早期乳児てんかん性脳症および Leigh 脳症様の 表現型を呈した DNM1L 関連脳症についての
iPS 細胞を用いた病態解析
ざ は きよ たか
座 波 清 誉
(成長発達臨床医学専攻)
防衛医科大学校
平成 30 年度
目 次
第 1 章 緒言 1 頁
第 2 章 DNM1L 遺伝子変異をもつ Leigh 脳症患者の同定と
その臨床的特徴 3 頁
第 3 章 DNM1L 遺伝子変異をもつ Leigh 脳症患者の病理学的特徴 6 頁 第 4 章 DNM1L 変異患者由来細胞のミトコンドリア形態・動態異常 8 頁
第 5 章 DNM1L 変異患者由来細胞における呼吸鎖酵素活性
および酸素消費速度測定 11 頁
第 6 章 DNM1L 変異患者由来の iPS 細胞を用いた
神経細胞、筋細胞への分化誘導 13 頁
第 7 章 考察 18 頁
第 8 章 結論 20 頁
謝辞 21 頁
単語・略語説明 22 頁
引用文献 24 頁
図表 27 頁
1
第1章 緒言
Leigh 脳症とは 1951 年に Leigh により報告された亜急性壊死性脳症であり、
2 歳以前に筋緊張低下、発達遅滞、けいれんなどで発症し、典型的には高乳酸血 症や高ピルビン酸血症を認める[1]。古典的には脳病理学的な診断であり、脳幹・
基底核・小脳を中心に両側対称性の海綿状変性、空胞変性、脱髄、グリオーシス などが認められる[2]。
近年では、画像診断技術の進歩により、脳画像検査にて診断される症例が多く なっている。頭部 MRI(Magnetic Resonance Imaging)画像検査の T2 および FLAIR(Fluid Attenuated Inversion Recovery)撮像にて、脳幹部、基底核に 左右対称性の高信号病変を呈することが特徴である。
原因遺伝子としては、ミトコンドリア電子伝達系酵素である complexⅠ~Ⅴに 関わる核遺伝子やミトコンドリア内に存在し、ピルビン酸をアセチル CoA に変 換するピルビン酸脱水素酵素複合体(Pyruvate Dehydrogenase Complex;
PDHC)の核遺伝子異常などが知られている[3]。
また、 Leigh 脳症の約 20-30%は、ミトコンドリア内に存在するミトコンドリ
ア遺伝子変異によることが知られている。 Mitochondrial DNA (mt DNA)は環
状 DNA で、 16569 塩基対からなり、イントロンはなく全てエクソンからなって
いる。mt DNA T8993G などの点変異が報告されており、母系遺伝の形式をと る[4]。有効な治療法はなく、発症後数年で死に至る予後不良な疾患である。
一方で、早期乳児てんかん性脳症、いわゆる大田原症候群(Early Infantile Epileptic Encephalopathy;EIEE)は、1976 年に大田原により報告された新生 児 ~ 早 期 乳 児 期 に 発 症 す る 予 後 不 良 の て ん か ん 性 脳 症 で あ り 、 脳 波 で
Suppression-Burst (SB)を示すのが特徴である[5]。神経伝達物質放出の際、分
泌小胞とシナプス前膜への融合に関与する STXBP1 (syntaxin-binding protein 1)遺伝子など、多くの原因遺伝子が報告されている[6]。
ミトコンドリアは ATP 産生に関わる重要な細胞内小器官であり、細胞の増殖 やアポトーシスにおいて、重要な役割を担っている。細胞周期や酸化ストレスに 対応し、細胞内で分裂、融合を繰り返していることが知られている。融合には mitofusin-1、 mitofusin-2 や Optic atrophy-1 (OPA1)が関わっており、分裂に は DNM1L(Dynamin 1 like)が関わっている[7]。
DNM1L はミトコンドリアの分裂に関わる GTP ase であり、ミトコンドリア
数の増加に需要な役割を果たしている。また、損傷を受けたミトコンドリアを切
り離し、オートファジーとしての自己処理を行う過程で、アポトーシスへと導く
マイトファジーにも関わっている。すなわち、ミトコンドリアの質的維持に重要
である。
2
Leigh 脳症と EIEE の合併は非常に稀であるが、両方の表現型を呈した 6 か
月男児例を経験し、その原因解明のために、エクソーム解析を行った。
その結果、ミトコンドリア分裂に関わる重要な核遺伝子である DNM1L に変異 があることを見出した。
本研究の目的は、DNM1L 関連脳症の病態を明らかにすることである。
DNM1L 変異を持つ患者より採取した線維芽細胞から疾患 iPS 細胞を樹立し、
神経細胞や骨格筋細胞へ分化誘導し、正常 iPS 細胞と比較検討することで、病 態解明を進めることとした。
なお、本研究はヒト検体を用いるため、防衛医科大学校倫理委員会の承認(受
付番号 1275「先天性免疫不全症の遺伝子解析研究同意患者を対象にしたヒト
iPS 細胞を用いた病態解明」 )(受付番号 1204「疾患特異的 iPS 細胞を用いた創 薬・疾患研究」)を得て iPS 細胞樹立、及び機能解析を実施した。
検体採取に際しては、対象者の保護者に研究内容を文書と口頭により説明し、署
名同意を得た。また、共同研究施設である京都大学 iPS 細胞研究所(CiRA)と
の共同研究契約「ヒト患者 iPS 細胞を用いた先天性免疫不全症の病態解明」に
基づき、疾患 iPS 細胞を CiRA より供与された。
3
第2章 DNM1L 遺伝子変異をもつ Leigh 脳症患者の同定とその臨床的特徴
第1節 背景
当初、脳波所見と特徴的なてんかん発作である spasms から EIEE と診断され た 6 か月男児を経験した。 EIEE の原因遺伝子について解析を進めたところ、 EIEE の既知遺伝子変異は認められなかったため、更なる原因遺伝子の解明のために、
エクソーム解析を行うこととした。
第2節 対象および方法
(1)対象
末梢血より抽出した患児およびその両親の genomic DNA(以下、gDNA)を解 析した。てんかん性脳症の原因遺伝子解析に関する同意を取得し、当初は EIEE の既知遺伝子についての解析を進め、既知遺伝子変異が同定できない場合は、エ クソーム解析に進む方法を取った。
(2)エクソーム解析
Sure Select Human All Exon v4 (Agilent Technologies、Santa Clara 、CA)
を使用して gDNA からエクソン領域を捕捉し、101bp の対端読み取りを有する Illumina HiSeq2000(Illumina、San Diego、CA)で配列決定した。全エクソン 配列決定(WES: Whole Exsome Sequencing)のデータを処理し、変異型の呼び出 し方法および変異型注釈は既報の方法に従って実施した[8]。 dbSNP135 データベ ースあるいは in-house の 575 コントロールデータベースに 1%以上、見出され た変異体は除外した。
(3)サンガーシークエンス
DNM1L 変異の検証は患児および両親の gDNA を用いてサンガーシークエンスに
て行われた。遺伝子のエクソン、およびエクソン・イントロン境界域を含む部分 を Polymerase Chain Reaction(以下 PCR)法にて増幅した。PCR 反応物をシー クエンス反応液として調整し、Dye Terminator 法により増幅反応を行った。シ ークエンス反応物を精製した後に、 Genetic Analyzer で遺伝子配列を決定した。
第3節 結果
(1)遺伝子解析結果
高いカバレッジで WES が患者に対して実施された。RefSeq 遺伝子における タンパク質コード領域の平均リード数は 227 であり、標的コード配列の 94.5%
が 20 以上のリード数が得られた。Single Nucleotide Polymorphism (以下
4
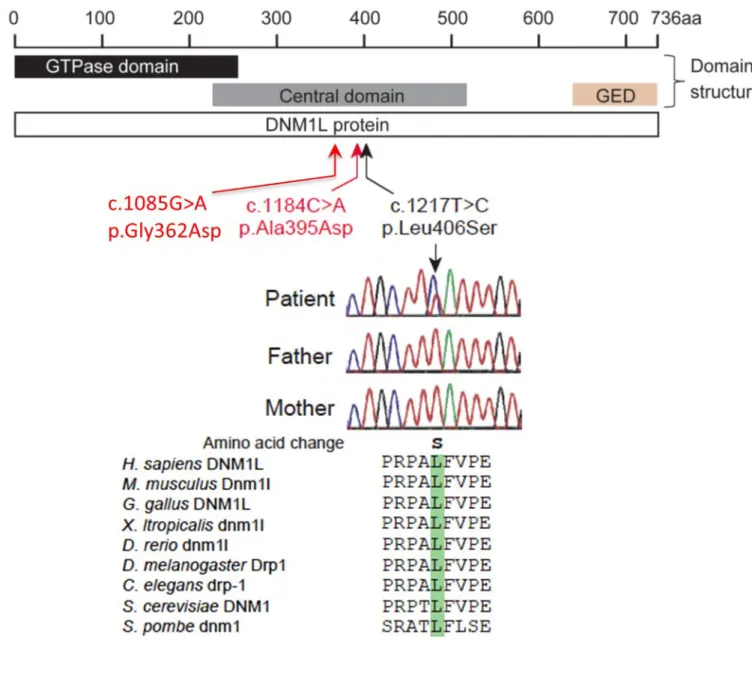
SNP)を除いた 223 の候補遺伝子の中で、DNM1L(NM_012062.4:c.1217T> C、
p.Leu406Ser)のミスセンス変異に注目した。患者とその両親の gDNA を用いた
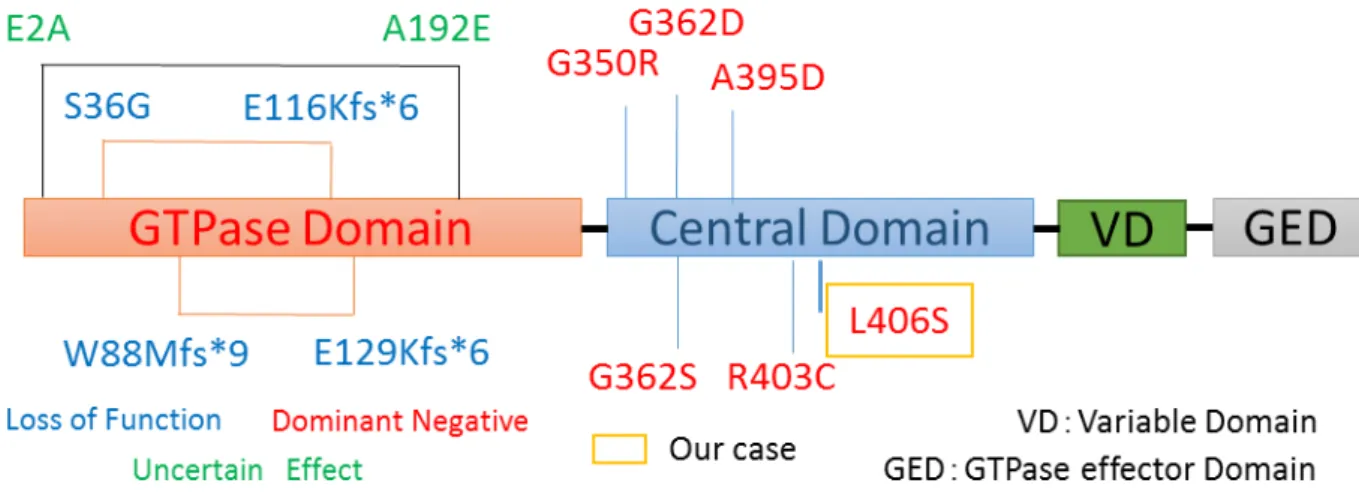
サンガーシークエンスでは、患者の DNM1L 変異が両親に見つからなかったこと が明らかになり、変異が de novo であることが示された(図 1) 。この突然変異 は、575 の in-house コントロールやその他のエクソームデーターベースに登録 されていなかった。また p.Leu406Ser は、真核生物の中で高度に保存されたア ミノ酸残基を置換し、SIFT(http://sift.jcvi.org/、スコア 0.00)、PolyPhen2 (http://genetics.bwh.harvard.edu/pph2 /、スコア 0.976)および Mutation Taster(http://www.mutationtaster.org/、スコア 0.999)などのコンピュー タプログラムによる変異予測では、damaging と予測された。p.Leu406Ser およ び以前に報告された p.Ala395Asp および p.Gly362Asp の突然変異は、DNM1L の Central Domain に集中していた(図 1) 。
(2)臨床的特徴
我々の症例は、出生時より低身長、低体重を認め、 1 か月時にも哺乳不良、体 重増加不良を認めたが、血液検査、脳 MRI では異常を認めなかった。生後 6 か月
時に spasms が出現し、発育不良および著明な筋緊張低下を認め、固視・追視も
認めなかった。血液検査では明らかな異常はなく、髄液検査にて軽度の乳酸、ピ ルビン酸の上昇を認めた。
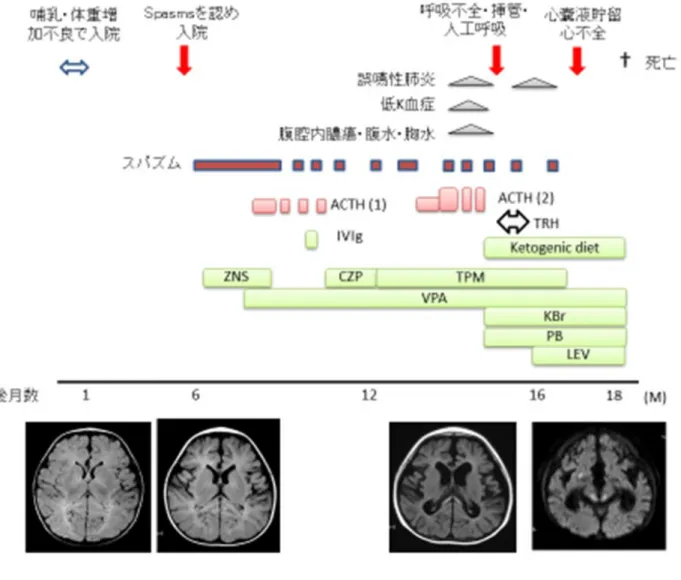
頭部 MRI では前頭葉の軽度の萎縮と、脳梁の菲薄化を認めたが、 Leigh 症候群 に特徴的な基底核・脳幹の信号異常は認めなかった。脳波検査にて SB パターン を示したため、EIEE と診断し、種々の抗てんかん薬、ACTH, ケトン食などで治 療を行ったが、難治に経過した。頭部 MRI では進行性の大脳萎縮を認めたが、脳 幹部の信号異常は生後 17 か月まで認めなかった。1 歳を過ぎてから肺炎や尿路 感染症などが頻発し、その後呼吸不全、心不全も出現し、生後 18 か月で永眠さ れた。 (図 2)
第4節 考察
エクソーム解析の結果、 DNM1L 遺伝子の Central Domain に de novo の missense 変異(p.Leu406Ser)を認めた。現在までに DNM1L 遺伝子の Central Domain の変 異は本症例を含め 7 例の報告がある。 (図 3)いずれも Dominant Negative 効果 によるミトコンドリアやペルオキシソーム形態異常を来すことが示されている。
DNM1L 変異に伴うミトコンドリア形態・動態異常については、第 3 章にて報告す
る。本症例と過去に報告された DNM1L 遺伝子の Central Domain に missense 変 異を持つ 6 例との臨床経過を比較した表を示す。 (表 1)DNM1L 変異による EIEE
および Leigh 脳症と診断された症例はなく、本症例が初めての報告である。
5
近年報告された DNM1L 変異に伴う小児てんかん性脳症は、共に感染を契機と したてんかん重積状態を来し、筋緊張低下を示す点が共通している[9]。また、
頭部 MRI の所見では、進行性の大脳萎縮は共通しているが、基底核の高信号病 変については、出現部位や出現時期は異なっている[9]。症例 5,6 は共に感染を 契機にてんかん重積状態を来し、筋緊張低下や退行を認めている点は Leigh 脳 症の臨床経過と合致する。しかし、2 症例共にてんかんの発症が 4 歳、5 歳であ り、過去の報告にある乳児期と比べ、遅い点が異なる[9]。
DNM1L 変異に伴うてんかんの種類や脳波の特徴などは、報告が少なく、脳波に
おける SB が DNM1L 変異に特徴的であるとは言えない。しかし、呼吸鎖酵素Ⅳ欠
損による EIEE の患児で SB を認めた報告[10]があり、本症例と類似している。
また、UBA5 機能異常による早期乳児てんかんの症例[11]、バルビツール中毒や 頭部外傷による広範囲な大脳皮質、皮質下白質障害では SB を示している。
DNM1L 変異が感染やてんかん発作を契機に、脳に重篤なダメージを起こし得る
危険因子であると考えられる。 DNM1L 変異によりミトコンドリアの分裂異常が生 じるが、ATP 需要度が高まった際に、分裂してミトコンドリアの数を増加させ、
ATP 需要に対応する反応が障害されることで脳に広範なダメージを与えると予 想される。
第5節 小括
本症例は、 DNM1L 変異により、ミトコンドリアやペルオキシソームの形態異常 を来すことが予想された。DNM1L 変異に伴う小児てんかん性脳症を来しており、
我々の症例は脳波検査により、 SB を示し、 spasms を伴う EIEE の臨床像を呈し、
難治に経過した。 DNM1L 遺伝子の Central Domain の missense 変異の報告は本症 例を含めて、 7 例あるが、致死的な乳児発症型[12、13]から、比較的軽症な幼児 期型[9]まで幅広い表現型を呈する。感染症などを契機に退行を示し、神経学的 予後は悪い点は共通しており、潜在的なミトコンドリア機能異常が示唆された。
6
第 3 章 DNM1L 遺伝子変異をもつ Leigh 脳症患者の病理学的特徴
第1節 背景
DNM1L 遺伝子変異による病理学的な変化は、末梢神経のミエリン化不全や海馬
ニューロンにおける好酸球性細胞内封入体の報告がある[13]。しかし、脳を含む 全身の臓器の病理学的な解析は行われていない。今回、我々は患児における剖検 を行い、病理学的解析を行うことができた。
第2節 対象および方法
患児のご両親より同意を頂き、死因究明のために剖検を行った。全身臓器に加 え、脳についても病理解剖を行った。
第3節 結果
(1)全身臓器に関する病理解剖
心臓は、重量 105.5g と肥大し、左心室、右心室共に拡張が認められた。組織 学的には一部の心筋細胞に大小不動や空胞性変化が認められた。右心不全に伴 うと考えられる胸膜及び、心膜滲出液の貯留、肺、肝臓、腎臓および脾臓の鬱血 が認められた。
肝臓には小葉中心性壊死、脾臓でも壊死が確認され、左心不全による低酸素血 症を反映していると考えられた。先天性泌尿生殖器異常として、小陰茎、膀胱壁 微小嚢胞、尿路嚢胞、左重複尿管を認めた。脾臓、肺胞壁、心筋、腎間質、およ び肝静脈洞に好中球浸潤を認め、敗血症を示唆する所見であった。
(2)筋病理解剖
Hematoxylin-Eosin(HE) 染 色 , modified Gomori Trichrome (mGT ) 染 色 , Succinate dehydrogenase(SDH) 染 色 で 異 常 を 認 め ず 、 Cytochrome c Oxidase(COX)欠損線維は認めなかった。剖検時の骨格筋病理では、ミトコンドリ ア病を疑わせる明らかな異常所見は認めなかった。 (図 4)
(3)脳神経病理解剖
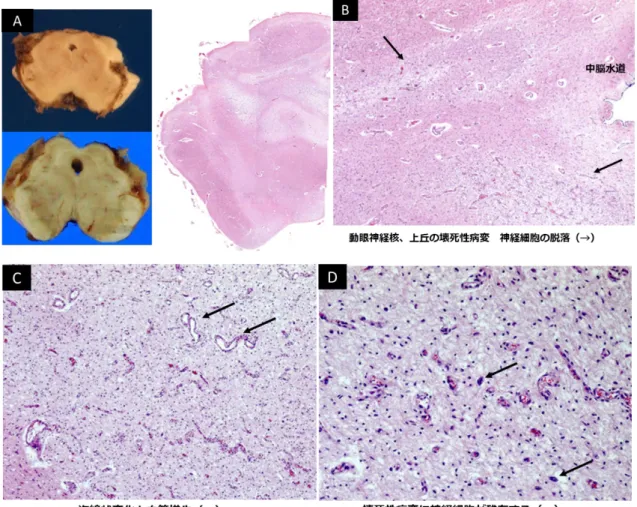
中脳の肉眼像、HE 弱拡像では、中脳水道周囲、動眼神経核、黒質の神経脱落 を認めた。強拡像では間質の粗造と血管の増生、一部の神経細胞の残存が認めら れ、Leigh 脳症に特徴的な亜急性壊死性脳症を呈していた。 (図 5)
第4節 考察
心筋炎の所見が二次的な変化であるのかどうかは不明だが、マウスにおける
7
DNM1L 変異による心筋炎の報告はあり、関連性が示唆される[14]。骨格筋病理に
ついては剖検時の筋組織であり、今までの報告同様、 COX 欠損症などに特異的な 筋病理変化は認めなかった[15]。脳神経病理では、亜急性壊死性変化を認め、
Leigh 脳症に特徴的な所見であったが、DNM1L 遺伝子の Central Domain での変 異による脳神経変化は報告がなく、今後の症例の蓄積が必要である。
DNM1L 遺伝子の GTPase Domain での機能喪失型変異を認めた症例[13,16]は、
2 例あり、うち 1 例で剖検が行われた[13]。末梢神経や脊髄神経における脱ミエ リン化、海馬ニューロンへの好酸球性細胞内封入体を認めており、病理学的な変 化としては、本症例と一致しなかった。 DNM1L 遺伝子変異の領域や変異による効 果も異なっており、本症例とは異なる病態であると考えられた。
第5節 小括
DNM1L 変異に伴う脳病理学的な所見として、亜急性壊死性脳症を認めた。
DNM1L 遺伝子の Central Domain に変異を持つ患者において、脳病理学的な解析 が行われたのは本症例だけであり、今後の症例の集積が必要である。骨格筋では 特異的な所見は認めなかったものの、心筋炎の所見は認め、ミトコンドリア病に おける心筋症の報告があることから、関連性が示唆された。
8
第 4 章 DNM1L 変異患者由来細胞のミトコンドリア形態・動態異常
第1節 背景
DNN1L 変異によるミトコンドリアの形態異常は、ヒトの細胞では 2007 年に
Hans R.Waterham らにより初めて報告された[12]。
DNM1L はミトコンドリアの分裂に関わる GTPase である。ヒトにおけるミトコ
ンドリア分裂機序の詳細は解明されていないが、酵母菌を用いた解析では、ミト コンドリア周囲に重合体を形成し、 GTP の加水分解により、重合体の収縮が起こ り、ミトコンドリアが切断されることが報告されている[17]。
ATP 需要の増大など細胞内環境に応じて、ミトコンドリアの移動、分裂、融合 が制御されている[16、 18]。 DNM1L は、エネルギー産生が必要な状況や酸化スト レス下でのミトコンドリア数の増加に重要な役割を果たしている。また、DNM1L は損傷を受けたミトコンドリアを切り離し、オートファジーとしての自己処理 を行うマイトファジーの過程において、重要な役割を担っていると考えられ、ミ トコンドリアの質的維持に欠かせないと考えられている[19]。
第2節 対象および方法
(1)線維芽細胞でのミトコンドリア、ペルオキシソーム形態
患者より皮膚生検を行い樹立した線維芽細胞を用いた。患者由来の線維芽細 胞を培養し、免疫染色を行った。4%パラホルムアルデヒドで固定し、次いで 2%スキムミルク中で非特異的反応をブロックした。
ペルオキシソームは、一次抗体として、抗 Pmp70、ペルオキシソーム膜マー カータンパク質である(SAB4200181; Sigma-Aldrich Co.,St. Louis,MO)を使 用し、二次抗体として Alexa Fluor 568 標識ヤギ抗ウサギ IgG 抗体(A11019;
Life Technologies,Carlsbad,CA)を用いて蛍光標識した。
ミトコンドリアは、培養液で 50nM に調製した MitoTracker Orange CMTMRos
(Invitrogen,Waltham,MA)と線維芽細胞を 37℃で 30 分間インキュベートし、
標識した。観察は蛍光顕微鏡を用いて行った。
(2)正常 DNM1L 遺伝子の導入実験
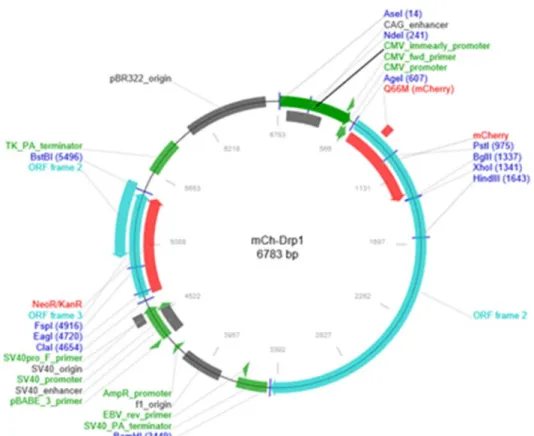
患者由来線維芽細胞に正常 DNM1L を含むプラスミド mCh-Drp1(plasmid#
49152; Addgene 、Cambridge、MA)を用いて、遺伝子導入実験を行った。(図 6) 遺伝子導入方法は Lipofectamine®2000(Thermo Fisher Scientific, Waltham, MA, USA)を用いたリポフェクション法を用いた。
遺伝子導入が成功した線維芽細胞は m Cherry を発現するため、蛍光顕微鏡下で
同定ことが可能となる。続いて、その線維芽細胞のミトコンドリア形状を確認し
9
た。ミトコンドリアの染色については、培養液で 200nM に調製した Mito-Tracker Green FM(Cell Signaling TECHNOLOGY, Danvers, MA)と線維芽細胞を 37℃で 30 分間インキュベートし、標識した。観察は蛍光顕微鏡(キーエンス、大阪)
を用いて行った。
(3)線維芽細胞におけるミトコンドリア動態
正常コントロール並びに患者由来の線維芽細胞を培養し、ミトコンドリアを 染色して観察した。染色については、200nM に調製した Mito-Tracker Green FM
(Cell Signaling TECHNOLOGY, Danvers, MA)と線維芽細胞を 37℃で 30 分間イ ンキュベートし、標識した。観察は蛍光顕微鏡(キーエンス、大阪)を用いて行 った。観察方法としては、タイムラプス撮影(1 枚/5 秒、2 分)を行い、2 分間 でのミトコンドリアの動態変化を確認した。正常コントロール、患者由来の線維 芽細胞内のミトコンドリアを任意で 10 か所選定し、各移動速度を測定した。各 速度を用いて、統計学的解析を行った。
第3節 結果
(1)線維芽細胞でのミトコンドリア形態
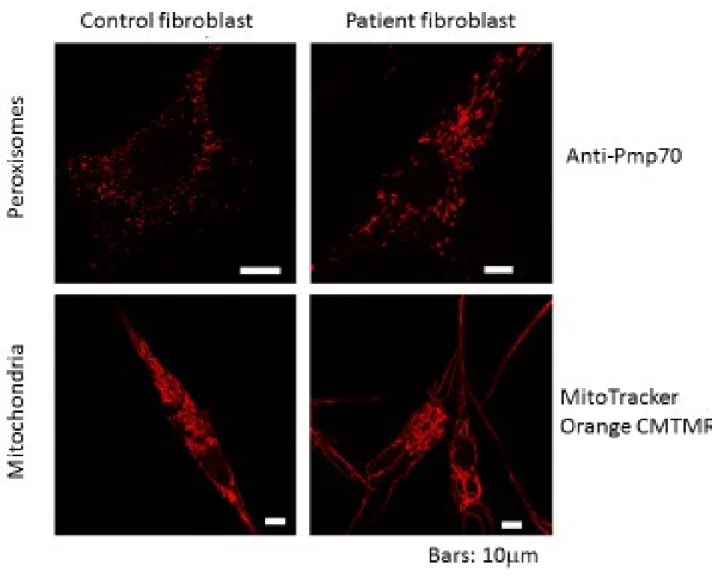
患者の線維芽細胞におけるオルガネラの免疫染色により、ペルオキシソーム およびミトコンドリアの分裂に欠陥があることが判明した。 患者のペルオキシ ソームは著しく変化し、線状構造を呈した。また、ミトコンドリアは細長く伸長 し、巨大な管状構造を示した。 (図 7)
(2)正常 DNM1L 遺伝子の導入実験
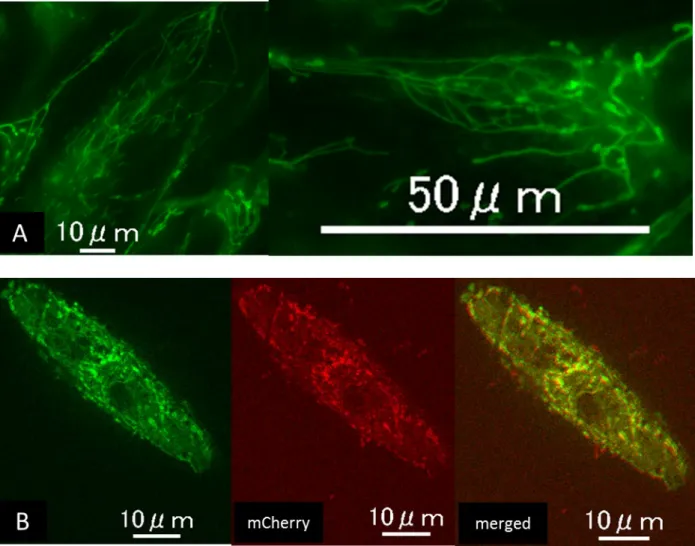
患者由来の線維芽細胞に正常 DNM1L を transfection させ、 overexpression さ せた。Transfection により、m Cherry の発光が確認できた線維芽細胞を確認し たところ、ミトコンドリアの形状が正常化し、顆粒状の構造を認めた。 (図 8)
(3)線維芽細胞におけるミトコンドリア動態
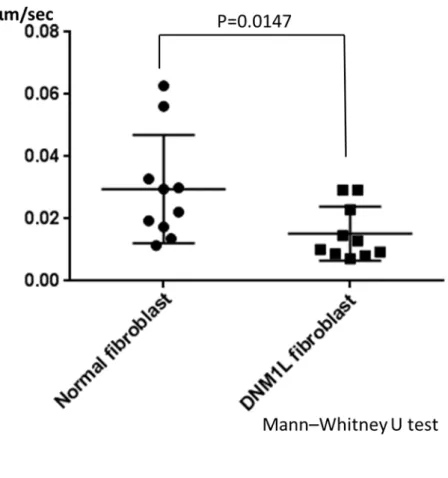
正常コントロール細胞でのミトコンドリア平均移動速度は 0.03μm/s であ った。患者由来の細胞でのミトコンドリア平均移動速度は 0.015μm/s であり、
半減していた。 (図 9、 10)統計学的解析の結果、 Mann–Whitney U test で両群に 有意差を認めた。 (P=0.0147)
第4節 考察
患者由来線維芽細胞にはミトコンドリアおよびペルオキシソームの形態異常
があり、患者由来線維芽細胞に正常の DNM1L を強制発現させるとミトコンドリ
10
アの形態異常が正常化した。
患者における DNM1L 遺伝子 c.1217T>C、p.Leu406Ser 変異は、過去に報告され た c.1184C>A、p.Ala395Asp および c.1085G> A、p.Gly362Asp などと同様に、
Dominant Negative 効果によるミトコンドリア分裂障害をきたしていると考え
られた。
DNM1L 変異を持つ線維芽細胞におけるミトコンドリア移動速度は、正常コント
ロールに比較し、半減していた。ミトコンドリアの分裂障害における動態低下を 反映していると考えられた。
第5節 小括
DNM1L 変異(c.1217T>C、 p.Leu406Ser)により、ミトコンドリアとペルオキシ ソームの形態が伸長、管状構造を示した。正常 DNM1L を発現させると形態は正 常化しており、 DNM1L 変異によるミトコンドリア分裂異常により、伸長、管状構 造を呈することが示された。また、 DNM1L 変異によりミトコンドリア動態が障害 され、ミトコンドリアの移動速度も低下していた。
11
第 5 章 DNM1L 変異患者由来細胞における呼吸鎖酵素活性および
酸素消費速度測定
第1節 背景
Leigh 脳症の患者由来の線維芽細胞や筋組織におけるミトコンドリア呼吸
鎖酵素複合体(以下、MRC:Mitochondrial Respiratory Chain Complex)活性 は、正常コントロールと比較し低下していることが知られている。特に電子伝 達系に関わる酵素 complexⅠ~Ⅴに関しては、遺伝子変異により各 complex 活 性が低下することが報告されている[20、 21]。しかし、MRC 活性の低下を認め ないミトコンドリア病も存在しており、病態解明に難渋することも多い。
近年、細胞を培養した状態で経時的にエネルギー代謝経路である解糖系やミ トコンドリアによる好気呼吸の状態を解析できる細胞外フラックスアナライ ザーを用いた研究が進んでいる[22]。実際、 Leigh 脳症においては MRC 活性が 低下していないものの、酸素消費速度(以下、 OCR:Oxygen Consumption Rate)
が低下しており、潜在的なミトコンドリア機能低下が示唆されている[23]。
我々の症例でも、ミトコンドリア機能を解析するために、線維芽細胞と筋組 織を用いた MRC 測定と培養線維芽細胞を用いた OCR 測定を行った。
第 2 節 対象および方法
(1)呼吸鎖酵素活性測定
線維芽細胞、及び筋組織をホモジナイズ処理し、電子伝達系酵素である
complexⅠ~Ⅴについて、complex 毎に測定した。培養皮膚線維芽細胞および
剖検骨格筋における MRC の活性を、既報の吸光度計を用いた方法により測定 した[20]。各酵素活性は、クエン酸合成酵素(CS)および複合体 II 活性に対 する相対比として評価した。線維芽細胞および剖検骨格筋の MRC 活性を測定 し、20%の CS 比で MRC 活性の有意な低下と判定した。
(2)酸素消費速度測定
酸素消費速度(OCR:Oxygen Consumption Rate)については、細胞外フラッ クスアナライザー(Agilent Technologies, Santa Clara, CA)を用いて解析 した。これにより、ミトコンドリアによる好気呼吸の状態を、細胞に対して無 侵襲・高感度に経時的計測が可能である。細胞培養環境内に半閉鎖的微小環境 を作ることで、迅速かつ高感度に計測が可能になっている。培養線維芽細胞を 用いて、ミトコンドリアの酸素濃度(pmol/min) ・水素イオン濃度(mpH/min)
の変化をルミネッセンス法により検知し、OCR を算出した。
12
第3節 結果
(1)呼吸鎖酵素活性測定
皮膚線維芽細胞では MRC 活性は正常であった。剖検筋組織では複合体Ⅰ、Ⅲ、
Ⅳ活性が低下していた。 (表 2)
(2)酸素消費速度測定
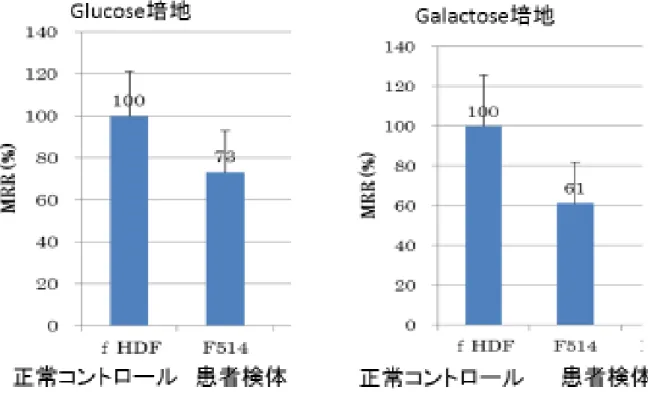
皮膚線維芽細胞を用いた解析では、 Glucose 培地において、正常コントロール と比較したところ、患者由来線維芽細胞では 27%の OCR 低下を示した。また Galactose 培地においては、39%の OCR 低下を認めた。 (図 11)
第4節 考察
線維芽細胞での MRC 活性は正常であり、筋組織での MRC 活性は低下していた が、剖検時の筋組織由来であり、正確な評価は困難であった。 DNM1L 変異による 病態はミトコンドリア呼吸鎖酵素の異常ではなく、ミトコンドリアの分裂障害 であり、MRC 活性に異常は認めないと考えられる。
DNM1L 変異の他症例では、MRC 活性は線維芽細胞、筋組織ともに正常との報告
[12、18]が多く、やはりミトコンドリアの電子伝達系における ATP 産生能異常
ではなく、分裂障害によるミトコンドリア数の低下による ATP 欠乏をきたして いると考えられる。
線維芽細胞における OCR 測定では、Glucose 培地、Galactose 培地両方におけ る解析で OCR の低下を認め、 MRC だけではわからなかったミトコンドリア機能低 下を示唆する所見であった。他の変異を持つ線維芽細胞を用いて、 OCR を測定し 再現性があるかどうか確認する必要がある。
第 5 節 小括
DNM1L 変異患者においては、電子伝達系酵素である complexⅠ~Ⅴそのものに
変異はなく、従来の MRC 活性測定では明らかな異常は確認できなかった。しか し、生細胞である線維芽細胞の OCR では低下を認めた。1例だけでの結果では あるが、DNM1L 変異によるミトコンドリア機能異常を評価するためには、OCR 測 定が有用であると考えられた。
13
第 6 章 DNM1L 変異患者由来の iPS 細胞を用いた 神経細胞、筋細胞への分化誘導
第1節 背景
DNM1L 変異によるてんかん性脳症や筋緊張低下の機序は不明である。本症例
におけるてんかん性脳症、および筋緊張低下は DNM1L 変異による神経細胞や筋 細胞における、ミトコンドリア機能低下に伴う ATP 欠乏が原因ではないかと推 察した。そこで、患者由来の iPS 細胞を樹立し、神経細胞、筋細胞に分化誘導 させ、in vitro で病態を再現することを試みた。
第2節 対象および方法
(1)iPS 細胞の樹立
第 3 章にて得られた患者由来の線維芽細胞から iPS 細胞を樹立した。共同研 究を行っている京都大学 iPS 細胞研究所(CiRA)にて樹立した DNM1L 変異を持 つ iPS 細胞を用いた。コントロールとしての正常 iPS 細胞は(Phenocell SAS, evry cedex, Paris, FRANCE)を使用した。
(2)iPS 細胞の培養、継代
初回の培養液としては、維持培養培地である Stem Fit AK02N(味の素、東 京)に約 1/1000 量の Y-27632(10mM)を加えたものを使用した。CiRA のプロ トコールに従い、Laminin-511 E8(i-Marix)でプレコーティングした 6well に iPS 細胞 13000 個/well で播種した。Stem-Fit+Y 培地は 1.5ml/well とし た。37℃、CO
2インキュベータ-で培養した。翌日からは Y-27632 の入っていな い維持培養培地に交換した。70-90%コンフルエントとなる 7 日前後まで培養 を行った。正常コントロール群と DNM1L 変異群で、検鏡(明視野)及び Day4、
Day7 での細胞数を測定し、増殖速度を比較検討した。
(3)iPS 細胞のドーパミン産生神経細胞への分化誘導
Sendai Virus(SeV)を用いて、iPS 細胞へ分化遺伝子を導入した。
SeV の F 遺伝子をゲノムから欠失させ、非伝播型に改変したベクターを用い た。SeV18/TSΔF ベクターはセンダイウイルス Z 株に由来する動物細胞用発現 ベクターで、HN 遺伝子、M 遺伝子に温度感受性変異を導入し、且つ P 遺伝子、
L 遺伝子にも持続感染型センダイウイルスベクター由来の変異を導入してい る。外来遺伝子は、SeV/TSΔF ベクターの 3´端に導入している。 (図 12)
外来遺伝子として、Neurogenic transcription factor(神経細胞分化遺伝
子)を挿入している。具体的なプロトコールは Quick-Neuron
TMDopaminergic
14
SeV Complete Kit(Elixirgen Scientific,Baltimore,MD)に従って行った。
(図 13)
(4)iPS 細胞の骨格筋細胞への分化誘導
Sendai Virus(SeV)を用いて、iPS 細胞へ分化遺伝子を導入した。
SeV の F 遺伝子をゲノムから欠失させ、非伝播型に改変したベクターを用い た。SeV18/TSΔF ベクターはセンダイウイルス Z 株に由来する動物細胞用発現 ベクターで、HN 遺伝子、M 遺伝子に温度感受性変異を導入し、且つ P 遺伝子、
L 遺伝子にも持続感染型センダイウイルスベクター由来の変異を導入してい る。外来遺伝子は、SeV/TSΔF ベクターの 3´端に導入している。 (図 12)
外来遺伝子として、Myogenic transcription factor(筋細胞分化遺伝子)
を挿入している。具体的なプロトコールは Quick-Muscle
TMSkeletal SeV Complete Kit(Elixirgen Scientific,Baltimore,MD)に従って行った。
(図 14)
(5)分化細胞の免疫染色
(ⅰ)ミトコンドリア
ミトコンドリアの染色として、ドーパミン産生神経細胞では、Day12、骨格 筋細胞は Day6 に 200nM の Mito-Tracker Green FM(Cell Signaling
TECHNOLOGY, Danvers, MA)と 37℃で 30 分間、共培養する。観察は染色蛍光顕 微鏡システム(キーエンス、大阪)で行った。
(ⅱ)ドーパミン産生神経細胞
ド-パミン産生神経細胞では Day13 の時点で、1 次抗体反応として、
Microtubule Associated Protein 2(MAP2) Antibody(Thermo Fisher Scientific, Waltham, MA) 、Tyrosine Hydroxylase(TH)Antibody(Gene Tex, Irvine, CA)を用いた。
二次抗体反応は Alexa Fluor 555 goat anti-mouse(Invitrogen , Waltham, MA)Alexa Fluor 488 goat anti-rabbit(Invitrogen , Waltham, MA)を用い た。観察は染色蛍光顕微鏡システム(キーエンス、大阪)で行った。
(ⅲ)骨格筋細胞
骨格筋細胞は、Day7 の時点で、1 次抗体反応では、Myosin Heavy Chain
(MHC)Anibody(R&D systems, Minneapolis, Miami, FL)を用いて、二次抗
体反応は Alexa Fluor 555 goat anti-mouse(Invitrogen, Waltham, MA)
15
Alexa Fluor 488 goat anti-rabbit(Invitrogen, Waltham, MA)を用いる。観 察は染色蛍光顕微鏡システム(キーエンス、大阪)で行った。
分化効率の測定のため、Day7 の時点で免疫染色を行い、DAPI 陽性、MHC 陽性 の紡錘型細胞を骨格筋分化細胞と定義した。骨格筋細胞数を 24well 中の 1well すべてを観察し、細胞数を測定した。正常群と DNM1L 変異群で細胞数を測定し 播種細胞数で除して、2 群間の分化効率を比較した。
第3節 結果
(1) (2)iPS 細胞の培養、継代
疾患 iPS 細胞では、正常 control に比べ、増殖速度は約 1/2 であった。正常 iPS 細胞は day2-3 頃からコロニーを形成するのに対し、DNM1L 変異 iPS 細胞は day6-7 からコロニーを形成し始める。 (図 15)
コンフルエントに達するのに正常 iPS 細胞が 7 日程度であるのに対し、DNM1L 変異を持つ iPS 細胞では、14 日程度を要した。
培養細胞数の推移は正常 iPS 細胞に比較し、 DNM1L 変異 iPS 細胞では、増殖速 度が低下しているのが確認できた。 (図 16)
(3)iPS 細胞のドーパミン産生神経細胞への分化誘導
軸索の伸長は、正常コントロールより遅延していた。正常 iPS 細胞由来のド ーパミン産生神経細胞は day5 以降に軸索の伸長が確認できたが、 DNM1L 変異 iPS 細胞由来のドーパミン産生神経細胞では、 day7 以降に軸索の伸長が認められた。
(図 17)
(4)iPS 細胞の骨格筋細胞への分化誘導
骨格筋細胞への分化誘導実験では、分化速度に大きな差は認められなかっ た。また、筋細胞の大きさや分布についても差は見られなかった。
(5)分化細胞の免疫染色 (ⅰ)ミトコンドリア
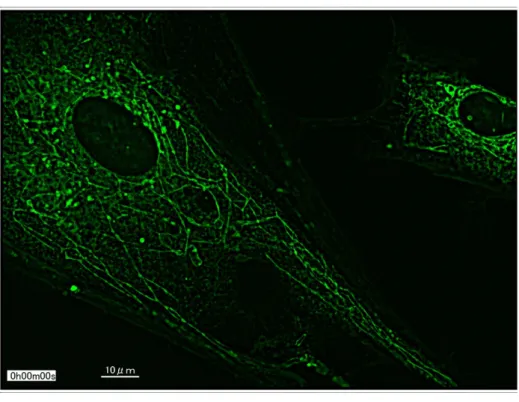
各分化細胞で、ミトコンドリアの形状を蛍光顕微鏡で観察した。正常 iPS 由 来細胞が顆粒状なのに対し、 DNM1L 変異 iPS 細胞由来の分化細胞では一部、線状 に延長した像が確認できた。 (図 18)
(ⅱ)ドーパミン産生神経細胞
正常コントロールと比較して、 DNM1L 変異 iPS 細胞由来のドーパミン産生神経
細胞において、神経細胞間のシナプス形成が乏しく、神経細胞の軸索伸長も短か
16
った。DNM1L 変異由来の神経細胞では、細胞質における MAP2 発現が低下し、一 部で細胞質より核内の DA 発現量が高かった。(図 19、20)
(ⅲ)骨格筋細胞
正常 iPS 細胞由来骨格筋細胞と DNM1L 変異 iPS 細胞由来骨格筋細胞では、
大きな差異は認めなかった。 (図 21)正常コントロール iPS 細胞の分化効率が 約 0.5%であるのに対し、 DNM1L 変異 iPS 細胞では、 約 0.06%と低下を認めた。
(図 22)
第4節 考察
iPS 細胞自体の増殖にも ATP は必要であり、DNM1L 変異を持つ iPS 細胞の増殖 速度が低下しているのは、ATP が欠乏していることが原因だと考えられた。
DNM1L 変異由来の神経細胞では、軸索の伸長速度は正常コントロールに比較し
て低下していた。マウスの海馬細胞における樹状突起、シナプス形成に DNM1L が 重要な役割を果たしているとの報告[24]もあり、 DNM1L 変異による ATP 欠乏状態 から、必要な ATP が十分に供給できずに、軸索伸長障害やシナプス形成障害が 起こっていると考えられた。 DNM1L 遺伝子の central domain の missense 変異で の軸索形成不全についての報告はない。しかし、神経系の成長発達の遅れは報告
[12、 18]があり、重症度に差はあるものの中枢神経障害が進行し、てんかん性脳
症の病態に至ると考えられた。また DNM1L 変異由来の神経細胞では、細胞質に おける MAP2 発現が低下し、一部で細胞質より核内の DA 発現量が高かった。ド ーパミン産生神経細胞の発育障害が示唆された。神経細胞内でのドーパミンの 異常蓄積などははっきりしなかった。神経変性疾患であるパーキンソン病では、
酸化ドーパミンのミトコンドリアやリソソームへの蓄積が神経細胞の障害へつ ながっている[25]との報告もあり、今後は、ミトコンドリアやリソソーム内への 酸化ドーパミンの蓄積についても解析を進めていく予定である。
患者由来の iPS 細胞から分化した骨格筋細胞は、正常由来の骨格筋細胞と比 較して明らかな差異は認めなかった。しかし、分化効率は低下していた。骨格筋 におけるミトコンドリアは個体の運動に伴い、ミトコンドリア生合成を促進し、
細胞への ATP 供給を増加させていることが知られている。マウスモデルでは加
齢に伴い OPA1 や DNM1L などミトコンドリア分裂、融合に関わるタンパクの発現
が増加することが知られている[26]。また、骨格筋の維持には、ミトコンドリア
の機能維持、つまり酸化ストレスなどにより損傷を受けたミトコンドリアを切
り離し、ミトコンドリアの質を維持する「マイトファジー」が重要である。実際
に、マウスにおけるマイトファジーの観察において、運動により筋細胞内でマイ
17
トファジーが亢進し、マイトファジーが繰り返されることで筋組織の質が向上 するとの報告がある[27]。
DNM1L 変異により、マイトファジーが障害され、ミトコンドリアの質が維持で
きず、筋組織損傷が進行し、筋力低下につながっている可能性がある。今後は、
マイトファジー機能を評価するために、酸化ストレス下でのミトコンドリアの 動態や形態変化の観察、心筋細胞などを用いた病態解明を進めていく予定であ る。
第5節 小括
DNM1L 変異を持つ患者由来の iPS 細胞は正常コントロールに比べ、約 1/2 程度
の増殖速度の低下を認めた。
DNM1L 変異を持つ iPS 細胞由来のドーパミン産生神経細胞は、正常と比べてシナ
プス形成や軸索伸長が障害されていた。また骨格筋細胞へ分化誘導実験では、正 常コントロールでの分化効率が約 0.5%であるのに対し、DNM1L 変異を持つ iPS 細胞では約 0.06%と低下を認めた。
神経細胞における軸索延長には ATP が必要であるが、 DNM1L 変異に伴う ATP 欠
乏、ミトコンドリアの機能障害により、軸索の伸長やシナプス形成の障害をきた
していると考えられた。また、マイトファジー障害によるミトコンドリアの質的
不安定性から、筋細胞維持が障害され、筋力低下につながっていると考えられた。
18
第 7 章 考察
生後 6 か月時に、脳波所見での SB と特徴的なてんかん発作である spasms か ら EIEE と診断した男児を経験した。てんかんは難治に経過し、精神運動の発達 は著明な遅れを認め、 18 か月時に永眠された。剖検を行い、 Leigh 脳症に特徴的 な亜急性壊死性脳症の病理像を呈していた。てんかん性脳症を呈する点や感染 症により退行、てんかんの増悪を認める点は Leigh 脳症と類似していた。しか し、EIEE と Leigh 脳症の合併は稀であり、原因究明のためにエクソーム解析を 行った。その結果、ミトコンドリア分裂に関わる GTPase である DNM1L の central domain に de novo の missense 変異(c.1217T>C、p.Leu406Ser)を同定した。
既報と同様に患者の線維芽細胞ではミトコンドリア、ペルオキシソームの管 状、伸長といった形態異常を認めた。
現在まで、DNM1L 遺伝子の Central Domain に missense 変異を認める DNM1L 関連脳症は、本症例を含めて 7 例の報告があるが、発症時期や重症度には多様 性があった。しかし、感染症などを契機に退行を示し、てんかん性脳症など神経 学的予後が悪い点は共通しており、潜在的なミトコンドリア機能異常が示唆さ れた。
ミトコンドリアは ATP 必要時に分裂し、 数を増やし、 ATP 需要に対応している。
また、損傷を受けた ATP 産生の質が低下してしまったミトコンドリアを切り離 し、オートファジーとしての自己処理を行うマイトファジーを行っている。
DNM1L 変異により、ミトコンドリアの分裂が障害されることで、数の低下による
ATP 欠乏および質の低下による ATP 産生能低下がもたらされたと考えられる。
DNM1L 変異を持つ線維芽細胞で、巨大で伸長したミトコンドリア形態異常に加
え、動態変化であるミトコンドリア移動速度の低下を認めた。また、DNM1L 変 異を持つ線維芽細胞では、酸素消費速度も低下しており、ミトコンドリア機能異 常による ATP 産生障害が示唆された。
筋緊張低下や中枢神経障害の原因を究明するために、患者由来の線維芽細胞 より iPS 細胞を樹立し、ドーパミン産生神経細胞、骨格筋細胞への分化誘導実 験を行った。 iPS 細胞の増殖速度低下、ドーパミン産生神経細胞でのシナプス形 成障害や軸索伸長障害、骨格筋細胞への分化効率低下は、DNM1L 変異による ATP 産生障害が原因と考えられた。 DNM1L 変異による ATP 産生障害による神経細胞や 筋細胞の発達・機能障害が示唆された。
また、筋細胞維持には、ミトコンドリアの質的維持が必要だが、 DNM1L 変異に よるマイトファジー障害により、筋組織損傷が進行し、筋力低下を認めた可能性 がある。分化細胞のミトコンドリア形状は一部で、DNM1L 変異を反映する管状、
伸長といった形態異常も認めた。
19
ATP 産生障害を定量的に解析するためには、酸化ストレス下での各分化細胞に
おける ATP 動態を確認する必要がある。今後、 ATP 変化を代用できるプローブを
用いて、定量的な解析を進めていく予定である。また、マイトファジー機能を評
価するために、酸化ストレス下でのミトコンドリア動態を観察し、ミトコンドリ
アがリソソームと結合した際に、蛍光強度が増大する試薬キットを用いてマイ
トファジーの定量的な解析を進めていく予定である。
20
第 8 章 結論
臨床的に SB の脳波を示す EIEE および Leigh 脳症の病理像を呈した患児の原 因遺伝子が DNM1L であることを見出した。
患者由来の線維芽細胞を用いて DNM1L 変異に伴うミトコンドリア、ペルオキ シソームの分裂障害により、巨大で伸長した形態異常を認めた。ミトコンドリア の動態変化である移動速度の低下も認めた。
DNM1L 変異を持つ線維芽細胞では、呼吸鎖酵素活性は正常であったが、酸素消
費速度は、低下していた。ミトコンドリア分裂障害によるミトコンドリア機能異 常が示された。
DNM1L 変異を持つ iPS 細胞の増殖速度が低下し、ドーパミン産生神経細胞のシ
ナプス形成や軸索伸長は障害され、骨格筋細胞への分化効率が低下しており、
ATP 産生障害が原因と考えられた。
各分化細胞でのミトコンドリアにおいても、一部で巨大で伸長した形態異常
が確認できた。
21
謝辞
本稿を終えるにあたり、全般にわたりご指導を賜りました防衛医科大学校小 児科学講座 野々山恵章教授、松本浩講師に深甚なる謝意を表します。
患児の病態究明に向け多大なるご協力をいただいた共著者の皆様、 iPS 細胞樹立 および分与を頂いた京都大学 iPS 細胞研究所の齋藤潤准教授に深謝いたします。
最後に、幼くして亡くなった患児と病態解明のために様々なご協力を頂いた患
児のご家族に改めて深く感謝いたします。
22
単語・略語説明
iPS 細胞(induced pluripotent stem cell:人工多能性幹細胞)
体細胞に山中 4 因子(Oct3/4、Sox2、Klf4、c-Myc)を導入して樹立される ES 細胞様の多能性幹細胞。2006 年に山中伸弥教授の研究グループにより世界で初 めてマウス体細胞を用いて樹立された。現在では、ヒト体細胞由来の細胞を用い た研究が進んでおり、再生医療、遺伝性疾患の病態解明や新薬開発などに幅広く 応用されている。
SNP(Single Nucleotide Polymorphism)
ある生物種集団のゲノム塩基配列中に一塩基が変異した多様性が見られ、その 変異が集団内で 1%以上の頻度で見られる時、これを一塩基多型; SNP と呼ぶ。
ヒトゲノム上で最も多く存在する多型であり、平均 1000 塩基に 1 か所の頻度で 認められる。
リポフェクタミン法
真核生物に効率に拡散を導入する方法の 1 つで、リン脂質などから構成される 陽イオン性リポソームと核酸の複合体を細胞に取り込ませ、一過性の遺伝子発 現を起こす。
HE 染色
HE 染色は組織の形態を観察する目的で細胞核、細胞質を染色する方法 。 細胞及び組織構造の全体像を把握する為に行う。染色と標本の管理がよければ 永久保存ができるメリットがある。
mGT(modified Gomori Trichrome)染色
筋線維を青緑色に有髄神経は赤色に染まる染色法で、主に神経筋疾患の診断に 用いられる。ミトコンドリアは赤染し、ミトコンドリアミオパチーにおいては異 常に増加したミトコンドリアを表出することができる。増殖し、集積した繊維は 赤色ぼろ繊維(ragged-red fiber)と表現される。
SDH(succinate dehydrogenase)染色
ミトコンドリア酵素であるコハク酸脱水素酵素を染色することで、ミトコンド
リアのみを特異的に染出する。赤色ぼろ繊維(ragged-red fiber)をより明確に
染出することが可能である。
23
COX(cytochorome c oxidase)染色
ミトコンドリア電子伝達系の酵素異常を検出する際に重要な染色法で、ミトコ ンドリアが存在する場所が顆粒状に染出され、酵素欠損を認める筋組織では全 体的に低染色性を示す。全く酵素活性を認めない線維が散在することがあり、部 分欠損を示す。慢性進行性外眼筋麻痺症候群(Chronic Progressive External Ophthaldeficiency;CPEO)に特異的な所見であるが、その他のミトコンドリ ア病などでも認められることがある。
Y-27632
細胞内透過性を有する ATP 拮抗的に作用する強力な ROCK(Rho-associated coiled-coil forming kinase)特異的阻害剤で、 Ca
2+受容体アゴニストの強力な阻 害剤として働く。
幹細胞における生存率向上に働くことが知られており、ヒト ES 細胞やヒト iPS 細胞の細胞分散時の細胞死を抑制、凍結保存後の生存率向上する効果が知られ ている。
Sendai Virus
パラミクソウイルス科レスピロウイルス属のウイルス。 1 本鎖 RNA を持ち、マ ウスなどを宿主とする。ヒトへの病原性の報告もなく、培養細胞内での増殖も良 好であり、遺伝子導入ベクターとして広く利用されている。F 遺伝子を欠失さ せ、 N 遺伝子上流に外来遺伝子を挿入するタイプのベクターが良く用いられる。
マイトファジー
障害を受けたミトコンドリアを選択的にオートファジー分解する機構。
障害を受けて機能低下、機能不全に陥ったミトコンドリアにパーキンソン病の
原因因子である PINK1、 Parkin が局在化することが知られており、 DNM1L も
ミトコンドリアの切断時に関与していると考えられている。
24
引用文献
[1] Leigh D. Subacute necrotizing encephalopathy in an infant. J Neurol Neurosurg Psychiatry 1951;14:216-21
[2] Kretzschmar, H. A., DeArmond, S. J., Koch, T. K., Patel, M. S., Newth, C. J. L., Schmidt, K. A., Packman, S. Pyruvate dehydrogenase complex deficiency as a cause of subacute necrotizing encephalopathy (Leigh disease). Pediatrics 79: 370-373, 1987.
[3] Koopman WJ, Willems PH, Smeitink JA.Monogenic mitochondrial disorders.N Engl J Med. 2012 Mar 22;366(12):1132-41. doi:
10.1056/NEJMra1012478.
[4] S. Lebon, M. Chol, P. Benit, C. Mugnier, D. Chretien, I. Giurgea, et al.
Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency.J Med Genet, 40 (2003), pp. 896-899
[5] Ohtahara S,Ishida T,Oka E, Yamatogi Y, Inoue H, Karita S, Ohtsuka Y.
On the specific age dependent epileptic syndrome: the early-infantile epileptic encephalopathy with suppression-burst. No to Hattatsu 8: 270- 279, 1976.
[6] Saitsu H, Kato M, Okada I, Orii KE, Higuchi T, Hoshino H,et al. STXBP1 mutations in early infantile epileptic encephalopathy with suppression- burst patternEpilepsia. 2010 Dec;51(12):2397-405. doi: 10.1111/j.1528- 1167.2010.02728.x. Epub 2010 Sep 30.
[7] Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases.Hum Mol Genet. 2009 Oct 15;18(R2):R169-76. doi: 10.1093/hmg/ddp326. Review
[8] Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013;45:445-9.
[9] Fahrner JA, Liu R, Perry MS, Klein J, Chan DC. A novel de novo
dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am J Med Genet A 2016; 170: 2002–11.
[10] Molinari F.J Mitochondria and neonatal epileptic encephalopathies with suppression burst. Bioenerg Biomembr. 2010 Dec;42(6):467-71. doi:
10.1007/s10863-010-9323-6.
[11] Mignon-Ravix C, Milh M, Kaiser CS, Daniel J, Riccardi F, Cacciagli P,
Nagara M, Busa T, Liebau E, Villard L. Abnormal function of the UBA5
25
protein in a case of early developmental and epileptic encephalopathy with suppression ‐ burst.Hum Mutat. 2018 Apr 16. doi:
10.1002/humu.23534. [Epub ahead of print]
[12] Waterham HR, Koster J, van Roermund CW et al. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 2007: 356:1736–
1741.
[13] Yoon G, Malam Z, Paton T, Marshall CR, Hyatt E, Ivakine Z, et al.Lethal disorder of mitochondrial fission caused by mutations in DNM1L. J Pediatr 2016; 171: 313–16.e1–2.
[14] Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy.PLoSGenet.2010.24;6(6):e1001000.doi:10.1371/journa l.pgen.1001000.
[15] Hallmann K, Kudin AP, Zsurka G, Kornblum C, Reimann J, Stüve B et al. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy.Brain. 2016 Feb;139(Pt 2):338-45.
doi: 10.1093/brain/awv357. Epub 2015 Dec 17.
[16] Nasca A, Legati A, Baruffini E, Nolli C, Moroni I, Ardissone A, et al.
Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum Mutat 2016; 37: 898–903.
[17] Jonathan R. Friedman, Laura L. Lackner, Matthew West, Jared R.
DiBenedetto, Jodi Nunnari and Gia K.Voelt. ER Tubules Mark Sites of Mitochondrial Division.2011.Science 334 (6054), 358-362.
[18] Vanstone JR, Smith AM, McBride S, Naas T, Holcik M, Antoun G, et al.
DNM1L-related mitochondrial fission defect presenting as refractory epilepsy.Eur J Hum Genet. 2016 Jul;24(7):1084-8. doi:
10.1038/ejhg.2015.243. Epub 2015 Nov 25.
[19] Nunnari J, Suomalainen A. Mitochondria: in sickness and in health.
Cell. 2012 Mar 16;148(6):1145-59. doi: 10.1016/j.cell.2012.02.035.
[20] Kirby DM1, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR.Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder.Neurology. 1999 Apr 12;52(6):1255-64.
[21] Piekutowska-Abramczuk D, Assouline Z, Mataković L, Feichtinger RG,
Koňařiková E, Jurkiewicz E,et al. NDUFB8 Mutations Cause
Mitochondrial Complex I Deficiency in Individuals with Leigh-like
Encephalomyopathy. Am J Hum Genet. 2018 Mar 1;102(3):460-467.
26
doi: 10.1016/j.ajhg.2018.01.008. Epub 2018 Feb 8.
[22] Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA,et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria.PLoS One.
2011;6(7):e21746. doi: 10.1371/journal.pone.0021746. Epub 2011 Jul 25.
[23] Ogawa E, Shimura M, Fushimi T, Tajika M, Ichimoto K, Matsunaga A,et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: a study of 106 Japanese patients.J Inherit Metab Dis. 2017 Sep;40(5):685-693. doi: 10.1007/s10545-017-0042-6.
Epub 2017 Apr 20.
[24] Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004; 119: 873–87.
[25] Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease.Science. 2017 .
[26] Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence.Cell Metab. 2017 Jun 6;25(6):1374-1389.e6. doi:
10.1016/j.cmet.2017.04.021. Epub 2017 May 25.
[27] Rhianna C. Laker, Joshua C. Drake, Rebecca J. Wilson, Vitor A. Lira, Bevan M. Lewellen,Karen A. Ryall et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exerciseinduced mitophagy. Nat Commun. 2017 Sep 15;8(1):548. doi:
10.1038/s41467-017-00520-9.
27
表 1 DNM1L 遺伝子 の Central D omain に変異を持つ患者の臨床像
28
表
2
皮膚線維芽細胞・剖検筋組織におけるミトコンドリア呼吸鎖複合体酵素活性CS
:クエン酸合成酵素 酵素活性低下基準(線維芽細胞:C S r at io
、CO
Ⅱr ati o 40
%以下、筋組織:C S r ati o
、CO
Ⅱr at io 30
%以下)29
図 1 DNM1L 遺伝子解析
30
図 2 臨床および治療経過
31
図 3 現在までに報告されている DNM1L 変異
Bars:100μm 図 4 剖検筋病理画像(上腕二頭筋)
A:HE 染色 B:mGT 染色 C:SDH 染色 D:COX 染色
32
図 5 脳神経病理標本 HE 染色
(A:肉眼像、B:×60、C:×150、D×150)
33
図 6 mCh-DRP1 プラスミドの構造
mCh-Drp1(plasmid#49152; Addgene , Cambridge, MA)
34
図 7 ペルオキシソームとミトコンドリアの免疫染色
35
図 8 ミトコンドリアの Mito GREEN FM による染色
A:DNM1L 変異患者のミトコンドリア
B: 正常 DNM1L 遺伝子導入後のミトコンドリア
36
A
B
図 9 Mito GREEN FM によるミトコンドリア染色
A:DNM1L 変異患者のミトコンドリア B: 正常コントロールのミトコンドリア
37
図 10 ミトコンドリアの移動速度の比較 試料:線維芽細胞
38
図 11 細胞外フラックスアナライザーによる酸素消費速度の測定 試料:皮膚線維芽細胞
図 12 SeV ベクター構造
39
図 13 Quick-NeuronTM Dopaminergic SeV Complete Kit プロトコール
図 14 Quick-MuscleTM Skeletal SeV Complete Kit プロトコール
40
図 15 iPS 細胞の増殖(左:正常 iPS 細胞、中央、右:DNM1L 変異 iPS 細胞)
図 16 iPS 細胞の増殖速度の比較
41
正常 iPS 細胞
DNM1L 変異 iPS 細胞
図 17 ドーパミン産生神経細胞への分化過程 (Day4 ×10、 Day7,10,12 ×40)
42
ドーパミン産生神経細胞
骨格筋細胞
図 18 ミトコンドリア染色( Mito-Tracker Green FM )油浸レンズ× 100
正常 iPS 細胞 DNM1L 変異 iPS 細胞
正常
iPS
細胞DNM1L
変異iPS
細胞43
正常
iPS
細胞由来(D AP I
、MA P2
、D A
)D N M 1L
変異iPS
細胞由来(D A PI
、M AP 2
、D A
) 図19
ドーパミン産生神経細胞の免疫染色(×40
)B ars :50
μm
44
D N M 1L
変異iPS
細胞由来(D A PI
、M AP 2
、TH
)正常