PTEN 遺伝子変異による原発性免疫不全症 Activated PI3K-delta like syndrome の発見
つ じ た ゆ き
辻田 由喜
(成長発達臨床医学専攻)
防衛医科大学校
平成28年度
目 次
第1章 緒言 1頁
第2章 PI3KCD遺伝子変異をもつAPDS患者の同定とその臨床的特徴 3頁
第3章 PTEN遺伝子変異をもつAPDS様免疫不全症患者の同定 8頁 第4章 PTEN変異患者細胞におけるPTEN発現の解析 11頁
第5章 PTEN変異およびPIK3CD変異患者リンパ球におけるAKT/mTOR/S6 シグナル伝達経路のリン酸化解析 14頁
第6章 PTEN変異患者とPIK3CD変異患者の免疫学的検討 18頁
第7章 総括 22頁
第8章 結論 24頁
謝辞 25頁
単語・略語説明 26頁
引用文献 31頁
図表 34頁
第1章 緒言
Activated PI3 kinase (PI3K) delta syndrome (APDS)は、2012年に初めて発見さ れた原発性免疫不全症候群であり、反復性下気道感染症、肝脾腫、多発性リンパ節腫 脹、CD4 陽性リンパ球の減少、クラススイッチしたメモリーB 細胞の減少などを特
徴とする 1), 2)。PI3K-delta を構成するサブユニットの一つ、p110δをコードする
PIK3CD 遺伝子の、常染色体優性遺伝形式をとる機能獲得変異(単語・略語説明 28
ページ参照)がAPDS の原因である。さらにこの遺伝子の変異は高IgM症候群、原 発性硬化性胆管炎の原因ともなり3)、B細胞性悪性腫瘍とも関連がある4), 5)。PIK3CD 遺伝子変異によるAPDS は APDS1 と分類され、PI3K-deltaを構成する別のサブユ ニットp85αをコードする遺伝子PIK3R1の機能喪失変異(単語・略語説明28ペー ジ参照)により発症するAPDS6), 7)をAPDS2と分類する。p110δはPI3Kの触媒ド メインであり、p85αがその作用を調整する。APDS1、APDS2双方の患者において、
p110δの機能亢進が認められている。
リンパ球において、PI3K/AKT/mTOR(単語・略語説明26, 29ページ参照)のシ グナル伝達経路は、細胞生存、増殖、成長、代謝に不可欠である 8)。PI3K は、イノ シトールリン脂質のリン酸化を行う酵素である。T細胞受容体、B細胞受容体、サイ トカインレセプターなどの受容体にリガンドが結合することでPI3Kの活性化が生じ、
3,4-イノシトール2リン酸(PIP2) (単語・略語説明29ページ参照)を活性型の3,4,5- イノシトール3リン酸(PIP3)(単語・略語説明29ページ参照)に変換し、次いでPIP3 はAKTをリン酸化して活性化させる。PI3Kによって活性化されるPIP3/AKT/mTOR のシグナル伝達経路は、生体内における細胞の生存、周期、糖代謝、あるいはタンパ ク合成など様々な生理活性に関わる細胞内シグナルを担っている。
一方、phosphatase and tensin homolog (PTEN)によって、AKTは不活化される。
これは、PTENによるPIP3のPIP2への脱リン酸化作用によることが、マウスモデ ルにおいてもヒトにおいても証明されている 9), 10)。そのため、PTEN の機能喪失は AKT/mTOR 経路を活性化させる方向に働く 11)。PIK3CD または PIK3R1 の変異に よりPI3Kの機能が亢進しAPDS1またはAPDS2が発症するように、PTENの機能
たため、我々はPTENの機能喪失変異がAPDS 類似の病態を発症しうるのではない かと考えた。
本研究では、まずわが国における原発性免疫不全症候群患者の中で、PIK3CD遺伝 子変異による APDS 患者の特徴を明らかにする検討を実施した。次いで原因不明の 原発性免疫不全症候群患者に対しexome解析(単語・略語説明27ページ参照)を行 って、ヘテロ接合型(単語・略語説明27ページ参照)PTEN遺伝子変異をもつ患者 を2名見出した。 PIK3CD機能獲得変異によるAPDS1患者、またはPIK3R1機能 喪失変異によるAPDS2 患者のように、この 2名の PTEN 遺伝子変異患者はAPDS 類似の所見を示した。PTEN の機能喪失変異が、リンパ球において AKT/mTOR/S6 のシグナルの亢進を引きおこすことを示し、APDS 類似の免疫不全症、APDS-like immunodeficiencyの原因となりうることを本研究で提唱する。
なお、本研究はヒト検体を用いるため、防衛医科大学校倫理委員会の承認(受付番 号438「先天性免疫不全症の遺伝子解析研究」および、受付番号566「原発性免疫不 全症の早期診断法の確立に関する研究」)を得て実施した。検体採取に際しては、対 象者もしくはその保護者に研究内容を文書と口頭により説明し署名同意を得た。
第2章 PIK3CD変異をもつAPDS患者の同定とその臨床的特徴
第1節 背景
APDSは、PIK3CDまたはPIK3R1遺伝子の常染色体優性またはde novo(単語・
略語説明27ページ参照)のヘテロ接合型変異で発症する。PI3K-delta を構成する分 子p110δのN334K、E525KまたはE1021K という3か所の機能獲得型変異は、さ まざまな組織で発現しているPI3K-alphaのがん関連機能獲得型変異と構造的に同じ 部位の変異である2)。海外からのAPDSの報告では、患者のほとんどがwhole exome 解析で発見された大家系の症例であった1), 2)。
本疾患は2013 年に初めて同定され海外から報告された新しい免疫不全症であり、
国内での患者の発症状況および臨床的特徴は不明であった。一方で、わが国において 原発性免疫不全症候群の患者の多数例を対象としたPIK3CDやPIK3R1遺伝子の変 異検索は行われていない。そこで、原発性免疫不全症候群患者の中から国内での
APDS1の患者を同定し、その臨床的特徴を明らかにすることとした。
第2節 対象および方法
(1) 対象
防衛医科大学校小児科と東京医科歯科大学小児科において原発性免疫不全症の免 疫学的検討の依頼を受け、末梢血3~5mlをEDTA加血液として入手できた患者のう ち、抗体産生不全(低ガンマグロブリン血症を伴っているもの、または罹患済感染症・
ワクチンに対して有効な特異抗体を産生できないもの)を呈し、遺伝子解析の同意を 得られている156 例を対象として、保存検体を用いてPIK3CD遺伝子変異を解析し た。
本疾患の報告後に、理化学研究所かずさDNA研究所および東京大学医科学研究所 にて今まで行われていたexome 解析例の結果を見直し、PIK3CD遺伝子の変異をも つことが判明した孤発性の患者4例においても、保存検体を用いて PIK3CD 遺伝子 変異を解析し確証を得た。これら4名の患者では、両親・同胞においても遺伝子解析 の承認と検体を得て PIK3CD 遺伝子変異を解析し、家族には変異が無いことも合わ せて確認した。
よび、東京医科歯科大学小児科で PIK3CD 遺伝子変異があらたに同定された孤発性 の患者2例について各主治医に問い合わせ、遺伝子変異の詳細と臨床情報について情 報提供を得た。
すべての患者情報は匿名化で得た上で、これらの情報をもとに、国内の PIK3CD 変異に伴うAPDS患者の状況を明らかにした。Germ-lineのPIK3CD 変異がみられ た患者については、家族の遺伝子検査の実施承認が得られた場合は、両親・同胞につ いても患者と同一の変異の有無を検索した。
(2) genomic DNAの抽出
末梢血1検体当たり 200μl をサンプル量とし、QIAamp DNA Micro キット (Qiagen,Hilden,Germany)を用いてgenomic DNAを溶出量 100 µl で抽出した。
得られたDNAはすべてQubit フルオロメ-ター (Invitrogen, Carlsbad, California, USA) またはGene Quant pro (GE Healthcare, Little Chalfont, Buckinghamshire, England) を用いて濃度を測定した。
(3) PIK3CD遺伝子シークエンス
①PCR法による各遺伝子の増幅
抽出した各患者由来genomic DNA につき、PIK3CD遺伝子の3か所のAPDS原 因変異部位を含む部分をpolymerase chain reaction (以下PCR) 法によって増幅し た。プライマーは、既報を参照して設計し作製した2)。PCR反応物は、High Fidelity PCR Master (Roche, Basel, Schweiz)を用いて調整し、Veriti® サーマルサイクラー (Applied Biosystems、以下 ABI,Foster,USA)を用いて、PCR 法による増幅反応 を行った。PCRの反応条件は、初期変性94℃5分間後、35サイクル(熱変性94℃ 60 秒,アニーリング60℃ 60秒, 伸長反応72℃ 60分)繰り返した後、72℃ 10分間で 最終伸長としてPCRを実施した。次いで、反応液を1.5%アガロースゲルにて電気泳 動し、PCR産物を確認した。
②ダイターミネーター法によるダイレクトシークエンス
精製したPCR産物をシークエンス反応液 [PCR精製物1μl、BigDye®
Terminator v1.1 (ABI) 4μl, BigDye® Terminator Sequencing Buffer (ABI) 2μl , Primer (10μM) 0.5μl、H2O 12.5μl] として調整し、Dye Terminator法により、
Gene Amp® PCR system 9700 (ABI, Foster,USA )を用いて増幅反応を行った。PCR の反応条件は初期変性96℃1分間を行った後、40サイクル (熱変性96℃10 秒、ア ニーリング50℃5秒、伸長反応60℃4分) 繰り返した後、60℃4分間で最終伸長とし た。
シークエンス反応産物は Millipore Multiscreen (Millipore, Billerica, USA) 、 Sephadex TM G-50 Superfine (Sigma-Aldrich, St. Louis, USA) を用いて精製した後、
Hi-Diホルムアミドにより希釈し、3500 Genetic Analyzer (ABI, Foster,USA ) に より遺伝子配列を決定した。
第3節 結果
(1)遺伝子解析結果
156例の抗体先生不全を伴う免疫不全症患者のうち、8例(5.1%)にPIK3CD遺伝子 変異を検出した。うち5名はPIK3CD遺伝子E1021K変異を持ち、家族は変異を持 たないことが確認された孤発例であった。他の3例はPIK3CD遺伝子E525A変異を 持ち、この3例は同一家系から見出された家族症例であった。E1021K変異は既報と 同じ変異であったが、E525A変異は他に報告のない新規変異であった。Exome解析 で判明していた4 例の遺伝子変異は、4例とも PIK3CD E1021K変異であり、家族 は変異を持たないことが確認された。富山大学小児科および東京医科歯科大学小児科 への問い合わせで確認した症例3例は、すべてPIK3CD E1021K変異を持つ孤発例 であった。このように、E1021K 変異を持つ患者12 例の家族では、検査実施の同意 を得た上で家族解析がなされ、PIK3CD 遺伝子 E1021K 変異は認めないことが確認 された。したがってこれら12例については、PIK3CD E1021K変異はde noveで生 じたと考えられた。
(2)臨床的特徴
我々が把握した国内のPIK3CD 遺伝子変異をもつAPDS 患者の臨床的特徴を表1 に示した。また、今回初めて PIK3CD 変異が見つかり、当院で詳細なリンパ球の機
のKindred D ~ Kindred Fに、臨床的特徴を表2に示す(患者P5 ~ P9)。発症年齢 は0歳4ヶ月から8歳で、平均は2.5歳であった。入院加療を要する下気道感染症な どの細菌感染症を93%で、ヘルペス属のウイルス(主にEpstein-Barr (EB)ウイルス およびサイトメガロウイルス)や真菌に対する日和見感染症を 47%の患者で認めた。
リンパ組織腫脹は80%の患者で伴っておりリンパ節腫脹をその全例で認めたが、さら に肝脾腫を47%の患者で、消化管粘膜下リンパ組織腫脹に伴う血便などの消化管症状 を 47%の患者で認めた。血球減少は 40%で認められ、脾腫による脾機能亢進や、自 己抗体産生に伴う血小板減少性紫斑病や溶血性貧血などの影響が考えられた。リンパ 腫の発症が15例中2例で確認され、それぞれ小腸のMALT型リンパ腫とEBウイル ス関連Bリンパ腫が1例ずつであった。低ガンマグロブリン血症を40%で、高IgM 血症を73%で伴っていた。これらの結果は表1に示すように、海外からの報告と同様 であった。
第4節 考察
今回の検討により、国内の免疫不全症患者における PIK3CD 変異による APDS1 患者の頻度、変異の部位と型が判明した。さらに、国内のPIK3CD変異患者15名の 詳細な臨床情報が明らかとなった。国内のPIK3CD 変異APDS 患者は孤発例がほと んどであり、家族例がほとんどである海外の既報告とは異なっていた1), 2)。海外から の報告は、多数の免疫不全症患者が発症している数家系を対象にExome解析を行っ た結果APDSを見出したという研究内容の報告であるため家族例がほとんどであり、
今回の国内患者の解析は家族歴のない多くの免疫不全症患者を対象に行ったため孤 発例が多いという、患者背景の違いが存在した可能性がある。また、E1021K変異を もつ患者が大多数であり、既報にあるE525K変異、N334K変異症例は存在しなかっ た。一方、E525Aという新規変異を 1家系 3 例に見出した。いずれの変異を持った 患者でも、国内患者の臨床症状は海外からの報告と同じく細菌感染症、リンパ組織腫 脹、高IgM血症などを特徴としていた1), 2)。
この解析の中で、原因不明の免疫不全症患者で APDS 様の臨床症状を認めながら
もPIK3CD 遺伝子変異をみとめなかった症例も存在したことから、PIK3CD近傍の
シグナル伝達関連遺伝子の変異により、APDS様の免疫不全症を呈する患者が存在す ることが示唆された。
第5節 結論
国内における PIK3CD 変異による APDS 患者 15 名の詳細な臨床症状を明らかに した。孤発例12名はいずれもE1021K変異を有し、すべてがde novo変異であった。
また、1家系 3 名はE525A 変異を有していた。これらの患者の臨床症状は国外の既
報と同様であった。PIK3CD 変異はみられないが APDS 様の免疫不全症を呈する症 例が存在し、PIK3CD近傍のシグナル伝達関連遺伝子の異常が発症に関わっている可 能性が推測された。
第3章 PTEN遺伝子変異をもつAPDS様免疫不全症患者の同定
第1節 背景
PIK3CD遺伝子変異による APDS1 は、本邦においても患者の存在が確認された。
さらにPIK3R1遺伝子変異によってもAPDS2を発症することが報告され6), 7)、我々 の解析した原因不明の免疫不全症患者の中にも2例のAPDS2患者が見出された(デ ータ示さず)。しかしAPDS類似の臨床症状を示しながらPIK3CD、PIK3R1変異の 確認されない患者も多く存在し、これら以外の近傍の遺伝子の変異により APDS と 同様の機序で免疫不全症を発症する可能性が考えられた。そこで、原因不明の免疫不 全症患者の中から新規遺伝子変異によるAPDS類似免疫不全症を見出すため、exome 解析を行った。
第2節 対象および方法
(1)対象と検体採取
防衛医科大学校小児科および東京医科歯科大学小児科に遺伝子変異検索の依頼が あった先天性免疫不全症症例 77 名において、両大学倫理委員会の承認を得た上で本 人または保護者から文書で同意を得て、末梢血全血3~10mlを採取した。遺伝子解析 の同意の得られた患者家族についても同時に末梢血を採取した。血液検体から第2章 と同様の方法でgenomic DNAを抽出した。
(2)Exome解析
抽出したgenomic DNAをSureSelectXT Human All Exon V4およびA5 kit、ま たは TruSeq Exome Enrichment kit をもちいてライブラリーを作製し、Illumina HiSeq 2000またはHiSeq 1000を用いてエクソーム解析を行った。得られたリード データから、dbSNP version 131、in-house SNP データベース、および Human Genetic Variation Database (単語・略語説明27ページ参照) に報告されているもの と同じ変異を除いた(図2)。
(3)候補遺伝子シークエンス
第2章2節と同様の方法により、Exome 解析で得られた候補遺伝子の変異を確認
した。
(4)臨床所見
有意な遺伝子変異が検出された症例の臨床所見について、患者の主治医および診療 録から情報を得た。
第3節 結果
Exome解析を行った原因不明の免疫不全症患者77 名中 2名(2.6%)で PTEN遺伝 子の変異を見出した。
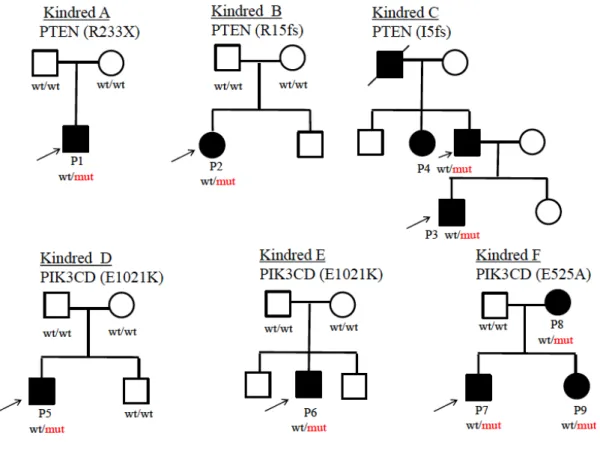
図1の家系Aに示すP1 は、非血縁間の健常な両親から出生した3歳男児であり、
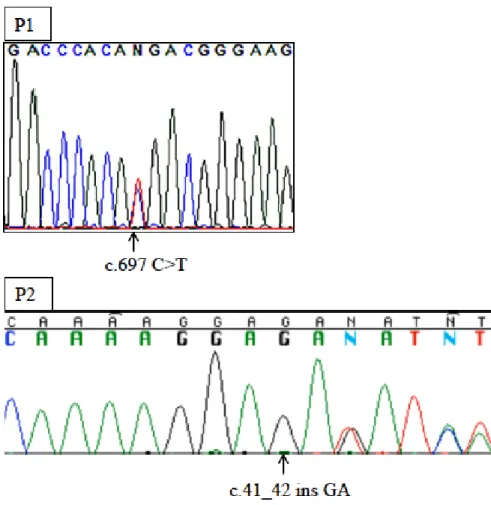
PTEN c.697C>T(R233X)というナンセンスのヘテロ接合型変異を有していた。図1 の家系 B に示す P2 は、非血縁間の健常な両親から出生した 14 歳女児で、PTEN c.41-42insGA(R15fsX9)というフレームシフトによるストップコドンへのヘテロ接合 型変異が認められた。P1、P2 ともに、変異はサンガー法シークエンスにて再確認さ れた(図3)。また、両家系とも両親に変異はなく、de novo変異であった。また、P1、 P2ともにPIK3CDおよびPIK3R1に変異を認めなかった。
PTEN遺伝子変異をもつ患者P1、P2ともに、繰り返す感染症、リンパ組織の腫脹 を認めた。さらにP1 は高IgM 血症を示し、これらの臨床症状は APDS患者と共通 するものであった(表3)。
第4節 考察
PTEN 遺 伝 子 は 腫 瘍 抑 制 因 子 と し て 同 定 さ れ 、PTEN 過 誤 腫 症 候 群(PTEN hamartoma syndrome)(単語・略語説明28ページ参照)の患者はPTEN遺伝子変異に ともなう過誤腫発症をはじめとした様々な症状が認められる 12)。また、Cowden syndrome (CS:単語・略語説明 26 ページ参照) 患者の 80%、 Bannayan–Riley– Ruvalcada syndrome (BRRS: 単語・略語説明26ページ参照) 患者の60%、 Proteus syndrome (単語・略語説明30ページ参照) 患者の20%、 Proteus-like syndrome (単 語・略語説明29ページ参照) 患者の50%にPTEN遺伝子の変異を伴っていることが 報告されている12)。過去に免疫不全症を伴ったCS、BRRS、PS患者の報告例は存在
最近、PTEN遺伝子変異の確認されたCS の患者で複合免疫不全(単語・略語説明 27 ページ参照)を発症している症例の報告がなされた18)。また、今回見出されたP1の変 異は、CSおよびBRRS患者で報告されている変異と同一のものであった19), 20)。
PTENはPIP3を脱リン酸化してPIP2へと変換する酵素作用をもち、全身の細胞 で発現している。PTENの機能が喪失するとPIP3の産生が亢進すると考えられ、こ れはPI3Kの触媒ドメインp110δの機能亢進と同じようなAPDS様の病態を引き起 こし、免疫不全症の原因となり得ることが推測された。
今回の PTEN 遺伝子変異を持つ免疫不全症患者の同定と、germ-line での PTEN 遺伝子変異によるPTEN 過誤腫症候群患者における複合免疫不全症合併の報告18)か ら、PTEN の機能喪失変異が APDS 様の免疫不全症の原因であるという仮説が成り 立つ。この仮説の証明のためには、PTEN変異を有する患者のリンパ球を用いた機能 解析の実験が必要と考えられた。
第5節 結論
APDS類似の免疫不全症状を示す2名のPTEN変異患者を同定した。
第4章 PTEN変異患者細胞におけるPTEN発現の解析
第1節 背景
今回我々が発見した、免疫不全症をともなう PTEN 遺伝子変異患者において、
PTEN の機能低下が APDS 類似の機序で免疫不全症を発症しているとの仮説に基づ くならば、患者リンパ球におけるPTEN発現は低下している可能性がある。
これを検証するため、患者のもつPTEN変異がリンパ球におけるPTEN発現にど のように影響を及ぼしているか、正常検体と比較をして評価をすることとした。
第2節 対象と方法
(1) 対象
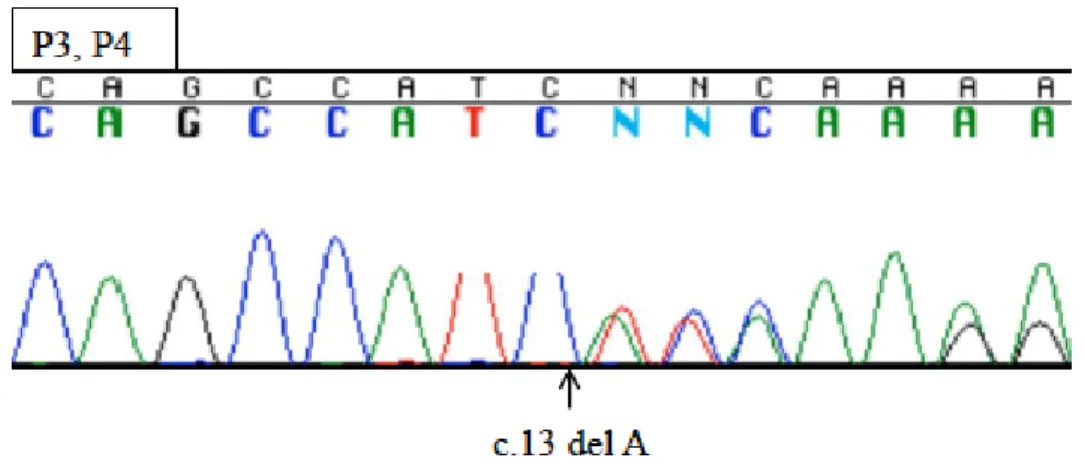
前章で同定した PTEN 変異をもつ患者2名(P1, P2)に加え、図1家系 C に示す PTEN変異患者P3、P4を対象とした。P3、P4は、家族性の反復性甲状腺腫瘍と消 化管ポリポーシスの発症により、家族性Cowden症候群(CS)と診断されており14), 15)、 PTEN遺伝子の変異(c.13Adel, p.I5fsX)を前章の方法で確認した(図4)。P3およびP4 ともに免疫不全症を示唆する症状は伴っておらず、リンパ球数、リンパ球サブセット、
血清免疫グロブリン値は全て正常であり(表 4)、ウイルス抗体価も正常であった。正 常コントロールは、健常人で同意の得られた3名とし、いずれも末梢血3~10mlを採 取した。
(2)PTEN蛋白のウエスタンブロッティングによる検出
① 活性化リンパ球の作成
採取した末梢血はLymphoprepTM (Axis-Shield、Oslo、Norway)を用いた比重遠心 法により末梢血単核球 (peripheral blood mononuclear cells; PBMC)に分離した。細 胞を 2 回洗浄し、10%ウシ胎児血清含有 RPMI-1640 メディウム(Gibco, Carlsbad, USA)で2×106 cells/mlにPBMCを懸濁し、IL-2含有固層化抗CD3フラスコ(TLY Culture kit25: Lymphotec, Tokyo, Japan)にて48時間培養し、活性化リンパ球を作 製した。
② ウエスタンブロッティング
得られた活性化リンパ球2×106 cellsに対し、プロテアーゼインヒビターカクテル (Thermo Fisher Scientific, Waltham, USA)とフォスファターゼインヒビターカクテ ル(Electrophoresis GmbH, Heidelberg, Germany)を添加した RIPA バッファーを 100ul加え、細胞を溶解した。得られた細胞溶解液は、4℃で15分間、15000rpmで 遠心し、上清を回収した。回収した溶解液は 98℃で 5 分間インキュベートをした。
得られた蛋白抽出液の濃度はすべてQubit フルオロメ-ター (Invitrogen, Carlsbad, USA)で測定し、各サンプル蛋白量25μgを用いてドデシル硫酸ナトリウムーポリア ク リ ル ア ミ ド ゲ ル 電 気 泳 動(sodium dodecyl sulfate-poly-acrylamide gel electrophoresis、SDS-PAGE)を行った。泳動後、蛋白をゲルからポリフッ化ビニリ デン(polyvinylidene fluoride、PVFD)膜に転写し、8%スキムミルクでブロッキング を行い、ラビットポリクローナル抗PTEN抗体(Cell Signaling Technology, Danvers, USA) とラビットポリクローナル抗GAPDH抗体(Abcam, Cambridge, USA)を1時 間室温にて反応させた。その後PVFD 膜を洗浄後、horseradish peroxidase (HRP) 標識抗ラビット二次抗体を室温で1時間反応させた。反応後洗浄し、化学発光法を利 用してPTEN蛋白をLAS-4000 mini (FUJI FILM, Tokyo, Japan)で検出した。得ら れたPTEN蛋白とGAPDH蛋白の発現について、その発光度をMulti-Gauge (FUJI FILM, Tokyo, Japan)を用いて数値化し、PTEN蛋白の発光度を内因性コントロール
蛋白 GAPDH の発光度で除した値をもって各患者および健常コントロールの PTEN
蛋白発現の値とし、それぞれの健常コントロールに対する患者の蛋白発現の割合を評 価した。
第3節 結果
ウエスタンブロッティングによる、活性化リンパ球由来 PTEN 蛋白の発現解析で は、PTEN 変異をもつ患者(P1-P4)では健常人と比較して、明らかに発現が低下して いた(図 5)。P1 の PTEN 蛋白バンドのシグナル強度は、健常コントロールのわずか 11.3%であった(図5)。患者P2、P3、P4のPTEN蛋白バンドのシグナル強度は、健 常コントロールのそれぞれ74%、64%、75%であった(図5)。
第4節 考察
PTEN R233X変異をもつP1、PTEN R15fsX9変異をもつP2、PTEN I5fsX変 異をもつ P3、P4 において、リンパ球における PTEN 蛋白の発現は健常と比較して 低下していた。これらの結果から、P1、P2、P3、P4 のもつ PTEN 遺伝子変異は、
PTEN 蛋白の発現を低下させることが判明し、それに伴って患者細胞では PTENの 機能が低下していると考えられた。
第5節 結論
PTEN R233X 、R15fsX9、I5fsX 変異をもつ患者リンパ球において、PTEN蛋白 の発現は低下していた。
第5章 PTEN 変異および PIK3CD 変異患者リンパ球における AKT/mTOR/S6 シグナル伝達経路のリン酸化解析
第1節 背景
PI3K活性の亢進は、AKT/mTOR/S6 経路の異常な活性化を誘導し、APDS1 およ びAPDS2発症を引き起こすことが報告されている1), 2), 6)。実際に、これらの患者で は、AKT Ser473(pAKT)、リボゾーマル蛋白S6 Ser235およびSer236(pS6)のリン 酸化亢進が確認されている1), 2)。
我々は、PTEN遺伝子変異をもち、APDSによく似た臨床症状を示す原発性免疫不 全症患者を2例見出した。これらの患者の細胞では、PTENの発現が低下しており、
PTENの機能喪失変異であると予想されることが前章で示唆された。PTEN変異によ り生じたPI3K活性の異常亢進がPIP3産生を増加させ、APDSと同様の機序で免疫 不全症の原因となっているならば、患者リンパ球において AKT/mTOR/S6 経路の異 常な活性化が誘導されているものと考えた。
そこで、PTEN 変異患者と PIK3CD変異による APDS 患者のリンパ球における、
AKT Ser473(pAKT)、mTOR Ser2448(pmTOR)、リボゾーマル蛋白S6 Ser235
およびSer236(pS6)のリン酸化を解析し、正常コントロールと比較することとした。
第2節 対象と方法
(1)対象
PTEN変異患者は、前々章で同定した2名の免疫不全症患者(P1, P2)に加え、前章 で解析した免疫不全の病歴をもたない家系Cに示す患者P3、P4を対象とした。
PIK3CD変異をもつAPDS患者は、第2章で同定されたPIK3CD E1021K変異を もつ2例(P5、P6)と、PIK3CD E525A変異をもつ家族患者3例(P7〜P9)の解析を行 った。
健常コントロールは、患者の健常な家族および健常成人とした。検体は末梢血
5~15mlを採取した。全例で文書による同意を得て検体を採取した。
(2)フローサイトメトリーによるAKT(Ser473)リン酸化解析
採取した末梢血はLymphoprepTM (Axis-Shield、Oslo、Norway)を用いた比重遠心 法により末梢血単核球 (PBMC)に分離した。細胞を2回洗浄し、RPMI-1640メディ ウム(Gibco, Carlsbad, USA)に2×104cells/mlの濃度でPBMCを懸濁した。細胞懸 濁液を等分にわけ、一方にはp110δ阻害薬であるIC87114を10μM添加し、抗CD3 抗体(BD Pharmingen, San Diego, USA)および抗CD19抗体(BioLegend, San Diego, USA)を 加 え 20 分 37℃ で イ ン キ ュ ベ ー ト し た 。 そ の 後 Cytofix Buffer(BD Pharmingen)を用いて細胞固定を行った。固定後、PERMⅢ buffer(BD Pharmingen) で懸濁して氷上 30 分間インキュベートして細胞膜透過処理を行い、抗リン酸化 AKT(Ser473)-Alexa Fluor 647色素結合抗体(Cell Signaling Technology, Danvers, USA)で細胞内染色を行った後、フローサイトメトリーで解析した。
(3)ウエスタンブロッティングによるリン酸化解析
採取した末梢血は、前章と同じ方法で細胞を調整し、活性化リンパ球を作成した。
得られた活性化リンパ球から前章と同じ方法でタンパクを抽出し、一次抗体としてラ ビットモノクローナル抗リン酸化 AKT 抗体(Cell Signaling Technology, Danvers, USA)、ラビットモノクローナル抗AKT抗体(Cell Signaling Technology)、ラビット ポリクローナル抗GAPDH抗体(Abcam, Cambridge, USA)、ラビットモノクローナ ル抗リン酸化S6抗体(Cell Signaling Technology)を用いてウエスタンブロッティン グを行った。
(4)マルチプレックスアッセイによるリン酸化解析
採取した末梢血は、前章と同じ方法で細胞を調整し、活性化リンパ球を作製した。
得られた活性化リンパ球 1×106 cells に対し、MILLIPLEX MAP kit(Millipore, Billerica, USA) 細胞融解バッファーにプロテアーゼインヒビターカクテル(Thermo Fisher Scientific, Waltham, USA)と フ ォ ス フ ァ タ ー ゼ イ ン ヒ ビ タ ー カ ク テ ル (Electrophoresis GmbH, Heidelberg, Germany)を添加し調整した溶液を100μl加 え、細胞を溶解した。得られた細胞溶解液は、4℃で10分間、12,000rpmで遠心し、
フィルターを通して不純物を取り除いたのち回収した。回収した蛋白は、Luminex
の発現を測定し、各検体内在性コントロール蛋白 GAPDH の発現で標準化した値を 比較検討した。
(5)統計解析
すべてのデータは、Studentのt検定、Welchのt検定、またはPeasonのカイ二 乗検定による解析を行い、平均値±標準誤差により表示した。
第3節 結果
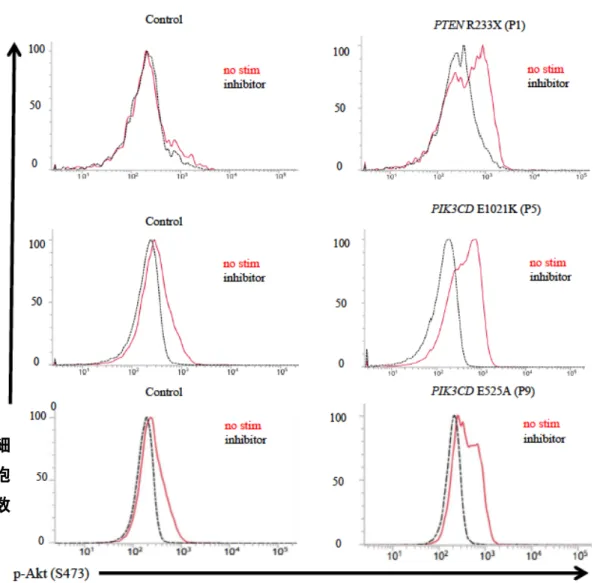
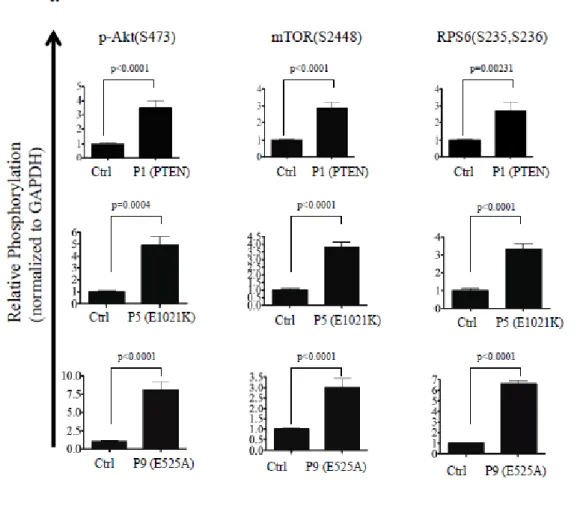
CD19陽性B細胞において、フローサイトメトリーによるAKT(Ser473)リン酸 化解析を行ったところ、PTEN変異患者から分離した細胞では、健常コントロールの 細胞と比較して、定常状態でpAKTのリン酸化が亢進していた(図6)。PIK3CD遺伝 子E1021K変異およびE525A変異をもつ患者から分離したB細胞においても、同様 の所見を得た(図6)。この pAKT の上昇は、PTEN変異患者細胞では p110δ阻害薬 を加えることで正常化したが、健常コントロール細胞では p110δ阻害薬添加による 変化はみとめられなかった(図6)。PIK3CD 変異患者細胞においては、臨床症状の 軽いE525A変異患者P8においてはp110δ阻害薬添加による有意な変化は検出され なかったが(データ示さず)、それ以外の患者ではp110δ阻害薬を加えることでリン 酸化の亢進が正常化した。
活性化T 細胞を用いたウエスタンブロッティングでは、AKTタンパクの発現は、
健常人および PTEN変異患者、PIK3CD変異患者で同等であった。しかしながら、
pAKT の発現は、全ての PTEN 変異患者(P1~P4)で健常コントロールと比較して著 明に亢進していた(図7)。PIK3CD変異患者の活性化T細胞においても、E1021K変
異、E525A変異ともに pAKT 発現は亢進しており、これは既報と同様の結果であっ
た1) 2)。活性化T細胞におけるpS6の発現についても、全PTEN変異患者(P1〜P4)
で亢進していた(図7)。
活性化T細胞を用いたマルチプレックスアッセイでは、全ての PIK3CD変異患者 におけるpAKT、pmTOR、pS6の発現は、コントロールと比較して亢進していた(図 8-1、8-2)。PTEN 変異患者においては、免疫不全症をもつ P1 の pAKT、pmTOR、 pS6の発現は、コントロールと比較して有意に亢進していた。同じく免疫不全症をも つP2では、pmTOR、pS6の発現は、コントロールと比較して有意に亢進しており、
pAKT の発現では有意差は認めないもののコントロールと比較して高い傾向にあっ た。免疫不全症を認めないP3、P4では、P3におけるpAKTの発現はコントロール と比較して有意に亢進しており、それ以外についても有意差は認めないものの、患者 において高い傾向にあった(図8-1、8-2)。
第4節 考察
以上の結果から、PIK3CD の機能獲得変異と同じように、PTEN の機能喪失変異 患者のリンパ球においても AKT/mTOR/S6 シグナル伝達経路の異常な活性化を生じ ていることが確認された。このことから、PTEN 変異患者においては、PIK3CD 変 異患者と同様の機序で免疫不全症が引き起こされていると推測された。
第5節 結論
PTEN 変 異 患 者 リ ン パ 球 で は 、PIK3CD 変 異 患 者 リ ン パ 球 と 同 様 に 、 AKT/mTOR/S6経路の異常活性化をみとめた。
第6章 PTEN変異患者とPIK3CD変異患者の免疫学的検討
第1節 背景
前 章 で PTEN 変 異 患 者 で は PIK3CD 変 異 患 者 と 同 様 に リ ン パ 球 に お け る
AKT/mTOR/S6 シグナルの異常な活性化を見出した。これが APDS と同様の機序で
免疫不全症を引き起こしていると考えられたが、PTEN変異患者における免疫学的解 析の詳細な報告は存在せず、APDSとの免疫学的所見の異同の詳細は不明である。
そこで、免疫不全症をもつ PTEN 変異患者の臨床症状および免疫学的評価を行い PTEN変異患者とPIK3CD変異患者の異同点を明らかにすることとした。
それぞれの患者の主治医および診療録から臨床情報の入手を試みた。
また、患者末梢血リンパ球の表面マーカー染色によりリンパ球分画を詳細に検討し、
比較検討することとした。
さらに、正常な T 細胞新生能、B 細胞新生能を評価するために、T-cell receptor excision circles (以下 TREC) (単語・略語説明 30 ページ参照) 、signal joint kappa-deleting recombination excision circles (以下sjKREC) (単語・略語説明28ペ ージ参照)について、我々が以前開発した T 細胞、B 細胞の新性能測定法を用いて解 析をすることとした21), 22)。TRECはT細胞新生能を、sjKRECはB細胞新生能をそ れぞれ反映するため、この測定値を用いて患者のリンパ球新生能を評価することがで きる。
得られた結果を比較することで、PTEN機能不全による免疫不全症と PIK3CD 機 能獲得変異によるAPDSの類似点、相違点を明らかにすることとした。
第2節 方法
(1)対象
PTEN 遺伝子変異患者 4 名(P1~P4)および PIK3CD 変異患者 5 名(P5~P9)より EDTA加末梢血3~5mlを入手した。健常コントロールは、健常な家族または健常成 人で同意を得られた7名とした。全例文書による同意を得て検体を採取した。
(2)臨床情報
患者の主治医および診療録から、後方視的に臨床情報を得た。
(3)細胞の調整
得られた末梢血検体を1×Lysis Buffer (BD, Franklin Lakes, USA) と混合し、室 温で15分間静置することで赤血球を溶解した。溶解後、室温で1700 rpm、5分間遠 心し、細胞上清を廃棄することを2回繰り返し、末梢血由来顆粒球および単核球を得 た。
(4)フローサイトメトリーによるリンパ球サブセット解析
(3)で得られた細胞を氷上でRound Bottom Tubes (BD, Franklin Lakes, USA) に加え、フローサイトメトリー用の抗体を用いて20~25分間反応させて染色した。
抗体は、FITC、PE、 PC5、 Per-CP、またはAPCでラベルされたanti-CD3 (clone:
SK7, BD)、 anti-CD4 (clone: LeuTM-3a+3b, BD) 、anti-CD8 (clone: Leu-2a, BD,)、 anti-CD16 (clone: Leu-11c, BD)、 anti-CD19 (clone: J4.119, Beckman Coulter, Inc.
Brea, USA)、 anti-CD21 (clone: BL13, Beckman Coulter, Inc.)、 anti-CD24 (clone: ALB9, Beckman Coulter, Inc.)、 anti-CD25 (clone: 2A3, BD)、 anti-CD27 (clone: 1A4CD27, Beckman Coulter, Inc.)、 anti-CD38 (clone: HB7, BD)、 antiCD56 (clone: N901, Beckman Coulter, Inc.) 、 anti-CD127 (clone:
HIL-7R-M21, BD)、 anti-CD45RA (clone: L48, BD)、 anti-CD45RO (clone: UCHL1, BD)、 anti-TCR V alpha24 (clone: C15, Beckman Coulter, Inc.)、 anti-TCR V beta 11 (clone: 56C5.2, Beckman Coulter, Inc.)、 anti-IgD (clone: BD)、 anti-IgM (clone: IA6-2, G20-127, BD)を用いた。細胞を染色後、フローサイトメトリー (FACS caliber および FACS verse, BD) を用いて解析した。
(5)リアルタイムPCRによるTREC、sjKRECの測定 第2章の方法に従い、genomic DNAを抽出した。
我々の既報に基づきプライマーと蛍光標識プローブを作製し、リアルタイム PCR (単語・略語説明 30 ページ参照) による TREC、KREC および内在性コントロール
タンダードサンプルの段階希釈系列の増幅曲線から作製される検量線をもとに算出 した。PCRは、Light Cycler 480 II(Roche, Basel, Schweiz)を用いて、50℃ 2分、
95℃ 10分の初期ステップの後、40サイクル (95℃ 15秒、60℃ 1分)で実施した。
リアルタイム PCR で得られた TREC、sjKREC および RNaseP の測定値は、1
µgDNA あたりに換算して最終的な定量値とし、定量値 1.0x102未満を陰性とみなし
た。
第3節 結果
本研究第2章にて見出されたPIK3CD変異による5名のAPDS患者の臨床所見の 特徴は表2に示した。また、PTEN変異をもつ免疫不全症患者2名および免疫不全症 のない家族性CS患者2名の臨床的特徴は表3に示した。さらにこれらの患者におけ る免疫学的所見を表4に示し、免疫不全症を伴う患者に共通する所見を表5にまとめ た。
反復感染をPTEN変異患者では全例、PIK3CD変異患者では80%に認めた。リン パ組織腫脹はどちらの患者も全例で伴っていたが、リンパ腫を合併した患者は今回解 析した中には存在しなかった。高IgM血症を伴っていた患者はPTEN変異では2例 中1例、PIK3CD変異では5例中2例であり、低IgG血症を伴っていた患者はPTEN 変異患者・PIK3CD 変異患者とも1例(それぞれ 50%、20%)であった。末梢血リ ンパ球の解析では、リンパ球数の減少が PTEN 変異患者、PIK3CD 変異患者とも 1 例で認められ、T細胞のCD4陽性細胞、CD8陽性細胞の比率の逆転がそれぞれPTEN 変異患者の1例、PIK3CD変異患者の2例で認められた。
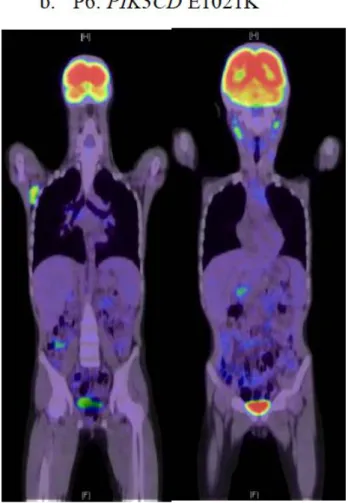
PET (単語・略語説明28ページ)はAPDS患者においてグルコース取り込みの増加 を見出しリンパ組織増殖の検出に有用であることが報告されており 2)、P1(PTEN 変 異患者)およびP6(PIK3CD E1021K変異患者)にて評価を行いその所見を比較した。
図9に示したように、全身性のリンパ組織腫脹を反映するグルコース取り込みの増加 が、両患者とも頸部、腋窩、および腹腔内リンパ節と脾臓で検出され、同様のリンパ 組織の腫脹を反映しているものと考えられた。
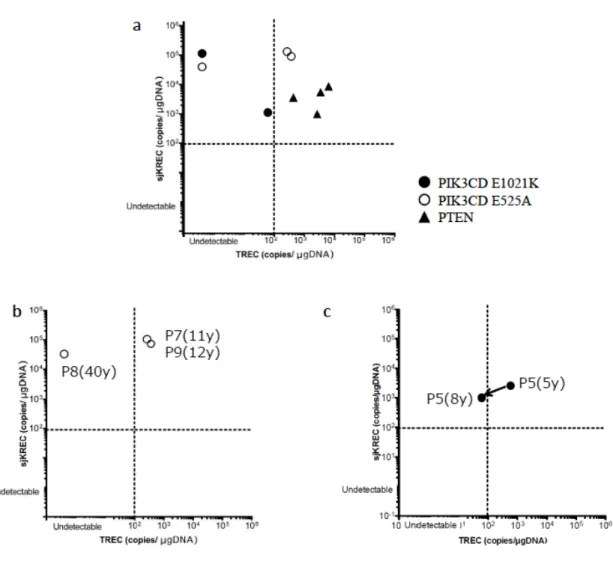
PTEN 変異患者では、全患者において、TREC および sjKRECは正常であった。
PIK3CD変異患者では、sjKRECは全患者正常であったが、TREC低値を示す患者が 存在した。P5、P6、P8はTREC陰性sjKREC陽性であった(図10a、表6)。さらに、
P5は5歳時にはTREC正常であったが、8歳時には陰性になっていた(図10c)。P7、 P8、P9は同じPIK3CD E525A変異を持つ家族患者であるが、最年長のP8のみTREC が陰性であった(図10b、表6)。
第4節 考察
PTEN変異患者の臨床症状は、APDS患者と臨床症状、リンパ球サブセットとも類 似点が多いことが判明した。
また、PIK3CD変異によるAPDS患者は年齢を経るとTRECが低下していくこと が予想されたことから、PTEN 変異にともなう免疫不全症患者の TREC 値について も同様の経過をとる可能性があると思われた。PTEN 変異患者の TREC 値について は、さらに経過を追い評価すべきであると考えられた。
第5節 結論
PTEN変異に伴う免疫不全症の臨床症状は、PIK3CD変異に伴うAPDS に類似点 が多いことが判明した。
第7章 総括
今回、PTEN 変異をもつ 2 名の免疫不全症患者(P1、P2)を見出した。これら 2 名 の患者の臨床症状は APDS 患者の表現型に酷似していた。両変異患者とも、繰り返 す感染症と全身性のリンパ組織腫脹を認め、P1はリンパ球減少、CD4/CD8比の逆転、
高IgM血症を伴い、P2は低IgG血症のため当初CVID(単語・略語説明27ページ 参照)と診断されていた(表3)。
APDSはPIK3CD変異またはPIK3R1変異によるPI3Kの機能亢進により発症す
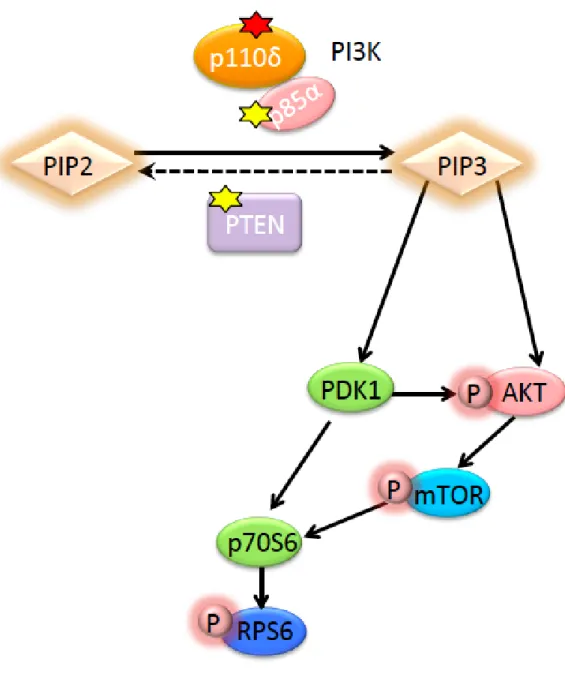
る1), 2)。PTENはPIP3をPIP2に変換する9), 10)。対照的に、PI3KはPIP2をPIP3
に変換する1)。このように、PTENもPI3K もPIP3 を介したシグナル伝達に関わる ため、PTEN 機能喪失変異が APDS 類似の免疫不全症を発症させる可能性は十分に 考えられた(図 11)。そのため、リンパ球の AKT/mTOR/S6 シグナル伝達経路の評価 をこれら4名のPTEN機能喪失変異患者で行い、P1、P2の2名のPTEN変異患者 ではAPDS患者と同様にAKT、mTOR、S6のリン酸化が亢進していることが確認さ れた。すなわち AKT/mTOR/S6 経路の異常な活性化を示していると考えられた。さ らにP1およびP2は、exome解析でPIK3CD、PIK3R1、その他免疫関連遺伝子に 病的変異のないことが確認されていた。以上から P1 と P2 の免疫不全症の原因は PTEN変異であり、PTENの機能喪失変異が APDS 類似の機序で免疫不全症を引き 起こしたと考えられた。
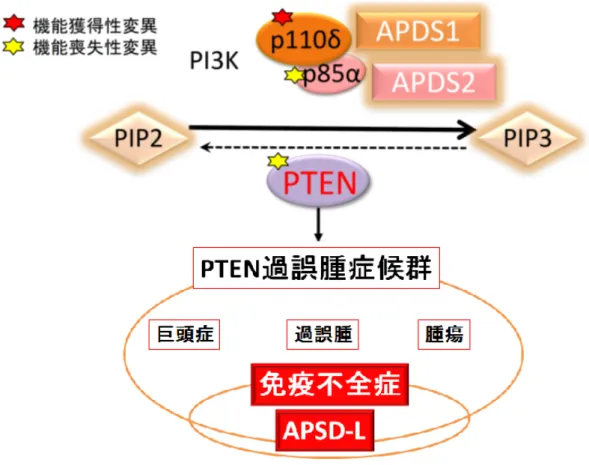
PTEN 遺伝子変異によりその機能が損なわれると、巨頭症や多発性の過誤腫を引き
起こし、甲状腺、子宮、乳腺等に良性・悪性の腫瘍を発症するリスクが高くなること が知られており、このような病態を PTEN 過誤種症候群 (単語・略語説明 28 ペー ジ)とよぶ 12)。しかし、PTEN 変異患者が APDS 様の免疫不全症、APDS-like immunodeficiency (APDS-L)をも発症するということは、今回我々が初めて見出した
(図12)。
免疫不全症のないPTEN変異患者(P3、P4)においても、免疫不全症のある PTEN 変異患者(P1、P2)と同様にリンパ球における異常な AKT/mTOR/S6 経路の活性化が 認められた。それにも関わらず、一部の PTEN 変異患者で免疫不全症を伴っていな い理由は不明であるが、以下の2つの可能性を推測している。まず第1に、PTEN変 異に伴って発症する免疫不全症は、不完全浸透である可能性がある。第2に、同家系
である P3、P4 のもつ PTEN 変異が免疫不全症に関連しない変異であるという可能 性である。これらを解明するためには、PTENの機能喪失変異に伴う免疫不全症患者 のさらなる解析と臨床情報の蓄積が必要である。
第8章 結論
PTEN変異による原発性免疫不全症APDS-Lを発見した。
APDS-L 患者の免疫学的所見は APDS 患者に類似しており、患者リンパ球では
APDSと同様にAKT/mTOR/S6の異常なリン酸化亢進が認められた。
謝辞
本研究を行うにあたり、全般にわたりご指導を賜りました防衛医科大学校小児科学 講座 野々山恵章教授に謹んで感謝申し上げます。また、ご指導賜りました先生方、
貴重な症例をご紹介くださった先生方、そして検体採取にご同意くださいました患者 様およびその保護者の方々に心より感謝いたします。
防衛医科大学校小児科学講座 関中 佳奈子 先生
加藤 環 先生 関中 悠仁 先生 座波 清誉 先生 賀佐希美子 様 冨田香織 様
東京医科歯科大学小児・周産期地域医療学 講座 今井耕輔先生
東京医科歯科大学小児科学講座 森尾 友宏 先生 葉 姿汶 先生 満生 紀子 先生 岡野 翼 先生 高島 健浩 先生 金兼 弘和 先生 高木 正稔 先生 広島大学小児科学講座
小林 正夫 先生 岡田 賢 先生 浅野 孝基 先生 隈病院外科
小林 薫 先生 かずさDNA研究所
岐阜大学小児科学講座
加藤 善一郎 先生 大西 秀典 先生 静岡こども病院免疫アレルギー科
木村 光明 先生 滋賀医科大学小児科学講座
丸尾 良浩 先生 國津 智彬 先生 京都大学腫瘍生物学講座
小川 誠司 先生 吉田 健一 先生
名古屋大学小児科学講座 小島 勢二 先生
奥野 友介 先生 村松 秀城 先生 東京大学医科学研究所ヒトゲノム 解析センター 宮野 悟 先生
白石 友一 先生 千葉 健一 先生
田中 洋子 先生 Division of Allergy and Immunology,
Boston Children’s Hospital
Ms. Nicole Luche 昭和大学藤が丘病院小児科
単語・略語説明 AKT:
セリン/スレオニンキナーゼでありProtein Kinase B(PKB)の別称。細胞外刺 激により、PI3Kなどのキナーゼを介して活性化されリン酸化AKTとなり、細 胞死、細胞増速などのシグナルを伝達するシグナル伝達物質である。過剰活性 化はさまざまな悪性腫瘍の病因となる。
APDS: Activated PI3 kinase delta syndrome
2012 年に初めて発見された原発性免疫不全症候群。原因遺伝子は PIK3CD、 PIK3R1。PI3Kを構成するサブユニットの一つ、p110δの機能獲得変異により 発症する。反復性下気道感染症、肝脾腫、多発性リンパ節腫脹、CD4 陽性リン パ球の減少、クラススイッチしたメモリーB細胞の減少などを特徴とする。
BRRS: Bannayan–Riley–Ruvalcaba syndrome (Bannayan–Riley–Ruvalcaba 症候群)
まれな過誤腫疾患で、多発性皮下脂肪腫、大頭、血管腫を生じる。常染色体 優性遺伝形式をとるが、散発例も報告されている。
CD: cluster of differentiation(クラスター分類)
白血球やその他の細胞が細胞表面に発現している、糖タンパクなどでできた さまざまな分子(表面抗原)。この分子の違いにより、細胞の違いを詳細に識別 することができる。
CS: Cowden syndrome(Cowden症候群)
消化管ポリポーシスが発症する常染色体優性遺伝形式をとる疾患。顔面小丘 疹、四肢角化性丘疹、口腔粘膜乳頭腫などを併発し、乳腺、甲状腺、泌尿器科 腫瘍の合併率が高い。カウデン病、多発性過誤種症候群ともよばれる。
CVID: common variable immunodeficiency(分類不能型免疫不全症)
抗体産生異常を主体とする先天性免疫不全症。B 細胞数は正常から低値で多 彩な臨床症状を呈し、未だに原因が不明という点から暫定的に分類された疾患 群である。25,000〜66,000人に1人発症し、原発性免疫不全症の中で最も発症 頻度が高い。
de novo変異
親から受け継いだ遺伝子変異ではなく、ある個体において新しく発生した遺 伝子変異のこと。生殖細胞(精子または卵子)における突然変異や、胚形成早期の 受精卵それ自体に生じた突然変異。
Exome解析
DNA の塩基配列のうち、Exon 部分のみを網羅的に捕捉するプローブを用い て、Exon部分のみに断片化されたDNAを抽出し、このExon部分配列のみを、
次世代シークエンサーにより網羅的に解析する実験技術。
ヘテロ接合型変異
両親から受け継いだ一対の対立遺伝子のうち、片方の対立遺伝子のみで変異 を認め、異なる種類の遺伝子を受け継いだ場合のこと。
HGVD: Human Genetic Variation Database (日本人変異データベース)
2013 年 11 月から公開されている、日本人のゲノム情報データベース。既知 変異データベースに含まれていない、日本人特異的な変異を多く含む。
複合免疫不全
T細胞の数または機能の異常により、細胞性免疫不全症を呈する疾患。T細胞 機能不全があれば、B 細胞による適切な抗体産生も障害されるため、液性免疫 不全も合併する。
機能獲得変異
遺伝子変異の結果、遺伝子産物が構造変化することによって、生理状態では みられない新しい機能を獲得すること。
機能喪失変異
遺伝子変異の結果、遺伝子産物が構造変化することによって、その機能が失 われる変異のこと。
KREC: kappa-deleting recombination excision circle
B 細胞の発生過程において、重鎖の有効な遺伝子再構成ができると、次にκ 鎖の再編成が始まる。その際κ鎖のVJ再構成が無効の場合、そのκ鎖を発現さ せないためにκ鎖定常領域の Cκを含む領域が染色体から切り出される。この 切り出された環状DNAがsignal joint (sj)KREC、染色体内に残存した部分が coding joint (cj)KRECである。
PET: positron emission tomography(陽電子放射断層撮影)
陽電子検出を利用したコンピューター断層撮影技術。糖代謝レベルの上昇を 検出することにより、中枢神経系の代謝レベルの観察や、腫瘍の診断などに利 用されている。
PTEN hamartoma syndrome (PTEN過誤腫症候群)
PTEN 遺伝子の変異によって発症し、過誤腫の発症を特徴とする。常染色体 優性遺伝形式をとる。CS、BRRS、PS、Proteus様症候群のうち、PTEN遺伝 子の変異が確認された患者が含まれる。
PID: primary immunodeficiency disease(原発性免疫不全症)
先天的に免疫系のいずれかの部分に欠陥がある疾患の総称。後天的に免疫力 が低下するHIV感染症などに伴う後天性免疫不全症症候群とは区別される。
PIP2: Phosphatidylinositol 3,4-bisphosphate(ホスファチジルイノシトール 3,4-ビスリン酸)
細胞膜に微量に存在する構成成分の一つ。細胞内シグナル伝達に関する分子 で、ホスファチジルイノシトールのイノシトールの 3,4 位 OH 基がリン酸エス テル化された化合物。
PIP3: Phosphatidylinositol 3,4,5-triphosphate(ホスファチジルイノシトール 3,4,5三リン酸)
PIP2がリン酸化されて生じる細胞膜にある化合物。AKTなどに代表されるさ
まざまなタンパク質を集合させ、下流シグナル伝達経路を活性化させるセカン ドメッセンジャーとしての機能をもつ。
PI3K: Phosphatidylinositol 3-kinase(ホスファチジルイノシトール3—キナー ゼ)
各種成長因子、サイトカインあるいはインスリンなどに活性化され、細胞質 から細胞膜へと移動して、3,4-イノシトール2リン酸(PIP2)を活性型の3,4,5-イ ノシトール3リン酸(PIP3)に変換する。リンパ球では、p110δと p85αから構 成されるPI3Kδが主に発現している。
mTOR: mammalian target of rapamycin
セリン/スレオニンキナーゼの一種。細胞内シグナル伝達に関与する。インス リンや他の成長因子、栄養・エネルギー状態、酸化還元状態など細胞内外の環 境情報を統合し、転写、翻訳などを通じて、それらに応じた細胞のサイズ、分 裂、生存などの調節に中心的な役割を果たす。
Proteus like symdrome(プロテウス様症候群)
PSの診断基準は満たさないものの、PSの臨床的特徴を顕著に示す患者。
PS: Proteus syndrome(プロテウス症候群)
手または足の巨大発育、四肢非対称、足底過形成、血管腫、リンパ管腫、表 皮母斑、大頭、骨化過剰および長官骨の過度成長によって特徴づけられる過誤 腫性症候群。
リアルタイムPCR
ポリメラーゼ連鎖反応 (PCR) 増幅産物をリアルタイムにモニタリングする 解析技術。PCRにより1サイクルごとにDNAが2倍になっていく増幅の様子 を蛍光により検出し、増幅曲線からDNAの定量を計算する。
S6: ribosomal protein S6 (リボソーマル蛋白S6)
細胞質リボゾームの40Sサブユニットの構成タンパク質。p70S6キナーゼの 基質であり、成長因子やマイトジェンなどによるシグナル伝達でp70S6キナー ゼの活性化が生じてS6のリン酸化を誘導し、タンパク質合成を制御する。
TREC: T-cell receptor excision circle
T細胞受容体遺伝子再構成の過程でゲノムDNAから切り出される環状DNA。 一度生成されると、増殖せず、細胞分裂に伴い希釈されるため、T 細胞新生能 を反映する。複合免疫不全症では責任遺伝子ならびに表現型は異なるものの新 生T細胞数の低下という共通した特徴を有するため、TRECsの欠損あるいは減 少を呈する。
引用文献
1. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science 2013;342:866-71.
2. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol 2014;15:88-97.
3. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of function mutations of PIK3CD as a cause of primary sclerosing cholangitis. J Clin Immunol 2014;35:11-4.
4. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol 2014;34:272-6.
5. Kracker S, Curtis J, Ibrahim MAA, Sediva A, Salisbury J, Campr V, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase δ syndrome. J Allergy Clin Immunol 2014;134:233-6.
6. Deau M-C, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene.
J Clin Invest 2014;124:3923-8.
7. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al.
Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med 2014; 211: 2537–47.
8. Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol 2003;3:317-30.
9. Maehama T, Dixon JE. The Tumor Suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998;273:13375-8.
10. Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Nat Acad Sci USA 1998;95: 15406-15411.
11. Stambolic V, Suzuki A, la Pompa de JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative Regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998;95:29-39.
12. Eng C. PTEN: One gene, many syndromes. Hum Mutat 2003;22:183-98.
13. Ruschak PJ, Kauh YC, Luscombe HA. Cowden's disease associated with immunodeficiency. Arch Dermatol 1981;117:573-5.
14. Riley HD Jr, Smith WR. Macrocephaly, pseudopapilledema and multiple hemangiomata. A previously undescribed heredofamilial syndrome.
Pediatrics 1960;26:293-300.
15. Halevy S, Sandbank M, Pick AI, Feuerman EJ. Cowden's disease in three siblings: electron-microscope and immunological studies. Acta Derm Venereol 1985;65:126-31.
16. Amer M, Mostafa FF, Attwa EM, Ibrahim S. Cowden's syndrome: a clinical, immunological, and histopathological study. Int J Dermatol 2011;50:516-21.
17. Hodge D, Misbah SA, Mueller RF, Glass EJ, Chetcuti PA. Proteus syndrome and immunodeficiency. Arch Dis Child 2000;82:234-5.
18. Browning MJ, Chandra A, Carvonaro V, Okkenhaug K, Barwell J.
Cowden's syndrome with immunodeficiency. J Med Genet 2015;52:856-9
19. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al.
Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997;16:64-7.
20. Marsh DJ, Dahia PLM, Zheng Z, Liaw D, Parsons R, Gorlin RJ, et al.
Germline mutations in PTEN are present in Bannayan-Zonana syndrome.
Nat Genet 1997;16:333-4.
21. Morinishi Y, Imai K, Nakagawa N, Sato H, Horiuchi K, Ohtsuka Y et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. J Pediatrics 2009;155:829-33.
22. Nakagawa N, Imai K, Kanegane H, Sato H, Yamada M, Kondoh K, et al. Quantification of k-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J Allergy Clin Immunol 2011;128: 223-25.
図1 PTEN変異およびPIK3CD変異を持つ患者家系図
黒の四角および丸は、変異が確認または予想される患者を示す。P1-P9 は本 文中の各患者を表す。
図2 患者P1のexome解析データの解析アルゴリズム
全単一塩基変異と挿入欠失変異データの解析を行った。各フィルタリング段 階に記載されている数字は、各段階の候補変異の数である。PTEN 以外に
B3GALT2 と PIEZO2 が最終候補遺伝子となったが、両遺伝子とも免疫系に関
係しないため、疾患発症には関与しないと考えられた。
図3 P1およびP2のPTEN遺伝子のDNAシークエンス波形 矢印が各患者のPTEN遺伝子変異部位を示す。
図4 P3およびP4のPTEN遺伝子のDNAシークエンス波形 矢印が患者のPTEN遺伝子変異部位を示す。
PTEN/GAPDH 0.11 1.0 0.74 1.0 0.64 0.75 1.0
図5 PTEN蛋白の発現
活性化T細胞抽出蛋白を用いて、PTEN変異患者P1~P4および健常コント
ロールのGAPDHとPTENの発現をウエスタンブロッティングで検出した。
数値は、内因性コントロールであるGAPDHの発現度でPTEN蛋白の発現度を 除したものであり、同時に行った健常コントロールの値を1.0とし、各患者の相 対値を示している。
図6 フローサイトメトリーによる細胞内AKTリン酸化解析
患者および健常コントロールにおける、定常状態のB細胞のリン酸化AKT(赤 線)と、p110δ阻害薬で処理した時のB細胞のリン酸化AKT(黒線)。横軸は
抗p-Akt抗体による染色の強度を示し、縦軸は細胞数を示す。
細 胞 数
p-AKT/AKT 1.0 4.9 1.0 1.4 1.0 1.3 1.2 1.0 2.9 1.0 2.8 1.0 2.3 3.4 4.4
p-S6/GAPDH 1.0 2.9 1.0 1.4 1.0 1.1 1.2 1.0 1.5 1.0 1.6 1.0 1.2 1.5 4.9
図7 AKTとS6リン酸化
各患者(P1~P9)および健常コントロール(Ctrl 1, Ctrl 2, Ctrl 3/4, Ctrl5, Ctrl6, Ctrl 7-9)活性化 T 細胞から抽出したタンパクの、リン酸化 AKT(Ser473)
〔p-Akt(S473)〕、AKT、リン酸化リボゾーマルプロテインS6〔p-S6(S235,236)〕、
GAPDHの発現をウエスタンブロッティングで示す。
数値は、リン酸化AKTの発現をAKTの発現度で除したものと、内因性コント ロール蛋白 GAPDH の発現度でリン酸化リボゾーマルプロテイン S6 蛋白の発 現度を除したものであり、同時に行った健常コントロールの値を1.0とし、各患 者の相対値を示している。
図8-1 P1、P5、P9のAKT、mTOR、リボゾーマルプロテインS6のリン酸化 免疫不全症をもつ PTEN 変異患者 P1、PIK3CD E1021K 変異患者 P5、 PIK3CD E525A変異患者P9および健常コントロール活性化のT細胞から抽出 したタンパクの、リン酸化 AKT(Ser473)〔p-Akt(S473)〕、リン酸化 mTOR
〔p-mTOR(S2448)〕、リン酸化リボゾーマルプロテインS6〔p-S6(S235,236)〕 の発現について、それぞれ内因性コントロール蛋白GAPDH の発現で標準化し た値を示す。各検体6~15回の実験を行いその平均を示している。各コントロー ルの値を1.0とし、各患者の値はコントロールに対する相対値を示す。
図8-2 P2~P4、P6~P8のAKT、mTOR、リボゾーマルプロテイン S6のリン 酸化
免疫不全症をもつPTEN変異患者P2およびもたない患者P3、P4、PIK3CD E1021K変異患者 P6、PIK3CD E525A変異患者 P7、P8および健常コントロ ー ル 活 性 化 の T 細 胞 か ら 抽 出 し た タ ン パ ク の 、 リ ン 酸 化 AKT(Ser473)
〔p-Akt(S473)〕、リン酸化mTOR〔p-mTOR(S2448)〕、リン酸化リボゾーマル プロテインS6〔p-S6(S235,236)〕の発現について、それぞれ内因性コントロー
ル蛋白 GAPDH の発現で標準化した値を示す。各検体 6~15 回の実験を行いそ
の平均を示している。各コントロールの値を1.0とし、各患者の値はコントロー ルに対する相対値を示す。
図 9 18F-fludeoxyglucose を投与した後撮影された P1 および P6 の PET 画像。
a. P1において、頸部、腹腔内リンパ節および脾臓組織へのグルコース取り 込み亢進が認められる。
b. P6において、頸部、腹腔内、腋窩リンパ節におけるグルコース取り込み 亢進が認められる。
図10 免疫不全症患者のTREC、KRECの値
a. PIK3CD変異患者およびPTEN変異患者のTREC、sjKREC値。患者は 以 下 の よ う に 分 類 さ れ た 。TREC(+)/KREC(+):P1—P4、P7、P9。 TREC(-)/KREC(+):P5、P6、P8
b. PIK3CD E525A変異をもつ家系において、11歳のP7 と12歳のP9は TREC(+)/KREC(+)であるが、40歳のP8はTREC(-)/KREC(+)である。
c. PIK3CD E1021K変異をもつP5は5歳時にはTREC(+)/KREC(+)であっ たが、8歳時にはTREC(-)/KREC(+)であった。
図11 PTEN機能喪失変異とAPDSについて模式図
PIK3CDおよびPIK3R1変異によるPI3Kの機能亢進は、PIP2からPIP3産 生を促進し、AKT、mTOR、リボゾーマルプロテインS6(RPS6)タンパクのリン 酸化を亢進させる。同様に、PIP3 を PIP2 へ変換する PTEN の機能喪失は、
AKT、mTOR、S6タンパクのリン酸化を亢進させる。
図12 PTEN変異とAPDS類似免疫不全症 (APDS-L) について模式図 PTEN の機能喪失により発症する PTEN 過誤腫症候群では、巨頭症、過 誤腫、さまざまな腫瘍発生という症状を呈する。それらに加え、APDS類似 の免疫不全症 (APDS-L) を発症することを今回証明した。
Angulo. Science, 20131) Lucas. Nature Imm. 20132)
(n = 17) (n = 9)
細菌感染 14 (93) 17 (100) 9 (100)
日和見感染 7 (47) 4 (24) NI.
リンパ組織腫脹 12 (80) NI. 9 (100)
リンパ節腫脹 12 (80) NI. NI.
肝脾腫 7 (47) 10 (59) 5 (56)
消化管症状 7 (47) NI. 6 (67)
血球減少 6 (40) NI. 3 (33)
汎血球減少 1 (7) NI. NI.
貧血 1 (7) NI. NI.
血小板減少 5 (33) NI. NI.
リンパ腫 2 (13) 1 (6) 2 (22)
MALT リンパ腫 1 (7) NI. NI.
EBウイルス関連Bリンパ腫 1 (7) NI. 2 (22) 低ガンマグロブリン血症 6 (40) NI. 4 (50)* 高IgM血症 11 (73) 14 (82) 3 (38)* NI. :no information
*8例中の割合(1例情報なし)
表1 国内および海外のPIK3CD変異患者の臨床症状 臨床症状 本検討(n = 15)
症例数 (%)