用いて、高い変換効率を達成できるため太陽電池を 低コスト化出来る可能性を持つ。化合物薄膜太陽光発電では、導電基板の上に光吸収層、バ ッファー層、透明電極等の複数の層が積層した構造を持つ。これらの内、バッファー層は安 価な非真空プロセスで作製されているが、低コスト化には光吸収層と透明電極を非真空プロ セスで作製する必要がある。また、その材料も安価で無毒な新規材料の利用が課題となって いる。 第1 章では、本論文の研究の背景及び目的と概要について記述した。 第2 章では、安価で無毒な元素から構成される硫化錫(SnS)ナノシートのワンポット合成と 評価を行った結果について記述する。厚さ 数十 nm、幅 数 μm の異方性を持つ、斜方晶型結 晶の SnS ナノシートをワンポット合成した。また、250~300°C、0~60 分の間で合成温度と 合成時間の影響についても検討した。XRD 測定と SEM による観察から、何れの温度でも合 成時間10 分までにシートの成長が終わっている事が分かった。Raman スペクトルから、これ らのSnS ナノシートは SnS2やSn2S3のような副生成物の存在しない単相で得られたことが示 された。TEM による制限視野電子回折像の観察から、電気特性に優れた a, c 軸方向にシート が広がっていることが分かった。シートの形と結晶面の比較から、シートの端には{101}面と {100}面及び{001}面が露出し、{101}面の成長が特に遅いことが示唆された。ナノシートが得 られた原因として b 軸に垂直な面が他の結晶面に比べ安定であること、オレイルアミンが表 面修飾している可能性があること、シートの端に原料供給が集中して起きている可能性があ ることの3 つが示された。拡散反射スペクトルの測定から、Tauc プロットを算出しバンドギ ャップを求めると、直接遷移のバンドギャップが約1.3 eV となることが分かり、一般的な SnS と同じ値であることが確認された。また、光電気化学測定により価電子帯のエネルギー位置 を0 V vs. SCE と決定した。真空準位に対するエネルギーに換算すると、計算予測と一致する 4.7 eV であることが分かった。また、価電子帯のエネルギー位置とバンドギャップより伝導 帯のエネルギー位置を見積もると、バッファー層に通常用いられているCdS を用いると、SnS との接合ではクリフを形成し効率が悪化することが示唆され、バッファー層材料の変更が必 要なことが分かった。 第3 章では、安価な元素から構成される Cu2ZnSnS4 (CZTS)の及び Cu2ZnSnSe4 (CZTSe)ナノ 粒子の合成と評価を行った結果について記述する。これまでに報告の無い、試薬の乾燥等の 多段階のプロセスを必要としないワンポットでの合成を試みた。CZTS の合成では、100°C で はCuS が生成したが、150°C 以上の温度で CZTS を合成することに成功した。TEM の観察か ら、粒径を約7-20 nm の間で制御できることが分かった。CZTSe の合成では 100°C から CZTS の合成に成功した。CZTS のバンドギャップは 1.5 eV と求められ、化合物薄膜太陽電池の光 吸収材料に適している事が確かめられた。 第4 章では、塩素を含む電解浴から電解析出法により作製した酸化亜鉛薄膜の物性評価を
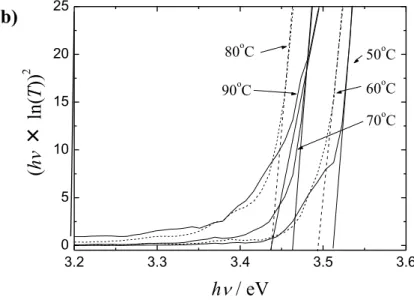
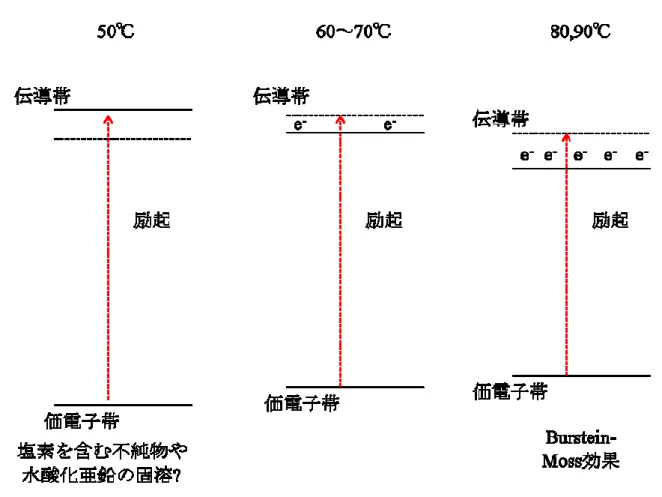
行った結果について記述する。特に、温度を変えて電析した酸化亜鉛薄膜の結晶構造、光学 的、電気的特性を測定した。XRD 測定より、電析で作製した酸化亜鉛は、ウルツ鉱型の六方 晶結晶構造を持ち、c 軸が基板に配向していることが分かった。電析で作製した酸化亜鉛は、 純粋な酸化亜鉛に比べ結晶の格子サイズが大きくなることが分かった。この格子サイズの増 加は塩素を含む不純物や、水酸化亜鉛によって引き起こされていることが示唆され、電析温 度が上昇すると、結晶格子のサイズが小さくなり、不完全な脱水種のような不純物の減少が 示された。また、光学特性の評価より、電析で作製した酸化亜鉛は純粋な酸化亜鉛より大き なバンドギャップを持つことが分かった。また、バンドギャップは電析温度が低いほど大き くなった。電気特性評価より、温度上昇によりキャリア密度が増加することが分かり、バン ドギャップの増加が、低温では不純物の固溶による価電子帯や伝導帯のエネルギー位置の変 化により引き起こされ、高温度では、Burstein-Moss 効果により起きていることが示唆された。 XPS の測定から、温度上昇により塩素のドープ量が減少し、酸素空孔が増加していることが 示唆された。また、電析温度60~80°C で、抵抗率の低い緻密な膜が得られることが分かった。 以上の結果より、化合物薄膜太陽電池の光吸収材料と透明電極を非真空プロセスで作製で きることが示された。
目次 第1 章 序論 1-1. 本研究の背景 ・・・・・・・・・・・・・・1 1-1-1. 多様なエネルギー源の必要性 ・・・・・・・・・・・・・・1 1-1-2. 太陽電池の種類と課題 ・・・・・・・・・・・・・・2 1-1-3. 化合物薄膜太陽電池の材料 ・・・・・・・・・・・・・・5 1-1-4. 非真空プロセスの種類と特徴 ・・・・・・・・・・・・・・6 1-1-5. 印刷型太陽電池 ・・・・・・・・・・・・・・9 1-1-6. 透明電極 ・・・・・・・・・・・・・・10 1-2. 本研究の目的 ・・・・・・・・・・・・・・11 1-3. 本論文の構成 ・・・・・・・・・・・・・・12 第2 章 硫化錫ナノシートの合成と物性評価 2-1. 諸言 ・・・・・・・・・・・・・・20 2-2. 実験方法 ・・・・・・・・・・・・・・23 2-2-1. SnS ナノシートの合成方法 ・・・・・・・・・・・・・・23 2-2-2. SnS ナノシートの物性評価 ・・・・・・・・・・・・・・23 2-3. 結果と考察 ・・・・・・・・・・・・・・25 2-4. 結論 ・・・・・・・・・・・・・・38 第3 章 Cu2ZnSnS4及びCu2ZnSnSe4ナノ粒子の合成と特性評価 3-1. 諸言 ・・・・・・・・・・・・・・41 3-2. 実験方法 ・・・・・・・・・・・・・・43 3-2-1. CZTS, CZTSe ナノ粒子の合成 ・・・・・・・・・・・・・・43 3-2-2. CZTS, CZTSe ナノ粒子の評価 ・・・・・・・・・・・・・・43 3-3. 結果と考察 ・・・・・・・・・・・・・・44 3-3-1. CZTS ナノ粒子の合成 ・・・・・・・・・・・・・・44 3-3-2. CZTSe ナノ粒子の合成 ・・・・・・・・・・・・・・52 3-3-3. 太陽電池の作製 ・・・・・・・・・・・・・・55 3-4. 結論 ・・・・・・・・・・・・・・57 第4 章 酸化亜鉛薄膜の電解析出法による作製と特性評価 4-1. 諸言 ・・・・・・・・・・・・・・59 4-2. 実験方法 ・・・・・・・・・・・・・・60 4-2-1. 酸化亜鉛薄膜の電析 ・・・・・・・・・・・・・・60 4-2-2. 酸化亜鉛薄膜の評価 ・・・・・・・・・・・・・・61 4-3. 結果と考察 ・・・・・・・・・・・・・・62
4-4. 結論 ・・・・・・・・・・・・・・84 第5 章 総括
5-1. 本論文内容のまとめ ・・・・・・・・・・・・・・87
5-2. 今後の展望 ・・・・・・・・・・・・・・88
1 第 1 章 序論 1-1. 本研究の背景 1-1-1. 多様なエネルギー源の必要性 人類が火の利用を始めたおよそ 50 万年前から、その後の農業への水力、風力の利用 に見られるように、文明の発展にはエネルギーが不可欠であった。16 世紀以降に石炭・ 石油の利用が始まると、エネルギーの 消費量は飛躍的に増加し、人類の文明は大きく 成長を遂げた[1]。 このようにエネルギーの消費量と成長には密接な関係が知られ、図 1-1(参考文献 2 を 元に作製)に示す日本における最終エネルギー消費量と GDP の関係にも、その傾向は見 られる。このような膨大なエネルギーを供給し続けるには大量のエネルギー源が必要 となる。 しかし、2011 年の東京電力福島原子力発電所の事故以降は、電力が火力発電によっ て賄われ、石油の使用量が大幅に増加した。一方で、図 1-2(参考文献 3 を元に作製)に 示すガソリンの卸売価格の上昇に見られる ように、近年では石油の価格が著しく上昇 している。また、新興国の発展によるエネルギー需要の増加によって、石油価格の増 加は今後も続く可能性がある。その他にも、2013 年のシリアの化学兵器問題に見られ るように、安価な石油の安定供給は必ずしも保証されたものでは ない。そのため、エ
2 ネルギー源の多様化が必要となる。 1-1-2. 太陽電池の種類と課題 エネルギー源の一つとして 太陽電池による太陽光発電が知られている。近年では固 定価格買い取り制度により普及が後押しされている 。逆にこの施策は、何らかの補助 無しでは普及が困難であることを示している。住宅用の太陽光発電の適正な価格は 37 ~46 円/kWh となり[4]、東京電力の従量電灯料金である約 25 円に比べ 1.5 倍以上の価 格となっている。そのため、太陽光発電が自発的に普及するには、現状の技術では困 難であり、石油価格のさらなる高沸や太陽電池のさらなる低コスト化が必要となる。 1970 1980 1990 2000 2010 0 4 8 12 16
Tot
al
e
ne
rg
y
co
ns
um
pt
io
n
/ 10
18J
Year
0 100 200 300 400 500 600Re
al
G
D
P /
1
0
12Y
en
図 1-1. 日本における最終エネルギー消費量と実質 GDP の推移3 2000 2002 2004 2006 2008 2010 2012 2014 0 20 40 60 80 100
Who
les
ale
pr
ice
of
reg
ul
ar g
aso
lin
e
/ y
en
d
m
-3Year
図1-2. レギュラーガソリンの卸価格の推移 表 1-1 に太陽電池の種類を示す(参考文献 4 を元に作製)。太陽電池の種類は大きく二 種類に分けることが出来る。一つは有機材料を用いた 有機系太陽電池であり、もう一 つは無機材料を用いた無機系太陽電池である。有機系太陽電には有機薄膜型と色素増 感型の二種類があり、多様な色を採用できデザイン性に優れることや、安価で軽量な プラスチック基板が利用できるために曲面にも用いることが出来ること、低い光強度 でも発電効率が低下しないことから、身の回りの物への一体化や室内での使用に向い ていると考えられる[5]。一方で、変換効率や耐久性が低いことが課題となっている。 無機系太陽電池では、耐久性が高いことが特徴として挙げられ[6]、住宅用やメガソ ーラーのような大規模発電に向いていると考えられる。しかし、 現在最も普及が進ん4
でいる結晶シリコン(Si)太陽電池では、既に効率、耐久性共に高く、低コスト化の余地
は少ない。また、部材となるシリコンに 99.9999%の高純度を必要とし、かつ約 1000°C
と高い温度の作製プロセスが必要となる。薄膜シリコンでは、低温で作製できるとい
うメリットがあるが、変換効率の大幅な向上が課題となっている。 それに対して、化
合物薄膜太陽電池は、Cu(In,Ga)Se2 (CIGS)や CdTe を用いて約 20%の変換効率が達成さ
れているが[7]、既存のプロセス技術等を用いながらも、多様な材料を利用することが 可能となっているため、大幅な低コスト化の余地があると考えられる。 表 1-1. 太陽電池の分類 大分類 太陽電池の種類 特徴 無機系太陽電池 結晶シリコン ・技術が確立されている ・変換効率と耐久性が高い ・低コスト化の余地が少ない 薄膜シリコン ・アモルファスや微結晶シリコン ・大面積化が可能 ・効率が低い 化合物薄膜 ・CIGS、CdTe が実用化されている ・様々な材料を用いることができる 有機系太陽電池 色素増感 ・低コスト化の可能性がある ・デザイン性が高い ・変換効率、耐久性が低い 有機薄膜
5
1-1-3. 化合物薄膜太陽電池の材料
化合物薄膜太陽電池は図 1-3 に示すように、導電基板の上に光吸収層、バッファー
層、高抵抗層、透明電極の積層した構造を持つため[8,9]、それぞれの層に安価な材料
を用いる必要がある。
高抵抗層や透明電極の材料は無ドープの ZnO や、Al をドープした ZnO が安価な材
料として用いられ、高い効率を示している。 これまで、バッファー層の材料には CdS が用いられてきたが、近年、Zn 系の材料を 用いたバッファー層の利用が進んでいる。 現在、光吸収層には CIGS や、CdTe が用いられている。これらの材料は吸収係数が 高いために光吸収層を薄膜化することができ、少ない使用原料で作製することができ るというメリットがある。他方で、In、Ga、Te の希少性や Cd の毒性が将来的に問題 となる可能性がある[10]。そのため、材料に無毒で安価なものを用いることにより太陽 電池の低害化や低コスト化が狙えると考えられる。現在注目を集めて いる化合物半導 体材料として CIGS 太陽電池に含まれる高価な In と Ga を、安価な Zn や Sn に置き換 え た Cu2ZnSn(S,Se)4 や[8,11-17] 、 二 種 類 の 安 価 な 元 素 の み で 構 成 さ れ る 硫 化 錫 (SnS)[18-20]が知られており、それぞれ変換効率 11.1%と 2.46%と低く開発の余地があ る [20,21]。他にも起電力は確認されていないが、極めて豊富に存在する鉄を用いた FeS2の研究も行われている[22-24]。
6 図1-3. 化合物薄膜太陽電池の構成 1-1-4. 非真空プロセスの種類と特徴 太陽電池の低コスト化には、用いる材料の他に作製プロセスが重要になる。現在、 化合物薄膜太陽電池のバッファー 層を除く各層は真空プロセスで作製されているが、 装置が高価であるために初期投資が大きくなり 作製する太陽電池のコスト増加の一因 となっている。そのため、真空装置を用いず、溶液中で材料を作る非真空プロセスが 低コスト化に有利であると考えられる。全ての層を非真空プロセスで作製すること に より、大幅な低コスト化を達成可能となる。さらに、非真空プロセスでは、前述した Cu2ZnSn(S,Se)4や SnS、FeS2を様々な手法で作製可能である[25-40]。
7 非真空プロセスの種類は、表 1-2 の一例に示すように、多くの方法が知られている。 そのため、化合物薄膜太陽電池の各層の作製には、それぞれの特徴に応じた方法を選 択することが低コスト化に有効であると考えられる。図 1-3 に示した各層の内、バッ フ ァ ー 層 は 化 学 浴 析 出 法 に よ り 作 製 さ れ 、 高 い 効 率 が 得 ら れ る こ と が 示 さ れ て い る [41-43]。 光吸収層には、吸収係数が 104程度の材料を用いる場合、数 μm の厚さと高い純度が 必要となるため、成長の速い 作製方法や、不純物が含まれにくい作製方法が適してい る。表 1-2 に示した方法の内、薄膜を作製する方法は、原料の金属塩に含まれる対イ オンが不純物として膜中に取り込まれ、効率を低下させることが知られている。一方、 ナノ粒子を作製する方法では、ナノ粒子の表面に有機分子が吸着しているものの、洗 浄により取り除くことが可能であり、7~10%の高い効率が得られている。 透明電極は、抵抗を下げることが目的となり、 粒界の数が極めて多くなるナノ粒子 を用いた方法に比べ、薄膜を作製する方法が適している。中でも電解析出法は 基板に 導電性が必要となるが、緻密な膜が高速で作製できるため適していると考えられる。
8 表1-2. 非真空プロセスの種類と特徴 非真空プロセスの種類 特徴 形態 化学浴析出法 ・基板を選ばない ・緻密な膜が得られる ・成長が比較的遅い 薄膜 電解析出法 ・緻密な膜が得られる ・基板に導電性が必要 ・成長が比較的速い ゾル-ゲル法 ・多種の材料作製可能 ・基板に耐熱性が必要となる場合が多い ・膜に溶媒の抜けた穴ができる 水熱合成 ・結晶性の高い材料が得られる ・耐熱耐圧容器が必要 ・原料に水溶性が必要 粒子 液相化学合成法 ・分散性の良いナノ粒子が得られる ・装置が簡易 ・粒子表面に有機物が吸着している 逆ミセル法 ・粒径のそろったナノ粒子が得られる ・原料に水溶性が必要
9 1-1-5. 印刷型太陽電池 ナノ粒子を用いる方法は、別に作製したナノ粒子を、印刷技術により塗布して焼成 するだけで光吸収層となるため[44,45]、生産性に優れた高速なプロセスで太陽電池を 作製することが可能となる。 ナノ粒子は数 nm~数十 nm 程度のサイズを持つ固体である。金属や半導体のナノ粒 子は、量子サイズ効果によりバルクには見られない光吸収やバンドギャップの拡大と いった、ユニークな光学的、電気的特性を示すことが知られている[46]。金属ナノ粒子 では局在表面プラズモン共鳴により光の吸収が起きることが知られ、太陽電池等の光 吸収材料としても研究されている[47,48]。また、サイズや形状により吸収波長が変化 することが知られている[49-52]。半導体のナノ粒子では、量子サイズ効果によりバン ドギャップの拡大や[53-56]、1 個の光子で 2 個の励起が生成される多重励起子生成が起 きることが知られている[56]。以上のような特性に加え、ナノ粒子は溶液中でブラウン 運動を行うため、適切な分散媒や添加剤を加えることにより沈殿を防ぐことができる。 そのため、ナノ粒子が均一に分散した懸濁液はインクとして用いることができる。近 年、ナノ粒子懸濁液をインクとして用いた研究が注目され始め、半導体のナノ粒子を 用いた太陽電池の他に[44,45,57-59]、インジウム錫酸化物(ITO)ナノ粒子を用いた透明 導電膜等が例として挙げられる[60-64]。しかし、従来の手法に比べて変換効率や電気 伝導率が低くなっているため、その改善が課題となっている。
10
1-1-6. 透明電極
一般に、透明電極には ITO や ZnO が用いられる。ZnO は材料が安価であり低コスト
化に有利であると考えられる。酸化亜鉛は透明導電膜やセンサー、発光材料への応用 が期待される材料であり、様々な手法で得ることが出来る[65-84]。また、溶液成長さ せた酸化亜鉛は、低温、未焼成にも関わらず 高い結晶性を得ることが出来る[85-88]。 さらに、作製条件により多孔質な構造を得ることも出来るため[89,90]、上述したデバ イスへの応用に応じて特性を制御できる可能性がある。 酸化亜鉛の電解析出は Lincot 等や伊崎等によって、それぞれ別の反応を用いた報告 が同時期に成されている[85-88]。また、過酸化水素の還元反応を用いた方法も報告さ れている[91,92]。これらの反応は式 1-1~1-4 のようになると考えられ、亜鉛イオンが 存在する水溶液中で、酸素や硝酸イオンを電気化学的に還元 させて生成した水酸化物 イオンにより、化学反応により水酸化亜鉛を作製する。その時、浴温を 70~80°C にし ておくことで水酸化亜鉛からの自発的な脱水を引き起こし、酸化亜鉛を析出させてい る[93,94]。
2H
O
4e
4OH
O
2 2 (1-1)
H
O
2e
NO
2OH
NO
3 2 2 (1-2)
2
e
2OH
O
H
2 2 (1-3)11
OH
ZnO
H
O
Zn
2OH
Zn
2
2
2 (1-4) 彼らの報告の後、電析温度や電位を変えた報告や[95,96]、亜鉛源の対イオンや支持電 解 質 を 変 え た 報 告[97-99] 、 ア ル ミ ニ ウ ム や ガ リ ウ ム の ド ー プ を 試 み た 報 告 の 他 に [100,101]、ポリマービーズ膜を用いた複合膜の報告[102]、等々のように、多くの研究 が行われてきた。 しかし、一般に透明導電膜では、非真空プロセスで作製したものは、真空プロセス で作製したものに比べて抵抗が高くなっている[60]。加えて、多くの場合に 300~400°C 程度の熱処理を必要とするため、太陽電池に積層した場合は熱処理が他の層に与える 影響により条件が複雑になる可能性がある。そんな中、塩素を含む電析浴から電解析 出法で作製した酸化亜鉛を CIGS 太陽電池の透明導電膜に用い、150°C の低温で焼成す ることにより、真空プロセスで作製した酸化亜鉛を用いたものに匹敵する変換効率が 報告され[103]、非真空で作製した材料が太陽電池の透明電極に利用できる可能性が示 された。 1-2. 本研究の目的 本研究の目的は、化合物半導体薄膜太陽電池に用いる材料の全てを、非真空プロセ スにより作製可能とする材料を合成・評価することである。バッファー層は非真空プ ロセスの技術が確立されていることから、 光吸収材料、透明電極について検討してい12 る。光吸収層の材料として低毒性で安価な材料から構成されている硫化錫(SnS)ナノシ ートの合成と評価を行い、化合物薄膜太陽電池の光吸収材料への応用 の可能性を探る ことを目的とした。また、透明電極材料として、電解析出法により酸化亜鉛を作製し、 その電気的、光学的な特性を評価し、透明電極への応用の可能性や、応用に必要な条 件を明らかにすることを目的とした。 1-3. 本論文の構成 本論文は全部で 5 章から構成される。始めに第 1 章となる本章で、研究の背景及び 目的を記述した。 第 2 章では、SnS ナノシートの合成と評価を行った結果について述べる。斜方晶結 晶構造を持つ SnS を電気特性の優れた a,c 軸方向に成長させた SnS ナノシートを合成 した。現在、SnS を用いた太陽電池では変換効率は 2.46%と低く、その原因として、SnS の伝導帯と価電子帯のエネルギー位置がバッファー層として利用されている CdS と合 わないことが挙げられている。そのため、SnS の価電子帯のエネルギー位置の決定を 試みた。 第 3 章では、安価な材料で構成されるもう一つの光吸収材料である CZTS のナノ粒 子を合成し、評価した結果について述べる。合成温度を変化させて粒径やバンドギャ ップを測定し、太陽電池の光吸収材料に用いるために適した合成条件の探索を行なっ
13 た。 第 4 章では、塩素を含む電解浴から電解析出法により作製した酸化亜鉛薄膜の物性 評価を行った結果について述べる。電解析出法により作製した酸化亜鉛は、化合物薄 膜太陽電池の透明電極に用いることが出来る 。そのために電気特性を評価し、電析条 件の検討を行った。 最後に第5 章では、本論文の全体の総括と今後の展望について記述した。 参考文献 [1] エネルギー白書 2006 年度版, 資源エネルギー庁 [2] エネルギー白書 2013 年度版, 資源エネルギー庁 [3] 石油製品卸価格調査結果推移表, 石油情報センター [4] NEDO 再生可能エネルギー技術白書, 新エネルギー・産業技術総合開発機構 [5] 桑野幸徳, 近藤道雄, 最新太陽光発電のすべて, オーム社, 2011.
[6] J.F. Guillemoles, Thin Solid Films 403-404 (2002) 405.
[7] M.A. Green, K. Emery, Y. Hishikawa, W. Warta, E.D. Dunlop, Prog. Phtovolt. Res. Appl. 21 (2013) 827.
[8] S. Delbos, Eur. J. Photovolt. 3 (2012) 35004. [9] H. Wang, Int. J. Photoenergy 2011 (2011) 801292.
[10] 完全図解周期表第 2 版 (ニュートンプレス, 東京, 2010 年) [11] K. Ito, T. Nakazawa: Jpn. J. Appl. Phys. 27 (1988) 2094.
[12] J. Seol, S. Lee, J. Lee, H. Nam, K. Kim: Sol. Energy Mater. Sol. Cells 75 (2003) 155. [13] T. Tanaka, T. Nagatomo, D. Kawasaki, M. Nishio, Q. Guo, A. Wakahara, A. Yo shida, H.
14
[14] H. Katagiri, Thin Solid Films 480-481 (2005) 426.
[15] K. Jimbo, R. Kimura, T. Kamimura, S. Yamada, W.S. Maw, H. Araki, K. Oishi, H. Katagiri, Thin Solid Films 515 (2007) 5997.
[16] T.K. Todorov, K.B. Reuter, D.B. Mitzi, 22 (2010) E156.
[17] S. Ahmed, K.B. Reuter, O. Gunawan, L. Guo, L.T. Romankiw, H. Deligianni, 2 (2012) 253.
[18] P. Sinsermsuksakul, J. Heo, W. Noh, A. S. Hock, R. G. Gordon, Adv. Energy Mater. 1 (2011) 1116.
[19] G. Biswajit, R. Rajarshi, C. Sumit, B. Pushan, D. Subrata, Appl. Surf. Sci. 256 (2010) 4328.
[20] P. Sinsermsuksakul, K. Hartman, S.B. Kim, J. Heo, L. Sun, H.H. Park, R. Chakraborty, T. Buonassisi, R.G. Gordon, Appl. Phys. Lett. 102 (2013) 053901.
[21] T.K. Todorov, J. Tang, S. Bag, O. Gunawan, T. Gokmen, Y. Zhu, D.B. Mitzi, Adv. En. Mater. 3 (2013) 34.
[22] D. Wang, Y. Jiang, C. Lin, S. Li, Y. Wang, C. Chen, C. Chen, Adv. Mater. 24 (2012) 3415. [23] C. Steinhagen, T.B. Harvey, C.J. Stolle, J. Harris, B.A. Korgel, J. Phys. Chem. Lett. 3
(2012) 2352.
[24] S. Seefeld, M. Limpinsel, Y. Liu, N. Farhi, A. Weber, Y. Zhang, N. Berry, Y.J. Kwon, C.L. Perkins, J.C. Hemminger, R. Wu, M. Law, J. Am. Chem. Soc. 135 (2013) 4412.
[25] H. Kou, X. Zhang, Y. Jiang, J. Li, S. Yu, Z. Zheng, C. Wang, 56 (2011) 5575. [26] Y. Cui, S. Zuo, J. Jiang, S. Yuan, J. Chu, Sol. En. Mater. Sol. Cells 95 (2011) 2136. [27] S.M. Pawar, B.S. Pawar, A.V. Moholkar, D.S. Choi, J.H. Yun, J.H. Moon, S.S. Kolekar,
J.H. Kim, Electrochim. Acta 55 (2010) 4057. [28] J.J. Scragg, P.J. Dale, L.M. Peter, 10 (2008) 639.
[29] H. Araki, Y. Kubo, A. Mikaduki, K. Jimbo, W.S. Maw. H. Katagiri, M. Yamazaki, K. Oishi, A. Takeuchi, 93 (2009) 996.
15
[30] S. Ahmed, K.B. Reuter, O. Gunawan, L. Guo, L.T. Romankiw, H. Deligianni, Adv. Energy Mater. 2 (2012) 253.
[31] V.A. Akhavan, B.W. Goodfellw, M.G. Panthani, D.K. Reid, D.J. Hellebusch, T. Adachi, B.A. Korgel, Energy Environ. Sci. 3 (2010) 1600.
[32] D.B. Mitzi, M. Yuan, W. Liu, A.J. Kellock, S.J. Chey, L. Gignac, A.G. Schrott, Thin Solid Films 517 (2009) 2158.
[33] M.M. Kamel, M.M. Ibrahim, J. Sol. Stat. Electrochem. 15 (2011) 683.
[34] M. Ichimura, K. Takeuchi, Y. Ono, E. Arai, Thin Solid Films 361-362 (2000) 98. [35] C. Gao, H. Shen, Thin Solid Films 520 (2012) 3523.
[36] T.H. Sajeesh, K.B. Jinesh, C.S. Kartha, K.P. Vijayakumar, Appl. Surf. Sci. 258 (2012) 6870.
[37] S.G. Hickey, C. Waurisch, B. Rellinghaus, A. Eychmüller, J. Am. Chem. Soc. 130 (2008) 14978.
[38] L. Ren, Z. Jin, W. Wang, H. Liu, J. Lai, J. Yang, Z. Hong, Appl. Surf. Sci. 258 (2011) 1353.
[39] L. Ren, Z. Jin, S. Cai, J. Yang, Z. Hong, Cryst. Res. Technol. 47 (2012) 461.
[40] K. Kravshyk, L. Protesescu, M.I. Bodnarchuk, F. Krumeich, M. Yarema, M. Walter, C. Guntlin, M.V. Kovalenko, J. Am. Chem. Soc. 135 (2013) 4199.
[41] T. Nakada, M. Mizutani, Jpn. J. Appl. Phys. 41 (2002) L165.
[42] K. Ramanathan, M.A. Contreras, C.L. Perkins, S. Asher, F.S. Hasoon, J. Keane, D. Young, M. Romero, W. Metzger, R. Noufi, J. Ward, A. Duda, Prog. Photovolt. Res. Appl. 11 (2003) 225.
[43] S. Siebentritt, Solar Energy, 77 (2004) 767.
[44] M.G. Panthani, V. Akhavan, B. Goodfellow, J.P. Schmidtke, L. Dunn, A. Dodabalapur, P.F. Barbara, B.A. Korgel, J. Am. Chem. Soc. 130 (2008) 16770.
16
Chem. Soc. 131 (2009) 12554.
[46] V.I. Klimov, Semiconductor and Metal Nanocrystals (Marcel Dekker, New York) p.421. [47] H. Tada, T. Mitsui, T. Kiyonaga, T. Akita, K. Tanaka, Nature Mater. 5 (2006) 782. [48] Y. Tian, T. Tatsuma, Chem. Comm. (2004) 1810.
[49] S. Link, M.A. El-Sayed, J. Phys. Chem. B 103 (1999) 4212.
[50] B.M.I. van der Zande, M.R. Bohmer, L.G.J. Fokkink, C. Schonenberger, J. Phys. Chem. B 101 (1997) 852.
[51] Y. Yu, S. Chang, C. Lee, C.R.C. Wang, J. Phys. Chem. B 101 (1997) 6661.
[52] M.B. Mohamed, K.Z. Ismael, S. Link, M.A. El-Sayed, J. Phys. Chem. B 102 (1998) 9370. [53] A.M. Smith, S. Nie, Acc. Chem. Res. 43 (2010) 190.
[54] M. Fujii, K. Nagasuna, M. Fujishima, T. Akita, H. Tada, J. Phys. Chem. C 113 (2009) 16711.
[55] T. Torimoto, H. Kontani, Y. Shibutani, S. Kuwabata, T. Sakata, H. Mori, H. Yoneyama, J. Phys. Chem. B 105 (2001) 6838.
[56] O.E. Semonin, J.M. Luther, S. Choi, H. Chen, J. Gao, A.J. Nozik, M.C. Beard, Science 334 (2011) 1530.
[57] T. Kameyama, T. Osaki, K. Okazaki, T. Shibayama, A. Kudo, S. Kuwabata, T. Torimoto, J. Mater. Chem. 20 (2010) 5319.
[58] V.A. Akhavan, B.W. Goodfellw, M.G. Panthani, C.Steinhagen, T.B. Harvey, C.J. Stolle, B.A. Korgel, J. Sol. Stat. Chem. 189 (2012) 2.
[59] S. Peng, S. Zhang, S.G. Mhaisalkar, S. Ramakrishna, 14 (2012) 8523.
[60] J. Lee, S. Lee, G. Li, M.A. Petruska, D.C. Paine, S. Sun, J. Am. Chem. Soc. 134 (2012) 13410.
[61] M. Yarema, S. Pichler, D. Kriegner, J. Stangl, O. Yarema, R. Kirchschlager, S. Tollabimazraehno, M. Humer, D. Häringer, M. Kohl, G. Chen, W. Heiss, ACS Nano 6 (2012) 4113.
17
[62] T. Sasaki, Y. Endo, M. Nakaya, K. Kanie, A. Nagatomi, K. Tanoue, R. Nakamura, A. Muramatsu, J. Mater. Chem. 20 (2010) 8153.
[63] D. Ito, K. Masuko, B.A. Weintraub, L.C. McKenzie, J.E. Hutchison, J. Nanopart. Res. 14 (2012) 1274.
[64] E.N. Dattoli, W. Lu, MRS Bulletin 36 (2011) 782.
[65] S. Zhou, X. Zhang, X. Meng, K. Zou, X. Fan, S. Wu, S. Lee, Nanotechnology 15 (2004) 1152.
[66] X. Li, X. Xu, Z. Quan, J. Guo, H. Wu, G. A. Gehring, J. Appl. Phys. 105 (2009) 103914. [67] M. Kemell, F. Dartigues, M. Ritala, M. Leskelä, Thin Solid Films 434 (2003) 20.
[68] X. Han, K. Han, M. Tao, ECS Transactions, 25 (2010) 93.
[69] W. Liu, F. Xiu, K. Sun, Y. Xie, K. L. Wang, Y. Wang, J. Zou, Z. Yang, J. Liu, J. Am. Chem. Soc. 132 (2010) 2498.
[70] A. Tsukazaki, A. Ohtomo, T. Onuma, M. Ohtani, T. Makino, M. Sumiya, K. Ohtani, S.F. Chichibu, S. Fuke, Y. Segawa, H. Ohno, H. Koinuma, M. Kawasaki, Nature Mater. 4 (2005) 42.
[71] N. Kouklin, Adv. Mater. 20 (2008) 2190.
[72] X. Han, K. Han, M. Tao, ECS transactions 25 (2010) 93.
[73] J. Nomoto, M. Konagai, K. Okada, T. Ito, T. Miyata, T Minami, Thin Solid Films 518 (2010) 2937.
[74] W.W. Wilson, A. Yamada, M. Konagai, K. Takahashi, Jpn. J. Appl. Phys. 30 (1991) L441. [75] W.W. Wilson, A. Yamada, M. Konagai, K. Takahashi, Jpn. J. Appl. Phys. 33 (1994) L283. [76] R Groenen, J Löffler, P.M Sommeling, J.L Linden, E.A.G Hamers, R.E.I Schropp, M.C.M
van de Sanden, 392 (2001) 226.
[77] Ki Cheol Park, Dae Young Ma, Kun Ho Kim, Thin Solid Films 305 (1997) 201.
[78] H. Zhu, E. Bunte, J. Hüpkes, H. Siekmann, S.M. Huang, Thin Solid Films 517 (2009) 3161.
18
[79] H. Sato, T. Minami, Y. Tamura, S. Takata, T. Mouri, N. Ogawa, Thin Solid Films 246 (1994) 86.
[80] K. Govender, D. S. Boyle, P. B. Kenway, P. O’Brien, J. Mater. Chem. 14 (2004) 2575. [81] M. Suscavage, M. Harris, D. Bliss, P. Yip, S.Q. Wang, D. Schwall, L. Bouthillette, J.
Bailey, M. Callahan, D.C. Look, D.C. Reynolds, R.L. Jones, C.W. Litton, Mater. Res. Soc. Symp. Proc. 537 (1999) G 3.40.
[82] L. Li, K.S. Hui, H.W. Park, D.H. Hwang, S. Cho, S.K. Lee, P.K. Song, Y.R. Cho, H. Lee, Y.G. Son, W. Zhou, Mater. Lett. 68 (2012) 283.
[83] N.S. Kumar, K.V. Bangera, C. Anandan, G.K. Shivakumar, J. Alloys Compd. 578, (2013) 613.
[84] T. Pauporté, I. Jirka, Electrochim. Acta 54 (2009) 7558. [85] S. Peulon, D. Lincot, J. Electrochem. Soc. 145 (1998) 864. [86] S. Peulon, D. Lincot, Adv. Mater. 8 (1996) 166.
[87] M. Izaki, T. Omi, J. Electrochem. Soc. 143 (1996) L53. [88] M. Izaki, T. Omi, Appl. Phys. Lett. 68 (1996) 2439.
[89] H.E. Belghiti, T. Pauporté, D. Lincot, Phys. Stat. Sol. A 205 (2008) 2360. [90] B. Cao, W. Cai, H. Zeng, Apll. Phys. Lett. 88 (2006) 161101.
[91] T. Pauporte, D. Lincot, J. Electrochem. Soc. 148 (2001) C310. [92] T. Pauporte, D. Lincot, J. Electroanal. Chem. 517 (2001) 54.
[93] S. Otani, J. Katayama, H. Umemoto, M. Matsuoka, J. Electrochem. Soc. 153 (2006) C551.
[94] K. Murase, H. Tada, T. Shinagawa, M. Izaki, Y. Awakura, J. Electrochem. Soc. 153 (2006) C735.
[95] A. Goux, T. Pauporté, J. Chivot, D. Lincot, Electrochem. Acta 50 (2005) 2239. [96] S. Chatman, L. Emberley, K.M. Poduska, Apll. Mater. Interf. 1 (2009) 2348.
19
23 (2008) 085013.
[98] J. Rousset, E. Saucedo, D. Lincot, Chem. Mater. 21 (2009) 534.
[99] L. Xu, Y. Guo, Q. Liao, J. Zhang, D. Xu, J. Phys. Chem. B 109 (2005) 13519. [100] M. Kemell, F. Dartigues, M. Ritala, M. Leskelä, Thin Solid Films 434 (2003) 20. [101] X. Han, K. Han, M. Tao, ESC Transact. 25 (2010) 93.
[102] D. Ramírez, H. Gómez, D. Lincot, Electrochem. Acta 55 (2010) 2191.
20
第 2 章 硫化錫ナノシートの合成と物性評価
2-1. 諸言
太陽電池の低コスト化のために は希少元素を含まない材料を用いた非真空の作製プ
ロ セ ス が 必 要 と な る 。 現 在 、 化 合 物 半 導 体 薄 膜 太 陽 電 池 の 材 料 と し て
Cu(In,Ga)(S,Se)2(CIGS)、CdTe が実用化されているが、高価な In,Ga,Te や有害な Cd を
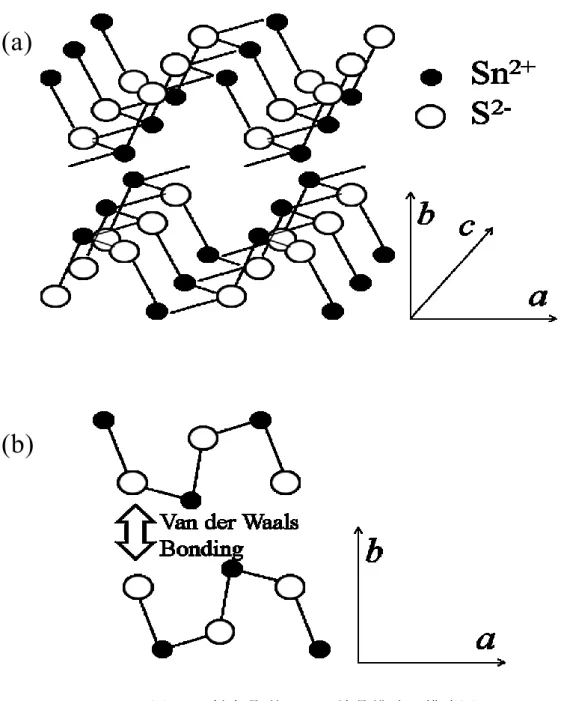
含むため、価格の高沸や環境への影響が懸念されている。その 問題を克服するために 埋蔵量が豊富な毒性の低い元素で構成される材料として Cu2ZnSnS4[1-5]や、FeS2[6]、 SnS[7-9]が注目されている。その中でも SnS は錫、硫黄の二種類の元素でのみ構成され ているために非常に組成の制御が容易な材料である。また、低コスト化に有利な非真 空方法で作製することが可能であり、これまでに電析[10,11]や CBD[12]、スプレー法 [13]の他ナノ粒子合成[14-16]等の様々な作製報告がなされている。 硫化錫(SnS)は高いキャリア移動度(~15 cm2 V-1 s-1)や高い光吸収係数(104~)及び、バ ンドギャップの値 1.1 eV、1.3 eV を間接遷移、直接遷移に対してそれぞれ持つため、 光吸収材料に適した物質である[7,10,17]。さらに、錫の割合によって p 型及び n 型のど ちらにもなることが出来る[18,19]。SnS の結晶構造は正方晶型や斜方晶型が知られて いる[20-22]。Herzenbergite 鉱型の SnS は、格子定数が a = 4.334、b = 11.200、c = 3.987 Å となる斜方晶型の結晶構造を持つ歪んだ岩塩構造として知られている。図 2-1 に斜方
21 晶型 SnS 結晶の模式図を示す。この結晶構造は、a,c 軸方向に Sn と S が共有結合した 平面構造の層が、b 軸方向に分子間力で結合し、積層した構造をしている。そのために a,c 軸方向の優れた電気特性を持ち[7,21] a,c 軸方向への異方成長した粒子を用いたデ バイスは、高い電気特性を示すことが期待される。また、このような 2 次元のナノ構 造を持つ材料は特異な電気的[23,24]、光学的特性[25]を示すことが知られている。SnS では、これまでに 2 種類の 2 次元構造が報告されている。一つは Zhang らが報告した b 軸方向に異方成長したナノシートである[26]。もう一つは Deng らが報告した、a,c 軸方 向に異方成長した SnS ナノリボンであり、(100)面と(001)面が露出した構造を持ってい る[27]。一般に露出結晶面は、その面上への同種、異種物質のバッファー層の析出や、 イオンや分子の吸着のような様々な現象に影響を与えることが知られている[28]。その ために本章では、(101)面が支配的に露出した SnS ナノシートをワンポットで合成し、 その特性評価を行った。また、太陽電池等のデバイス作製に有用な情報となる価電子 帯と伝導帯の位置を光電気化学測定により決定した。
22
(a)
(b)
図2-1. 斜方晶型 SnS の結晶構造の模式図
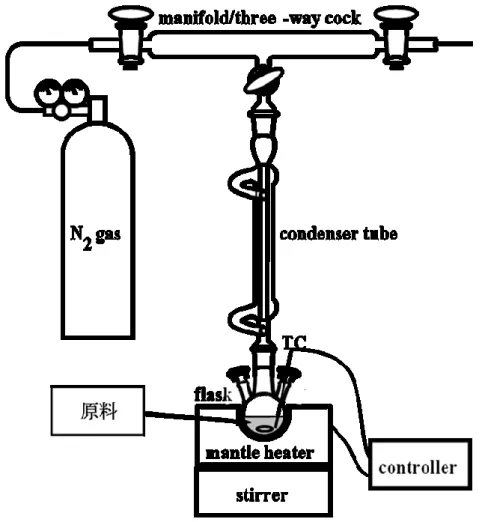
23 2-2. 実験方法 2-2-1. SnS ナノシートの合成 SnS ナノシートはオレイルアミンの熱浴中で合成した。図 2-2 に合成装置の模式図を 示す。三口フラスコに硫黄 0.5 mmol、オレイルアミン 10 mmol を加え 5 分間撹拌し硫 黄を溶解させた。その後、酢酸スズ(IV)0.5 mmol を加え、容器内を窒素により置換し た。600 rpm で撹拌しながらマントルヒーターにより 250~300°C まで昇温した後、60 分間保持した。この時、昇温後 0、10、30、60 分後に反応溶液からシリンジによりサ ンプルを抽出した。得られたサンプルに 30 ml の 2-プロパノールを加え、3500 rpm、3 分の遠心分離により SnS ナノシートを分離した。 2-2-2. SnS ナノシートの物性評価
分離したSnS ナノシートの結晶構造解析を XRD (Rint-Ultima II, Rigaku)により行った。
形 態 を 走 査 型 電 子 顕 微 鏡(SEM)(S-4800, Hitachi) 、 透 過 型 電 子 顕 微 鏡 (TEM)(JEM-210, JEOL)により観察した。光学特性を拡散反射スペクトル(U-4000, Hitachi)により測定し
た。ドロップキャストにより ITO 基板上に SnS ナノシートの製膜を行い、光電極とな
る SnS/ITO 電極を作製した。光照射下と暗状態下の Linear Sweep Voltammetry (LSV)
(HZ-5000, Hokuto Denko)により SnS/ITO 電極の光電気化学特性の評価を行った。電位
24
い、電解液には 0.1 M KCl 及び 10 mM Eu(NO3)3を含む水溶液を用いた。光電気化学測
定の光源には Xe ランプを用い、100 mW cm-2の光強度に 420 nm のシャープカットフィ
ルターを挿入したものを用いた。
25 2-3. 結果と考察 得られた SnS の XRD パターンを図 2-3 に示す。250°C、300°C 共に昇温後 0 分から、 斜方晶系のSnS の参照データ(JCPDS 39-0354)と一致したピークが得られ、Herzenbergite 鉱型 SnS の生成が確認された。250°C では昇温後の 10 分までは時間経過によって半値 幅が狭くなり、その後大きく変化しなかった。一方 300°C では昇温後 0 分からシャー プな回折ピークが得られ、時間経過による半値幅の変化は観測されなかった。同じ保 持時間で 250°C と 300°C を比較すると、300°C ではより狭い半値幅が得られた。これ は結晶性の向上や結晶サイズの増加を示唆している。また、XRD パターンからは SnS2 や Sn2S3が観察されなかった。しかし、SnS 薄膜の作製では SnS2やSn2S3が副生成物と して生成しやすく、XRD で検出されなかった SnS2やSn2S3がRaman スペクトルにより 検出出来ることが報告されている[29]。そのため、Raman スペクトルを用いて SnS2や Sn2S3の副生成物の存在の有無を確認した。
26 20 30 40 50 60 60 min 30 min 10 min
Inte
nsity / a.u
.
2
/ degree
0 min(a)
(2 10 ) (1 31 ) (0 40 ) (1 30 ) (1 11 ) (1 01 ) (0 21 ) (1 20 ) (1 10 ) 20 30 40 50 60 60 min 30 min 10 min 0 minInte
nsity / a.u
.
2
/ degree
(b)
(1 01 ) (2 10 ) (1 31 ) (0 40 ) (1 11 ) (1 30 ) (0 21 ) (1 20 ) (1 10 ) 図2-3. 合成した SnS ナノシートの XRD パターン、合成温度 250°C(a)、300°C(b)、昇温 し て か ら サ ン プ ル を 抽 出 す る ま で の 時 間 を 図 中 に そ れ ぞ れ 示 す 。 図 下 の 棒 グ ラ フ は JCPDS 39-0354 の斜方晶型 SnS のピークの位置と強度を示す。27 SnS ナノシートの Raman スペクトルを図 2-4 に示す。165,191,218 cm-1にピークが観 測された。単結晶の SnS では、b(ac)b-の偏光で測定された B3gの振動モードが 164 cm-1 に、また、b(aa)b-及び b(cc)b-の偏光で測定された Agの振動モードが 192,218 cm-1にピ ークとして現れることが知られている[40]。得られた結果は、良く一致し、SnS の生成 が確認された。また、SnS2、Sn2S3のピークとしてそれぞれ知られている 312,307 cm-1 にはピークが観測されず[7]、合成した SnS ナノシートには SnS2,Sn2S3が含まれていな いことが分かった。 100 150 200 250 300 350 SnS2 Sn3072S3 312 218 192 164 300oC 250oC
In
te
nsi
ty
/
a.u
.
Ramanshift / cm
-1 図2-4. SnS ナノシートの Raman スペクトル。307、312 cm-1の破線はそれぞれ Sn 2S3、 SnS2のピーク位置を示す。SnS は合成時間 10 min のサンプルを用いた。28 合成した SnS ナノシートの SEM による形態観察像を図 2-5 に示す。250°C では、昇 温後 0 分から幅がおよそ 200 nm、厚さがおよそ 15 nm の丸みを帯びた多角形のナノシ ートが得られ、異方成長が起きていることが確認された。10 分後には幅が約 1 μm、厚 さが 30-50 nm の歪んだ四角形や、その角が欠けた多角形の形状が観察され、シートが 成長していることが分かった。30 分以降は 10 分後とシートの形や幅、厚さに大きな変 化は見られず、昇温後 10 分で成長が止まっていることが分かった。この 0~10 分の間 の SnS ナノシートの成長は、XRD 測定から予測される結果と一致した。昇温後 10 分 で成長が止まった原因としては原料の枯渇が考えられる。一方、300℃では、昇温後 0 分で幅が 2-3 μm、厚さが 30nm 程度の歪んだ四角形や角の欠けた多角形の SnS ナノシ ートが得られた。10 分以降は形や幅、厚さに大きな変化は見られず、昇温直後にすで に原料の枯渇が起きていると考えられる。以上の結果から 250、300°C で合成した SnS ナノシートの比較は、合成時間 10 分で比較することとした。また、原料が枯渇した後 の SnS ナノシートを比較すると、300°C では 250°C に比べてシートの平均的な厚さが 薄く、幅が大きくなっていることが分かる。 SnS ナノシートを TEM により観察した結果を図 2-6 に示す。250°C では幅約 1 μm の 多角形のシートが観察され、300°C では幅約 3 μm の多角形のシートが観察された。こ れらのナノシートの制限視野電子線回折像を観察した結果、250、300°C 共にスポット 状の像が得られ、多角形状の SnS ナノシート一枚が一つの単結晶であることが分かっ
29 た。また、晶帯軸は 250、300°C 共に[010]と求められ、斜方晶構造の a,c 軸方向に異方 成長したナノシートになっていることが分かった。これは参考文献 26 とは異なる成長 方向となっている。図 2-6a,c の矢印で示した部分の高分解能 TEM 像より、250、300°C 共に格子間隔が0.293 nm の格子縞の 2 つが約 95°で交差している様子が観察され、(101) 面と(10-1)面であることが分かった。さらに、300°Cでは格子間隔が 0.217 nm の(200) 面と(101)面が約 47°で交差している様子が観察された。これらの結晶面とナノシート の形状を比較すると、ナノシートの各辺にそれぞれ{101}面及び{100}面が平行に配列 していることが分かる。また、各辺の大きさを比較すると、{101}面に平行な辺がより 大きくなっていることが分かった。これは{101}面の成長が{100}面に比べて遅いこと を示唆している。
30
(a)
(b)
(c)
(d)
図 2-5. 各温度で合成した SnS ナノシートの SEM 像。250°C(a,b)、300°C(c,d)、昇温 後からサンプルを抽出したまでの合成時間は、それぞれ 0 分(a,c)、10 分(b,d)とした。 図中の埋め込みに SnS ナノシートの断面図とシートの厚さを示す。31
(a)
(b)
(c)
(d)
(e)
(f)
図2-6. 250°C(a,b)と 300°C(c,d)で合成した SnS ナノシートの TEM 像。図中の埋め込み
はSAED パターンを示す。図 2-6a,c の矢印の部分の高分解 TEM 像(b,d)、図中に面間
隔と各面が交差する角度を示す。SnS ナノ粒子が付着した SnS ナノシートの端部の TEM 像(e)及び高分解能 TEM 像(f)、SnS ナノシートの格子縞に加え、ナノ粒子の格子 縞が観察された。
32 溶液中における結晶の異方成長は、一般的に次の 3 つの理由で起きると考えること が出来る。1 つ目は物質由来の異方成長である。成長中の結晶で露出している各結晶面 の成長速度が大きく異なる場合は、異方性を持つ粒子が得られる[30]。2 つ目は表面吸 着種の影響である。特定の結晶面にイオンや有機分子が吸着することにより 、表面エ ネルギーの低下や原料供給の阻害を引き起こ す。その結果、その結晶面の成長を促進、 または抑制することで異方成長が起きる[31]。3 つ目は原料供給の影響である。特定の 結晶面にのみ原料を供給し成長させることにより異方成長が起きる[32]。 このSnS ナノシートの異方成長では、上記の 3 つ全てが影響している可能性がある。 斜方晶型 SnS の結晶は、b 軸に垂直な面では、わずかに突き出た表面の Sn イオンから 孤立電子対が露出しており、ダングリングボンドを持つ他の面に比べ て表面エネルギ ーが低く安定であると考えられている[33,34]。そのため、b 軸方向の成長が遅く、a,c 軸方向への異方成長が起きると考えられる。合成に用いたオレイルアミンは、ナノ粒 子へ吸着することが知られている[35]。b 軸に垂直な面に吸着することにより b 軸方向
への成長を抑制し、a,c 軸方向への異方成長が起きる可能性がある。図 2-6e,f に示す TEM
像より、SnS ナノシートに数 nm 程度の SnS ナノ粒子が付着した様子が観察された。こ
のナノ粒子は 180°C から生成し、時間経過により減少することから、オストワルド熟
成により SnS ナノシートの原料になっていると考えられる。さらに、SnS ナノシート
33 子が観察された。このことから、SnS ナノ粒子がシート上を表面拡散し、シートの端 で溶解し a,c 軸方向に SnS ナノシートを異方成長させた可能性が考えられる[36]。 SnS ナノシートの拡散反射スペクトルを図 2-7a に示す。SnS ナノシートは希釈剤に 混合するとシートの破断等の可能性が考えられるため、反射剤に薄くまぶして測定を 行った。250、300°C 共に 1100 nm 程度から緩やかに反射率が減少し、900 nm 程度で反 射率が約 30 %となり減少が止まった。900 nm で反射が大きくなった理由は、測定した SnS サンプルの表面がフラットでは無いためと考えられる。拡散反射スペクトルの結 果から Kubelka-Munk (K-M)変換により作製した Tauc プロットを図 2-7b に示す。
34 900 1000 1100 1200 1300 1400 1500 0 20 40 60 80 100
(a)
300oC 250oCR
ef
lecta
nce
/ %
Wavelength / nm
1.20 1.24 1.28 1.32 1.36 0.0 0.4 0.8 1.2 1.6 2.0(b)
300oC 250oC(hv
F(R
∞))
2photon energy / eV
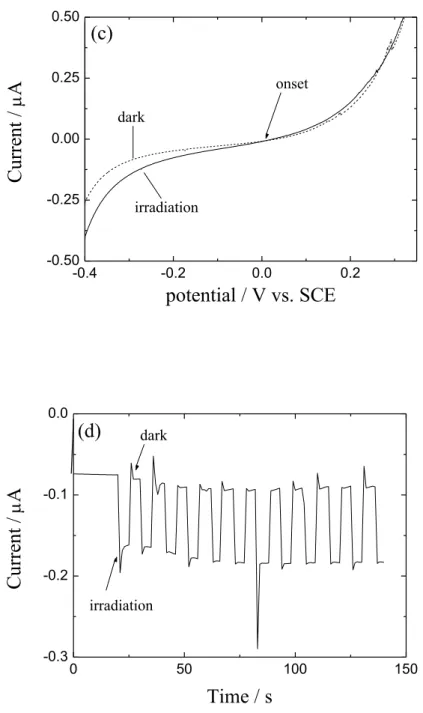
図 2-7. SnS ナノシートの拡散反射スペクトル(a)、拡散反射スペクトルを K-M 変換 して求めた Tauc プロット(b)、図中に SnS の合成温度を示す。35 約 1.31~1.34 eV の間は、フォトンエネルギー(hv)に対してフォトンエネルギー × K-M 関数の二乗((hv × F(R∞))2)が直線関係を示すことから、直接遷移の吸収であること が分かった。外挿した接線と x (hv)軸の交点からバンドギャップエネルギーを見積もる と約 1.29 eV となり、バルクで報告されている値(1.3 eV)とほぼ一致することが分かっ た[7]。 合成したSnS ナノシートを用いた SnS/ITO 光電極の LSV 測定の結果を図 2-8 に示す。 暗状態下と光照射下の電流-電位(I-V)特性を比較すると、アノード分極(+側への分極) 時は特性に変化は見られなかったが、カソード分極時にカソード電流の増加が観測さ れた。この結果から、SnS ナノシートが p 型半導体であることが確認された。また、-0.35V の定電位を印加して断続的に光を照射した時の電流を測定した結果 、光照射による明 確なカソード電流の増加が観測された。光電流の立ち上がりの電位からフラットバン ド電位を求めると、約 0V となった。一般に、p 型半導体のフラットバンド電位は価電 子帯下端に近いため価電子帯の下端の電位に近似することが出来る[37]。そのため、光 学測定から求めたバンドギャップを用いること により、SnS ナノシートの価電子帯と 伝導帯の上端と下端の電位はそれぞれ 0V、-1.3V と見積もられた。この価電子帯下端 の電位は、NHE の値を真空準位に対して 4.5 ± 0.1eV とすると[38]、飽和カロメル電 極の電位+0.241V vs. NHE より、参照文献 [39]で報告されている真空準位に対する価電 子帯の電位 4.7 eV と良い一致を示すことが分かった。
36 -0.4 -0.2 0.0 0.2 -2 -1 0 1 2 onset irradiation dark
Cu
rr
en
t /
A
potential / V vs. SCE
(a)
0 50 100 150 -0.8 -0.6 -0.4 -0.2 0.0Current
/
A
Time / s
dark irradiation37 -0.4 -0.2 0.0 0.2 -0.50 -0.25 0.00 0.25 0.50 dark irradiation
C
urre
nt /
A
potential / V vs. SCE
onset(c)
0 50 100 150 -0.3 -0.2 -0.1 0.0 irradiation dark(d)
Cu
rr
en
t /
A
Time / s
図 2-8. 各温度で合成した SnS ナノシートを用いた SnS/ITO 電極の Linear sweep
voltammogram (a,c)、図中に示すように破線と実線はそれぞれ暗電流と光電流を示 す。-0.35 V vs. SCE の定電位で光を断続照射した時の電流応答 (b,d)
38 2-4. 結論 1-3μm の幅と 30-50nm 程度の厚さの 2 次元構造を持つ SnS ナノシートの合成に成功 した。このナノシートは斜方晶の結晶構造を持ち、a,c 軸方向へ異方成長していること が分かった。また、側面にはこれまでに報告されていない(101)面が優先的に露出して いることが分かった。直接遷移に対するバンドギャップ エネルギーが約 1.29 eV と求め られ、太陽電池の光吸収材料に適した値であることが分かった。光電気化学測定より SnS ナノシートは p 型半導体であり、価電子帯と伝導帯の電位がそれぞれ 0 V, -1.3 V vs. SCE と見積もられた。これらの価電子帯、伝導帯の端の電位は太陽電池を作製する際 のバッファー層等を選択する際に重要な指針となる。 参考文献
[1] K. Ito, T. Nakazawa, Jpn. J. Appl. Phys. 27 (1988) 2094.
[2] J. Seol, S. Lee, J. Lee, H. Nam, K. Kim, Sol. Energy Mater. Sol. Cells 75 (2003) 155. [3] T. Tanaka, T. Nagatomo, D. Kawasaki, M. Nishio, Q. Guo, A. Wakahara, A. Yoshida, H.
Ogawa, J. Phys. Chem. Solids 66 (2005) 1978. [4] H. Katagiri, Thin Solid Films 480-481 (2005) 426.
[5] K. Jimbo, R. Kimura, T. Kamimura, S. Yamada, W.S. Maw, H. Araki, K. Oishi, H. Katagiri, Thin Solid Films 515 (2007) 5997.
[6] C. Steinhagen, T.B. Harvey, C.J. Stolle, J. Harris, B.A. Korgel, J. Phys. Chem. Lett. 3 (2012) 2352.
39
(2011) 1116.
[8] G. Biswajit, R. Rajarshi, C. Sumit, B. Pushan, D. Subrata, Appl. Surf. Sci. 256 (2010) 4328.
[9] P. Sinsermsuksakul, K. Hartman, S.B. Kim, J. Heo, L. Sun, H.H. Park, R. Chakraborty, T. Buonassisi, R.G. Gordon, Appl. Phys. Lett. 102 (2013) 053901.
[10] M.M. Kamel, M.M. Ibrahim, J. Sol. Stat. Electrochem. 15 (2011) 683.
[11] M. Ichimura, K. Takeuchi, Y. Ono, E. Arai, Thin Solid Films 361-362 (2000) 98. [12] C. Gao, H. Shen, Thin Solid Films 520 (2012) 3523.
[13] T.H. Sajeesh, K.B. Jinesh, C.S. Kartha, K.P. Vijayakumar, Appl. Surf. Sci. 258 (2012) 6870.
[14] S.G. Hickey, C. Waurisch, B. Rellinghaus, A. Eychmüller, J. Am. Chem. Soc. 130 (2008) 14978.
[15] L. Ren, Z. Jin, W. Wang, H. Liu, J. Lai, J. Yang, Z. Hong, Appl. Surf. Sci. 258 (2011) 1353.
[16] L. Ren, Z. Jin, S. Cai, J. Yang, Z. Hong, Cryst. Res. Tech. 47 (2012) 461.
[17] A.E. Abdelrahman, W.M.M. Yunus, A.K. Arof, J. Non-Cryst. Sol. 358 (2012) 1447. [18] M. Ristov, G. Sinadinovski, I. Grozdanov, M. Mitreski, Thin Solid Films 173 (1989) 53. [19] C. Huang, Y. Lin, C. Chuang, C. Liu, Y. Yang, J. Alloy Compd. 553 (2013) 208.
[20] E.C. Greyson, J.E. Barton, T.W. Odom, small 2 (2006) 368.
[21] W. Albers, C. Haas, H.J. Vink, J.D. Wasscher, J. Appl. Phys. 32 (1961) 2220. [22] L.A. Burton, A. Walsh, J. Phys. Chem. C 116 (2012) 24262.
[23] K. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang, S.V. Dubonos, I.V . Grigorieva, A.A. Firsov, Science 306 (2004) 666.
[24] S. Pedetti, B. Nadal, E. Lhuillier, B. Mahler, C. Bouet, B. Abécassis, X. Xu, B. Dubertret, Chem. Mater. 25 (2013) 2455.
40
(2011) 936.
[26] Y. Zhang, J. Lu, S. Shen, H. Xu, Q. Wang, Chem. Comm. 47 (2011) 5226.
[27] Z. Deng, D. Cao, J. He, S. Lin, S. M. Lindsay, and Y. Liu, ACS Nano 6 (2012) 6197. [28] X. Yu, Q. Meng, T. Luo, Y. Jia, B. Sun, Q. Li, J. Liu, and X. Huang: Sci. Rep. 3 (2013)
2886.
[29] N.R. Mathews, H.B.M. Anaya, M.A. Cortes-Jacome, C. Angeles-Chavez, J.A. Toledo-Antonio, J. Electrochem. Soc. 157 (2010) H337.
[30] M. Suscavage, M. Harris, D. Bliss, P. Yip, S.-Q. Wang, D. Schwall, L. Bouthillette, J. Bailey, M. Callahan, D. C. Look, D. C. Reynolds, R. L. Jones, and C. W. Litton, Mater. Res. Soc. Symp. Proc. 537 (1999) G 3.40.
[31] Y. Yin, A.P. Alivisatos, Nature 437 (2005) 664.
[32] H. El Belghiti, T. Pauporté, D. Lincot, Phys. Stat. Sol. A 205 (2008) 2360. [33] W. Tremel, R. Hoffmann, Inorg. Chem. 26 (1987) 118.
[34] D. Avellaneda, M.T.S. Nair, P.K. Nair, J. Electrochem. Soc. 155 (2008) D517. [35] S. Mourdikoudis, L.M. Liz-Marzán, Chem. Mater. 25 (2013) 1465.
[36] K. Govender, D. S. Boyle, P. B. Kenway, P. O’Brien, J. Mater. Chem. 14 (2004) 2575. [37] T. Sasamura, T. Osaki, T. Kameyama, T. Shibayama, A. Kudo, S. Kuwabata, T. Torimoto,
Chem. Lett. 41 (2012) 1009.
[38] A.J. Bard, L.R. Faulkner, Electrochemical Methods (Wiley, New York, 2001) 2nd ed., p. 749.
[39] L.A. Burton, A. Walsh, Appl. Phys. Lett. 102 (2013) 132111.
[40] H.R. Chandrasekhar, R.G. Humphreys, U. Zwick, M. Cardona, Phys. Rev. B 15 (1977) 2177.
41 第 3 章 Cu2ZnSnS4及び Cu2ZnSnSe4ナノ粒子の合成と特性評価 3-1. 諸言 第 2 章では光吸収材料として SnS について述べた。本章では、安価、無毒な元素を 用いるもう一つの材料である Cu2SnZnS4(CZTS)のについて検討した。また、CZTS と固 溶させることによりバンドギャップを制御するため に用いられる Cu2ZnSnSe4(CZTSe) についても同様に検討した。 化合物薄膜太陽電池の材料として、現在最もよく用いられているのは CuInSe(CIS、2
Eg = 1.0 eV)、および In を一部 Ga で置換した四元混晶の Cu(In,Ga)Se(CIGS、E2 g = 1.0-1.7 eV)である。特に CIGS 太陽電池は、20.3%の変換効率が報告されている[1]。直接遷移 半導体で光吸収係数が大きく2 μm 程度の薄膜でも太陽光を吸収可能、経年劣化がない、 耐放射線性に優れるなどの非常に優れた特性を持つ 。これらの理由から、活発に研究 開発が行われ、既に実用化が始まっている。CIS 系太陽電池の課題は、希少元素である In の資源量にあると指摘されている。最近の液晶ディスプレイの生産量増加に合わせ て In の生産量は急速に増大しており、今後の ITO 透明電極基板の需要増大やそれに伴 うインジウム価格の高騰が懸念される。 CZTS は近年開発が始まった材料で、上記の CIS 系に結晶形態が似るが、希少元素の In を Zn と Sn で置換することにより得られる化合物半導体である。CZTS は 1.4-1.5 eV
42 の禁制帯幅と、104 cm-1以上の光吸収係数を持ち、その構成元素が地殻中に豊富(Cu:75 ppm, Zn:80 ppm, Sn:2.5 ppm, S:260 ppm)に存在し、安価であるのことが特長である[2]。 これまでに原子ビームスパッタ法、電子ビーム蒸着・硫化法、同時蒸着法による CZTS 薄膜の作製について報告されている。これらの物理的手法を用いて CZTS をセルに用 いたときの変換効率は 8.5%が豊田中央研究所から報告されている[1]。一方、電析法や 還 元 法 な ど の 化 学 的 手 法 を 用 い た 作 製 方 法 も い く つ か 報 告 さ れ て い る[3-6]。また、 CZTS は、Se を混ぜることによりバンドギャップを制御することが可能である。 現在 のところ液相合成で作製した Cu2ZnSn(S,Se)4(CZTSSe)ナノ粒子をセルに用いて、最高 で変換効率 11.1%が報告されている[7]。しかし、この手法はヒドラジンという反応性 の高い非常に危険な物質を使用しているため、大量生産は難しく、実用化 するのは困 難である。また、他の合成手法が数多く開発されているが 、二段階で反応させる手法 や減圧乾燥などの複雑な工程が入るため、大量合成には不向きである。これらの原因 は、CZTS 中の 3 つの金属原料の 3 分の 2 以上に酢酸塩を用いるため、金属塩に含まれ る多量の水分を除去する必要があるためである。 本研究では、デバイス作製時に問題となる塩素などを含まない金属塩を用いて 、減 圧などの複雑な工程を含まない手法により CZTS ナノ粒子と CZTSe ナノ粒子のを合成 を試みた。また、印刷型の化合物薄膜太陽電池の光吸収材料として用いるために特性 の評価を行った。
43 3-2. 実験方法 3-2-1. CZTS, CZTSe ナノ粒子の合成 CZTS 及び CZTSe ナノ粒子の合成には、第 2 章の図 2-2 の合成装置を用い、液相化 学合成法により合成した。三口フラスコに硫黄或いはセレン 0.8 mmol と、オレイルア ミン(C18H37N) 15 mmol を加え、5 分間撹拌して硫黄を溶解させた。その後、銅アセチ
ル ア セ ト ネ ー ト(Cu(ACAC)2) 0.4 mmol、亜鉛アセチルアセトネー ト(Zn(ACAC)2) 0.2 mmol、酢酸スズ(Sn(OAC)4) 0.2 mmol 及び、ジベンジルエーテル(C14H14O) 10 ml を加え た後、装置内を窒素(N2)で置換して 100-300°C の各温度まで昇温した。その後、30 分 間攪拌しながら保持した。得られたサンプル溶液に 2-プロパノールを加え、3500 rpm で 3 分間、遠心分離を行って、生成した粒子を分離した。 3-2-2. CZTS, CZTSe ナノ粒子の評価 得られた粒子をクロロホルムに分散させ、UV-vis-NIR 吸収スペクトルを測定した。 また、XRD、ラマンスペクトル測定及び、透過型電子顕微鏡(TEM)及び走査型電子顕 微鏡(SEM)、EDX により評価した。
44 3-3. 結果と考察 3-3-1. CZTS ナノ粒子の合成 反応温度を変えて作製した粒子の XRD パターンと、CuS、CZTS、閃亜鉛鉱型(立方 晶系)の ZnS の XRD パターンを図 3-1 に示す。100°C で合成した粒子は、CuS に由来 するピークが観察され、CZTS ナノ粒子が得られなかった。150°C 以上の合成温度では CZTS 或いは ZnS のピークに一致する結果が得られた。また、合成温度の上昇に伴い、 各ピークの半値幅が減少した。これはナノ粒子結晶性の向上、或いは粒径の増加が起 きたことが示唆している。図 3-2 に示すように、CZTS の結晶構造は、ZnS を 2 つ並べ たものに極めて似ており、格子定数には 0.3%程度の違いしか見られない。したがって、 CZTS と ZnS を XRD で判別するのは困難である。作製したナノ粒子が、CZTS か ZnS、 もしくはそれらの混合物である可能性があるため、ラマンスペクトル測定により、得 られた粒子を評価した。
45 CZTS ZnS SnS CuS
20
30
40
50
60
70
80
Inte
nsity
2
/ degree
図3-1. 各温度で合成したナノ粒子の XRD パターン Cu Zn Sn S Stannite型(Cu2ZnSnS4) Zn S 閃亜鉛鉱型(ZnS) 0.541 nm 0.541 nm 0.543 nm 1.085 nm 図3-2. ZnS と CZTS の結晶構造46 各温度で合成した粒子のラマンスペクトルを図 3-3 に示す。スペクトル全体にアモ ルファスであるかのようなノイズが見られる。これは測定試料がナノ粒子であり、結 晶径が小さいためであると考えられる。合成温度が 150°C 以上の時は、334~336 cm-1 にピークが観測され、バルクの CZTS と一致した[8]。このことから 150°C 以上で CZTS の粒子が得られることが確認された。また、ZnS のラマンスペクトルのピーク位置は 350 cm-1と報告されているが、どの温度で合成した粒子にも 350 cm-1にピークは検出さ れず、ZnS が混合していないことが分かった。100°C では XRD の結果同様、ピークが 475cm-1付近に観測され、CuS の生成が確認された。
47
300
350
400
450
500
In
te
nsi
ty
Raman shift / cm
-1300℃
250℃
200℃
150℃
100℃
図3-3. 各温度で合成した粒子のラマンスペクトル 100~300°C で合成したナノ粒子の TEM 像と粒径分布を図 3-4 に示す。100°C におい ては 1.2-11.2 nm、150°C では 2.9-31.4 nm、200°C では 2.8-33.3 nm、250°C では 3.2-25.2 nm、300°C では 2.5-45.3 nm の粒径分布が得られた。反応温度が高温になるにしたがっ て、平均粒径は 7.6 nm から 22.5 nm へと増加していく傾向が見られた。これら粒子の 生成メカニズムは、反応初期で、CuS の核が生成し、150°C で CuS 上に SnS および ZnS が成長する。この時、150°C という低温であるために粒子内での原子拡散が不十分で48 あり、粒子内での組成に分布ができる。さらに、温度をあげていくと、核発生と核成 長が同時に進行し、不均一核発生によって粒子が生成する。200°C から 250°C にかけ てはオスワルド熟成が起こり粒子のサイズが均一化される。さらに、300°C では、残 った原料が大きな粒子の表面にエピタキシャル成長したた めに粒径が増加したのだと 考えられる。 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm
a
b
c
d
e
0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nma
b
c
d
e
100°C
150°C
49 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm
a
b
c
d
e
0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nma
b
c
d
e
0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nm 0 5 10 15 20 25 0 10 20 30 40 50 F re qu en cy / % Diameter / nma
b
c
d
e
200°C
250°C
300°C
図3-4. 100-300°C で合成したナノ粒子の TEM 像と粒径分布 図中に合成温度を示す。50 合成した粒子のクロロホルム分散液の吸収スペクトルを図 3-5 に示す。吸収スペク トルより、ラマンスペクトル及び XRD から CuS と判別された反応温度 100°C で作製 した粒子は、700~1000 nm にかけて CuS 特有の光吸収が見られた。また、150°C でも 若干の吸収が見られ、CuS が僅かに残っている事が示唆された。一方、200°C 以上では ほぼ消えている。また、バルクの CZTS のバンドギャップは化学量論比の組成の場合 には 1.5 eV と報告されている。本研究にて得られた吸収の吸収端は 825 nm(1.5 eV)前 後にあり、報告されている値とほぼ一致した。 300 400 500 600 700 800 900 1000 100 150 200 250 300