■■ 1. はじめに Werner の配位説以来,中心金属イオンの酸化数およ び配位数の違いにより中心金属周りの配位子を選択する ことで様々な錯体が合成され,その構造が明らかにされ てきた。また,構造だけでなく,錯体のもつ色,磁性, 酸化還元能など多様な機能性も明らかにされてきた。私 が錯体の研究を始めた 1970–80 年代は有機金属錯体の勃 興期であり,日本から多くの研究者がアメリカやドイツ などの海外の大学に留学し,空気中で不安定な化合物を 取り扱う真空ラインや Schlenk 管,それにグローブボッ クスなどのテクニックを学び,日本でもその技術を用い た合成法で予期せぬ化合物群が次々に合成され,単結晶 X 線構造解析されていた時代であった。そんな中で,私 が初めに取り組んだのはイリジウム (I)- イソシアニド錯 体であった。アリルイソシアニドをもつ平面四配位の [Ir(CN-R)4]+錯体を合成したが,この錯体は [Rh(CN-R)4]+ 錯体1から予期していた橙から黄色ではなく,黒緑色を 示した。この [Ir(CN-R)4]+錯体は,光照射下で次第に黄 緑色に変化する2。ほとんど同時期に,イソシアニドの 置換基をアリル基からメチル基に変えた [Ir(CN-Me)4]+ 錯体が報告された3。この錯体は,最初青色であるが, 光にあてるとほとんど瞬時に橙色に変化することが報告 された3。この光反応は,平面四配位構造をもつイリジ ウム錯体が積み重なって Ir–Ir 間に結合をもつ集合体と なっており,光により集合体の Ir–Ir 結合が開裂する反 応であると結論された4。イリジウム (I) テトラキスイ ソシアニド錯体では,アリル基をメチル基に変えると光 反応が格段に速くなることがわかった。このイリジウ ム (I) イソシアニド錯体が示す Ir–Ir 金属間結合は同族の ロジウムイソシアニド錯体でも存在することがわかり5, 金属間結合の光開裂を利用した水素発生触媒へと展開し ていった。新しく錯体を合成する際には,配位子の置換 基をいろいろと変えて中心金属への影響を調べてみるこ とで機能性が顕著に変わることがあること,さらには金 属 – 金属間相互作用など新しい相互作用の導入により錯 体の集合化6が可能となること,さらには錯体の機能性 をより広い視点から考えることが重要であることをイリ ジウム錯体の研究で,錯体研究の入り口に立った駆け出 しの学生時代に学んだ。 1970 年代の後半に起こった中東戦争は世界にオイル ショックを引き起こし,石油に頼らない新しいエネルギ ー資源として太陽光から化学エネルギーを得ようとする 研究が盛んになっていた。そのような時代背景の中で, 1976 年に D. Whitten らにより発表された論文は衝撃的 であった7。それは,長鎖アルキル基をもつ [Ru(R 2bpy) (bpy)2]2+ (bpy = 2,2′- ビピリジン ) 錯体を酸化インジウム・ 酸化スズ (ITO) 電極に塗布した修飾電極を水に浸けて光 を照射すると,水が分解されて水素と酸素になるという 速報であった。非常に革新的なアイデアであったので, その後いくつかのグループで追試実験が行われたが,再 現性はとれなかった。しかしながら,私はこの研究に大 いに触発されて,ルテニウムを中心金属とする錯体の合 連絡先著者名:芳賀 正明 連絡先:112-8551 東京都文京区春日 1-13-27 中央大学研究開発機構 Tel: 81-3-3817-7401 Fax: 81-3-3817-1895 Corresponding Author: Masa-aki Haga E-mail: [email protected]
Address: 1-13-27 Kasuga, Bunkyo-ku, Tokyo 112-8551, Japan
Keywords: ruthenium, benzimidazole, redox-active, proton-coupled electron transfer, surface coordination chemistry, electrochemistry, molecular device
表面錯体ナノ化学の機能創成
Surface Coordination Nanochemistry Based on Functional Metal Complexes
中央大学研究開発機構 芳賀 正明
Research and Development Initiative, Chuo University Masa-aki Haga*
Received, November 12, 2020; Accepted, November 17, 2020; Published, November 30, 2020
Metal complexes exhibit distinctive optic, magnetic, and redox properties that often depend on the specific combination of metal ion and ligand(s), and both components are attractive building blocks for the construction of nanostructures on surfaces in a bottom-up approach. Surfaces usually represent a two-phase boundary (solid-liquid or liquid-air) that provides an attractive platform on which plays with metal complexes in order to design and modulate chemical functionality with potential applications in nanoscience and nanotechnology. Herein, this review focuses on the development of (i) molecular design of Ru complexes with proton-coupled electron transfer both in solution and on an ITO electrode surface, and (ii) surface modification based on redox-active Ru complexes with layer-by-layer (LbL) assembly toward molecular devices.
成化学をスタートさせた。そして,含窒素ヘテロ環配位 子であるベンズイミダゾール BIm が配位した錯体の酸 化還元電位や吸収および発光スペクトルを検討している 中で,BIm がルテニウムに対して光照射下でも安定な 配位子として働くことや7,BIm イミノ N–H 基が水素結 合形成や脱プロトン化ができるサイトとして働くなど多 様な機能性をもつ配位子として働くことがわかってきた 9–11。そこで,BIm 誘導体をもつ二核錯体の合成に展開 して,多電子移動できる系として,金属間相互作用の強 さを電気化学的手法を利用して研究した。研究の初めは, 混合原子価状態での金属間相互作用の強さを原子価間電 荷移動遷移(IVCT 帯)から決定する静的物性として捉 えていた9,12。その頃に,F. L. Carter らによる分子を構 成要素としてデバイスを創るという分子エレクトロニク スの概念を知り13,錯体を分子デバイスの構成要素に使 おうと考えた。多核金属錯体内での金属間電荷移動は分 子デバイスの分子内伝導と関連する重要な素過程であ り,これを外部刺激により動的に制御できれば分子スイ ッチとなる。電子伝導を外場でスイッチングできる分子 スイッチは,分子エレクトロニクスの中でも重要な基本 的な要素素子である。そこで,多核混合原子価錯体内で の原子価間電荷移動遷移を利用して,外部刺激による金 属間相互作用のスイッチングを研究した。しかし,溶液 中での多核錯体の電荷移動の評価だけでは分子デバイス と呼ぶには限界があると感じたので,固体表面に錯体の 動的物性変化を持ち込み,固体表面で動作する分子デバ イスの研究を進めた。金表面での自己組織化膜を用いた 電気化学的アプローチはフェロセンなど限られた錯体に ついてのみ研究されていた。そこで,機能性錯体を表面 に固定化してデバイスに応用する “ 表面錯体ナノ化学 ” として展開した。本総説では,(1) BIm 配位子の分子設 計とそれらの錯体のプロトン共役電子移動,(2) 表面錯 体化学へのアプローチ,(3) 表面錯体でのプロトン共役 電子移動とそのデバイス応用について述べる。 ■■ 2. ベンズイミダゾール BIm(含窒素配位子) BIm が配位子として働く場合,ピリジン配位子と比べ ると,(1) ピリジンが σ 供与性と π 受容性をあわせもつ のに対して,BIm はより強い σ 供与性はもつが,π 受容 性は小さいという特徴をもつ14。(2) BImは配位する際に, 中性の配位子としてだけでなくイミノ N–H プロトンが 解離してアニオン性配位子として,2 個の金属間を架橋 する架橋配位子としても働くことが可能で15,多様な配 位様式がとれる。最近ではプロトン化あるいは N- アル キル化したカチオン種であるベンズイミダゾリニウムの 脱プロトン化から生成する NHC カルベンが非常に安定 な有機金属錯体を生成することが報告されている。また, このような NHC カルベンは金表面に結合して自己組織 化膜を形成し,その金表面に対する結合は有機チオール 基より強固である16,17。このように多様な結合様式が可 能な BIm を分子内にもつ誘導体を配位子とするルテニ ウム単核錯体を合成した (Scheme 1)。これらの BIm 配 位子は,有機カルボン酸あるいは有機ニトリルと o- フ ェニレンジアミンとの縮合反応により合成でき,ベンゼ ン環のないイミダゾール配位子に比べると比較的容易に 得られる14。
Scheme 1. Chemical structures of mononuclear [Ru(bpy)2(L)]2+ complexes, where L = benzimidazole derivates.
また,これらの BIm 配位子をもつ単核ルテニウム錯 体は,BIm 基が金属イオンに配位することでイミノ N– H 基が脱プロトン化しやすくなり11,脱プロトン化する ことで別の金属イオンへの金属錯体配位子として働き, Scheme 2 に示したルテニウム二核錯体を生成する。こ れらの二核錯体では,BIm 架橋配位子がアニオン性であ ることから,Ru(II)–Ru(III) 混合原子価錯体を生成する 均化定数は非常に大きい。また,Ru(II)–Ru(III) 混合原 子価錯体では架橋配位子の HOMO π 軌道と Ru(III) dπ 軌 道が強く相互作用して,近赤外領域に強い IVCT 吸収帯 をもつこともわかった9。
Scheme 2. Chemical structures of dinuclear [{Ru2(bpy)4}(L)]2+ complexes bridged by L = benzimidazolate derivates.
配位した BIm のイミノ N–H 基は,脱プロトン化だけ ではなく,水素結合相互作用での水素供与基として働く ことができるので18,様々な外部刺激への応答サイトと しても機能する。電荷移動を制御する分子スイッチを構 築するために,BIm の脱プロトン化 N–H サイトを保持 した BIm 架橋配位子を合成した。(Scheme 3)
Scheme 3. Chemical structures of dinuclear Ru complexes with PCET reaction having bis-bidentate and bis-tridentate ligands containing benzimidazole substituents as a bridging ligand. All complexes have a 4+ charge, but are omitted here.
■■ 3. Ru-BIm 錯体のプロトン共役電子移動
BIm 配位子をもつ Ru-BIm 錯体は,BIm の N–H プロ トンによりブレンステッド酸として働く。中心金属が Ru(II) から Ru(III) に酸化されると,BIm の N–H プロト ンの酸性度が増し,脱プロトン化されやすくなる。ルテ ニウム中心の酸化が化学結合を隔てた BIm N–H の解離 を促進することから,Ru(III)–BIm 間での電荷の非局在 化が起こっていることがわかる。[Ru(bpy)2(PBimH)]2+の アセトニトリル – 水混合溶液中での電子移動とプロトン 移動の四つの錯体種間の平衡を Fig. 1 に示した。
Fig. 1 Square scheme of electron/proton equilibria in [Ru(bpy)2 (2-(pyridyl)benzimidazole)]. また,Ru-BIm 錯体の N–H 基は水素結合供与体として 働くことも可能である。たとえば,[Ru(bpy)2(PBimH)]2+ 溶液に水素結合受容体となるピリジン類やイミダゾール 類を加えると,水素結合受容体の pKaに依存して Ru(II/ III) 酸化電位がシフトする18。最近 Wenger らは,一連 の Ru- イミダゾール錯体の励起状態でのプロトン共役 電子移動 (PCET) 反応について詳細な研究を報告してい る19,20。我々は,この Ru-BIm 錯体構成ユニットを繋い だ混合原子価二核錯体での Ru–Ru 間相互作用のプロト ンによるスイッチングについて検討した。新たに合成し た BIm 架橋配位子をもつ二核錯体と略号は Scheme 3 に 示した。錯体 1 と 2 は骨格構造異性体であるが,錯体 1 では Ru–Ru 間はベンズイミダゾール環同士が C–C 結合 で繋がれた構造であるのに対して,錯体 2 ではピリジ ン環同士が繋がれた構造である。錯体 1 では,二段階 1 電子酸化に伴う酸化波がみられるが,脱プロトン化し た状態では二段の Ru(II)/Ru(III) 1 電子酸化電位の差がプ ロトン化状態に比べて大きくなり,脱プロトン化によ り Ru–Ru 間相互作用が増大することがわかった21。一 方,錯体 2 では脱プロトン化により Ru–Ru 間相互作用 は弱くなる22。構造異性体であるのに,両者でプロトン 化状態による Ru–Ru 間相互作用の強さの変化が逆の傾 向を示すのは興味深い22。これは,混合原子価状態にお いて架橋配位子部の π 軌道あるいは π* 軌道のどちらが 強く Ru(II) あるいは Ru(III) dπ 軌道と相互作用するかに よって決定されることによる。BIm の脱プロトン化は ベンズイミダゾール基の電子密度を上げるので,Ru(III) dπ 軌道とより強く相互作用して,混合原子価状態での Ru(III) dπ と架橋配位子 π 軌道 (HOMO) を介してのホー ル型電荷移動が重要な超交換相互作用のパスを与えるこ とになる。実験的には,錯体 1 の脱プロトン化により原 子価間電荷移動 (IVCT) 遷移が近赤外領域に強い強度を もって現れることから,脱プロトン化により混合原子価 状態での Ru(II)–Ru(III) 間相互作用が強く働くことがわ かる。三座架橋配位子をプロトン応答サイトにもつ錯体 3 および 4 の場合には,架橋部位の 4,4′- ビピリジンや ピリミジン基など LUMO の π* 軌道が低く,BIm 部位の 脱プロトン化により架橋部の電子密度は周辺の BIm–に 偏るので,二核金属間の相互作用は弱くなる23。この他 にも,架橋部位のプロトン化・脱プロトン化により混合 原子価状態での相互作用をスイッチできる錯体系が報告 されている24,25。このような一連の二核錯体は,金属間 の相互作用をプロトンという外部信号によりで制御でき るので,Scheme 4 のように一般化でき,分子デバイス におけるスイッチング機能の基本要素を備えていると考 えることができる。
Scheme 4. Schematic description of proton-induced molecular switching devices, where the bridging ligand acts as a proton-responsive site (Ref. 21).
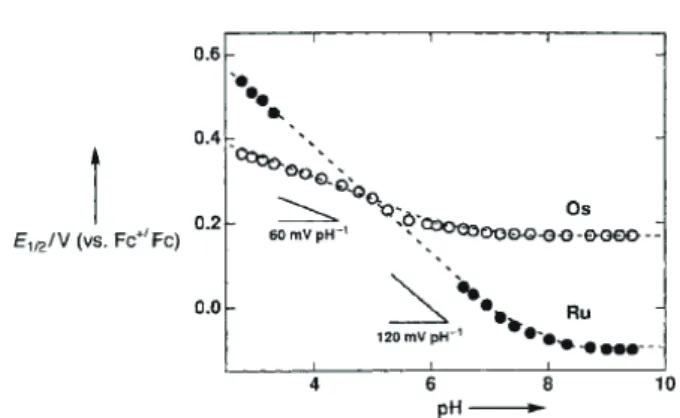
さらに,二核錯体からルテニウム・オスミウム混合金 属四核錯体に拡張した (Scheme 5)。同じ配位子をもつ場 合,中心金属がオスミウムの方がルテニウムより酸化さ れやすい。また同じ金属イオンの場合,周辺配位子に σ 電子供与性である BIm 基をもつ錯体の方が π 電子受容 性であるピリジンが配位した場合より酸化されやすい。 以上の事実を考慮して,中心にルテニウム,周辺にオス ミウムビス ( ビピリジン ) 部位をもち,それらを 2,2′- ビ ス ( ベンズイミダゾリル )-4,4′- ビピリジンで架橋した四 核錯体 5 を合成し,そのプロトン応答性を検討した26。 Fig. 2 に,錯体 5 のプールベ図を示した。この図で,pH 5 以下の酸性側では Ru(II/III) 過程に比べて Os(II/III) 過 程がより低電位側で酸化が起こり,pH 6 以上では逆転 して Ru(II/III) 酸化過程がより低電位で起こる。ここで, Os(II/III) 過程は 3 電子過程であり,一方 Ru(II/III) 過程 は 1 電子過程である点に注意してほしい。今,錯体 5 の 脱プロトン化体に 0 V の電位をかけると,1 電子酸化し た混合原子価状態 Ru(III)Os(II)3が生成する。ここにプロ トンを加えたときの回転ディスクボルタンメトリーでの Os(II/III) の 3 電子過程に 1 電子分のカソード電流が含 まれることから,プロトンの添加により Os(II) から中心 Ru(III) への分子内電子移動が誘起されて,Ru(II)Os(III) Os(II)2状態が生成することがわかった (Scheme 6)。さら に,プロトンを添加したときに生成する金属の酸化状態 の変化は,近赤外吸収スペクトルにおいて Os (III) d5錯 体に特徴的な dπ-dπ 遷移がプロトンを加えると 1900 nm と 2300 nm に観測されることからも明らかになり,プロ トンが RuOs3多核錯体内での分子内電子移動を誘起で きる外部信号として働くことが明らかになった26。
Scheme 5. Chemical structures of dendritic tetranuclear RuOs3 complex bridged by bis(benzimidazolyl)-4,4′-bipyridine (complex 5) (Ref. 26).
Fig. 2 Pourbaix diagram of dendritic tetranuclear RuOs3 complex (Complex 5) in CH3CN-buffer (1:1) at 25 °C (Ref. 26).
Scheme 6. Schematic drawing of proton-induced electron transfer reaction in one-electron oxidized RuOs3 complex.
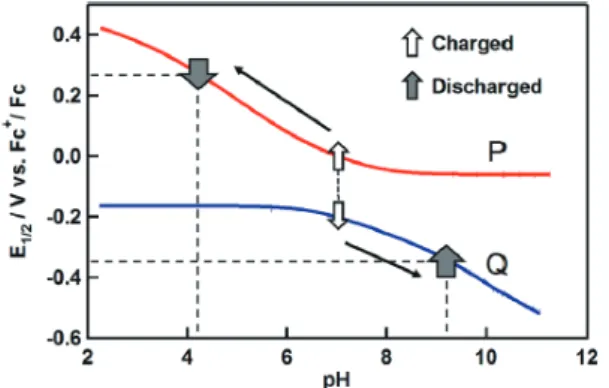
以上のように,混合原子価 RuOs3四核錯体のプール ベ図での Os(II/III) と Ru(II/III) 過程の電位 –pH プロット の曲線が交差することにより,プロトンによる分子内で の電子移動が引き起こされた。水が溶媒として働く生体 系では,プロトンは反応やエネルギー蓄積に非常に重要 な役割を演じている。たとえば,生体膜でのプロトン勾 配を利用した ATP 合成はその典型的な例である。言い 換えると,pH の差がエネルギーとして蓄えられている ことになる。プロトン共役電子移動可能な二つの錯体で の pH 差による電位変化をエネルギーとして蓄電する系 を,二つの錯体 6, 7 (Scheme 7) を用いて試みた27。錯体 6, 7 のプールベ図は Fig. 3 の実線で示される。錯体 5 で は二つの Ru と Os サイトの曲線が pH 5 付近で交差する が (Fig. 2),二つの錯体 6, 7 ではプールベ図でのそれぞ れの曲線の交差はない。しかし,二つの曲線は pH 7 付 近で互いに接近し,pH がそれ以上でも以下でも二つの 錯体の電位差は大きくなる。すなわち,pH 7 の中性か ら充電すれば,Fig. 3 の曲線に沿って電位差が生まれ, 同時に pH 差として蓄電されることになる。実際に,そ れぞれの錯体をナフィオン隔膜で隔てて,CH3CN/ 非緩 衝水溶液中で定電流法により 6 を酸化,他方の 7 を還元 して充電を行い,放電では極性を逆にして行った。充放 電に伴う各極の pH 変化を観測すると,Fig. 4 に示した ように,カソードの 6 側では充電に伴って pH の低下が みられ,放電では pH の上昇が観測された。一方のアノ ードの 7 側では,充電に伴う pH の上昇および放電での pH の低下が起こる。また,繰り返しの充放電に対しても, 溶液の pH は上昇・低下を繰り返す。この実験から,プ
ロトン共役電子移動を示す錯体の適当な組み合わせによ り,pH 差を電位差としてエネルギー蓄積できるレドッ
クス電池の原理を提案できた27。同じように,プロトン
勾配を電気エネルギーに変換した研究例はこれまでにも
いくつか報告されている28,29。
Scheme 7. Chemical structures of Ru complexes having proton-coupled electron transfer, 6 and 7.
Fig. 3 Pourbaix diagram for a solution redox battery based on a pair of Ru complexes 6 (P) and 7 (Q). The electric energy is stored as a pH gradient between the half-cells (Ref. 27).
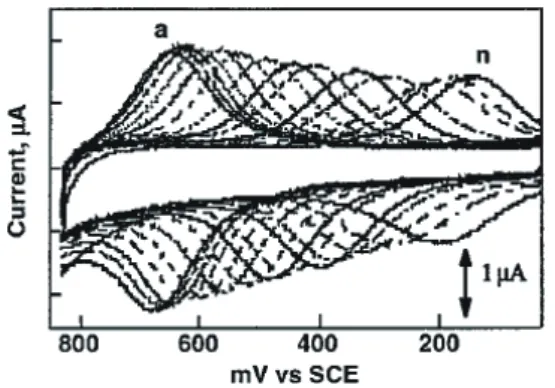
Fig. 4 The temporal change of cell voltage and pH for the cell composed of two solutions between 6 and 7 during charging/ discharging (Ref. 27). ■■ 4. 表面錯体化学へのアプローチ シリコンのフォトリソグラフィを用いたトップダウン 微細加工技術がナノメートルサイズに達し,光の波長限 界に近づきつつある。この限界を打破するために,サブ ナノメートルである原子・分子の基本ユニットから素子 を組み上げるボトムアップ技術として分子エレクトロニ クスが提案され,ナノテクノロジーを牽引する分野とし て様々な新しい研究やアイデアが展開されてきている 30。分子デバイスへの応用を考えると,分子を固体基板 上に固定集積化する必要がある。そこで,我々はこれま でに述べてきた溶液中で研究した錯体のプロトンスイッ チや混合原子価錯体の機能や物性などを基礎として,表 面への錯体分子の積層集積あるいは基板選択的配列や固 定錯体同士の配線,分子スイッチおよびメモリなど分子 素子の基本要素技術を表面錯体で機能発現できる分子シ ステムの構築を行った31–33。 4. 1. アンカー基による錯体の表面固定化と pH 応答性 表面に分子を固定する方法として,チオール基をもつ 分子の金表面での自己組織化膜 (SAM 膜 ) がよく知られ ている。我々は溶液でのプロトン共役電子移動を表面錯 体系に拡張するために,表面固定配位子として BIm の N–H 基をチオアルキル基に置換し,補助配位子に BIm N–H 基をもたせた錯体 8 を金表面に固定して,そのプ ロトン共役電子移動および表面での pKa値を溶液系と比 較した (Scheme 8)34。金電極表面に錯体 8 をオクタンチ オールで希釈した自己組織化膜について,溶液の pH を 1.33 から 7.35 まで変化させたときのサイクリックボル タモグラム (CV) を Fig. 5 に示した34。この CV の pH 変 化より得られるプールベ図から,錯体 8 の SAM 膜の二 段階の pKaは 6.0 と 7.8 であることがわかり,溶液中に 溶かした N- アルキル部位にメチル基をもつルテニウム 錯体の pKa (6.35, 7.87) とほとんど変わらない。金表面に 固定した有機カルボン酸の SAM 膜の pKa値は,プロト ンが解離すると表面部分が中性からカルボキシレートア ニオンになって隣の官能基部位の分子間静電反発が増え ることから,プロトン解離が抑えられ,pKa値は約 2 程 度増加することが報告されている。本錯体 SAM 膜の場 合には,錯体自身が電荷をもち,錯体周りを溶媒水が取 り囲む空間ができているので,溶液との pKaの差が少な いものと考えられる34。
Scheme 8. Chemical structures of Ru complexes with a long alkyl chain and anchor groups, 8 and 9.
Fig. 5 Cyclic voltammograms of self-assembled 8 diluted by octanethiol in different pH solution at a scan rate of 0.2 V/s. The pH values a–n (from left to right) are 1.33, 1.83, 2.21, 2.59, 3.10, 3.58, 4.09, 4.52, 4.86, 5.56, 5.83, 6.35, 6.88, and 7.35 (Ref. 34). ITO 薄膜からなる透明導電性ガラスは,レドックスを 伴う吸収スペクトル変化を分光電気化学としてモニター するのに有用な電極である。しかし化学気相成長 (CVD) 法で作製された ITO 電極は,結晶が凹凸のドメイン構 造をとり表面が粗いものが多く,表面に吸着した分子を 観察するのが難しいという難点があった。しかし,近年 有機 EL 素子の作成のために平滑性の高い ITO 基板(表 面粗さ ±1 nm 以下)が入手できるようになった。この ような ITO 基板を使えば,表面分子を AFM で観察する ことも可能であり,同時に表面錯体の吸着を吸収スペク トルでもモニターできる。そこで我々は,導電性がある 平滑な ITO 基板上での錯体の固定集積化の研究を行っ た。酸化物表面に化学吸着することが知られていたホス ホン酸基をアンカーとして選び,ビス ( ベンズイミダゾ リル ) ピリジン三座配位子の側鎖にアルキルホスホン酸 基を修飾した錯体 9 を合成し (Scheme 8),ITO 基板上へ の化学吸着性を CV でモニターし,その pH 依存性を調 べた。その結果,ホスホン酸基をアンカーとする錯体 9 は補助配位子が N- メチル基で保護されているので電 位のシフトはみられない。ただ,溶液の pH が 10 以上 になると,錯体は ITO 基板から脱着することがわかっ た。錯体 8 と 9 が化学吸着する際の固体基板への選択性 を調べた。錯体 8 と 9 の等モル混合溶液に金基板と ITO 基板を 3 時間浸漬した後,それぞれの基板の CV 測定を pH 6 の水溶液中で行ったところ,金基板では +0.36 V vs Ag/AgCl に Ru(II/III) の酸化波が観測され,錯体 8 が pH 6 でのプロトン共役電子移動のために負電位側にシフト したと考えられる。一方,ITO 電極では 0.70 V に Ru(II/ III) の吸着波が観測されると同時に +0.36 V に非常に小 さなピークが観測されることから,チオール基は選択的 に金基板に吸着する。一方,ITO 基板へのホスホン酸ア ンカー基とチオール基の吸着割合は約 9:1 の比であり, ホスホン酸基がより吸着しやすいことがわかった。 先に述べた錯体 3 で用いたテトラ ( ベンズイミダゾ リル )-4,4′- ビピリジンを架橋配位子にもち,両端にブ チルホスホン酸基をアンカーとする錯体 10 を合成し た (Scheme 9)35。錯体 10 は,ITO 電極表面に表面被覆量 0.80 × 10–10 mol cm–2 で吸着し,1 < pH < 10 の水溶液中で プロトン共役電子移動系として振る舞うことがわかっ た。さらに,錯体 10 を修飾した ITO 基板を薄膜分光電 気化学の電極として可視紫外吸収スペクトル変化を測定 した結果を Fig. 6 に示した。錯体 10 薄膜に +0.80 V の 電位を印加したときの差吸収スペクトルでは,Ru(II) 状 態でみられる 550 nm の MLCT 帯が消失し,750 nm 付 近に新たな LMCT 帯がみられる。さらに,錯体膜の電 圧印加時の安定性をみるために,+0.80 V と +0.3 V 間の パルス電位を繰り返し加えた。吸光度変化は繰り返しに 対して再現よく観測されることから,ITO 基板上の錯体 10 修飾膜は電圧印加に対して安定であることがわかっ た。また,アルキル鎖長は調節可能であるので,剛直な 中心錯体構造を多脚アンカー基で表面に固定する際には この柔軟性を利用できる。例えば,錯体 11 は 1,3,5- ト リス ( ターピリジル ) ベンゼンが架橋配位子として 3 個 のルテニウムを繋ぎ,補助配位子からそれぞれ 2 個のア ンカー基から外に突き出た三核錯体構造をもつ (Scheme
Scheme 9. Chemical structure of rod-shaped dinuclear Ru complex with a long alkyl chains and anchor groups 10.
Fig. 6 (a) The differential UV-vis spectra of complex 10 on an ITO electrode, (b) pulse sequence between +0.3 V and +0.8 V, and (c) the response of absorbance at 550 nm (Ref. 35).
10)36。中心のベンゼン環に結合するターピリジル基はベ ンゼン環平面に対して捻れた立体配座をとる。そのため に補助配位子であるビス ( ベンズイミダゾリル ) ピリジ ン三座配位子は必然的に中心のベンゼン環と平行な配座 をとることになり,6 個のアンカー基が自己組織的に固 体基板に吸着する場合には天蓋構造をもつことになって 表面錯体と基板の間に空孔が生じることになる。例えば, テトラチアフルバレン (TTF) はこの天蓋型錯体の空孔に 内包されるが,電位掃引により TTF が +0.18 V で酸化 されラジカルカチオンになると,錯体との静電反発によ り外の溶液中に放出される36。
Scheme 10. Chemical structures of canopied trinuclear Ru complex 11 where RH represents alkylphophonic acid group and schematic drawing of the conformation of 11 on an ITO surface (Ref. 36). 4. 2. 自立型アンカー配位子の分子設計 錯体の側鎖に直鎖アルキル基を導入した系では,錯 体を固体基板上に自己組織的に吸着させることはでき るが,吸着した分子の配向を規制することは難しい。 特に基板との電子移動を研究する場合には,たとえば Scheme 11 のようにアルキル基の配座の違いで電極と分 子との距離が大きく違うので,電子移動速度に違いが生 じる。また,第一層の分子層の上に第二層,第三層と表 面に逐次的に積層していこうとする場合には,第一層の 分子配列・配向が重要になる。そこで我々は,表面での 八面体錯体の金属まわりに Δ, Λ の立体異性体ができな いように C2v対称性をもつ三座配位子のビス ( ベンズイ ミダゾリル ) ピリジンを基本骨格とし,そのイミノ N–H 側鎖に,束縛回転により立体配向が決まり表面に多点吸 着できるように 4 個のホスホン酸基を導入した三座配位 子を合成した (Scheme 12)。この四脚アンカー三座配位 子は,Scheme 12 のように 2 個ずつのホスホン酸基が “ 鳥 の羽 ” のように両側に出ており,表面に 4 点で吸着でき る。そして,補助三座配位子は基板に対して垂直に配向 するので,補助配位子が金属イオンへの配位基をもつ場 合には,垂直方向への積層化に適していると予想される。 これまでにも,固体表面への自立型ホスホン酸基をもつ 多脚アンカー基が報告されている37,38。
Scheme 11. Two different orientations of Ru complex with a long alkydisulfide group (Ref. 32).
Scheme 12. Proposed molecular orientation of tetraphosphinic acid anchors on an ITO surface (Ref. 32).
4. 2. 1 自立型表面錯体の表面機能化 自立型四脚ホスホン酸アンカー基をもち,補助配位 子に様々な機能性を賦与した錯体 12, 13, 14 を合成した (Scheme 13)39,40。錯体 12, 13, 14 はいずれも ITO 電極,マ イカ,SiO2/Si などの表面に吸着するが,吸着するのに 最適な錯体溶液の pH が存在する。これはホスホン酸基 –P(O)(OH)2のイオン化度が基板表面に存在する水酸基 との結合反応が起こる条件に関わっているためだと考え られる。
Scheme 13. Chemical structures of self-standing Ru complexes with functional auxiliary ligands such as DNA intercalator group, metal coordination group, and PCET on an ITO surface (Ref. 39, 40).
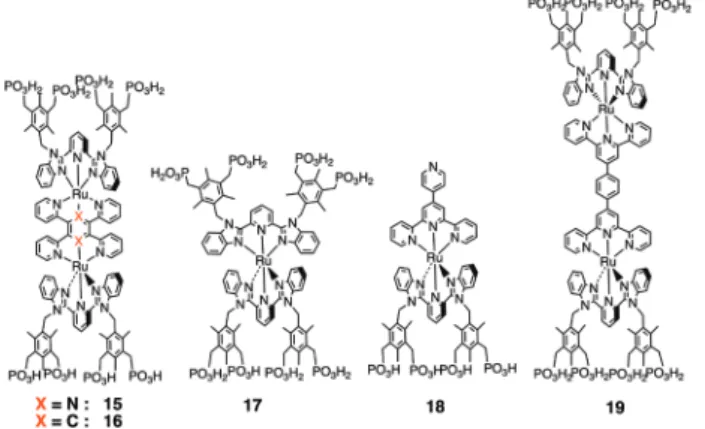
錯体 12 は,二重鎖 DNA の塩基対間にインターカレ ートすることが知られているアクリジン環を補助配位 子末端にもつ。この錯体を SiO2/Si 基板上にパターン化 した Au アレイ電極上に選択的に吸着させて,DNA 溶 液中から二重鎖 DNA を錯体へのインターカレーション により捕捉して,隣り合った二つの Au 電極間を DNA で繋ぐ DNA ワイヤリングを検討した39。二重鎖 DNA は Au アレイ電極上に固定された錯体に捕捉されるが, DNA の捕捉には,表面に固定する錯体濃度に最適値が あることがわかった。表面錯体が密に詰まると,DNA の表面への捕捉固定化率が下がることがわかった。これ は,DNA が錯体分子にインターカレートするためには, 分子の周りにある程度の隙間の空間が必要なためではな いかと考えている39。 錯体 13 は,末端にフリーのターピリジン基をもつの で,Fe(II) イオンと錯形成した後に,ビス ( ターピリジ ル ) ベンゼン架橋配位子とのさらなる錯形成を経て逐次 的に一次元の多核錯体形成が起こる。表面 Ru-Fe 二核錯 体の光電気化学応答で電圧パルスを印加した前後で光電 流の方向が逆転することから,電圧印加により書き込み, 光で読取りができるメモリデバイスとして動作すること を報告した40。 ITO 電極表面に固定した錯体 14 は,溶液の pH によ り pH 1–9 の範囲で Ru(II/III) 酸化電位が変化するプロト ン共役電子移動を示した。pH > 10 では,錯体は ITO 基 板表面からすべて脱着する。錯体 14 を最外層にもつヘ テロ積層膜では,溶液の pH 変化に応じて電極との電子 移動の方向を制御できる整流デバイスの電位チューニン グサイトとして働くことができる。 4.3. 錯体の表面積層化と機能 4.3.1. 表面錯体のナノ積層構造の作製 . 先に述べた表面固定された錯体単分子膜上に配位可 能なサイトが溶液側を向いて存在すれば,液中の金属イ オンと錯体単分子膜との錯形成が起こる。このような固 体基板上固定吸着した配位基をもつ錯体単分子膜(プラ イマー層と呼ぶこともある)に対し,金属イオンと錯体 ユニットの溶液を交互浸漬することにより,逐次的に錯 形成した積層膜を固体表面に作製できる。この方法は, Layer-by-Layer 法(以下 LbL 法と略す)と呼ばれ,この 方法によって様々な機能をもつ金属錯体を表面に任意の 積層数とユニットの組合せ順序で組み上げることができ る。また,錯体ユニットを選ぶことで,電位勾配や pKa 勾配を層内に付与できるので,表面での “ 配位プログラ ミング ” が可能となる利点がある41。たとえば,異なる 二種の錯体ユニット A, B を基板上に構築する場合,最 初に A を基板に固定したときには四積層膜構造でも基板 |-A-A-A-B,基板 B-A,基板 |-A-A-B-B,基板 |-A-B-A-B など七種類の組み合わせが可能となる。構成ユニッ トの組み合わせを変えることで,A, B の酸化還元電位が 違う場合には様々な電位勾配を作ることができ,電極と の電子移動の方向を制御することができる。さらに,光 感応性錯体を組み込んだ錯体ユニットを組み合わせれば, 光合成系の電子移動鎖のような光カスケード型電子移動 を起こせる電位傾斜型ナノ構造体の作製も可能となる42。 そこで,固体基板に自己組織化できる有機官能基を もつ分子をまずプライマー層として化学吸着させて,そ の上にナノネットワーク構造あるいは金属有機構造体 (MOF) を構築する方法がよく用いられる43,44。ここで, 表面での構造を区別するために,“ ネットワーク構造 ” の語は表面での逐次錯形成によって作製される膜がアモ ルファス構造をもつときに用い ,“MOF 構造 ” の語は規 則的なナノ結晶薄膜で XRD ピークを与える表面構造の 場合を指すときに用いることにする。我々は,Scheme 14 に示した自立型四脚アンカー配位子をもち,架橋配位子 で繋がれたロッド状で剛直な二核錯体の両端にそれぞれ 4 個のホスホン酸基をもつ錯体 15, 19,それと補助配位 子の異なる単核錯体 17, 18 を合成した。これらの錯体溶 液に平滑な透明導電性 ITO 電極基板を約 3 時間浸漬する だけで,錯体は自己組織的に吸着する。吸着量の時間変 化を CV の Ru(II/III) 波および UV-vis スペクトルの MLCT 帯からモニターすると,吸着は Langmuir の等温式に従 うことが確認できた。原子平坦面をもつサファイア基板 表面や平滑 ITO 表面を錯体の希薄溶液に短時間浸漬して AFM 観察すると,疎らに吸着した錯体分子がドット状 のモルフォロジーとして観測され,その高さは分子が垂 直に自立した高さとほぼ一致した (Fig. 7)。3 時間後には, 数十 μm のドメインをもつ構造で表面が均一に隙間なく 埋めつくされている様子が AFM により観察された。CV の Ru(II/III) 酸化波から得られた錯体の表面被覆量は,単 核,二核錯体にかかわらずほぼ (5 ± 0.5) × 10–11 mol cm–2で あることから,分子占有面積が自立型四脚アンカー配位 子で決まっていることがわかった。錯体 15–19 はいずれ も上端に金属イオンと錯形成可能なホスホン酸基やピリ ジル基をもつので,固体基盤を金属イオンと錯体ユニッ
トのそれぞれの溶液に交互浸漬すると錯形成が起こり, 逐次積層化によりネットワーク構造をもつ多積層膜を作 製することが可能である。ジルコニウム (IV) イオンと錯 体のホスホン酸基との錯形成を利用した錯体 15–17, 19 の 固体表面上への逐次積層膜の生成過程は,様々な物理化 学的測定手法により,たとえば CV の電気量,UV-vis ス ペクトルの吸光度,AFM のスクラッチ法による分子薄膜 の膜厚などの物理量としてモニターできる。いずれの物 理量も,基板の溶液への浸漬回数と直線関係が成り立ち, 逐次積層化の際に錯体膜は基板平面に対して層構造をと って成長していることがわかった31,45。
Scheme 14. Chemical structures of self-standing Ru complexes with metal coordination groups on an ITO surface.
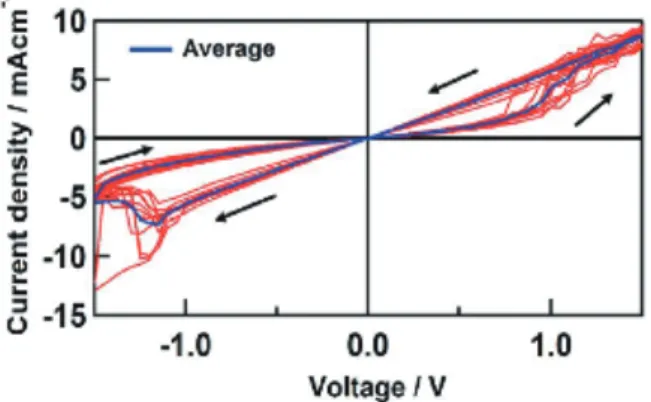
Fig. 7 AFM image of dinuclear Ru complex 19 on a flat ITO surface immobilized from a 1 mM solution of complex 19 for 10 min (left) and 25 mM solution for 3 h and their surface images (Ref. 32). 4. 3. 2 表面ナノ薄膜の分子伝導機能 平滑 ITO 電極表面に多点吸着した自立型錯体 15, 17, 18 の単層膜の伝導性についてコンダクティブ AFM (C-AFM) を用いて,Pt 上に ITO を蒸着した探針と修飾 ITO 電極との二端子法で測定した46。その結果,測定雰 囲気が低湿度の乾燥状態では,単核・二核錯体のどち らも電流 – 電圧特性(I–V 特性)は正負電位で対称とな り,単なる分子ワイヤとして動作することがわかった。 しかし,測定環境が高湿度条件になると,ルテニウム 二核錯体 15 の場合には I–V 特性が正側だけで大きな電 流が流れる整流性がみられるようになり,整流比 R (= |I (–V)|/|I(V)|) が数千程度の大きな値を示すことがわかっ た (Scheme 15)。ここで,|I(V)| はある電圧における順方 向あるいは逆方向の電流密度の絶対値を表す。再び測定 系の湿度が低くなると,元の左右対称の I–V 特性となり, 整流効果がみられなくなる。この伝導度のスイッチング は,単核錯体 17, 18 では観測されない。高湿度条件では, 径の小さな探針が二核錯体 13 に接すると探針周りによ り強く水分子を引き寄せ,錯体分子の探針側とより強く 相互作用して混合原子価状態に大きな非対称性を生じ, 分子軌道エネルギーと電極のフェルミ準位との間に大き なギャップが生まれ,その結果大きな整流比を与えるこ とが計算科学から示唆された46。このように,外部湿度 により整流性が ON/OFF するスイッチング現象は分子 として初めての例である。
Scheme 15. Schematic representation of humidity switching of complex 15 molecular junction on I–V plots (Ref. 46).
また,ITO 電極上に吸着した自立型ルテニウム錯体 19 は,電極上端のホスホン酸基と溶液中の錯体 19 のホス ホン酸基との間で水素結合が生じやすく,溶液を pH 6 にすると多重水素結合で連結された積層膜が時間ととも に表面で成長することがわかった。この ITO 上に成長し たルテニウム錯体積層膜の伝導度について明らかにする ために,上端電極として電導性高分子 PEDOT:PSS を用 いたサンドイッチ型二端子デバイスで I–V 特性を測定し た47。積層膜の膜厚と電流の関係を調べたところ,電流 すなわち電子移動速度の距離依存性の尺度となる減衰係 数 β 値は小さい値 (0.012 ~ 0.021) を示すことが明らかと なった47。錯体多積層膜系では膜厚変化に対して小さい β 値を示しても,ホスホン酸 – ホスホン酸水素結合で錯 体ユニット間が隔てられているために低伝導度である。 低伝導度にもかかわらず長距離電荷輸送が可能になる理 由を明らかにするために,密度汎関数法・非平衡グリー ン関数法を用いた計算科学から長距離電子輸送について 考察した。その結果,配位子の π 電子で覆われた金属中 心が「飛び石」となり,周辺配位子の π 軌道との混成を 通じて電荷が移動して電極間を透過する「飛び石」機構 (stepping-stone mechanism) で起こる新しい機構を提案す
ることができた47。逐次錯形成で作製されたターピリジ ン錯体ナノワイヤでも小さな β 値が報告されている48,49。 4. 3. 3. 表面ナノ積層膜の電気化学 ジルコニウム (IV) イオンとの錯形成により ITO 電極 上に錯体 15 を多積層化した多層膜について,積層数を 増加させていったときの CV および異なる掃引速度 (0.1 V/s と 1 V/s) の CV を Fig. 8 に示した。錯体 15 の多積層 膜は二段の 1 電子酸化波を示した。掃引速度が 0.1 V/s では,1 層から 20 層に積層数が増えてもピーク電位は 大きく変化せずに電流値が増加していくが,掃引速度 が 1 V/s では,積層数が増加するとともにアノードピー ク電位とカソードピーク電位の差が増加する。これは膜 厚が増加すると,層間をホッピングする電子移動速度 が律速となるためであると考えられる。Fig. 9 にはポテ ンシャルステップクロノアンペロメトリー (PSCA) で求 めたみかけの電子移動速度の対数と膜厚のプロットを示 した。その傾きから,電子移動速度の距離依存性の尺度 となる減衰係数 β は 0.014 Å–1 ( 錯体 15) および 0.010 Å–1 ( 錯体 16) となり,非常に小さい値をとる。このことは, 積層数が増えても電極から遠距離にある錯体層にまで電 子移動が起こることを示している50。電極表面での錯体 15 の多積層ネットワーク薄膜の Ru(II)/Ru(III) 酸化過程 では,錯体の正電荷が増加する。この表面での正電荷の 増加を補償し,膜内での電荷を中性に保つためにバルク 溶液からアニオンの膜内への拡散が起こる。このイオン の取り込みは,リチウムイオン二次電池で利用されてい るイオンの蓄電過程と同じである。そこで,錯体 15 を 65 層積層した薄膜である ITO||( 錯体 15)65の蓄電機能を 定電流法で評価した。その結果,電流 10 μA cm–2での最 大容量は 95.3 F g–1となり,一般的な炭素キャパシタと 比較しても見劣りしない性能を示した45。また,素早い 充放電が可能で,かつ 2000 回の充放電後でも 80% の容 量を保持できる優れたレドックスキャパシタとして働く ことが明らかになった。積層膜の場合には積層数を増や すことで蓄電容量を増やせるメリットがある。
Fig. 8 Cyclic voltammograms of complex 15 with increasing number of layers 1–2 layers at two scan rates 0.1 V/s (left) and 1 V/s (right).
Fig. 9 Plots for ln(kapp) as a function of the film thickness for ITO||(complex 15)n film (Ref. 50). 4. 3. 4. 表面ナノ積層膜のプロトン共役電子移動 先にも述べたが,二つの錯体のプロトン共役電子移動 での電位差と pKa差を利用すると,酸化還元に伴うプロ トン移動を引き起こすことができ,プロトン濃度差とし て蓄電することが可能である。そこで,自立型ホスホン 酸アンカー基をもち,電極表面でプロトン共役電子移 動が可能なベンズイミダゾリル N–H 基をもつ二核錯体 20, 21 を新たに合成した (Scheme 16)51。これらの錯体は, 先に述べた単核錯体 6, 7 を二核化し,表面積層化のため にアンカー部位としてホスホン酸基を両端に付けたもの である。ホスホン酸をエチル基で保護した錯体の 20Et は 4 個 の N–H 基 を も ち, そ の pKaは Ru(II) 状 態 で は 4.2, 5.3, 7.4, 8.5,また Ru(III) 状態では <2.2, 2.5, 3.8 であ った。一方,21Et の pKaは,Ru(II) 状態では 8.4, >11.0, また Ru(III) 状態では 5.2, 7.3, 9.1, 9.8 であった。また, 錯体 21 は Ru–C シクロメタル結合をもつので,近接し た 2 個の Ru(II/III) 1 電子酸化過程の電位は負電位側に 大きくシフトしており,2 個のルテニウムは Ru(III)― Ru(III) の酸化状態で単離される。さらに,錯体 21 のベ ンズイミダゾリル N–H 基の酸性度は低く,プロトンと して解離しにくい。一方の錯体 20 は Ru(II)-Ru(II) の酸 化状態で単離され,Ru(II) 状態では中程度の酸性度をも つ。しかし,酸化されて Ru(III) 状態となると,プロト ンは解離しやくなる。この二核錯体 20, 21 は両端にホ スホン酸基をもつので,ITO 電極上に Zr(IV) を介して レドックス活性な多積層膜として固定化できる。そこ で,2 個の錯体積層膜修飾電極,ITO|( 錯体 20)nと ITO| ( 錯体 21)n (n は積層数 ) を対向させて 0.1 M NaClO4で挟 み込んだ二電極デバイスを作成し,そのレドックスキャ パシタとしての動作性能について定電流法で検討した。 電流 10 μA cm–2で,積層数を 10 層から 50 層まで変えた ときの充放電特性を Fig. 10 に示した。非線形の充放電 曲線から,イオンの出入りだけでなく,電極上の錯体の レドックスが関与してプロトンを含むイオン移動が起こ っていることがわかった51。また,積層数が増えるにつ れて,比容量が増加していくことがわかった。比較のた めにベンズイミダゾリル N–H 部位を N–Me 部位に置換
した錯体 22 および 23 では,プロトン共役電子移動反応 は起こらない。この二つの錯体 22 と 23 からなる二電極 デバイスのキャパシタ容量は,錯体 20 と 21 の組み合わ せで得られる容量よりも 77% も減少することがわかっ た。これは,錯体 20 と 21 との電極間でのプロトン移動 が充放電に重要な役割をもつことを示している。そこで, プロトンがレドックスに応じて積層薄膜内を動くかどう かについて,分光電気化学薄層セル内に pH 指示薬であ る 5(6)- カルボキシフルオレセインを溶かすことにより 調べた。錯体 20 の積層膜での電解酸化の際に 555 nm の 錯体の MLCT 帯の消失時に観測される等吸収点 480 nm において,pH 指示薬の吸収変化を観察すると,薄膜の 酸化に伴う 480 nm の吸光度の減少が観測され,プロト ンが放出されていることが確認できた。また ITO 被覆 した電気化学水晶振動子マイクロバランス (EQCM) のチ ップ上に,錯体 20 とベンズイミダゾリル N–H 基をメチ ル保護した錯体 22 の積層膜を作製して,レドックスに 伴う質量の増減を観測した。EQCM の結果を比較して Fig. 11 に示した。錯体 22 では,メチル基で保護されて いるために,積層膜での酸化には式 2(ここで LMe は メチル化架橋基を示している)で示すように,表面での 電荷補償のためにバルクからアニオンの取り込みが主に 起こるので,表面での周波数減少,すなわち質量増加が 観測される。一方,溶液が中性の場合には,錯体 20 の 酸化に伴ってプロトン共役電子移動が起こり,プロトン が錯体から解離して錯体の電荷を補償するためにイオン の取り込みはない(式 1 ここで LH2はプロトン化架橋基, L は脱プロトン化した架橋基を表す)。このために表面 での周波数変化は小さくなったと解釈できる。 錯体 21 の PCET 酸化反応: Ru(II)2LH2 ⇄ Ru(III)2L + 2e– + 2H+ (式 1) 錯体 22 のイオン対生成を伴う酸化反応:
Ru(II)2LMe + 2ClO4– ⇄ Ru(III)2LMe(ClO4)2 + 2e– (式 2)
Scheme 16. Chemical structures of dinuclear Ru complexes having N–H sites and their N-methyl protected ones.
Fig. 10 Galvanostatic charge/discharge curves of ITO||(complex 20)n (the number of layers, n = 10, 20, 30, 40 and 50) (Ref. 51).
Fig. 11 EQCM frequency responses of ITO immobilized 10 layers film of (a) complex 22 and (b) complex 20 (Ref. 51).
以上より,ITO|( 錯体 20) と ITO|( 錯体 21)n (n は積層 数 ) の二端子デバイスでは,プロトン移動を介してプロ トンロッキングチェア型レドックスキャパシタとして動 作することが明らかになった。1000 回の繰り返しの充 放電では,80% のクーロン効率,容量は 60%を保持でき, 安定性もあることがわかった (Scheme 17)51。
Scheme 17. Schematic representation of proton-rocking-chair-type redox capacitor composed of two PCET Ru multilayer complexes, 20 and 21 (Ref. 51).
4 .4. 表面ヘテロ積層膜の電子機能 4. 4. 1. ヘテロ積層膜の光応答性メモリ シリコンデバイスの p 型ドーピングと p 型ドーピング した表面が接するときにできる pn 接合は,接合面にで きるエネルギー差のために,整流性をもつダイオードや 発光ダイオードや太陽電池など有用なデバイスとして利 用されている。そこで,これまでに積層膜構築に用いて きた電位が大きく異なる二種の錯体 15 と 16 を錯体ユニ ットとして基板上でヘテロ接合した積層薄膜を構築し
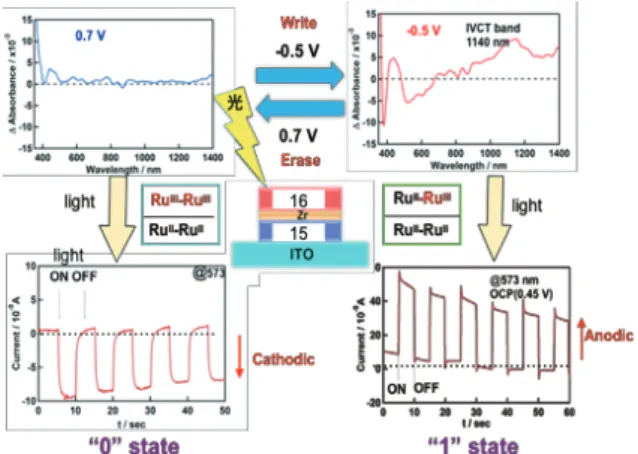
た52。ここで,錯体 16 は二段の 1 電子 Ru(II/III) 酸化を –0.37 V と +0.09 V vs Fc/Fc+に,さらに正側の +1.15 V に Ru(III/IV) 酸化を示す。一方錯体 15 は,Ru(II/III) 酸化を +0.83 V と +1.04 V に示し,–0.5 V 付近に架橋基の還元 による立ち上がりがみられる。両者の錯体間には大きな 電位差がある。そこで,先に述べた逐次積層法により平 滑 ITO 基板上にまず錯体 15 を n 層,その上に Zr(IV)– ホスホン酸基層を挟んで,次の層として錯体 16 の層を m 層積み上げて ITO||( 錯体 15)n/( 錯体 16)mのヘテロ積 層膜を作製した。Fig. 12 に錯体 15, 16 単層ならびにヘ テロ積層で n = 1, m = 1 および n = 2, m = 2 の CV を示し た。ここで,内層の錯体 15 が 2 層以上になると,外層 の錯体 16 から ITO 電極への直接電子移動はエネルギー 障壁のためにブロックされて起こらない。しかし,錯 体 15 内層の酸化が起こる電位まで電位掃引すると,錯 体 16 外層から一気に電子移動が起こり,Fig. 12 の CV 図の (*) で示した触媒電流が流れる。すなわち,ヘテロ 接合面の外層に電荷が貯まった状態を作り出し,内層に ホールができたときに一気に電子移動が起こる整流効果 を持たせることができる。ITO 電極上での薄膜であるの で,薄層分光電気化学によって各電位での UV-Vis-NIR スペクトルを測定することで,電極に加えた電位により 錯体 16 外層の電荷の有無を吸収スペクトル変化から確 認でき,二つの酸化状態を電圧印加により作り出すこと ができる (Fig. 13 上段 )。興味あることに,このヘテロ 接合積層膜の二つの異なる酸化状態に光照射して得られ る光電流の方向が,錯体 16 外層内の電子の有無(錯体 16 の酸化状態の違い)により,アノード電流あるいは カソード電流となり,電流方向が逆転する (Fig. 13 下段 )。 この ITO||( 錯体 15)n/( 錯体 16)mのヘテロ接合膜における 光電流の特性を利用すると,ヘテロ接合膜への –0.5 V 電圧印加で書き込み,錯体 16 外層に電子移動が起こり Ru(II)-Ru(III) 混合原子価状態となり,0.7 V の電圧印加 で外層に貯められた電子は酸化により除かれて Ru(III)-Ru(III) 状態になる。電圧印加による錯体 16 外層の電子 の有無を “0”“1” の二状態として書き込み,薄膜に光を 当ててその光電流の向き(正負)で読み出すことができ る光メモリとして動作する。
Fig. 13 Switching between two “0” and “1” states by applying potential pulses in heterolayer ITO||(complex 15)4|(complex 16)4.Vis-NIR spectra for two states (top) and corresponding photocurrent responses (bottom).
4. 4. 2 プロトンメモリスタ 最近,抵抗,キャパシタ,インダクタに次ぐ第四の 受動素子として,メモリスタが注目されている。メモ リスタは,Chua により 1971 年に磁束と電荷を関係づけ る新しい電子回路の構成要素として予測されたが53,デ バイスとして実証されたのは,2008 年の TiO2を挟んだ 二端子デバイスでの酸素欠陥のフィラメント形成による “ 8” 字型での抵抗スイッチングを示す実験であった54。 それ以来,酸化物だけでなく高分子材料など多くの材料 でも同様な “ 8” 字型の非線形な電圧電流ループが観測 されてきている。このように,電圧掃引に伴う電荷を記 憶して,それに伴う電流が非線形的に変化することで抵 抗が変化する素子を,一般的にメモリスタと呼んでいる。 特に最近では,メモリスタは神経回路のシナプスでの記 憶と関係づけて検討されることが多くなっている。そこ で,シナプスのイオン移動にヒントを得て,PCET を示 すルテニウム錯体積層膜電極でプロトン伝導する高分子 や MOF などを挟み込んだ二端子デバイスがメモリスタ 機能をもつのではないかと考えた。先に述べたプロトン ロッキングチェア型レドックスキャパシタでは,二つの 電極 ITO||( 錯体 20)nと ITO||( 錯体 21)nを非緩衝水溶液で 挟み込み,水をプロトン伝導の媒体とした。水に代わる 他のプロトン伝導物質を錯体間に挟みこめば,PCET 錯 体の pKaとプロトン伝導物質の pKaとが上手くマッチす れば膜間での pKa差によりプロトン移動を誘起できるは ずである。そこで,ITO 基板上にて,二つの錯体積層膜 電極 ITO||( 錯体 20)nと ITO||( 錯体 21)n間にプロトン伝 Fig. 12 Cyclic voltammograms of (a) ITO||(complex 16), (b)
ITO||(complex 15), (c) ITO||(complex 15)|(complex 16) and (d) ITO||(complex 15)2|(complex 16)2 (Ref. 52).
導を示す有機高分子ポリビニルピリジン (P4VP) (pKa = 4.4–5.2) を挟んだ二端子デバイス ITO|( 錯体 20)3/(P4VP)/ ( 錯体 21)3|ITO を作製した55。ここで,錯体 20/P4VP お よび錯体 21/P4VP の接合面ではピリジン基とベンズイ ミダゾリル N–H 基との間に水素結合相互作用があるこ とが CV から推定された。そして I–V 曲線は,期待した 通り “ 8” 字型のヒステリシスを示し,メモリスタとし て動作することがわかった (Fig. 14)55。初めの状態にお いて,このデバイスの二端子上では,錯体 21 は Ru(III) 状態,錯体 20 は Ru(II) 状態にある。この初めの状態で は,接合した三つの層,錯体 20,P4VP,錯体 21 とも にほぼ同じ pKaであり,プロトン勾配はないのでプロト ン伝導性のよくない OFF 状態である。しかし電圧掃引 により二端子間で Ru(II)/Ru(III) のレドックスが起こる と,P4VP を隔てた二つのヘテロ界面で大きなプロトン 勾配が生じ,錯体 –P4VP 内の水素結合を介してのプロ トン伝導が誘起されて,良伝導の ON 状態となり,メモ リスタ機能が発現したものと考えている (Fig. 15)。 4. 4. 3 表面錯体膜上にヘテロ接合した MOF 構築とイ オンゲート機能 金属有機構造体 (MOF) は金属イオンと有機分子によ り構成された多孔質材料であり,内部の空孔を利用した ガス吸蔵,触媒やセンサーデバイスなどへの応用が期待 されている。この MOF をデバイスに応用するために, 基板への MOF の固定成長が Fischer らによって報告さ れ,SURMOF とよばれるようになり研究例が増えつつ ある。プルシアンブルー (PB) は Fe–C≡N–Fe 架橋からな るジャングルジム構造をもち,内部にナノ細孔をもつ。 また,PB は鉄の酸化状態の変化により,還元されると Fe(II)-Fe(II) 状態のプルシアンホワイト (PW),酸化され ると Fe(III)-Fe(III) 状態のベルリングリーン (BG) に変化 する二段階のレドックス反応を行うことが知られてい る。さらに PB の酸化還元に伴い,ナノ細孔へのイオン の取り込み・放出が起こることから二次電池への応用も 検討されている。 最近我々は,ITO 基板上に作製した錯体積層膜を, K3[Fe(CN)6],塩化鉄 (III) の混合水溶液に浸漬するだけ で,修飾基板上に PB 結晶が結晶方位を揃えて自発的に 析出することを見出した56。Fig. 16 にせん断 SEM の時 間変化を示した。時間とともに基板から成長して 12 時 間で膜厚が約 180 nm になる。ITO 基板上のプライマー 層として錯体 15 を用いて作製した ITO||( 錯体 15)3/PB ヘ テロ接合膜の酸化還元挙動を CV 測定したところ,酸化・ 還元それぞれ 1 個のピーク電位だけが観測された。内 層の錯体表面濃度は外層の PB に比べて極めて小さいの で,観測される CV には PB だけの電流応答がみられる。 ITO 電極に直接結合した PB 膜の場合には二つの酸化還 元対のピークを示すが,ITO||( 錯体 15)3/PB ヘテロ接合 膜では PB の酸化波 1 個と還元波 1 個,それぞれ別のレ ドックス反応のピークが観測される (Fig. 17)。そこで, ヘテロ接合 ITO 電極での PB 層のイオンの取り込み量を EQCM 法で測定したところ,電位掃引に伴う二つのヒ ステリシスループをもつことがわかった56。これは,内 層の錯体 15 のエネルギーレベルと PB のエネルギーレ ベルとが重なり合わずに電子移動速度が遅くなり,整流 性が出るためだと考えられる。EQCM 上の修飾膜にパ ルス電位を印加すると,ITO||( 錯体 15)3/PB ヘテロ接合 膜では PB と同様に還元によるイオンの取り込み・酸化 によるイオンの放出が観測された。一方,錯体 16 をプ ライマー層とした PB 膜では,錯体 16 の電位は錯体 15 に比べて負電位シフトしているために,ITO||( 錯体 16)3/ PB ヘテロ接合膜では CV において PW/PB 間のみの酸化
Fig. 14 I–V plots of ITO||(complex 20)3/P4VP/(complex 21)3||ITO, corresponding 10 scans on the same device. The average of 10 different devices is also shown as a dark line (Ref. 55).
Fig. 15 Schematic representation of pKa gradient under the applied positive bias potential for ITO||(complex 20)3/P4VP/ (complex 21)3||ITO two terminal device. The numbers refer to the pKa values for the Ru(II) and Ru(III)complexes (Ref. 55).
還元波が観測され,また EQCM でも PW/PB 間でのイオ ンの取り込みのみの質量変化が起こる。パルス電位を印 加した場合にも,PW/PB 間でのイオン取り込みのみの 質量変化が観測された。これらの結果から,プライマー 層に用いる錯体の酸化還元電位すなわちエネルギーレベ ルが,PB ナノ結晶 ITO 間との電子移動の方向を決めて いることがわかる56。膜厚から考えると内層は数ナノメ ートル,PB の膜厚は数百ナノメートルである。以上の 結果は,ナノメートルサイズの錯体層のエネルギーレベ ルが,ナノ結晶と電極との電子移動のエネルギーバリア ーあるいは電子移動メディエーターとして働くことを示 している。つまり,伝導性 MOF のイオン取り込み制御 をヘテロ接合分子膜で行うことができることを示してお り,イオン移動を伴う電荷移動の制御に関して重要な知 見であると考えている。
Fig. 16 Cross-sectional SEM images of the growth of PB nanocrystals deposited on ITO||(complex 15)3 ITO electrode; immersion time = 3, 6, 12 h, and schematic drawing of layered structure (right) (Ref. 56).
Fig. 17 EQCM responses of ITO||(complex 16)3|(PB) (a) and ITO||(complex 15)3|(PB) (c) overlaid by cyclic voltammograms and EQCM responses to a potential pulse sequence of ITO||(complex 16)3|(PB) (b) and ITO||(complex 15)3|(PB) (d).
■■ 5. おわりに
ルテニウム -BIm 錯体の酸化還元電位はルテニウム周 りの配位子を変えることで大きく変わり,空気中室温で 安定な Ru(II), Ru(III), Ru(IV) など様々な酸化数もとれる ようになる。さらに,プロトン共役電子移動により電位 は溶液の pH により制御することも可能である。 溶液化学で研究して得られた錯体の基礎物性を表面錯 体に展開して,錯体を固体表面に吸着固定させることで ヘテロ接合など新たな複合機能を見つけ出すことができ る。たとえば,表面錯体の固体触媒機能,センサーやメ モリデバイスへの応用などが考えられ,他の領域との融 合領域に足を踏み入れることが可能となり,視点が変わ ることで研究テーマも大きく拡がる。私自身も錯体化学 から界面コロイド化学や表面科学などの基礎知識や界面 科学特有の観測法を勉強しながら,表面に吸着するアン カー配位子の設計や表面での機能発現のための補助配位 子や錯体を分子設計してきた。分子エレクトロニクス での単一分子計測法やデバイス評価などの研究は,様々 な分野の研究者に教えを請いながら共同研究できた。 Carter の論文13中の図に分子ワイヤの側鎖グループに混 合原子価錯体を導入することで,外部電場で主鎖の分子 伝導度をスイッチングできるモデル図があった。混合原 子価錯体がこのような制御グループに使える可能性があ るなら,それを実験的に研究してみようと思い,研究を 溶液から電極固体表面での錯体化学に展開してきた。今 後も,表面錯体は SURMOF などの研究を通してさらに 発展していくと思われる。錯体の表面での構造と表面機 能とがリンクした研究は,今後新しい機能の創成に重要 である。この分野がさらに発展するためには,固体表面 に作製した錯体集積体の構造の決定,そして錯体集積体 の関わる電極表面での電子移動に伴うイオンの取り込み や構造体内でのイオン・分子の動きなどの動的機能と構 造がどのように関係しているのかを知ることが大切だと 考えている。今後も若い研究者にはぜひ錯体が関わる融 合領域の面白さを感じてもらい,その醍醐味を味わって いただきたいと願っている。 ■■ 謝辞 本論文にまとめた研究成果は,主に科学研究費助成金 (KAKEN) および科学事業団学術研究振興資金の支援の もとに行ったものです。また,ここで報告した研究を遂 行することができたのは,三重大教育学部,分子科学研 究所,中央大理工学部で “Squeeze brain” で一緒に考え実 験を進めてくれた多くの学生諸君の努力の賜物であり, 彼らに深く感謝します。また,同時に助手・助教として 大学・研究所で学生指導や研究を支えてくれた文殊四郎
秀昭氏,豊智奈氏,小林克彰氏,金井塚勝彦氏,小澤寛 晃氏に謝意を表します。同時に論文の共著者となってい る多くの先生方にも感謝します。特に,私を錯体化学の 道に導いてくださった故木下達彦先生,森和亮先生,故 田中敏夫先生,故河上克彦先生,田中晃二先生,電気化 学では松村竹子先生,加納健司先生,錯体光化学では大 野健先生,野崎浩一先生,界面錯体化学の先駆者 山岸 皓彦先生に深く感謝申し上げます。また海外では,研究 室に受け入れていただき,滞在中たくさんのことを学ば せていただいた Prof. A. B. P. Lever, Prof. A. M. Bond, Prof. T. E. Mallouk にも謝意を表したいと思います。
最後に,長年私の研究生活を陰で支えてくれた亡き妻・ 芳賀昌子にこの論文を捧げます。
■■ 文献
1 J. W. Dart, M. K. Lloyd, R. Mason, J. A. McCleverty, J. Chem. Soc., Dalton Trans. 1973, 2039–2045.
2 K. Kawakami, M. Haga,T. Tanaka, J. Organometal. Chem. 1973, 60, 363–373.
3 W. M. Bedford, G. Rouschias, J. Chem. Soc., Chem. Commun. 1972, 1224.
4 G. L. Geoffroy, M. G. Bradley, M. E. Keeney, Inorg. Chem. 1978, 17, 777–779.
5 Y. L. Chen, K. Li, H. O. Lloyd, W. Lu, S. S. Y. Chui, C.-M. Che, Angew. Chem. Int. Ed. 2010, 49, 9968–9971.
6 C. L. Exstrom, D. Britton, K. R. Mann, M. G. Hill, V. M. Miskowski, W. P. Schaefer, H. B. Gray, W. M. Lamanna, Inorg. Chem. 1996, 35, 549–550.
7 G. Sprintschnik, H. W. Sprintshnik, P. P. Kirsch, D. G. Whitten, J. Am. Chem. Soc. 1976, 98, 2337–2338.
8 M. Haga, T. Tanaka, Chem. Lett. 1979, 863–864. 9 M. Haga, Inorg. Chim. Acta 1980, 45, L183–L184. 10 M. Haga, Inorg. Chim. Acta 1983, 77, L39–L41. 11 M. Haga, Inorg. Chim. Acta 1983, 75, 29–35.
12 M. Haga, A. M. Bond, Inorg. Chem. 1991, 30, 475–480. 13 F. L. Carter, Physica. D. 1984, 10, 175–194.
14 M. Haga, Benzimidazole Class Ligands. In Comprehensive Coordination Chemistry II, J. A. McCleverty, T. J. Meyer, Eds. Pergamon Press: 2003; Vol. 1, pp 3063–3071.
15 M. Haga, M. Ishizuya, T. Kanesugi, T. Yutaka, D. Sakiyama, J. Fees, W. Kaim, Indian J. Chem. 2003, 42A, 2290–2299. 16 S. Roland, X. Ling, M. P. Pileni, Langmuir 2016, 32, 7683–7696. 17 C. M. Crudden, J. H. Horton, M. R. Narouz, Z. Li, C. A.
Smith, K. Munro, C. J. Baddeley, C. R. Larrea, B. Drevniok, B. Thanabalasingam, A. B. McLean, O. V. Zenkina, Ebralidze, II, Z. She, H. B. Kraatz, N. J. Mosey, L. N. Saunders, A. Yagi, Nat Commun 2016, 7, 12654.
18 M. Haga, A. Tsunemitsu, Inorg. Chim. Acta 1989, 164, 137–142. 19 R. Hones, M. Kuss-Petermann, O. S. Wenger, Photochem.
Photobiol. Sci. 2013, 12, 254–261.
20 A. Pannwitz, O. S. Wenger, Phys Chem Chem Phys. 2016, 18, 11374–11382.
21 M. Haga, T. Anno, K. Kano, S. Yamabe, Inorg. Chem. 1991, 30, 3843–3849.
22 M. Haga, M. M. Ali, S. Koseki, K. Fujimoto, A. Yoshimura, K.
Nozaki, T. Ohno, K. Nakajima, D. J. Stufkens, Inorg. Chem. 1996, 35, 3335–3347.
23 K. Kobayashi, M. Ishikubo, K. Kanaizuka, K. Kosuge, S. Masaoka, K. Sakai, K. Nozaki, M. Haga, Chem. Eur. J. 2011, 17, 6954–6963.
24 H. Tannai, K. Tsuge, Y. Sasaki, Inorg. Chem. 2005, 44, 5206– 5208.
25 J.-P. Launay, Eur. J. Inorg. Chem. 2020, 329–341.
26 M. Haga, M. Ali, R. Arakawa, Angew. Chem. Int. Ed. 1996, 35, 76–78.
27 D. Motoyama, K. Yoshikawa, H. Ozawa, M. Tadokoro, M. Haga, Inorg. Chem. 2017, 56, 6419–6428.
28 K. Tsuge, M. Kurihara, K. Tanaka, Bull. Chem. Soc. Jpn. 2000, 73, 607–614.
29 X. Xie, G. A. Crespo, G. Mistlberger, E. Bakker, Nat. Chem. 2014, 6, 202–207.
30 Y. Wada, M. Tsukada, M. Fujihira, K. Matsushige, T. Ogawa, M. Haga, S. Tanaka, J. J. Appl. Phys. Part 1 2000, 39, 3835–3849. 31 M. Haga, T. Yutaka, Inorganic supramolecular architectures
at surfaces. In Trends in Molecular Electrochemistry, A. J. L. Pombeiro, C. Amatore, Eds. Marce. Dekker: 2004; pp 311–336. 32 M. Haga, K. Kobayashi, K. Terada, Coord. Chem. Rev. 2007, 251,
2688–2701.
33 H. Ozawa, M. Haga, Bull. Jpn. Soc. Coord. Chem. 2012, 60, 2–23. 34 M. Haga, H. Hong, Y. Shiozawa, Y. Kawata, H. Monjushiro, T.
Fukuo, R. Arakawa, Inorg. Chem. 2000, 39, 4566–4573. 35 M. Haga, T. Takasugi, A. Tomie, M. Ishizuya, T. Yamada, M. D.
Hossain, M. Inoue, Dalton Trans. 2003, 2069–2079.
36 K. Terada, K. Kobayashi, M. Haga, Dalton Trans. 2008, 4846– 4854.
37 R. S. Loewe, Ambroise, K. Muthukumaran, K. Padmaja, A. B. Lysenko, G. Mathur, Q. X. Li, D. F. Bocian, V. Misra, J. S. Lindsey, J. Org. Chem. 2004, 69, 1453–1460.
38 B. Long, K. Nikitin, D. Fitzmaurice, J. Am. Chem. Soc. 2003, 125, 5152–5160.
39 K. Kobayashi, N. Tonegawa, J. Hikida, S. Fujii, H. Nozoye, K. Tsutsui, Y. Wada, M. Chikira, M. Haga, Langmuir 2008, 24, 13203–13211.
40 K. Terada, K. Kanaizuka, V. M. Iyer, M. Sannodo, S. Saito, K. Kobayashi, M. Haga, Angew. Chem. Int. Ed. 2011, 50, 6287–6291. 41 H. Nishihara, Bull. Jpn. Soc. Coord. Chem. 2016, 67, 2–9. 42 芳賀正明,金井塚勝彦, 超分子金属錯体 三共出版 : 2009. 43 J. Liu, C. Woll, Chem. Soc. Rev. 2017, 46, 5730–5770. 44 L. Heinke, C. Woll, Adv. Mater. 2019, 31, e1806324.
45 V. Kaliginedi, H. Ozawa, A. Kuzume, S. Maharajan, I. V. Pobelov, N. H. Kwon, M. Mohos, P. Broekmann, K. M. Fromm, M. Haga, T. Wandlowski, Nanoscale 2015, 7, 17685–17692.
46 H. Atesci, V. Kaliginedi, J. A. Celis Gil, H. Ozawa, J. M. Thijssen, P. Broekmann, M. Haga, S. J. van der Molen, Nat Nanotechnol. 2018, 13, 117–121.
47 K. Terada, H. Nakamura, K. Kanaizuka, M. Haga, Y. Asai, T. Ishida, ACS Nano. 2012, 6, 1988–1999.
48 Y. Nishimori, K. Kanaizuka, T. Kurita, T. Nagatsu, Y. Segawa, F. Toshimitsu, S. Muratsugu, M. Utsumo, S. Kume, M. Murata, H. Nishihara, Chem. Asian J. 2009, 4, 1361–1367.
49 Z. Karipidou, B. Branchi, M. Sarpasan, N. Knorr, V. Rodin, P. Friederich, T. Neumann, V. Meded, S. Rosselli, G. Nelles, W. Wenzel, M. A. Rampi, F. von Wrochem, Adv. Mater. 2016, 28, 3473–3480.
![Fig. 1 Square scheme of electron/proton equilibria in [Ru(bpy) 2 (2- (2-(pyridyl)benzimidazole)]](https://thumb-ap.123doks.com/thumbv2/123deta/6762281.1161438/3.892.84.431.82.310/fig-square-scheme-electron-proton-equilibria-pyridyl-benzimidazole.webp)