九州大学学術情報リポジトリ
Kyushu University Institutional Repository
ゲノムデータ解析への演繹データベースの応用に関 する研究
佐藤, 賢二
https://doi.org/10.11501/3110980
A Study on Application of Deductive Database to Analysis of Genome Data
Kenji Satou
January 1996
Abstract
A deductive database is one of the promising frameworks for developing next generation database systems because it have both high performance of rela
tional database and flexibility on query description which logic programming system provides. Through inference using logic rules, it can derive facts which are not explicitly described in base facts.
As to research works on deductive database, theoretical aspects, e.g. rule rewriting for optimization of query evaluation, have been actively studied.
However, new application areas (e.g. engineering data on CAD), in which more complicated searches have to be executable than those in a relational database system, activated application studies of deductive database to the management of such data.
In this study, we focus on genome database which is a kind of scientific database. Unlike the case of engineering data generated through computa
tion, there are few studies on application of deductive database to genome data mainly generated as the experimental result of observation of biochem
ical phenomena. However, genome database is one of attractive application areas of deductive database because it has many features which could not be found in engineering database. For example, since genome data consist of a different variety of data and include erroneous data inevitably generated through experimental observations, search requests are often ambiguous ones for highly complex entities. In such a case, a database system is requested to perform approximate search (e.g. a similarity search) over heterogeneous data.
In this study, we focus on the application of deductive database to the analysis of a database of three-dimensional structure of protein as an example of genome databases.
The following results were obtained through this study:
• A prototype deductive database system, which stores data on three
dimensional structure of protein, was developed. Some real-world ex
amples could be described on the systetn, and they could be processed in short time with the well-known query optimization technique called the Magic Set method.
• By extending the prototype system, it was enabled to search for similar structures in proteins with simple query description. This extension
did not destroy denotationality of query description which is one of the advantages of deductive database. Queries in this extended system could also be optimized through the Magic Set method. As to the more extended version of similarity search function which automatically con1pute a closure of indirect similarity relationships among protein structures, cost of computation tended to be linear to the size of search space.
Acknowledgements
I would like to express 1ny deep thanks to Professor Kazuo Ushijima at Kyushu University for his sincere guidance, support, and constructive criti
Cism.
I also express n1y thanks to Professor Toshihisa Takagi at the Univer
sity of Tokyo. I started research works on deductive database systems from 1989 under his guidance. Many ideas of this thesis were suggested by him.
Moreover, he taught me how researcher should be.
Professor Satoru Kuhara at Kyushu University, who is in Graduate School of Genetic Resources Technology, was indispensable to this thesis. Without his valuable comments based on extensive knowledge about protein engineer
ing, it would be impossible to proceed with such a interdisciplinary research.
I would like to express my thanks to Professor Fumihiro Matsuo, Professor Akifumi Makinouchi, and Professor Ryuzo Hasegawa at Kyushu University who reviewed early versions of this thesis and gave me many valuable com
ments.
I wish to thank Professor Keijiro Araki at Nara Institute of Science and Technology, who was my supervisor when I was a graduate student of Kyushu University, for introducing me to the field of computer science.
I would like to thank Dr. Takahiko Suzuki at Kyushu University and Dr. Susumu Goto at Kyoto University for agreeing willingly to using their prototype deductive database system for my thesis.
I am grateful to Mrs. Emiko Furuichi at Fukuoka Won1en's Junior College for her collaboration. She managed remarkable amount of works on making preparations for system development and experiments. Through discussions with her, I learned many things on biological database, particularly, on Pro
tein Data Bank. I am also grateful to five more persons who collaborated with me to implement a prototype deductive database system PACADE de
scribed in this thesis: Mr. Hideki Takehara at Asahi Chemical Industry Co.,Ltd., lVliss Kyoko Takiguchi at Fujitsu Laboratories Ltd., Miss Yukiko Tsukamoto at Teijin Systems Technology Ltd., Mr. Akio Nishikawa at Ube College, and Mr. Shin 'ichi Hashimoto at Kyushu University.
I am also grateful to Dr. orihiro Sakamoto at Kyushu niversity, Asso
ciate Professor Hirofumi Amano at Kyushu University, Dr. Akira Suganuma at Kyushu University, and Associate Professor Masatoshi Arikawa at Hi
roshima City University for their encouragement.
Finally, I would like to thank all the people in the laboratories directed by Professor Ushijima, Professor Takagi, and Professor Kuhara.
Contents
1 Introduction
2 Preliminaries
2.1 Deductive Database System 2.1.1 Terminology ... . . 2.1.2 Query Evaluation ..
2.1.3 Top-down vs. Bottom-up . 2.1.4 The i\tlagic Set 1\!Iethocl . .
2.2 Scientific Databases and Genon1e Databases 2.3 PDB ... . . .
2.4 Related \tVorks . . . . . . . . . . . . .
3 Applicability of Deductive Databases to Search for Biological 1
5 5 5 7 9 9 12 13 1.5
Data 17
3.1 The PACADE Systen1 . . . 17
3.1.1 System Configuration with DEE . . 18
3.1.2 System Configuration with CORAL 19
3.1.3 Relations Stored in PACADE . 21
3.2 Exan1ples of Protein Structure Search . 3.2.1 Finite Pattern Search ... . 3.2.2 Another Finite Pattern Search . 3.2.3 Hydrophobic Clusters ... .
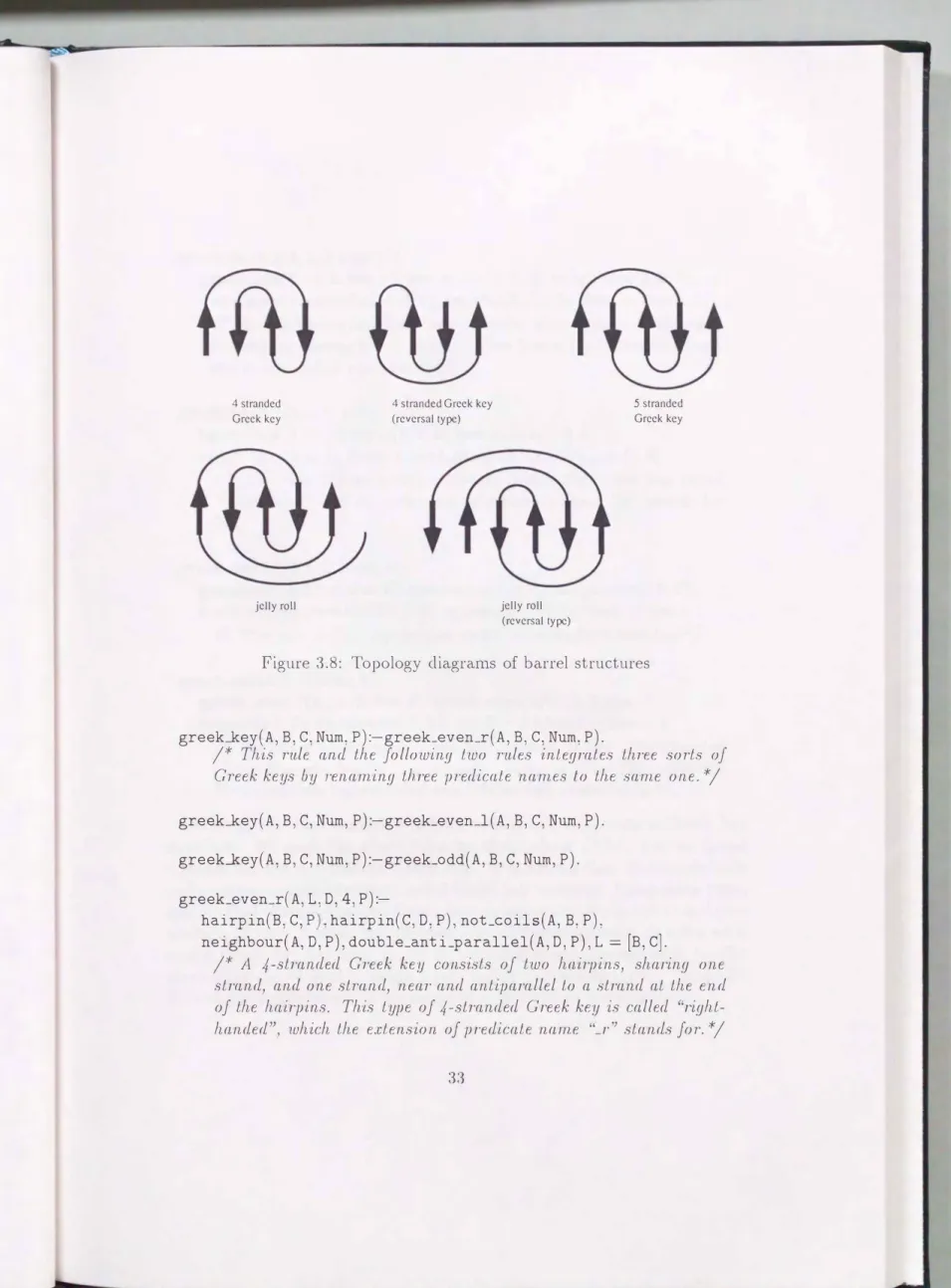
3.2.4 Chains of Electric Charges (Dipole i\tlon1ents) 3.2.5 Supersecondary Structures ... . 3.3 Perforn1ance Evaluation ... . 3.3.1 Chains of Electric Charges (Dipole i\tlon1ents) 3.3.2 n-stranded :Vleanders and Greek Keys ...
22 23 23 24 25 30 3.5 35 37
4 Similarity Search in a Deductive Database 4.1 Concept of Similar Structure Search . . . . . 4.2 Overview of a Search for Similar Structures .
4.2.1 Annotations in Rules and a Query 4.2.2 Algorithm
4.3 linplen1entation ... .
5 Applicability of Similarity Search Technique to Search for 47
48 .so .51 .53 .59
Si1nilar Structures in Proteins 61
5.1 Exa1nples of Searches for Similar Supersecondary Structures in Proteins . . . . .
5.1.1 Greek Key ... .
5.1.2 n-stranded Nleander . . . . . . . 5.2 Sin1ilarity Search for Arbitrary Structure 5.3 Discussion
.
. . . ..5.3.1 Co1nparisons with Other Approaches
.5.3.2 Extensions of Descriptive Ability of Prefixes .5.3.3 Utilization of \t\faste
(
Discarded)
Bindings5.3.4 Reduction of Prefixes ... .
61 61 6.5 67 70 70 71 72 72
6 Computation of Closures of Similarity Relationships 73 6.1 Closure of Similarity Relationships . . . 74 6.1.1 Regarding Answers as Closed Queries . . . 7 4 6.1.2 Semi-Naive Algorithm for Differential Evaluation 76 6.1.3 Optimizing Characterization Search Using Nlagic Set
i\lletho d . . . 78 6.2 System Configuration ... .
6.3 An Example of Indirect Similarity Search . 6.4 Perforn1ance Evaluation ... . 7 Conclusion
Bibliography
List of Publications
80 82 86
88 91 95
Chapter 1 Introduction
In 1970, Codd proposed the relational data model which represents a database as a set of tables
[
9]
. Two years later, he sho\ved a result that the information retrieval in a relational database and the inference in mathen1atical logic are related to each other[
1 0]
. This result implies that a search in a relational database can be carried out through the inference with a set of logical formulae. IVIoreover, it means that the expressive power of relational query languages can be extended by using logic as query language. In this way, a deductive database
[
17,3],
in which we can specify what should be retrieved in the forn1 of logic, was developed as extension of relational database.Deductive database inherits good features of relational database
(
e.g.data independence
)
. l\!Iany of query optimization techniques for relational database are also applicable to deductive database. Furthern1ore, logical query enables concise description of recursive relation and high speed cornputation of such relation in computers.
In the research field of deductive database, theoretical works have been active for two reasons 1
)
deductive database is based on mathen1atical logic, and2)
naive evaluation of deductive database is inefficient and query optimization technique for deductive database is an earnest desire. For example, algorithmic improvernent of the efficiency of query evaluation and mathemat
ical models of deductive database with negation have been actively studied.
However, in recent years, new application areas of database
(
e.g. engineering database
)
arose out of the original application area, that is, business data, in which flexible and efficient search is also desired. For this reason, studies on applicability of deductive database to the real-world problen1s inthese new areas have been activated (e.g. studies on CAD database
[
30])
.In such a study, high expressive power of deductive database was confirmed by using real data in an application area, and to n1anage area-dependent problems with which ordinary search facility could not deal, it is discussed how the framework of deductive database can be extended.
In this study, we focus on the application of deductive database to the analysis of a database of three-din1ensional structure of protein. Such database of molecular biology is generally called ge no·me database. Unlike to the case of engineering data, there are few studies on application of deductive database to genon1e data.
A biological term geno·me means a full set of genetic inforn1ation which a living thing holds. Since then vVatson and Crick elucidated structure of cleoxy ribonucleic acid(DNA
)
, molecular biology has been rapidly growing.As its growth, large amount of experimental data on sequence, structure and function of biological molecules
(
DNA, protein, etc.)
have been accun1ulatecl.The total amount of these geno·me data has been increasing exponentially, and it is indispensable for more growth of molecular biology to establish technology for flexible and efficient retrieval of massive genome data stored in con1puters. These situations gave rise to a interdisciplinary research area between molecular biology and computer science, called geno·me injo1matics or bioinformatics. This study is placed arnong other works in this research area.
There are various geno·me databases which store genome data. For exan1- ple, GenBank
[
8]
stores sequence data of DNAs, PIR[
4]
and SvVISS-PROT store sequence data of proteins, and so on. A database of three-dimensional structure of protein, on which we focus in this study, is called Protein Data Bank(PDB[
6])
. Data in PDB have been gathered by Brookhaven -ational Laboratory and distributed over the world in the form of text files. In comparison with experimental determination of a protein sequence, it is time consuming to experimentally determine the three-dimensional structure of a protein. Therefore, the number of PDB entries, each of which contains the experimental results of a specific protein, is smaller than the nun1ber of PIR or SvVISS-PROT entries. However, since a PDB entry contains three
dimensional coordinates of all aton1s in a protein, it is generally a larger file than a PIR or SvVISS-PROT entry file. As to total size of database, PDB is about 10 tin1es larger than then1.
and typical substructures ( seconda7'y slructure) as well as data on aton1ic coordinates ( terl'iary or lhree-cHme n . .,io nal structure). \Vhen we started this study, there were no flexible ways to search PDB data consisting of such h t
erogeneous data. Computer softwares which use PDB data were n1ainly to graphically display the shape of proteins (called "viewer';) or to refer known protein structures for designing new proteins (called "n1olecule n1odeling soft
ware"). Then, to find specific substructures in PDB data, rnolecular biolo
gists had to in1plen1ent how and what should be searched for by using pro
granlrning language (e.g. Fortran). To solve this problen1, it was needed to rnanage large amount of PDB data by database n1anagen1ent system (DB.\!IS) for high performance and provide flexible query language interface.
Furthermore, genon1e database is one of attractive application areas of deductive database because it has many features which could not be found in engineering database. For example, since genon1e data consist of a different variety of data and include erroneous data inevitably generated through ex
p rin1ental observations, search requests are often an1biguous ones for highly complex entities. In such a case, a database systen1 is requested to per
form approximate search (e.g. a similarity search) over heterogeneous data.
Though easy description of queries for con1plex entities is one of the strong points of deductive database, it was difficult to perfonn such an approxin1ate search on it.
The main purposes of this study are as follows:
• By applying deductive database technology to this problem provide n1olecular biologists to search PDB data flexibly and efficiently.
• Through this application, point out the features which deductive database should have in order to manage genome database.
• In1plement the needed extension of deductive database to have such features.
• Through experimental query description of real-world problen1s and perfonnance evaluations, show the applicability of deductive database to genon1e data.
Rest of this thesis is organized as follows. Chapter 2 gives terminol
ogy and basic concepts on deductive database and PDB. In chapter 3 our
prototype deductive database system PACADE
(
Protein Atomic Coordinate Analyzer with Deductive Engine)
is described in detail. Chapter 4 shows an extension of deductive database for similarity search. In chapter 5, son1e experin1ental results on the application of the sin1ilarity search technique to real-world problems are shown. In chapter 5, the similarity search function is extended to automatic computation of closure of similarity relationships.Finally, chapter 7 concludes this thesis.
Chapter 2
Preliminaries
2.1 Deductive Database System
2.1.1 Terminology
In this subsection, terminology of deductive database is explained by describ
ing the syntax of deductive database used in this study.
[constant]
A constant has one of the following forms:• a syrnbol denoted by a string begun with a lo'vver case alphabet
• a syrnbol denoted by a string consists of numeric characters
• a syn1bol denoted by a string whose first and last character is double quote
The second kind of constants are called nu·meric constants and the third kind of constants string constants in this thesis.
[variable J
A variable is denoted by a string begun with an upper case alphabet.
[function J
In general, the nan1e of a function is denoted by a string begun with a lower case alphabet. In this thesis however, only the following two types of functions are used:• A list is a function. The name of this function 1s described in ordinary Prolog style using
"['
and']'' .
• An arith1netic expression is a function. It consists of numeric constants and binary arithmetic operators (e.g. + ,
-
, *,/).
It is described in infix notation for easy understanding.[ tern1 J
A ter·m is defined recursively as follows.• A constant is a tenn.
• A variable is a term.
• A function
is a term, where
Ti ('i
= 1,... , m)
are terms. An arithmetic expression is also a tern1.
A function is a co·mplex tern1. A term is called a ground ter·m when there are no variables in the term.
[a torn J
An ato·m has a forn1where
p
is the predicate na·me (denoted by a string begun with a lovver case alphabet), andTi ('i
= l: . .. ,n, n can be0)
are terms.p(T1, T2,
. .•,
7�) is an n-ary predicate(
n can be0).
Besides the above ordinary predicates, there are special predicates called evaluable predicates. For exan1ple, binary arithmetic predicates which co1npose numerical forn1ulae (e.g. =,<,>) are evaluable predi
cates. An atom with such an arithmetic predicate is described in infix notation for easy understanding.
A ground ato·m is an atom without variables.
[fact J
A fact is a ground atom followed by a period.[rule]
A nde iswhere H is an a torn, and Li
(i
= 1 ..., k)
are atoms. H is the head of the rule, and L 1, L2, ... , L k is the body of the rule. Each L i is a body atom.A ground 'instance of a ruler is a rule obtained from r by substituting all variables in r with ground tern1s.
[database]
A(
deductive)
rial abase is a union of a set of rules and a set of facts. A set of facts is called e:r;le nsio nal database(ED B),
and a set of rules intensional database(!DB).
vVithout loss of generality, one can assume there are no predicates which occur ED B and a head of a rule in IDB. For this reason, without rnisun
derstanding, we can use the words
EDB
predicates and !DB predicates which refer to predicates in ED B and the others, respectively.[query]
A query to a deductive database is denoted by: -Q. where Q is an atom.[dependency graph]
The dependency graph for a database is a directed graph defined as follows.• For each predicate name in the database there is a node in the graph which has a one-to-one correspondence to the predicate.
• For each rule H0 : -L1, L2, ... , Lk., there is a directed edge from a node of the predicate of H0 to nodes of the predicate of Li
(i =
l, ... , k)
.[recursive rules]
A rule is recursive if the same predicate name appears in both the head and the body of the rule. A set of rules is mutually recursive iff head-predicates of the rules in the set compose a strongly connected component in the dependency graph. Rules in a mutually recursive set are recursive.2.1.2
Query Evaluation
Deductive database systen1 is one integrating the technologies of relational database system and logical inference system. The former technologies con
tribute to efficient handling of large amounts of data, and the latter offer a powerful query processing system to users.
The inference in a deductive database system, called query e·valuation, is perfonned by using facts, rules, and a query. For example,
[Fl]
distance(6 14 4.631, "4ape").[F2]
distance(l4, 1.S.S, 4.430, "4ape ').are facts,
[Rl]
cluster()(, Y, P):-clistance(X, Y, Dxy,P),
Dxy < .5.[R2]
cluster(X, Y, P):-chLster(X, Z,P),
cl-uster(Z,Y, P).
are rules and
[Ql]
:-cluster(X, Y, P).is a query. "distance" is a predicate and arguments are constants. Since the facts in deductive database can be regarded as tuples in a set of relations ( ta
bles) in relational database, the facts can be managed by relational database management system
(RDBi\IIS)
and retrieved at the need of inference systen1.R2 is recursive because it has the same predicate on both sides of implication.
From the facts and the rules, the inference systen1 can infer the following new facts which are not stored explicitly in relational database.
[F3]
cl1lster(6, 14, "4ape").[F4]
cluster(14,1.5.5
"4ape'').[F5]
cl·uster(6,1.5.5,
"4ape' ).If recursive rules are not included in the set of rules, the inference clone in deductive database systen1 can be replaced by SQL query. For exan1ple, if the relational database has following relation named distance, the query Q1 to
R1
is equivalent to the SQL query "select (code,r_num1,r_num2) from distance where dist < .S".I
r_numlI
r_nuin2I
distI
codeI
6 14 4.631 4ape
14 l.S.S 4.430 4ape
However, the query Q1 to the set of two rules
Rl
and R2 can not be replaced by any S QL query.In this point, a deductive database system excels ordinary relational database system with SQL. A user can execute a recursive search by vvriting rules recursively. Another excellence of a deductive database systen1 is the cleclarativeness of notation. A user need not to write procedural queries to get answers. These excellence give a deductive database systen1 the following two feature which are suitable for the search over three din1ensional data on proteins.
l. The knowledge concerning searches over three dimensional data of pro
teins can easily be written in denotational style and procedural pro
grams are not required.
2. A search for paths, the length of which is unknown, like dipole ·mo·menls shown in subsection 3.2.4, is feasible using recursive rules.
2.1.3 Top-down vs. Bottom-up
A query in a deductive database can be evaluated in two manners, i.e. top
clown and bottom-up. Typical method of top-down evaluation is called SLD
AL resolution, which guarantees termination of evaluation in definite Dat
alog databases by using tables of intermediate results. On the other hand, Botton1-up evaluation is based on the fixpoint operation and also guarantees tern1ination of evaluation in the same program class.
Today, bottom-up evaluation is n1ore popular than top-clown evaluation because:
• Fixpoint operation can be implemented easily.
• Some of optimization techniques are applicable, thus large an1ounts of facts can be handled efficiently.
For this reason, we adopted bottom-up evaluation in implen1enting our pro
totype system.
2.1.4 T he Magic Set Method
The 1\tlagic Set method
[
5]
is a rule rewriting technique which improves performance of bottorn-up evaluation. Assume a database composed of the following rules and facts:
(2-1) anc(X, Y):-par(X, Y).
(2-2) anc(X, Y):-par(�X", Y).
par(a,b). par(b,c). par(c,d). par(d,e).
par(d, f). par(j, b). par(j, i). par(i, c).
par(k,h). par(h,g). par(g,f).
Correspond to query
:
-anc(c, Y).a
Figure
2.1:
An Ancestor Relation For a query(2-3) :-anc(c,�Y).
naive evaluation would generate whole
anc
relations in the least model of the program(
Figure2.1).
However, some facts in the least model, such asanc(k,g)
oranc(a b),
have no relation to the query,The i\.tlagic Set method rewrites the rules into the following form.
(2-4) magicanc( c).
(2-6)
anc(X, Y):-mag·icanc(X), par( X,Y).
(2-7)
anc(X, Y):-·magicanc(X), par(�)(, X1), anc(�)(1, Y).Bottom-up evaluation of the rewritten database returns the san1e answer for the query
(2-3),
and does not generate anc facts that are not related to c.Fact
(2-4)
is a seed of n�agic facts. Rule(2-.S)
is a ·magic nde which is used to restrict the search space relevant to a given query.Rule Rewriting Procedure of the Magic Set Method
The SIPS In order to apply rnagic set rewriting procedure to a database, one n1ust supply the sideway s information passing strategy (SlPS)[5] for each body atom according to a given query.
In top-down evaluation like SLD-AL resolution, binding in a query : -anc( c,
Y).
is propagated during execution. For instance, at first call, the binding anc( c,Y)
is given to the head of rule(2-2).
The binding is propagated to the first atom, and evaluation ofpa·r(c
_\"l) binds variable �X'l to value d. As a result, the second body atom is bound to anc( d, Y). At next call, only d's ancestors are searched. Binding propagation from the head and/ or body aton1s to another body aton1 is called SIPS. SIPS for anc(XliY)
for rule(2-2)
can be written in the following form.(x r)
(2-8)
{anc(X,Y),
par(_)(, X1)} :::? anc(Xl, Y).This SIPS specifies that the binding for variableX1 in anc(_\1, Y) is propa
gated from the binding in the head anc(X, Y) and frorn
pa·r(X,
.. \:'1).Adorned Databases From binding in the query (constant c) and from the SIPS, predicate anc is adorned to ancbf that indicates the first argument of the predicate is bound and the second argument is free. The following is an adorned[5] version of the database.
ancbJ(X, Y) : -par( .. \:',
Y).
ancb1()(, Y) : -par(�X",�\1), anc61(X1, Y).
If there are rnore than one adornment for a predicate, the adorned database contains copy of rul · defining the predicate with different aclornn1ents.
Magic Rules and Rewritten Rules Then, the i\tlagic Set method gen
erates magic rules. The seed (2-4) is generated from constant c in the query.
Fron1 the SIPS (2-8), a 1nagic rule (2-5) is generated.
Final step is the rewriting of adorned rules. To propagate bindings to an adorned rule, a magic aton1 is added to the body. (2-G) and (2-6) are exan1ples of rewritten rules.
2.2 Scientific Databases and Genome Databases
There have been various scientific data in several fields of the earth (e.g., oceanography, climatology, geology), life (e.g., 1nolecular biology), chemistry, and space (e.g., astronomy, astrophysics) sciences. Fron1 n1any years ago, managen1ent of these data has been a known and critical problen1 because
1)
they have different characteristics from business data which RD B1VIS can manage successfully, and 2) unlike business data, so1ne characteristics of scientific data are don1ain specific because scientific data are subject to further domain specific analysis.
To discuss problen1s on the n1anagement of scientific data a workshop was held in
1990[7].
In the workshop, the following n1ain problen1s were addressed:1. Since scientific database holds a wide spectrum of kinds of data, n1eta
data associated with then1 n1ust be preserved and updated.
2. It is needed to describe hypothesis easily and corroborate them on scientific database.
3. It is needed to integrate the data management and domain-specific analysis environments.
4. Since there is the semantic gap between the relational model and what scientists need, it is needed to seek alternatives such as extending the re
lational paradigm, object-oriented database technology, extensible tool kits, and logic databases. It is also needed to consider alternatives to the relational n1odel for efficiently supporting temporal, spatial, in1age, sequences graph, and other more richly structured data.
5. Appropriate analysis operators within existing DB�IS, for manipulat
ing the kinds of data encountered in scientific applications, are needed.
6. Since there is heterogeneity in data and operational environrnents, stan
dardization of scientific data within disciplines is needed.
7. Standards for data citation are needed because the data used 1n the investigation should be located and examined precisely.
And two n1ore problen1s, i.e. scientific data are large an1ount and include erroneous ones, were also addressed.
As to these two problems and 2. 3. 4. and 5. above, genome database can be regarded as a type of scientific database. On the other hand, deductive database seems to be pronusing especially as to 2. above. Therefore, we chose PDB as an application and deductive database systen1 as a tool for managernent and analysis of scientific data.
2.3 PDB
PDB is an archival con1puter database of three-dimensional structures of pro
teins. The database contains bibliographic citations, prin1ary sequence and secondary structure information, as well as crystallographically determined aton1ic coordinates. Though infonnation is available on protein, DNA, RNA, virus and carbohydrate structures, we focuses on protein only.
Here is an exan1ple entry of PDB.
HEADER HYDROLASE (ENDORIBONUCLEASE) COMPND RIBONUCLEASE SA (E.C.3.t.4.8) SOURCE ( STREPTOl'IYCES $AUREOF ACIENS) AUTHOR J.SEVCIK,E.J.DODSON,G.G.DODSON REVDAT t t5-APR-92 tSAR 0
JRNL AUTH J.SEVCIK,E.J.DODSON,G.G.DODSON
t3-DEC-90 tSAR
JRHL TITL DETERMINATION AND RESTRAINED LEAST-•SQUARES JRHL TITL 2 REFINE�ffiNT OF THE CRYSTAL STRUCTURES OF JRHL TITL 3 RIBONUCLEASE SA AND ITS COl'IPLEX WITH
JRHL TITL 4 3' -•GUANYLIC ACID AT t. 8 ANGSTROl'IS RESOLUTION JRNL REF ACTA CRYSTALLOGR.,SECT.B V. 47 240 t99t
JRNL REFN ASTM ASBSDK DK ISSN Ot08-768t 622
REMARK t REMARK 2
REMARK 2 RESOLUTION. t.8 ANGSTROMS.
REMARK 3
REMARK 3 REFINEMENT. BY THE RESTRAINED LEAST-SQUARES PROCEDURE OF J.
tSAR 2 tSAR 3 tSAR 4 tSAR 5 tSAR 6 tSAR 7 tSAR 8 tSAR 9 tSAR to tSAR 11 tSAR t2 tSAR t3 tSAR t4 tSAR t5 tSAR t6 tSAR 17 tSAR t8
REMARK 3 KOMHERT AND W. HENDRICKSON (PROGRAM +PROLSQ+). THE R RE�IARK 3 VALUE IS 0.172. THE �IS DEVIATION FROM IDEALITY OF THE RENARK 3 BOMD LEMGTHS IS 0. 022 AHGSTRmiS. THE �IS DEVIATION FROM RENARK 3 IDEALITY OF THE BOND ANGLE DISTANCES IS 0.059 ANGSTROMS.
RE�IARK 4
tSAR 19 tSAR 20 tSAR 21 tSAR 22 tSAR 23 tSAR 24 tSAR 25 tSAR 26 RE�IARK 4 THE AVERAGE B VALUE FOR PROTEIN ATmiS IliJ "I'IOLECULE A IS
RE"I'IARK 4 17. 6h+2 AND IN �IOLECULE B 19. 2h+2. FOR WATER �IOLECULES RE"I'IARK 4 IT IS 35. 7A++2 AND FOR THE SULFATE ANION ATmiS IT IS RE�IARK
SEQ RES SEQ RES SEQ RES SEQ RES SEQ RES SEQ RES SEQ RES SEQRES SEQ RES SEQ RES SEQ RES SEQ RES SEQ RES SEQ RES SEQ RES SEQ RES FTNOTE FTNOTE HET FO�!UL FOR"I'!UL HELIX HELIX HELIX HELIX SHEET SHEET SHEET SHEET SHEET SHEET TURN TURN TURN TURN TURN TURN TURN TURN TURN TURN SSBOND SSBOliJD CRYSTt ORIGX1
4 22.5A++2. tSAR 27
1 A 96 ASP VAL SER GLY THR VAL CYS LEU SER ALA LEU PRO PRO tSAR 28 2 A 96 GLU ALA THR ASP THR LEU ASH LEU ILE ALA SER ASP GLY tSAR 29 3 A 96 PRO PHE PRO TYR SER GLliJ ASP GLY VAL VAL PHE GLH ASH tSAR 30 4 A 96 ARG GLU SER VAL LEU PRO THR GLN SER TYR GLY TYR TYR tSAR 31 5 A 96 HIS GLU TYR THR VAL ILE THR PRO GLY ALA ARG THR ARG tSAR 32 6 A 96 GLY THR ARG ARG ILE ILE CYS GLY GLU ALA THR GLH GLU tSAR 33 7 A 96 ASP TYR TYR THR GLY ASP HIS TYR ALA THR PHE SER LEU tSAR 34
8 A 96 ILE ASP GLM THR CYS tSAR 35
1 B 96 ASP VAL SER GLY THR VAL CYS LEU SER ALA LEU PRO PRO tSAR 36 2 B 96 GLU ALA THR ASP THR LEU ASH LEU ILE ALA SER ASP GLY tSAR 37 3 B 96 PRO PHE PRO TYR SER GLN ASP GLY VAL VAL PHE GLH ASH tSAR 38 4 B 96 ARG GLU SER VAL LEU PRO THR GLH SER TYR GLY TYR TYR tSAR 39 5 B
6 B 7 B
96 HIS GLU TYR THR VAL ILE THR PRO GLY ALA ARG THR ARG 96 GLY THR ARG ARG ILE ILE CYS GLY GLU ALA THR GLH GLU 96 ASP TYR TYR THR GLY ASP HIS TYR ALA THR PHE SER LEU 8 B 96 ILE ASP GLH THR CYS
1
1 PRO A 27 AND PRO B 27 ARE CIS PROLINES.
S04 A 97 5 SULFATE ANION 3 S04 04 Sl
4 HOH +240(H2 01) 1 Hl CYS A 7 LEU A 2 H2 PRO A 13 ASP A 3 H3 CYS B 7 LEU B 4 H4 PRO B 13 ASP B 1 St 3 TYR A 51 VAL A
11 5 ISOLATED TURN OF 3/10-HELIX 25 REGULAR ALPHA HELIX
11 5 ISOLATED TURN OF 3/10-HELIX 25 1 REGULAR ALPHA HELIX
57 0 2 St 3 ARG A 68 GLU A 74 -1 3 Sl 3 GLU A 78 THR A 82 -1 1 S2 3 TYR B 51 VAL B 57 0 2 S2 3 ARG B 68 GLU B 74 -1 3 S2 3 GLU B 78 THR B 82 -1 1 Tt ASH A 39 SER A 42 2 T2 SER A 48 TYR A 51 3 T3 THR A 59 ALA A 62 4 T4 ALA A 75 GLU A 78 5 TS ASP A 84 ALA A 87 6 T6 ASH B 39 SER B 42 7 T7 SER B 48 TYR B 51 8 T8 THR B 59 ALA B 62 9 T9 ALA B 75 GLU B 78 10 T10 ASP B 84 ALA B 87 CYS A 7 CYS A 96 2 CYS B 7 CYS B 96
64.900 78.320 38.790 90.00 90.00 1.000000 0.000000 0.000000
90.00 p 21 21 21 0.00000
8
tSAR 40 tSAR 41 tSAR 42 tSAR 43 tSAR 44 tSAR 45 tSAR 46 tSAR 47 tSAR 48 tSAR 49 tSAR 50 tSAR 51 tSAR 52 tSAR 53 tSAR 54 tSAR 55 tSAR 56 tSAR 57 tSAR 58 tSAR 59 tSAR 60 tSAR 61 tSAR 62 tSAR 63 tSAR 64 tSAR 65 tSAR 66 tSAR 67 tSAR 68 tSAR 69 tSAR 70 tSAR 71 tSAR 72
ORIGX3 0.000000 0.000000 1.000000 0.00000 1SAR 74
SCALE1 0.015410 0.000000 0.000000 0.00000 1SAR 75
SCALE2 0.000000 0.012770 0.000000 0.00000 1SAR 76
SCALE3 0.000000 0.000000 0.025780 0.00000 1SAR 77
nmr 1 H ASP A 1 45.106 12.961 8.943 1.00 22.78 1SAR 78
nmr 2 CA ASP A 45.321 12.380 7.624 1.00 37.66 1SAR 79
nmr 3 c ASP A 46.787 12.300 7.143 1.00 31.44 1SAR 80
nmr 4 0 ASP A 47.702 12.890 7.663 1.00 31.43 1SAR 81
nmr 5 CB ASP A 1 44.524 13.134 6.522 1.00 52.79 1SAR 82
ATmr 6 CG ASP A 43.989 12.323 5.322 1.00 61.59 1SAR 83 ATmr 7 OD1 ASP A 44.654 11.668 4.450 1.00 63.63 1SAR 84 ATmr 8 OD2 ASP A 42.699 12.365 5.220 1.00 65.39 1SAR 85
nmr 9 H VAL A 2 46.886 11.533 6.054 1.00 29.47 1SAR 86
nmr 10 CA VAL A 2 48.181 11.357 5.394 1.00 27.51 1SAR 87
nmr 11 c VAL A 2 48.237 12.464 4.328 1.00 31.76 1SAR 88
ATmi 12 0 VAL A 2 47.256 12.808 3.621 1.00 28.31 tSAR 89 ATmi 13 CB VAL A 2 48.270 9.889 4.917 1.00 33.86 1SAR 90 ATmi 14 CG1 VAL A 2 49.603 9.612 4.221 1.00 30.31 1SAR 91 ATmr 15 CG2 VAL A 2 47.944 8.890 6.025 1.00 30.84 1SAR 92
(
rest of atomic coordinates data are omitted.)
A PD B entry is a forn1atted text file. In each row, first 6 characters define what type of information are described in the row. For instance, rows which have SEQRES and ATOM are for the data on primary and tertiary structures, respectively. The rows which have HELIX, SHEET or TURN are for the data on secondary structure.
Today, PDB contains over 3 thousands of entries. However, we use only several hundreds of entries because:
• About 10 percent of PDB entries contain non-protein data
(
e.g. DNA)
.• In 1nany cases, some entries are for the same molecule. For example, they differ in experi1nental conditions
(
most typically, resolution of Xray diffraction analysis of crystallized moleculi
)
.• Son1e entries can not be used for structural search because they lack in structural data
(
e.g. data on secondary structure)
.2.4 Related Works
In proportion to the size of gen01ne data, a database system plays an in1- portant role in searches for large an1ounts of genome data. vVith growth in the nun1ber of PD B entries, research on relationships between structure and function of protein has also greatly advanced. lf the same sub-structure
can be found in a group of proteins with the san1e function for exan1ple enzymatic activity, then the sub-structure would play an in1portant role in function. To exan1ine such relationships, a well-designed database systen1 for protein structure analysis is needed and several prototypes were developed.
In 1982 K uhara et al. built a relational database system G Ef\ AS for primary sequences, at the Con1puter Center of Kyushu University[l5].
The BIPED project [13] led to developn1ent of a database to store and search for data in PDB. The database was 1nanaged by a con1n1ercial RDB;\IS called ORACLE and the main query language was SQL, a standard for rela
tional databases.
On the other hand, the con1n1on logic programming system, that is Pro
lag, works as a powerful query language for relational databases by regarding tuples in a relational rlatabase as Prolog's facts. In the field of biology, :.Vlorf
few and Todd experimented with the use of Prolog as a language to search protein data[18]. A serious atten1pt to use the data to search for secondary structure motifs was undertaken by Rawlings et al .[22]. The data in PDB were converted into Prolog's facts and held in n1en1ory. However, Prolog's one-tuple-at-a-time execution methods tend to be less than satisfactory when large amounts of facts are kept in disk storage (i.e. a relational database).
l\!Ioreover, a user of Prolog must write the control of a progran1 in order to guarantee termination and co1nputation of all answers.
vVith advances in the object-oriented n1odel, an object-oriented database for protein structure analysis was developed by Gray et al. [11]. The database can be queried using Prolog or the query language Daplex.
Chapter 3
Applicability of Deductive Databases to Search for Biological Data
In this chapter, design and in1plen1entation of PACAD , a prototype of de
ductive database systen1 to search for PDB data, are shown. Using some practical examples of query and PD B data stored in it, potential of P ACAD E is examined fron1 two points of view, that is, expressive power and perfor
nlance of deductive engine.
3.1 The PACADE System
A deductive database system can be regarded as a complex of an RDB..'VIS and a logical inference systen1. In the first stage of this study, we developed PACADE with a commercial RDBi\IIS INGR S and a deductive database DEE which had been developed by Suzuki,T. et al.[30]. As this study pro
ceeds we replaced INGRES with Sybase, then replaced Sybase and D E with EXODUS storage manager and CORAL deductive database system de
veloped by Ra1nakrishnan,R. et al. [21] so that we can easily put new program n1oclules into PACADE. In addition, this replacement enables FACADE sys
tenl to be distributed as free software.
3.1.1 System Configuration with DEE
Figure 3.1 shows the systen1 configuration of the PACAD systern with DEE.
vVe developed this system on Sun-3 workstation. This system consists of three
Extractor
-Deductive Engine DEE-
Bottom-up Evaluator
r+
INGRES� �;�:
srormer .... � ... --t·�� End user'---� �---�- �---�
r--
Protein Data
Bank files Database Rule files
Figure 3.1: System configuration of PACADE with DEE
sorts of files
(
Protein Data Bank file, Database, and Rule file)
and three main modules(
INGRES relational database management system, Extractor, and deductive engine DEE)
.[Protein Data Bank file J
The Protein Data Bank[
6]
file is a text file which contains various structural data(
Coordinates of atoms, secondary structure, etc.)
. To keep the database consistent, we eliminated the entries such as the entries ofDNA/RNA,
the entries which have no SEQR
ES records, and the entries whose ATOl\11 records have no data of residues.[Database J
The database is a disk volume storing data extracted from the[Rule Files J
The rule files are text files v\'hich contain rules used by the deductive engine DEE.[INGRES]
INGRES is a relational database managen1ent systern developed at the University of California. It creates relations and appends tuples by co1nn1ands fron1 Extractor. Receiving commands from the Deductive Engine, it searches the database, retrieves tuples, and returns then1 to the DeductiveEngine.
[DEE]
Deductive engine DEE consists of two submodules.(1)
Rule Transfonner This subrnodule gets a query from a user, reads rules fror11 the rule file and transforms a set of rules in the rule files to efficient ones, based on binding pattern of the query. The rule rewriting method is mainly based on the i\llagic Set n1ethod. vVe will describe this method in section2.4.
(2)
Botton1-up Evaluator This subrnodule obtains rules and a query from the transforn1er and evaluates then1 using a semi-naive evaluation n1ethod and returns an answer to the user. vVhen tuples in the databases are needed during the evaluation, this subn1odule retrieves the tuples by using IJ\GR S and translates then1 to facts. vVhen a user directly gives a query to I �GRES, the user rnust describe con1mands or procedural programs written in the conventional command form of the INGRES.[Extractor J
The Extractor reads the data from the Protein Data Base file and transfers then1 to INGRES to create relations and append tuples in the database.3.1.2 System Configuration with CORAL
Figure 3.2 shows the system configuration of the PACAD E system with CORAL. vVe developed this system on SPARCstation 10 workstation. The configuration of this system is basically the same as one in the previous sub
section except DEE and INGR S are replaced with CORAL and EXODUS, respectively.
[EXODUS]
EXODUS storage n1anager is a low-level database n1anagement systen1 developed at the University of vVisconsin. It creates relations and appends tuples to the database by commands from Extractor. ReceivingDeductive Engine CORAL
Database
End user
Figure 3.2: Systen1 configuration of FACADE with CORAL
con1mands from CORAL deductive engine, it searches the database, retrieves tuples, and returns them to CORAL.
[CORAL]
CORAL deductive engine has been developed at the University of vVisconsin[
21]
. This module perforn1s not only query evaluation evaluation but also rule rewriting for efficiency.This module gets a query from a user, reads rules fron1 the rule file and transfonns a set of rules into efficient ones, based on binding pattern of the query. The rule rewriting method is n1ainly based on the !vlagic Set n1ethocl.
Then, this 1nodule evaluates optimized rules and a query by using a semi
naive evaluation method and returns an answer to the user. vVhen tuples in the databases are needed during the evaluation, this module retrieves then1 fron1 the database through EXODUS.
3.1.3 Relations Stored 1n P ACADE
The relations stored in PACAD are in the form of facts. As this study proceeds, son1e in1provements were added to then1. For exan1ple, the facts concerning about secondary structur of protein were not considered in the first stage of this st udy
[
l6]
. Examples of the latest version of facts are as follows.source("RAT (RATTUS $RATTUS) ATYPICAL MAST CELLS FROM THE SMALL INTESTINE","3rp2").
;-t The string of the first argu·ment is the SOURCE record of the PDB file for prote-in ))3rp2'). */
author("R.REYNOLDS","3rp2").
/* The string of the first a7'g1t·ment is the AUTHOR record of the PDB file for pr·otein ))3rp2 )). One of the authors of this file is
))R.REY1VOLDS)). */
rernark("strings in REMARK record","3rp2").
/* The . tring of the fir·st arg·u·ment is the REi\tfARJ\� record of the PDB file for protein ))31p2 ). */
journal("strings in JRNL record","3rp2").
/* The string of the first argu·ment is the JR1VL record of the PDB file for protein )) 3rp2)). * /
coordinate(26,"ca",20,19.202,4.442,52.769,"a","3rp2").
/* The 26th ato·m in the protein ))3rp2 )) is )) ca )) and in the chain
'l a)). The coorchnales of the ato·m is {19.202 angstro·m) 4.442 angstr·o·m) 52.76.9 ang tro·m). */
arnino_acid(20,"val",25,31,"a","3rp2").
/*The 20th residue in the chain )) a)) of the protein ))31p2)) is valine
())val)))
and it consists of the ato·ms fro·m the 25th to the 31th.*/distance ( 2 0 , " a" , 2 4 , " a" , 11 . 7 , "3 rp 2" ) .
/* The distance between two residues) that i.'j) the 20th residue in the chain ) a)) and the 24th resid·ue in the chain )) a )) is 11.7 angstro·m in the protein ))3rp2)). */
secondary_structure( 2,strand, 2,"a" ,''3rp2").
/* The 2nd secondary structure in the chain ))a)) in the protein ))3rp2)) 'is strand type and consists of 2 residues. */
secondary_position( 2, 20 , 21 , "a", "3rp2").
/* The 2nd eco ndary structure in the chain )) a )) in the protein )3rp2) consists of the re. idues fro·m the 20th to the 21th. * / angle(2, "a", 38, "b", 30.454,29. 893,-101.428, -93.792, "3rp2'').
I* In the protein ''3rp2'', the an.fJular relat-ion betwee n two sec
ondary structures, that is, the 2nd secondary structllre in the chain ''a'' and Lhe 38th secondary structure in the chain ''b'', is e:rpressed by four para·meters. 30.454 angstr·om and 29.893 ang. tro·m are two distances. -101.428 and -93.79,.., are two angles ·varying fro·m -180 to 180 degrees. -t I
min_distance ( 2, "a", 38, "b", 37.268, "3rp2") .
I* The distance between two secondary struclttres, that is, the 2nd in the chain "a" and the 38th in the chain "b", is 37.268 angstro·m in the protein "3rp2". *I
hydrophobic_parameter("val", -1. 500).
I* The hydrophobicity of valine is -1.500. *I name_charge("val", "zero").
I* Valine is not char.r;ed. *I
PACADE contains three kinds of relations in the form of facts. The first kind of facts (source, author, remark, journal, coordinate and amino_acid) are directly extracted from PDB files. The second kind of facts (distance, secondary _structure, secondary _posit ion, angle and min_distance) are generated through some calculations. For exan1ple, Kabsh and Sander's
rnethod
[
l4]
was used for calculation of secondary _structure, secondary _position, angle and min_distance because these data on secondary structure are notalways included in Protein Data Bank files. The facts named distance are calculated from the data in ATO.YI records. If a protein has the n residues, the number of all distance facts between residues in it an1ounts to n x n.
Because of the limitation of diskspace, the facts of distance are restricted to those whose values of the 5th argun1ents are less than 1.5 angstron1. The third kind of facts (hydrophobic_parameter and name_charge) represent some sort of biological knowledge, which holds independent of PDB data, about characteristics of amino acids.
3.2 Examples of Protein Structure Search
In this section expressive power of PACAD is demonstrated through sorr1e example rules for structural search of proteins.
3.2.1 Finite Pattern Search
The following rule searches for trypsin-like catalytic triad
(
Ser-Ilis-Asp ).
Thistriad generates proteolytic activity in serine protease
[
23]
.catalytic_triad(X, Cx, Y, Cy, Z, Cz, P):
amino_acid(X, "ser", Sx, Ex, Cx, P), amino_acids(Y, "his", Sy, Ey, Cy, P),
amino_acid(Z, "asp", Sz, Ez, Cz, P) distance(X, Cx, Y, Cy, Dxy, P), distance(Y,Cy,Z,Cz,Dyz,P),Dxy < 5,Dyz < 5.
vVhen PACAD E receives the following query
:-catalytic_triad
(
X, Cx, Y, Cy, Z, Cz, "2ptn")
.it ret urns all triads of Ser-1-Iis-Asp in the protein "2ptn" in which the dis
tances both between serine and histidine and between histidine and aspirin are less than 5 angstron1. By keeping only one rule in a rule file, a user of FACADE can get such residues n1atching a specified pattern, without coding a particular program. In case that the constant "2ptn" in the above query is replaced by free variable, evaluation of the query means the search for all catalytic triads in all protein structures.
3.2.2 Another Finite Pattern Search
Biologists often find diversity in amino acid sequence an1ong some functional proteins
[
29]
, and even in such cases, a user of PAC AD E can retrieve the pattern by adding a few rules to the above exan1ple.cataly_triad_group(X, Cx, Y, Cy, Z, Cz, P):-
ser_group(X, "ser", Cx, P), his_group(Y, "his", Cy, P), asp_group(Z, "asp", Cz, P), distance(X, Cx, Y, Cy, Dxy, P), distance(Y,Cy,Z,Cz,Dyz,P),Dxy < 5,Dyz < 5.
ser_group(X, Xname, Sx, Ex, Cx, P):
name_charge(Xname,zero),amino_acid(X,Xname,Sx,Ex,Cx,P).
his_group(Y, Yname, Sy, Ey, Cy, P):
name_charge(Xname,plus),amino_acid(Y,Yname,Sy,Ey,Cy,P).
asp_group(Z, Zname, Sz, Ez, Cz, P):-
name_charge(Xname,minus) amino_acid(Z,Zname,Sz,Ez,Cz,P).
3.2.3 Hydrophobic Clusters
hydrophobic_connect(X, Cx, Y, Cy, P):-
amino_acid(X,Xname,Sx,Ex Cx,P),amino_acid(Y,Yname,Sy,Ey,Cy,P), hydrophobic_parameter(Xname, X val), X val < 2.0,
hydrophobic_parameter(Yname, Yval), Yval < 2.0, distance(X, Cx, Y, Cy, Dxy, P), Dxy < 6.0.
The above rule, used to search for hydrophobic clusters, is based on the following denotational and biological knowledge of the clusters.
(1)
Any residue with a value over2.0
in the hydrophobicity determined by Zinlffiern1an(1968)
is considered to be a hydrophobic residue.(2)
The pair of hydrophobic residues satisfying the condition that the distance between them is less than
6
angstro1n distant is considered to form hydrophobic connects.(3)
The group of residues linked by hydrophobic connects is considered to be hydrophobic cluster.The following logical query to this rule
:-hydrophobic_connect
(
X, Cx, Y, Cy, "4ape")
.is equivalent to the SQL query below, where r_nu·ml, c_narnel, r_nll·m2, c_nan�e2, code, riist, r_n?J:m, r_na·me and vallle are the attribute names of re
lations distance(r_nu·ml, c_na·mel, r_nll·m2, c_na·me2,code), a·mino_acid(r_nu·m, r_na·me,s_nu·m, e_nu·m, c_na·me, code) and hydTophobic_para·meter(r_na·me, vai'ue).
select r_nu·ml, c_na·mel, r_nu·m2, c_na·me2, code fro·m distance where code = 4 ape
and dist<
6. 0
and r_nu·m 1 in
(select r_nu·m from a·mino_acid wheTe code=4ape
and r_na·me in
and r_nu·m2 in
{select r_nu·m fro·m a·mino_acid where corle=4ape
and r_na·me in
{select T_na·me front hydrophob.Zc_para·meter where ualue<2.
0))
However, the logical query is n1ore simple and easy to understand. Fur
thermore, a logical query appears almost the same as facts while an SQL query is so mew hat like a pro grain.
vVe used the rule in the beginning of this subsection to search for hy
drophobic clusters in three acid proteases ( 4ape, 3app, 2apr). Acid proteases have hydrophobic don1ains around the active site and they tend to cleave be
tween hydrophobic amino acids. The hydrophobic clusters in three proteases are illustrated in Figure 3.3, Figure 3.4 and Figure 3.5. Each protease has four similar hydrophobic domains.
As another exan1ple, we searched for hydrophobic clusters in the five ser
ine proteases (lsbc, 2sbt, 2gch, 2sga, 2prk). vVe found no common clusters, however, in the group of subtilisin (lsbc and 2sbt), similar hydrophobic clus
ters were detected.
3.2.4
Chains of Electric Charges (Dipole Moments)
Dipole moment is one of important long range interactions among a1nino acids in protein. For example, existence of dipole mon1ent in a-helix enhances protein thern1ostability. If a user of PACADE wants to search for dipole n1oment in a protein, only it is needed to write the following rules specifying the recursive search for all dipole moments[l9].
dipole
(
X,Cx,Y,Cy,P)
:-name_charge
(
Xname, plus)
, name_charge(
Yname, minus)
:amino_acid
(
X,Xname,Sx,Ex,Cx,P)
,amino_acid(
Y:Yname:Sy:EY:Cy:P)
:distance
(
X,Cx,Y,Cy,Dxy,P)
,Dxy < 7.dipole_path
(
X, Cx, Y, Cy, P)
:-dipole(
X, Cx Y, Cy, P)
.dipole
(
X, Cx Y, Cy, P)
:-dipole
(
X Cx,21,Cz1,P)
,dipole(
22 Cz2 21 Cz1 P)
:dipole_path
(
Y, Cy, 22, Cz2, P)
.0
TyrD
PhED
Ile0
Pro0:
LeuFigure 3.3: Hydrophobic clusters found in an acid protease rhizopus
pepsin
(
2apr)
0
TyrD
PheD
Ile0
Pro0:
LeuFigure 3.4: Hydrophobic clusters found 1n an acid protease penicil
lopepsin(3app)
0
TyrD
Phe6
Ile0
Pro0:
LeuFigure 3.5: Hydrophobic clusters found 1n an acid protease endothia
pepsin