博士論文
新規 CSF-1R 低分子阻害薬と HCV ポリメラーゼ低分子阻害薬の
創薬研究
令和 2 年 2 月
池頭 和孝
岡山大学大学院
医歯薬学総合研究科
目次
要旨 1
略語表 2
第1章 序論
1-1 経口投与可能な低分子治療薬と生物学的製剤 4
1-2 慢性炎症性疾患 5
1-3 関節リウマチ 6
1-4 慢性C型肝炎 8
第2章 新規CSF-1R Type II 低分子経口阻害薬の創薬研究
2-1 緒言 11
2-2 GW2580をシード化合物とするCSF-1R Type II阻害薬の新規骨格の探索
19
2-3 化合物の合成 23
2-4 化合物のCSF-1R阻害に関する評価 28
2-5 化合物 (S)-23b のX線複合体結晶構造とキナーゼ選択性
39
2-6 考察 42
第3章 新規HCV RNA 依存型 RNAポリメラーゼ低分子経口阻害薬の創薬研究
3-1 緒言 45
3-2 化合物26をシード化合物とするHCVポリメラーゼ阻害薬の
新規骨格の探索 47
3-3 化合物の合成 50
3-4 化合物のHCVポリメラーゼ阻害に関する評価 56
3-5 考察 66
第4章 結論 69
Experimental Section 71
References 116
謝辞 121
1
要旨
本研究では慢性炎症性疾患である関節リウマチを対象とした colony-stimulating factor-1 receptor (CSF-1R) 低分子経口阻害薬と慢性 C 型肝炎を対象とした hepatitis C virus (HCV) RNA依存型RNAポリメラーゼ低分子経口阻害薬の創薬研究を行った。
慢性炎症性の自己免疫疾患である関節リウマチの治療薬としては抗 TNF-α 抗体を代表と する生物学的製剤とメトトレキサートやステロイド薬などの低分子経口薬が広く使用され ている。生物学的製剤では感染症の出現や薬価が高額であること,また,非経口投与による 利便性の低さが長期にわたる使用の障害となっている。メトトレキサートやステロイド薬 などの既存の低分子治療薬では薬効の低さや副作用の懸念が残されている。そのため,経済 性および利便性に優れた治療効果が高い低分子治療薬が望まれている。そこで,著者はCSF- 1Rを阻害することで関節リウマチに対する高い薬理作用が期待できると考えた。第2章に
て「新規CSF-1R Type II 低分子経口阻害薬の創薬研究」で経口投与可能な低分子阻害薬の
創出を目的に行った研究について述べる。キナーゼ選択性の高い CSF-1R Type II 阻害薬
GW2580 からキナーゼ選択性を維持したままオリジナル構造の取得と薬理活性向上を目的
に二つのステップに分けて構造変換を実施した。まず,L字型コンフォメーションに着目し た化合物設計によりユニークなアゼチジン骨格を見出した。次に,化合物とCSF-1Rとのド ッキングモデルを利用して置換基導入を行い,細胞での阻害活性向上と物理化学的性質の 改善を実施した。その結果,in vivo 評価で高い薬理活性を示す開発候補化合物 (S)-23bを見 出した。
慢性 C 型肝炎の有効な治療法は,注射剤のペグ化インターフェロンと核酸由来化合物リ バビリンとの併用治療であるが,治癒率が低い。また,投与期間中の重篤な副作用の発現に より治療を中断する症例が多かった。そのため,より有効性と安全性が高い経口投与可能な 抗HCV薬の開発は必須であった。そこで,第3章にて「新規HCV RNA依存型RNAポリ メラーゼ低分子経口阻害薬の創薬研究」でHCV RNAの複製を直接的に阻害できるHCVポ リメラーゼをターゲットとした経口投与可能な低分子阻害薬の創出を目的に行った研究に ついて述べる。著者が所属するグループで取得していたHCVポリメラーゼ低分子阻害薬で あるベンゾイミダゾール系化合物をもとに,細胞でのRNA複製阻害活性の向上を目的に二 つのステップに分けて構造変換を実施した。まず,ベンゾイミダゾール環とベンゼン環との 二面角に着目した化合物設計により四環性化合物を見出した。次に,化合物とHCVポリメ ラーゼとの複合体結晶構造を利用して,塩基性のピペリジン環を導入することで酵素阻害 活性およびHSAを添加したreplicon細胞でのRNA複製阻害活性が向上し,抗HCV経口治 療薬として起点となり得る化合物64を見出した。
以上のように,本研究では関節リウマチを対象としたCSF-1R 低分子経口阻害薬と慢性C 型肝炎を対象としたHCV RNA依存型RNAポリメラーゼ低分子経口阻害薬の起点となりう る新規骨格の化合物 (S)-23b と 64 を創出した。
2
略語表
Ac acetyl
ATP adenosine triphosphate
BINAP (2,2'- bis(diphenylphosphino)-1,1'-binaphthyl)
BMMCs bone marrow-derived macrophage cells
Bn benzyl
Boc tert-butoxycarbonyl
CSF-1R colony-stimulating factor-1 receptor
CIA collagen-induced arthritis
Compd compound
cPr cyclopropyl
Cy cyclohexyl
DFG motif aspartyl-phenylalanyl-glycyl motif
DMA N,N-dimethylacetamide
DME dimethoxyethane
DMF N,N-dimethylformaimde
DMRADs disease modified anti-rheumatic-drugs
DMSO dimethyl sulfoxide
dppf 1,1'-bis(diphenylphosphino)ferrocene
EC50 50% effective concentration
Et ethyl
ESI electrospray ionization
FBS fetal bovine serum
FT-IR fourier transform-infrared spectroscopy
HCV hepatitis C virus
hERG human ether-a-go-go related gene
HOBt 1-hydroxybenzotriazole
HPLC high performance liquid chromatography
HRMS high resolution mass spectrometer
HSA human serum albumin
HTS high throughput screening
IC50 50% inhibitory concentration
IL interleukin
iPr iso-propyl
JAK janus kinase
3
LPS lipopolysaccharide
mCSF-1 mouse colony-stimulating factor-1
MD molecular dynamics
Me methyl
MOM methoxymethyl
mp melting point
mRNA messenger RNA
Ms mesyl
MS mass spectrometry
NS non-structual
PEG-IFN PEGylated-interferon (PEG = polyethylene glycol)
PBS phosphate buffered saline
RBV ribavirin
PK pharmacokinetics
Ph phenyl
PO per os (oral administration)
Py pyridyl or pyridine
QOL quality of life
RNA ribonucleic acid
SBDD structure-based drug design
Sol. solubility
TFA trifluoroacetic acid
THF tetrahydrofuran
THP tetrahydropyranyl
TNF tumor necrosis factor
Ts tosyl
WSC (Water Soluble Carbodiimide)
1-ethyl-3-(3’-dimethylaminopropyl)carbodiimide hydrochloride
4
第 1 章 序論
1-1
経口投与可能な低分子治療薬と生物学的製剤
近年,数多くの生物学的製剤が開発され,様々な疾患を対象とした治療法が拡大してきた が,生物学的製剤に対する懸念点が指摘されている。1) Table 1に低分子治療薬と生物学的 製剤の長所と短所を示す。2) 生物学的製剤は特異性が高いのでoff-target の問題が少ないが,
患者の経済的負担が大きいことと投与ルートが限定されていることが欠点である。一方,低 分子治療薬はoff-target の作用に基づく安全性の懸念があるが,低価格で投与が簡便である ので経済性および利便性の利点により長期にわたる治療が可能となる。
また,生物学的製剤は生体内の分子を対象としているため,細菌やウイルスの働きを直接 阻害する作用は有さない。一方,低分子治療薬は宿主に存在しない細菌やウイルス由来のタ ンパク質などを標的とすることが可能で,低分子治療薬の利点の一つとなっている。このこ とは,低分子治療薬の中で抗菌薬などの感染症を対象とした治療薬の割合が多いことから も分かる。2)
Table 1. 低分子治療薬と生物学的製剤の長所と短所
低分子治療薬 生物学的製剤
長所
製造が安価(低価格)
投与が簡便(経口投与可能)
化学的安定性が高い
ウイルスや細菌に対する直接作用
特異性が高い
Off-target の懸念が低い
短所
予測できない問題(off-target への 作用,薬物動態に関する問題)
化合物の最適化に時間を要する
経済的な負担が大きい(高価格)
投与ルートが限定的(注射剤)
一つの分子を長時間抑制すること による懸念
アナフィラキシー反応の懸念
5 1-2
慢性炎症性疾患
炎症は内的・外的ストレスに対する生体防御反応であり,細菌やウイルスが引き起こす感 染性の炎症と生体内の成分で引き起こされる非感染性の自然炎症に分けられる。感染性の 炎症では細菌やウイルスの構成成分による刺激により防御反応が始まる。自然炎症では,細 胞がストレスにさらされたり,障害を受けることにより健常では細胞内に留まっている成 分が細胞外に出ることが刺激となって防御反応が始まる。
また炎症は,炎症の経過から比較的早期に収まる急性炎症と症状が短期で収束しない慢 性炎症に区別される。急性炎症は,細菌・ウイルスへの感染や外傷などにより引き起こされ,
1週間程度まで疼痛・腫脹・発熱などの症状が強く現れる。3a) 慢性炎症は免疫細胞をコント ロールするメカニズムの異常,ウイルスや細菌の感染の持続,および,花粉のような環境に ある抗原により引き起こされ,炎症症状が1週間以上長引き,症状が急性炎症より弱いこと が多い。3, 4)
免疫細胞をコントロールするメカニズムの異常による慢性疾患として,関節リウマチ5a, b) や多発性硬化症 5c) などの自己免疫疾患と喘息 6a) やアトピー性皮膚 6b) などのアレルギー 疾患が良く知られている。感染が持続する例としては,ピロリ菌,7a) HIV 7b) や肝炎ウイル ス7c) などがよく知られている。これら慢性疾患は長期間にわたって症状が継続し,時間が 経過すると患部組織の障害や構造変化に伴い,しばしば患部組織が有する機能に障害をき たすことが問題となる。3)
6 1-3
関節リウマチ
1-3-1 関節リウマチの疾患
自己免疫疾患は内在性および外来の抗原を起点とした生体の過剰または異常な免疫反応 の慢性化が引き起こす疾患である。4) 大部分の自己免疫疾患において,個々の発症原因や進 展のメカニズムについての十分な解明は進んでいない。治療法は疾患によって異なるが,免 疫異常が疾患の原因であることから,多くの場合はステロイド薬と免疫抑制剤が主要な治 療薬となってきた。近年は,抗TNF-α 抗体に代表される生物学的製剤が開発され,治療法 の選択肢が増えると共にその発症原因や進展のメカニズムに対する理解も少しずつ進んで きた。
自己免疫疾患の中でも関節リウマチは患者数が多い疾患の一つである。患者数は全世界
で2,370万人以上,5a) 日本国内で60–100万人以上5b) と推定されている。関節リウマチは
滑膜を病変の主座とする全身性慢性炎症性疾患であり,滑膜の慢性炎症により複数の関節 で疼痛,腫脹が持続する。また,長期に炎症が持続することによって軟骨の骨破壊,関節周 辺部の腱や靭帯の炎症性破壊が進行し,関節変形や機能障害をきたす。また,全身症状とし ては疲労,発熱,体重減少などが報告されている。このような長期にわたる関節疼痛や徐々 に進行する骨破壊や関節変形,また発熱などの全身症状による患者の生活活動および QOL (quality of life)の低下は著しい。さらに,これらに伴う社会的経済的問題による精神的苦痛 が加わって鬱病などの精神障害をきたす例もある。8)
滑膜炎症は,活性化した免疫細胞が病変部に浸潤して,それらの細胞から産生されるIL-
6 や TNF-α などの炎症性サイトカインによって引き起こされる。骨破壊は免疫細胞から産
生されたサイトカインによって活性化された破骨細胞および繊維芽細胞様滑膜細胞が重要 な役割を担っていることが報告されている。5a)
1-3-2 関節リウマチの治療薬
関節リウマチの経口治療薬としては,メトトレキサートに代表される疾患修飾性抗リウ マチ薬(DMARDs: disease-modifying anti-rheumatic drugs)とステロイド薬が使用されること が多い。9) また近年,JAK (Janus kinase) 阻害薬であるトファシチニブが承認されているが,
感染症や発がんリスクの上昇などの安全性の懸念が残されている。10) 現在,世界中の製薬 企業が,トファシチニブに続く安全性の高いJAK阻害薬の開発を実施している。
生物学的製剤としては抗TNF-α抗体,11a) 抗IL-6受容体抗体11b) およびCTLA4-Ig11c) が 承認され,治療に広く使用されている。また,IL-17, 12) GM-CSFRα12b) に対する抗体の臨床 開発が現在行われている。
7
1-3-3 新規関節リウマチ低分子治療薬の創出の意義
DMARDsは疼痛および腫脹を改善するが,効果不十分例や効果減弱が認められる。また,
DMARDsは関節破壊進行の抑制作用が十分でない。さらに,DMARDsは遅効性であるため,
効果発現までの患者のQOLの改善が不十分と言える。そのために長期の服薬が必要である が,DMARDs に懸念される消化器症状,血液障害および肝障害,並びにステロイド薬で懸 念される骨粗鬆症等の副作用は処方の中止に繋がり,治療を困難にしている。代表的な DMARDsの有害事象をTable 2に示す。8b,9)
Table 2. DMARDsの有害事象
一般名 有害事象
メトトレキサート 過敏症,血液障害,肝障害,腎障害,間質性肺炎,胃腸障害,
催奇形
D-ペニシラミン 皮疹,血液障害,味覚障害,重症筋無力症
ロベンザリット 胃腸障害,腎障害,肝障害 オーラノフィン 下痢,皮疹,口内炎,血液障害
ブシラミン 皮疹,胃腸障害,血液障害,肝障害,腎障害,間質性肺炎 ミゾリビン 血液障害,間質性肺炎,胃腸障害,肝障害,皮疹,催奇形 アクタリット 腎障害,肝障害,皮疹,白血球減少
サラゾスルファビリジン 胃腸障害,皮疹,血液障害,肝障害 Ref. 8b) 順天堂医学 2002, 48, 192–196. を改変して使用した。
一方,TNF-αおよびIL-6などのサイトカインの機能を選択的に抑制する生物学的製剤が 関節炎を速やかに改善することが明らかになった。しかし,感染症や投与時の重篤なアレル ギー反応が出現するなどの副作用も報告されている。11) さらに,薬価が高額であること,
非経口投与による利便性の低さが長期にわたる使用の障害となっている。以上の事から,疼 痛および腫脹を速やかに改善すると共に,関節破壊の進行を抑制し,安全性,利便性および 経済性の点で既存の治療薬を上回る経口投与可能な治療薬が望まれている。
著者は,炎症と骨形成の両方に関与する CSF-1R をターゲットとして選抜して関節リウ マチを対象とした低分子阻害薬の創出を目的に研究を行った。第2章で論述する。
8 1-4
慢性
C型肝炎
1-4-1 C型肝炎の疾患とC型肝炎ウイルス
C型肝炎13a, b) は血液・体液を介して体内にC型肝炎ウイルス(HCV : hepatitis C Virus)
が侵入することで発症する。HCVの感染者は世界で7,100万人,13a) 日本だけでも190-230 万人以上13b) と推定されている。
HCV の感染では,重症型の急性肝炎(劇症肝炎)になることは稀である。急性肝炎の症 状が強いほど自己の免疫応答によってHCVの排除が行われるが,初期感染者の70%程度は 感染が遷延化して持続感染へ移行する。また更に持続感染者の70%程度(初期感染者の50%
程度)が慢性肝炎へと移行する。慢性肝炎の状態が長期間経過することで,肝硬変や肝癌へ と進行する (Figure 1)。13c, d)
HCVは非A非B型肝炎の原因として発見されたフラビウイルス科ヘパシウイルス属に属 する一本鎖RNA ウイルスであり,14) 塩基配列により1–6 の遺伝子型に分類され,さらに サブタイプのa, b に分類される。世界で最も多い遺伝子型は1型で,日本では1b型が多い と報告されている。13b)
HCVは肝細胞に侵入して,細胞内で1本鎖RNAゲノムを複製して増殖する。HCV RNA は宿主の遺伝子に組み込まれることなく,mRNA として働き,ゲノムにコードされている 幾つかの非構造タンパク質を生成させる。非構造(NS)タンパク質には,NS2/NS3 間にコ ードされるNS2プロテアーゼ,NS3-4にコードされる NS3-4プロテアーゼ,NS5Aにコー ドされる膜に結合するリン酸タンパク質そして NS5B にコードされる HCV RNA 依存型 RNA ポリメラーゼ(HCV ポリメラーゼ)等が存在する。15)
Figure 1. C型肝炎の病態経過
9 1-4-2 慢性C型肝炎の治療薬
研究を実施していた10年程度前の有効な治療法は注射剤のペグ化インターフェロンα
(PEG-IFNα)と核酸由来化合物リバビリン(RBV)との併用治療13d) だけに限られており 治癒率が低い状況であったが,現在では非構造タンパク質を標的とした経口投与可能な低 分子治療薬およびこれらの複合剤が承認されており,慢性C型肝炎の治療の幅は広がって きている。代表阻害薬としてNS3-4プロテアーゼ阻害薬(アスナプレビル,グラゾプレビ ル),NS5A阻害薬(ダクラタスビル,レジパスビル),NS5B 非核酸型阻害薬(ベクラブ ビル)の化学構造をFigure 2 に示す。13d, 15, 16)
Figure 2. HCV非構造タンパク質を標的とした治療薬の化学構造と阻害薬名
10
1-4-3 新規慢性C型肝炎治療薬の創出の意義
研究を開始した当時,有効な治療法は注射剤(週一回の皮下注)のPEG-IFNαとRBVと の併用治療でありHCV 1型の治癒率は50% 程度と限られていた。また,投与期間は48週 と長期にわたり,その間に重篤な副作用の発現などにより治療を中断する症例が多い状況 であった。13d) PEG-IFNαとRBVの有害事象をTable 3 に示す。
Table 3. PEG-IFNαとRBVの有害事象 治療薬 有害事象
PEG-IFNα 発熱,倦怠感,食欲不振,抑うつ
RBV 貧血,白血球減少,消化器症状
以上の事から,より有効性と安全性が高く,短期間で治療可能であり経口投与可能な抗 HCV 薬の開発は必須であった。また,ウイルスが変異することを考慮すれば治療は多剤併 用が必要になることが予想され,より錠数が少ない低コストの薬剤が望まれていた。
そこで著者は,HCV ポリメラーゼをターゲットとした低分子阻害薬の創出を目的に研究 を行った。第3章で論述する。
11
第2章 新規 CSF-1R Type II 低分子経口阻害薬の創薬研究
2-1
緒言
2-1-1 CSF-1R (Colony-stimulating factor-1 receptor)
滑膜炎に関与する炎症性の単球・マクロファージ,および,骨びらんに関与する破骨細胞 は起源を同一とする前駆細胞から分化誘導される。Colony-stimulating factor-1 receptor(CSF-
1R)は,これらの共通する前駆細胞に発現している。17) CSF-1R はクラスIII 受容体型チ
ロシンキナーゼドメインを有するレセプターであり,そのリガンドは colony-stimulating factor-1(CSF-1 または,macrophage colony-stimulating factor (M-CSF))18) とinterleukin-34(IL-
34)19) である。CSF-1Rはリガンドと結合することで二量体を形成し,自己リン酸化する事
により活性化され,細胞内にシグナルを伝達する。活性化されたCSF-1Rは単球・マクロフ ァージ系細胞の分化,増殖,生存,遊走を刺激する。この刺激でインターロイキンなどの炎 症性メディエーターを産生し,各種の免疫細胞を分化,増殖及び活性化する。これらの活性 化した免疫細胞は関節リウマチなどの炎症性病態に関与する。17, 20) また,CSF-1Rのシグ ナル伝達は破骨前駆細胞から成熟破骨細胞への分化も促進する。20) 破骨細胞は骨破壊及び 骨吸収を促進して骨粗鬆症だけでなく関節リウマチの骨変形において重要な役割を担う。
このように,CSF-1R の細胞内への刺激を阻害することで通常の抗炎症薬とは異なり疼痛や 腫脹だけでなく骨変形・関節破壊を同時に抑える効果が期待できる。したがって,CSF-1R 阻害薬は関節リウマチの治療に非常に有益であると考えられている (Figure 3)。
Figure 3. 関節リウマチを対象としたCSF-1R 阻害薬の期待される作用
CSF-1R (緑色)とリガンドであるCSF-1, IL-34(ピンク色)が結合することで発現する作用とCSF-1Rの
リン酸化を阻害することで期待される薬理作用(赤字)を模式的に示した。
12 2-1-2 キナーゼ Type II 阻害薬
キナーゼは,ヒト体内に500種類以上存在することが知られ,それぞれが細胞内シグナル 伝達に重要な役割を担っている。キナーゼはATP のリン酸基をセリンやチロシン等のアミ ノ酸残基の水酸基に転移させることで細胞内にシグナルを伝達する。すべてのキナーゼ間 でATP を認識する方法が保存されており,活性部位に結合する阻害薬はキナーゼ選択性を 獲得するのが難しいとされる。21a) そのため,多くのキナーゼ阻害薬では標的以外のキナー ゼを阻害することによる副作用が報告されている。以上の理由により,キナーゼ阻害薬の創 薬研究,特に長期の服薬を必要とする慢性炎症性疾患を対象としたキナーゼ阻害薬の創薬 研究では,標的とするキナーゼに対する選択性が重要なポイントとなる。
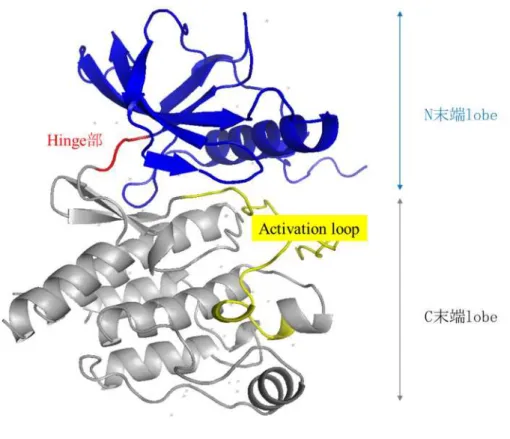
キナーゼの構造はN末端lobeとC末端lobeに分けられ,その間にhinge部が1本のルー プ構造として存在する (Figure 4)。N末端lobeとC末端lobeに挟まれた疎水性ポケットに
ATPがhinge部と水素結合を形成しながら結合してリン酸基の転移反応が進行する。21a, b)
Figure 4. キナーゼの典型的な構造
CSF-1RのX線複合体結晶構造(PDB ID: 3LCD)のタンパク質構造を使用して作図した。22, 23) CSF-1RのN 末端lobe(residues: 558–663)を青色,Hinge部(residues: 664–666)を赤色,activation loop (residues: 796–825) を黄色, C末端lobe(residues: 667–924)の内activation loopを除いた領域を灰色でcartoon表示した。
13
キナーゼ阻害薬はType I ~ Type IVの四つのタイプに分けられる。Type I 阻害薬はキナ ーゼのATP結合部位に結合する化合物,Type II 阻害薬はATP結合部位からactivation loop が移動して形成される疎水性ポケットまでの領域に結合する化合物,Type III阻害薬は ATP結合部位近傍のアロステリック部位に結合する化合物,Type IV阻害薬はそれ以外の アロステリック部位に結合する化合物である。21a, 24) 以下,本研究に関連するType I阻害
薬とType II阻害薬について説明する。
キナーゼには活性型と不活性型のコンフォメーションが存在する。活性型と不活性型の コンフォメーションの構造をFigure 5 22, 25) に示す。
キナーゼは細胞内でactivation loop のリン酸化,脱リン酸化を介して活性型と不活性型の コンフォメーションを行き来しており,Figure 5-a, b に示す様に活性型と不活性型コンフ ォメーションで黄色で表記した activation loop の位置が大きく異なる。hinge 部近傍の
activation loop が開始する位置にDFG(Asp, Phe, Gly)モチーフがキナーゼ共通に存在して
いる。キナーゼ活性を発現する活性型コンフォメーションではDFGモチーフがin(DFG-in 型),キナーゼ活性が阻害される不活性型コンフォメーションではout(DFG-out型)となる
(Figure 5-c, d)。21a, 26) DFG-out型コンフォメーションを取ることで,フェニルアラニンが 大きく移動し,フェニルアラニンのベンジル基が結合していた ATP結合部位の奥側の空間 に疎水性ポケットが新たに形成される。Type I 阻害薬は活性型のDFG-in 型コンフォメーシ ョンのATP結合部位に結合している。ATP結合部位のアミノ酸残基の相同性はキナーゼ間 で比較的高いので,高いキナーゼ選択性を有する Type I 阻害薬を見出すことは一般的に難 しいと考えられている。一方,Type II 阻害薬はDFG-out型コンフォメーションのキナーゼ
に対して hinge 部からフェニルアラニンが移動して形成した疎水性ポケットまでの領域に
結合する。この疎水性ポケットはキナーゼにより形状が異なるので,Type II 阻害薬は一般 的に高いキナーゼ選択性を示す。また,Type II 阻害薬はDFG-out型に構造変化したタンパ ク質に結合するので,化合物とタンパク質との結合と解離が遅くなる(会合速度定数(kon
値)と解離速度定数(koff値)が共に小さくなる。)。つまり,化合物とタンパク質とが結合 するための時間は長く要するが,一回結合すると解離するためにも長い時間を要し,その結 果として化合物とタンパク質との結合時間は長くなり(residence time が長くなる),生体内 での薬理活性に有利に働くことが期待できる。21a, 27) また,不活性型コンフォメーションの キナーゼは競合基質であるATPとの親和性が低下していることからも,Type II阻害薬は酵 素評価活性より生体内での薬理活性が高くなることが期待できる。27c)
14
Figure 5. キナーゼの活性型コンフォメーションと不活性型コンフォメーションの構造
(a) 活性型コンフォメーション・DFG-in型のCSF-1RとPfizer化合物のX線複合体結晶構造(PDB ID: 3LCD)。
22, 23) Activation loop (residues: 796–825) を除くCSF-1Rを灰色,activation loopを黄色のcartoonで表示し,
Pfizer化合物の炭素原子を緑色,窒素原子を青色,酸素原子を赤色でstick表示した。(b) 不活性型コンフォ
メーション・DFG-out型のCSF-1RとGW2580のX線複合体結晶構造。23, 25) Activation loop (residues: 796–
825) を除くCSF-1Rを灰色, activation loopを黄色のcartoonで表示し,GW2580の炭素原子を緑色,窒素 原子を青色,酸素原子を赤色でstick表示した。(c) 図aのATP結合部位を拡大した図。CSF-1Rの表面を灰 色で表示し,DFG(Asp796, Phe797, Gly798)モチーフの炭素原子を黄色,窒素原子を青色,酸素原子を赤
色でstick表示した。Pfizer化合物の炭素原子を緑色,窒素原子を青色,酸素原子を赤色でstick表示した。
(d) 図bのATP結合部位を拡大した図。CSF-1Rの表面を灰色で表示し,DFG(Asp796, Phe797, Gly798)
モチーフ炭素原子を黄色,窒素原子を青色,酸素原子を赤色でstick表示した。GW2580の炭素原子を緑色,
窒素原子を青色,酸素原子を赤色でstick表示した。
15
2-1-3 既知CSF-1R 阻害薬の課題と本研究の目的
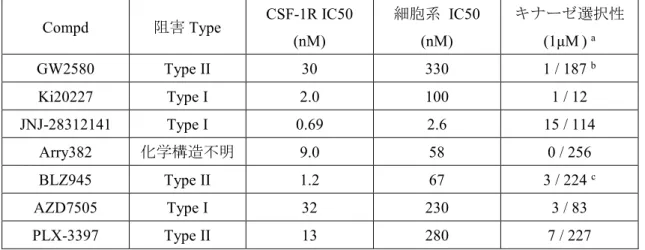
研究の当初より,GW2580,25) Ki20227,28) JNJ-28312141,29) Arry382,30) BLZ945,31) AZD7505,32)
PLX-3397 33) (Figure 6) など多くのCSF-1R 低分子阻害薬の報告34) はあったが,上市さ
れている低分子化合物は未だにない。その原因として細胞での薬理活性とキナーゼ選択性 の低さが考えられた。各阻害薬の文献中の阻害活性値とキナーゼ選択性をTable 4 にまとめ た。
Figure 6. 報告されている代表CSF-1R 阻害薬の化学構造25, 28, 29, 31, 32, 33)
16
Table 4. CSF-1R阻害薬の薬理活性情報 25, 28, 29, 30, 31, 32, 33)
Compd 阻害Type CSF-1R IC50
(nM)
細胞系 IC50 (nM)
キナーゼ選択性 (1μM) a
GW2580 Type II 30 330 1 / 187 b
Ki20227 Type I 2.0 100 1 / 12
JNJ-28312141 Type I 0.69 2.6 15 / 114
Arry382 化学構造不明 9.0 58 0 / 256
BLZ945 Type II 1.2 67 3 / 224 c
AZD7505 Type I 32 230 3 / 83
PLX-3397 Type II 13 280 7 / 227
a キナーゼ選択性についてGW2580とBLZ945以外の化合物は,1 μM での「CSF-1R以外の阻害活性を示 したキナーゼ数 / CSF-1R以外の評価したキナーゼ数」を示した。b 5 μM でのキナーゼ選択性。c 10 μM で のキナーゼ選択性。
報告されている CSF-1R 阻害薬の中でキナーゼ選択性が高い化合物の一つが GW2580 25) である。GW2580はDFG-out型のCSF-1R に結合するキナーゼType II阻害薬であり,187 キナーゼに対するキナーゼパネル評価で TrkA 以外の 186 キナーゼに阻害を示さずにキナ ーゼ選択性が高いことが報告されているが,薬理活性に関しては細胞での阻害活性が低い
(cell IC50 = 0.33 μM)。25) また,GW2580は芳香族性NH2基を二つ,合計四つの水素結合し 得る水素原子を有している。水素結合し得る水素原子数の多さは,経口吸収性の観点からリ ード化合物の性質としての懸念点となる。また,芳香族性 NH2基には変異原性の毒性が懸 念される。35) そこで著者はGW2580の結合様式とそれに伴うキナーゼ選択性を維持したま ま,芳香族性NH2基の構造を回避したオリジナル構造の取得と細胞およびin vivo 評価での 薬理活性向上を目的に研究を実施した。
17 2-1-4 GW2580とCSF-1R の複合体結晶構造
新規CSF-1R阻害薬を分子設計するにあたり,既知のCSF-1R阻害薬であるGW2580と
CSF-1R の複合体構造情報に着目した。Figure 7にGW2580とCSF-1RのX線複合体結晶
構造25) を示す。
O O N
N NH2
H2N
O
A B
C
GW2580 b
Figure 7. GW2580とCSF-1R とのX線複合体結晶構造
(a) GW2580とCSF-1R とのX線複合体結晶構造。23, 25) CSF-1Rを灰色で示し,CSF-1RのThr663, Glu664, Tyr665, Cys666, Asp796, Phe797, Gly798 の炭素原子,窒素原子,酸素原子,硫黄原子はそれぞれ白色,青色,
赤色,オレンジ色のstickで表示した。GW2580 の炭素原子,窒素原子,酸素原子はそれぞれ緑色,青色,
赤色のstickで表示した。GW2580とCSF-1Rとの水素結合は水色の破線で表示し,原子間距離を表記した。
(b) GW2580 の化学構造。ジアミノピリミジン環をring A,中央のベンゼン環をring B,メトキシベンジル
基をring Cとして青字でA, B, Cで表記した。
18
GW2580は,そのring A とring Bがメチレン基を軸にねじれ,L字型コンフォメーショ
ンをとっている。この特徴的なコンフォメーションは,hinge部から疎水性ポケットまで化 合物が結合する際に必要な形状であると考えられる。
Ring Aのジアミノピリミジン環は,CSF-1Rのhinge部(Glu664, Tyr665, Cys666)とその 隣のアミノ酸残基(Thr663)と四つの水素結合を形成している。また,ring Bのメトキシ 基とベンジルオキシ基の二つの酸素原子がDFG(Asp796, Phe797, Gly798)モチーフの
Asp796の主鎖のNHと二つのパターンの水素結合で相互作用し,DFGモチーフをout型の
コンフォメーションで固定化している。一般的なType II阻害薬では一つの水素結合で DFGモチーフと相互作用していることが多く,26) このようにDFGモチーフと二つのパタ ーンの水素結合を獲得していることは非常に特徴的であり,GW2580のキナーゼ選択性が 高い要因の一つと考察できる。
GW2580のメトキシベンジル基ring C はDFG-out型コンフォメーションによって
Phe797のベンジル基が移動して形成された疎水性ポケットを占有し,CSF-1Rと疎水性相
互作用を形成している。GW2580のring C は,CSF-1Rの疎水性ポケットの奥まで占有し ていて,これ以上大きな置換基の導入は困難と考えられる。
このようにGW2580は典型的なType II 阻害薬の結合様式でCSF-1Rと結合しているた め高いキナーゼ選択性を示していると考えられる。
19
2-2 GW2580
をシード化合物とする
CSF-1R Type II阻害薬の新規骨格の探索
GW2580をシード化合物として新規のCSF-1R Type II阻害薬を見出すために,著者は二つ
のステップで作業を行う戦略を立てた。ステップ1としてGW2580の複合体構造情報を参
考にDFG-out型のCSF-1Rに結合する新規骨格を創出し,ステップ 2 として新規骨格の複
合体構造情報を利用して効率的な置換基導入を実施することによる薬理活性向上と物性改 善を実施する計画を立てた。
ステップ1 化合物の剛直化を利用した新規骨格の創製
GW2580のキナーゼ選択性には,Asp796 の主鎖のNHと二つのパターンの水素結合を形
成しているring BとDFG-out型コンフォメーションにより形成された疎水性ポケットを占 有しているベンジル基ring Cが重要である。そこで,キナーゼ選択性は維持しつつGW2580 から新規骨格を探索する戦略として,GW2580のring Bとring C の構造を保持したまま,
そのring B とring A を連結しているメチレン基からring A部分の構造変換を実施すること
を考えた。
GW2580のring A とring B が形成している特徴的なL字型コンフォメーションを取り得
る構造として,Figure 8に示したアゼチジン,ピロリジン,および,ピペリジンの飽和複素
環がring Bに直接結合した構造を設計した。立体的に嵩高い飽和環が直接ring Bに結合す
ることで飽和環とring Bとの結合(Figure 8 中の赤色で示した結合)の回転が,GW2580の メチレン基との回転より抑制されるため,化合物のコンフォメーションが剛直化してタン パク質との親和性が向上すると予想した。
それぞれ三つ構造(I, II, III)を GW2580のX線複合体結晶構造に簡易的に重ね合わせた
図をFigure 9に示す。Hinge部側(紙面奥側)のタンパク質表面と衝突しないようなコンフ
ォメーションで,構造I, II, IIIをGW2580にring Bおよび赤く表示した結合とで重ね合わ せた。Figure 9-b,Figure 9-d,Figure 9-fから,アゼチジン構造Iが周りのタンパク質と衝 突することなく最もよくフィットするが,ピロリジン構造 II とピペリジン構造 III はタン パク質と衝突することが推測された。この結果から,アゼチジン環を足掛かりにしてCSF-
1Rのhinge部と結合する構造を探索することにした。
Figure 8. L字型コンフォメーションの骨格変換の設計
矢印の左側にシード化合物GW2580のring Aとring Bの部分構造の化学構造を示した。矢印の右側に設計 した構造I, II, III を示した。
20
Figure 9. 化合物I, II, IIIとGW2580とCSF-1Rとの複合体結晶構造の重ね合わせ図 23, 25)
(a) GW2580とCSF-1RとのX線複合体構造に化合物I を重ね合わせた図。CSF-1Rの表面を灰色で示し,
GW2580の炭素原子,窒素原子,酸素原子はそれぞれ緑色,青色,赤色でstick表示した。化合物Iの炭素
原子と窒素原子はマゼンタ色と青でstick表示した。GW2580と化合物Iのring Bのベンゼン環とFigure 8 の赤色で表示した結合を重ね合わせた。(b) 図aの化合物Iをsphere表示した。(c) GW2580とCSF-1Rとの X線複合体構造に化合物II を重ね合わせた図。CSF-1Rの表面を灰色で示し,GW2580の炭素原子,窒素 原子,酸素原子はそれぞれ緑色,青色,赤色でstick表示した。化合物IIの炭素原子と窒素原子はマゼンタ 色と青でstick表示した。GW2580と化合物IIのring Bのベンゼン環とFigure 8の赤色で表示した結合を 重ね合わせた。(d) 図cの化合物IIをsphere表示した。(e) GW2580とCSF-1RとのX線複合体構造に化合 物III を重ね合わせた図。CSF-1Rの表面を灰色で示し,GW2580の炭素原子,窒素原子,酸素原子をそれ ぞれ緑色,青色,赤色でstick表示した。化合物IIIの炭素原子と窒素原子はマゼンタ色と青でstick表示し た。GW2580と化合物IIIのring Bのベンゼン環とFigure 8の赤色で表示した結合を重ね合わせた。(f) 図 eの化合物IIIをsphere表示した。
21
キナーゼの hinge 部は,ATP のアデニン環の NH2とピリミジン環の一つの窒素原子とで 二つの水素結合を形成して ATP と結合する。キナーゼの hinge 部と結合する化合物の部分 構造には少なくても一つの水素結合アクセプターが必要であり,平面構造が好まれること が良く知られている。キナーゼのhinge部と結合する部分構造として良く知られている構造 をFigure 10 に示す。36)
Figure 10. 代表的なhinge部と結合する部分構造
CSF-1R のhinge 部と水素結合を形成する必要があるが,新たな水素結合を獲得するため
には適切な距離と角度に水素結合ドナーとアクセプターを配置する必要があり,論理的な 化合物設計は難度が高い。そこで,hinge部と結合する代表的な部分構造の中から水素結合 アクセプターを一つ有する単純な環構造のピリジン環を用いて,アゼチジン骨格Iを足掛か
りにして hinge 部と相互作用する水素結合アクセプターの位置を幅広く探索することにし
た。化合物設計のイメージ図をFigure 11に示す。
Hing region
Figure 11. ピリジン環を利用した化合物設計のイメージ図
ピリジン環とCSF-1Rのhinge部との水素結合を青色の破線で,ピリジン環とアゼチジン環との探索する 結合を波線で,L字型コンフォメーションを形成するアゼチジン環とring B(水色)のベンゼン環の結合 を赤色で表示した。
22
ステップ2 複合体構造情報を利用した置換基導入による薬理活性向上と物性改善
ステップ1でring Bからhinge部と結合する新規母核を創出した後に,結合ポケット周辺
に置換基を伸長して薬理活性向上と物理化学的性質を改善することを考えた。置換基を導 入する時には,新規母核の化合物とCSF-1Rとの複合体構造情報を参考に効率的に置換基の 導入を実施することにした。
ステップ 1 およびステップ 2 の新規骨格の探索において,合成する目的化合物の構造の 一般式をFigure 12 に示す。
Figure 12. 目的化合物の化合構造の一般式
R1, R2 は置換基,Xはアミド結合,メチレン基,直接結合のいずれかを示す。ピリジン環に対するアゼチ ジン環とR2の置換位置は固定されない。
23 2-3
化合物の合成
本章で合成,評価した化合物はScheme 1–3に示すルートで合成した。37)
化合物9a–h, 11a,b, 13a,b, 16a,bはScheme 1に示すルートで合成した。37) 4-ヨード-2-メト キシフェノール (1) を出発原料とし,4-エチルベンジルクロリド (2) を炭酸カリウム存在
下N,N-ジメチルホルムアミド(DMF)中でベンジル化して化合物 3 を得た。化合物 3 に
対し,N-Boc-3-ヨードアゼチジン (4) からN,N-ジメチルアセトアミド(DMA)中にて調整 したジンケート試薬 5 38) をPd触媒存在下でDMA中にて根岸カップリング反応39) を行い 化合物 6 を得た。
本スキーム中では,Pd 触媒系としてはbis(triphenylphosphine)palladium (II) dichlorideを使 用しているが,Pd(OAc)2, Sphos (di-cyclohexyl(2’,6’-dimethoxy-[1,1’-biphenyl]-2-yl)phosphine)触 媒系40) も使用できる。開発後期での100 gスケール合成時の条件検討の結果ではPd(OAc)2,
Sphos 触媒系を使用した方が収率は良好であった。
ジンケート試薬 5 は N-Boc-3-ヨードアゼチジン (4) と亜鉛からリチウムクロリド存在
下,41) DMA 中で調整した。ジンケート試薬 5 は THF などのエーテル系溶媒より DMA
中で安定であったことから,試薬の調整と根岸カップリング反応の反応溶媒としてDMAを 使用した。
化合物 6 からエタノール中メタンスルホン酸による脱 Boc 化にて中間体 7 のメタンス ルホン酸塩を得た。なお,この反応は溶媒としてイソプロパノールも利用可能である。HCl での脱 Boc 化反応はアゼチジン環が開環した副生成物が生成して低収率となったため,使 用する酸と溶媒を予備的に検討した。その結果,最も収率が良好であったメタンスルホン酸 とエタノールを使用した。収率が良好であった理由として,加える酸の等量をコントロール 可能であること,加熱が可能であること,メタンスルホン酸塩の結晶が析出して生成物が反 応系外に出ることでオーバーリアクションが進行しにくいことが考えられる。
中間体 7 と対応するカルボン 8 をWSC•HClとHOBt•H2Oを使用してカップリング反応 を行い化合物9a–jを得た。また,中間体 7 と対応するピリジルメチルクロリド 10 をアル キル化して化合物 11a,b を得た。対応するピリジルブロミド 12 と中間体 7 を Pd(OAc)2,
BINAP触媒系で塩基として炭酸セシウムを使用してアミノ化42) して化合物13a,bを得た。
16a,bは,それぞれ9i,jから 2-ブロモエタノール (15) (もしくはエチレンカーボナート
(14))を炭酸カリウム存在下,DMF中にてアルキル化することで得た。
24 Scheme 1
25
16a,c, 18, 19a,b, 20a–g は化合物9i,kを出発原料として Scheme 2 に示すルートで合成し た。37b, c)
化合物18, 19a,bは中間体9kから2ステップで合成した。フェノール性水酸基に対応する
アルキルハライドもしくはアルキルトシレートを炭酸カリウム存在下DMF中でエーテル化 するか対応するアルコールと光延反応によりエーテル化し,酸性条件でのアセタール基の 脱保護,もしくは,エステル基のアルカリ加水分解をすることにより18, 19a,bを得た。
化合物20a–gは,中間体 9i, 9kから3ステップで合成した。化合物 9i, 9kのフェノール 性水酸基に炭酸カリウム存在下 DMF 中で 2-ブロモエタノールもしくはエチレンカーボネ ートをアルキル化し16a,c を得た。16a,cの水酸基をメタンスルホニルクロリドとトリエチ ルアミンでTHF中メシル化し,対応する環状アミンを1-プロパノール中で反応させてアミ
ン化合物20a-gを得た。
26 Scheme 2
27
化合物 23a–e は化合物 21a,b を出発原料としてScheme 3に示すルートで合成した。37b,
c) 化合物 21 のベンジルアルコールをメタンスルホニルクロリドとトリエチルアミンでメ シル化後,得られたメシレートに対して対応するアルコール 22a–c を水素化ナトリウム存 在下DMF中でエーテル化し,必要ならアセタール基を酸性条件で脱保護して化合物23a–e を得た。
Scheme 3
28
2-4
化合物の
CSF-1R阻害に関する評価
2-3で合成した化合物の各種薬理評価を日本たばこ産業・生物研究所にて依頼して実施し た。評価結果をTable 1 ~ Table 3に示した。37a, b)
ステップ1 化合物の剛直化を利用した新規骨格の創製
GW2580 の 4-メトキシベンジルオキシ基の代わりに酵素阻害活性が同等で酸性条件に安
定な 4-エチルベンジルオキシ基を利用して,アゼチジン環の窒素原子上に様々な結合を介
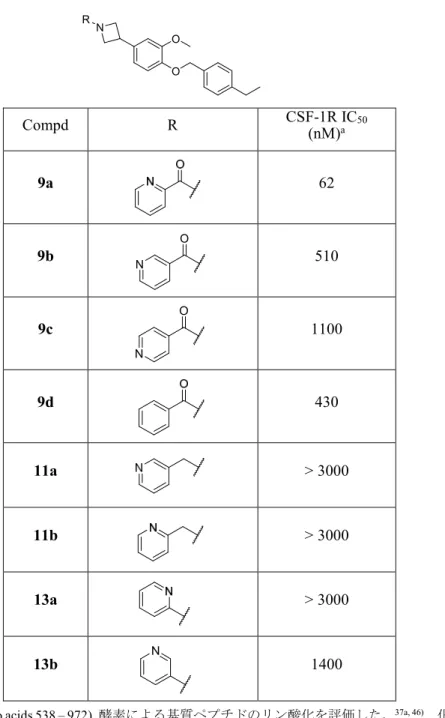
してピリジン環を導入した化合物のCSF-1R酵素評価の結果をTable 1に示した。37a) アゼチジン環とピリジン環をアミド結合で繋いだ化合物 9a–d,メチレン基で繋いだ化合
物11a,b,直接結合した化合物13a,bを評価した。その結果,アミド結合で繋いだ化合物群
9a–dがCSF-1R阻害活性(以後,酵素阻害活性)を示し,2-ピコリルアミド化合物 9a が最
も高い酵素阻害活性を示した(IC50 = 62 nM)。その他のピコリルアミド化合物 9b, c では酵 素阻害活性が減弱した(9b: IC50 = 510 nM, 9c: IC50 = 1100 nM)。驚いたことにピリジン環を 有しないベンゾイル化合物 9d でも酵素阻害活性を示した(IC50 = 430 nM)。一方,アゼチ ジン環とピリジン環をメチレン基で介したピリジルメチル化合物 11a,b や直接ピリジン環 がアゼチジン環に結合した化合物 13a,b では酵素阻害活性が大幅に減弱した。
29
Table 5. 化合物 9a–d, 11a,b, 13a,bのCSF-1R阻害評価結果
Compd R CSF-1R IC50
(nM)a
9a 62
9b 510
9c 1100
9d 430
11a > 3000
11b > 3000
13a > 3000
13b 1400
aヒトCSF-1R (amino acids 538 – 972) 酵素による基質ペプチドのリン酸化を評価した。37a, 46) 化合物の阻害 活性は50% 阻害する濃度をIC50 として表記した。IC50は3回の実験の平均値を示した。
N O
N O N
O O R
N
30
9a がGW2580と同様にDFG-out型CSF-1Rに結合していることを確認するために,CSF-
1R変異タンパク質に対する阻害活性を評価することにした。CSF-1Rを含むクラスIII受容 体型チロシンキナーゼファミリーでは,activation loop の7番目のアスパラギン酸をバリン に変異させることでキナーゼが DFG-out 型コンフォメーションが取れなくなり,常に活性 型コンフォメーションを取ること(constitutively active)が知られている。43) つまり,この 変異タンパク質にはType II阻害薬が結合できなくなる。
CSF-1RのAsp802をValに変異させたD802V変異タンパク質を生物研究所で作製して酵
素阻害活性を予備的に評価した。その結果,9aはGW2580と同様にD802V酵素に対して阻 害活性を示さなかった(IC50 = >30 μM)。この結果から9a はGW2580と同様にDFG-out型
CSF-1Rに結合していることが示唆された。
また,hinge部との相互作用様式に関しては,ピリジン環を有しないベンゾイル化合物9d でも酵素阻害活性を示したことから,カルボニル基の酸素原子が水素結合アクセプターと して機能してCSF-1Rのhinge部と結合していると考察した。
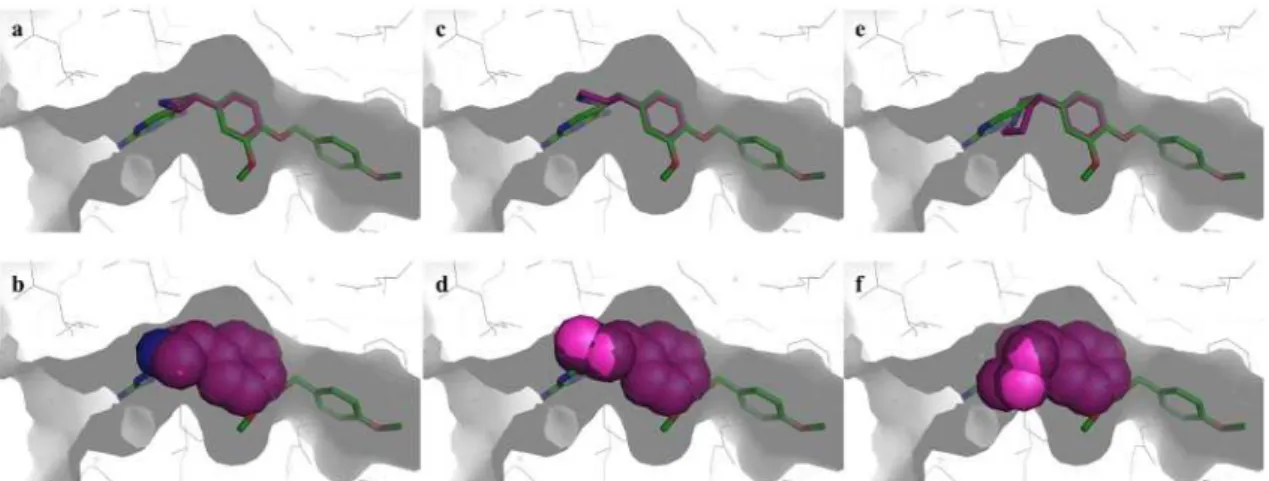
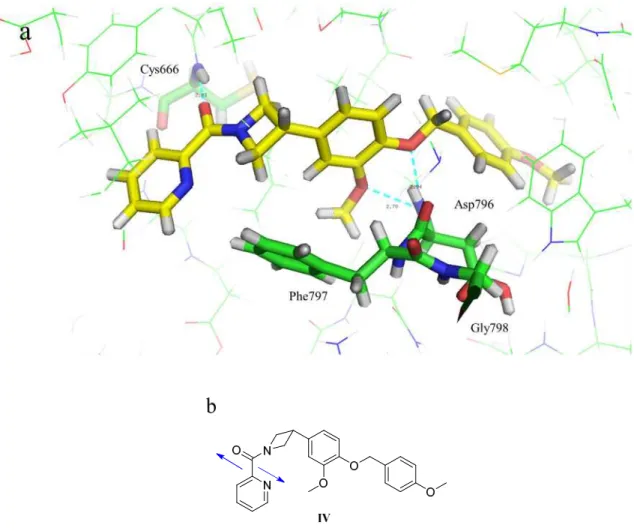
これらの結果を考慮して,GW2580の X 線複合体結晶構造情報を基に化合物 IV のドッ キングモデルを日本たばこ産業・計算機化学チームに依頼して作製した。得られたドッキン グモデルの図をFigure 13に示す。37a)
31
Figure 13. 化合物 IVとCSF-1Rとのドッキングモデルの図
(a) GW2580とCSF-1R とのX線複合体結晶構造25) を基にGlide(Schrödinger Suite 2017)のSP docking protocol を用いて作製した化合物IVとCSF-1R ドッキングモデル図。23, 37a)CSF-1Rの炭素原子,窒素原子,
酸素原子,硫黄原子はそれぞれ緑色,青色,赤色,オレンジ色で示し,Cys666, Asp769, Phe797, Gly798は
stick表示した。化合物IVの炭素原子,窒素原子,酸素原子はそれぞれ黄色,青色,赤色のstickで表示し
た。化合物IVとCSF-1Rとの水素結合は水色の破線で表示し,数値は重原子間距離を示した。(b) 化合物 IVの化学構造。青色の矢印はカルボニル基の酸素原子とピリジン環の窒素原子の静電的反発を示した。
ドッキングモデルでは,ベンゼン環とアゼチジン環がL字型コンフォメーションを形成 して化合物IVがDFG-out型のCSF-1Rと結合している。CSF-1Rのhinge部Cys666の主鎖 のNHとは化合物設計時に想定していたピリジン環上の窒素原子ではなくアミド結合のカ ルボニル基の酸素原子が水素結合している。
32
また,Figure 13-bに青矢印で示したようにアミド結合カルボニル基の酸素原子とピリジ ン環上の窒素原子とが静電的に反発することにより,2-ピコリル構造がC=Oとピリジン環
のC=Nが180°になる様に平面コンフォメーションに固定化されている。
このように,当初の化合物設計とは異なり,ピリジン環の窒素原子は水素結合アクセプタ ーとしてではなく 2-ピコリルアミド構造を平面コンフォメーションに剛直化するために働 いていた。
GW2580がhinge部と四つの水素結合で結合していたのに対して,見出したアゼチジン骨
格ではhinge部と形成している水素結合が一つであり,エンタルピー的には不利である。し
かし,ring Bとアゼチジン環との立体障害によるコンフォメーションの剛直化と2-ピコリル
アミド構造の静電的な反発によるコンフォメーションの剛直化によってエントロピー的に は有利となり,同等の酵素阻害活性を示したと考察した。Hinge部と一つの水素結合で同等 の活性を有する構造を見出せたことは,結果的に経口剤としての物理化学的性質および毒 性的に懸念がある芳香族性 NH2基構造を回避することに繋がった。GW2580 の一つの欠点 を解決したことになる。
このように化合物が活性コンフォメーションに剛直化されることにより CSF-1R との親 和性が向上して,GW2580とは全く異なるユニークな新規アゼチジン骨格を見出した。
ステップ2 複合体構造情報を利用した置換基導入による薬理活性向上と物性改善
見出した新規アゼチジン骨格の化合物 9a は著者がシードとした GW2580 と同等の酵素 阻害活性であり,薬理活性は不十分であると考えられたので,酵素,細胞,および,in vivo での薬理活性向上を目的に作製したドッキングモデルを利用して置換基の導入を実施した。
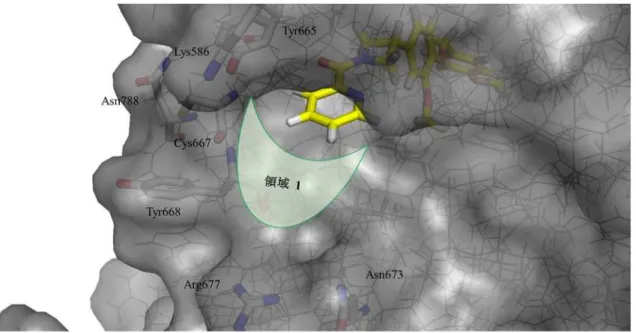
作製したドッキングモデルに置換基を伸長する領域1を表示した図を Figure 14 に示す。
タンパク質が占有していない空間として領域 1 が存在する。領域 1 周辺には,Lys586, Tyr665, Cys667, Tyr668, Asn673, Arg677, Asn788などの極性アミノ酸残基がある。しかし,こ れら極性アミノ酸残基は,結合している化合物IVから距離がある,もしくは,溶媒側に完 全に露出しているので多くの水分子と水素結合を形成していると考えられる。化合物がこ のようなアミノ酸残基と相互作用を獲得するためには,アミノ酸残基と水素結合している 水分子を排除しなければならない。この水の排除に要する脱水和エネルギーにより結合エ ネルギーが損失してタンパク質との親和性が向上しない可能性が高い。したがって,著者は 領域 1 方向への置換基の展開は特定のアミノ酸残基との相互作用を狙わずに,様々な性質 の置換基の導入を検討することで薬理活性向上と物性改善を目指した。
33
Figure 14. 化合物 IVとCSF-1Rとのドッキングモデル図と領域1の位置 23, 37a)
GW2580とCSF-1RとのX線複合体構造25) を基に作製した化合物IVとCSF-1R ドッキングモデルに領域
1を加えた図。CSF-1Rの表面を灰色で表示し,CSF-1RのLys586, Tyr665, Cys667, Tyr668, Asn673, Arg677,
Asn788の炭素原子,窒素原子,酸素原子,硫黄原子はそれぞれ白色,青色,赤色,オレンジ色のstickで表
示した。化合物IVの炭素原子,窒素原子,酸素原子はそれぞれ黄色,青色,赤色のstickで表示した。領 域 1を薄緑色で示した。
Figure 14 から領域1方向へ置換基を伸長するためには,2-ピコリル構造のピリジン環上
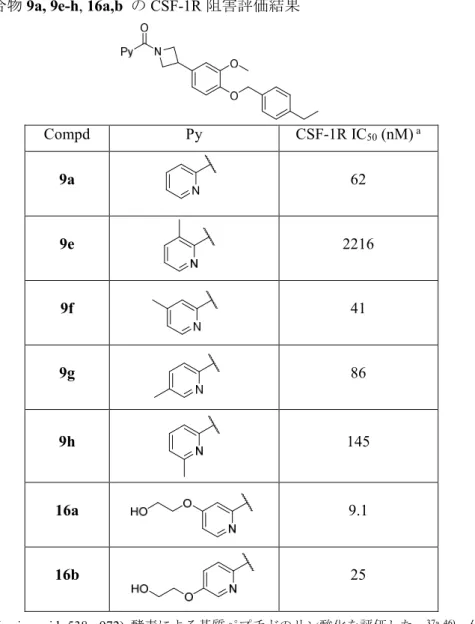
に置換基導入が可能と考えられた。ピリジン環上にメチル基とエチレングリコール基を導 入した化合物の酵素阻害評価を実施した。得られた結果をTable 6に示す。37a)
まず,ドッキングモデルと同様に空間が空いていることを確認するためにピリジン環上 にメチル基を導入した。4-メチル置換体では酵素阻害活性が若干向上し(9f, IC50 = 41nM),
5-メチル体では酵素阻害活性が若干低下した(9g, IC50 = 86 nM)。一方,6-メチル体(9h, IC50
= 145 nM)では数倍の酵素阻害活性の低下,3-メチル体(9e, IC50 = 2216 nM)では大幅な酵
素阻害活性の低下を示した。ピリジン環の3-位,6-位へ導入したメチル基がタンパク質と衝 突したため酵素阻害活性が低下したと考察している。特に,ピリジン環 3-位は作製したド ッキングモデル(Figure 13)ではhinge部へ向かう方向であるので,化合物9e の酵素阻害 活性が大幅に低下したことはドッキングモデルが正しいことを示唆した。
4-位(9f),5-位(9g)へのメチル基の導入は許容された事から,化合物とタンパク質の結
合様式を大きく変化させずに4-位および5-位への置換基の導入が可能だと考察した。一方,
4-位および5-位へのメチル基の導入で大幅な酵素阻害活性向上に至らなかった理由として,
34
脂溶性の置換基を導入したことで領域 1 のタンパク質表面の水分子をエンタルピー的に不 安定化させたと考察した。そこで,親水性官能基を導入してタンパク質表面の水分子と水素 結合ネットワークを形成し,エンタルピー的に安定化させることで酵素阻害活性が向上す ると考えた。親水性の官能基として末端に水酸基を有するエチレングリコール基を導入し た化合物を設計した。4-位置換体(16a, IC50 = 9.1 nM)は9aから大幅に酵素阻害活性が向上 した。5-位置換体(16b, IC50 = 25 nM)も酵素阻害活性は向上したが,16aより向上幅は小さ かった。この結果より置換基を導入する位置はピリジン環の 4-位が最適であることと,親 水性置換基の導入による水分子の安定化で酵素阻害活性が向上することを確認した。
Table 6. 化合物9a, 9e-h, 16a,b のCSF-1R阻害評価結果
Compd Py CSF-1R IC50 (nM) a
9a 62
9e 2216
9f 41
9g 86
9h 145
16a 9.1
16b 25
aヒトCSF-1R (amino acids 538 – 972) 酵素による基質ペプチドのリン酸化を評価した。37a, 46) 化合物の阻害 活性は50% 阻害する濃度をIC50 として表記した。IC50は3回の実験の平均値を示した。
N
35
次に,Figure 15に示したアゼチジン構造の一般式を基本骨格として細胞およびin vivo での薬理活性を向上させることを目的に,ピリジン環4-位のR2に導入する親水性置換基 の検討を行った。
R1 はエチル基(Et),シクロプロピル基(cPr)で固定した。メトキシ基が酸性条件で分 解しやすいこと,R1が導入されているベンジルオキシ基の4-位以外の2-位や3-位への置換 基の導入はドッキングモデルの疎水性ポケットの形状から困難と考えられること,4-位へ 導入する置換基はメトキシ基,Et基,cPr基より嵩高い置換基の導入はドッキングモデル の疎水性ポケットの大きさから困難と考えられることから,R1をEt基とcPr基で固定し た。
Figure 15. アゼチジン骨格の化学構造の一般式
検討する置換基をR1, R2 として表記した。
In vivo にて所望の薬理作用を得るためには,細胞での阻害活性だけでなく良好な PK
(pharmacokinetics)プロファイルを有する必要がある。そのためには,溶解度,膜透過性,
代謝安定性などの薬剤としての物理化学的性質を調整しなければならない。In vivo での薬 理作用を示す化合物を見出すため,細胞での阻害活性と物理化学的性質の両立を目指し,R2 について様々な性質の極性官能基を検討した。
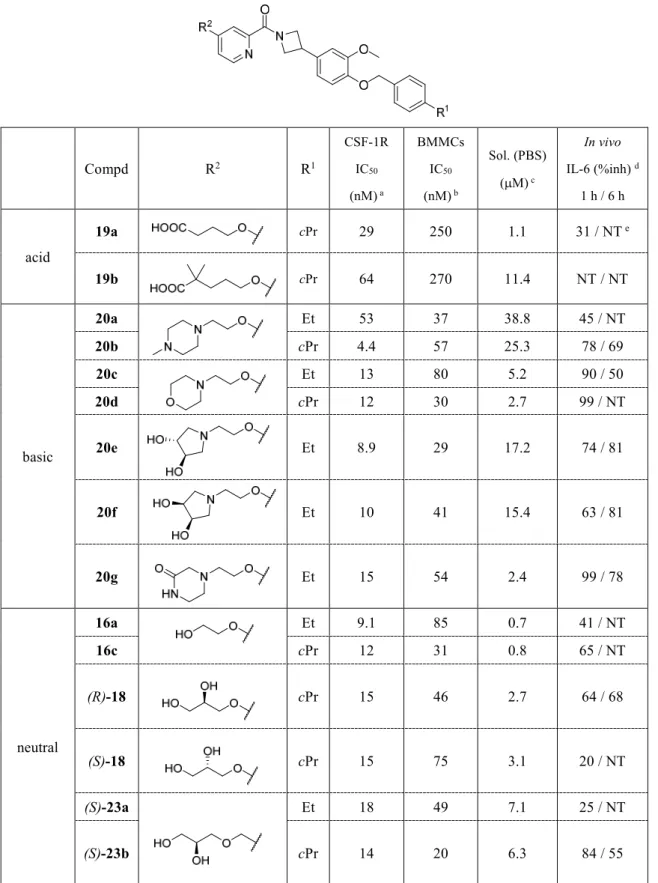
酸性官能基(19a, b),塩基性官能基(20a-g),水酸基を含む中性官能基(16a,c, 18, 23a-e)
を有する化合物を設計した。合成した化合物の酵素阻害評価(CSF-1R IC50),マウス骨髄由 来マクロファージ細胞評価(BMMCs IC50),経口投与によるin vivoでのマウスLPSチャレ ンジモデルのIL-6産生抑制作用(in vivo IL-6 %inh.)を日本たばこ産業・生物研究所へ評価 を依頼し,PBS(phosphate-buffered saline)バッファーへの溶解度(Sol.(PBS))測定を日本た ばこ産業・薬物動態研究所へ評価を依頼して実施した。評価結果をTable 7に示す。37b)
36
Table 7. CSF-1R 阻害活性,細胞での阻害活性,PBSへ対する溶解度,in vivoでの薬理活性
に対する置換基R2 の影響
Compd R2 R1
CSF-1R IC50
(nM) a
BMMCs IC50
(nM) b
Sol. (PBS) ( M) c
In vivo IL-6 (%inh) d
1 h / 6 h
acid
19a cPr 29 250 1.1 31 / NT e
19b cPr 64 270 11.4 NT / NT
basic
20a Et 53 37 38.8 45 / NT
20b cPr 4.4 57 25.3 78 / 69
20c Et 13 80 5.2 90 / 50
20d cPr 12 30 2.7 99 / NT
20e Et 8.9 29 17.2 74 / 81
20f Et 10 41 15.4 63 / 81
20g Et 15 54 2.4 99 / 78
neutral
16a Et 9.1 85 0.7 41 / NT
16c cPr 12 31 0.8 65 / NT
(R)-18 cPr 15 46 2.7 64 / 68
(S)-18 cPr 15 75 3.1 20 / NT
(S)-23a Et 18 49 7.1 25 / NT
(S)-23b cPr 14 20 6.3 84 / 55