2015年度(平成27年度) 修士学位論文
ファージ感染機構解明のための LPS 生合成に関与する 遺伝子を欠失した大腸菌変異株の作成法の研究
三重大学大学院生物資源学研究科 博士前期過程
生物圏生命科学専攻 生命機能科学講座 生理活性化学教育研究分野
清水 陽介
1
目次
第 1 章 緒論
第1節 バクテリオファージX174および感染における スパイクタンパク質の重要性
4
第2節 リポ多糖(LPS)の構造について 6
第3節 研究の背景および目的 9
第 2 章 本論
第1節 E.coli C株のゲノムDNAに存在するgalE遺伝子配列の同定 12
第1項 E.coli C株のゲノムDNA抽出 14
第2項 E.coli C株のゲノムDNAに存在するgalE配列同定のための
プライマー設計およびPCRによるgalE配列増幅実験 15
第2節 Degenerate PCR法によるE.coli C株のゲノムDNAに存在する galE配列の同定
16
第1項 E.coli C株に存在するgalE配列増幅のための
Degenereteプライマーの設計およびDegenerate PCR
17
第2項 PCR遺伝子断片のシークエンス解析 21
第3節 糖転移酵素を欠損させたE.coli F2513株変異体の作製法の検討 22 第1項 PCRを用いたE.coli F2513株のwaaWおよびwaaT配列の確認 23
2
第2項 標的遺伝子の相補的配列を含む薬剤耐性遺伝子断片の作製 27 第3項 L-Arabinose添加によるE.coli F2513株の増殖に与える影響 30 第4項 コンピテントセルの作製および形質転換 31 第5項 エレクトロポレーションを用いたpKD46による
E.coli F2513株の形質転換 33
第6項 エレクトロポレーション法によるwaaT欠失
E.coli F2513株変異体の作製 34
第 3 章 結論
35第 4 章 実験の部
第1節 E.coli C株のゲノムDNAに存在するGALE遺伝子配列の同定 39
第1項 E.coli C株のゲノムDNA抽出 39
第2項 E.coli C株のゲノムDNAに存在するgalE配列同定のための
プライマー設計およびPCRによるgalE配列増幅実験 40 第2節 Degenerate PCR法によるE.coli C株のゲノムDNAに存在する
galE配列の同定 41
第1項 E.coli C株に存在するgalE配列増幅のための
Degenereteプライマーの設計およびDegenerate PCR 41 第2項 PCR遺伝子断片のシークエンス解析 42 第3節 糖転移酵素を欠損させたE.coli F2513株変異体の作製法の検討 45 第1項 PCRを用いたE.coli F2513株のwaaWおよびwaaT配列の確認 45 第2項 標的遺伝子の相補的配列を含む抗生耐性遺伝子断片の作製 47
3
第3項 L-ArabinoseがE.coli F2513株の増殖に与える影響 49 第4項 コンピテントセルの作製および形質転換 50 第5項 エレクトロポレーションを用いたpKD46による
E.coli F2513株の形質転換 52
第6項 エレクトロポレーション法によるwaaT欠失
E.coli F2513株変異体の作製 53
謝辞
54参考文献
554
第 1 章 緒論
第1節 バクテリオファージX174および感染における スパイクタンパク質の重要性
バクテリオファージX174は1959年に発見され、有機体の中ではじめて完全 な塩基配列が確認され、古くからモデル生物として研究が行われている 1, 2)。
X174は正20面体構造をしており、12箇所の頂点にスパイクと呼ばれるタンパ
ク質(Gタンパク質およびHタンパク質から形成)が存在している(Figure 1-1)。 このウイルスは、大腸菌やサルモネラ菌などの腸内細菌科のラフ株を宿主細菌 としており、宿主細菌に対する感染の初期過程において、①可逆的吸着②不可 逆的吸着③DNAの挿入という3つの段階を経ると考えられている3)(Figure 1-2)。
X174 がこの 3 段階を起こす際、スパイクタンパク質が以下のように重要な役
割を担うと考えられている3~5)。
①「可逆的吸着」
宿主細菌の細胞表面に存在するリポ多糖(LPS)を認識し、吸着する。
②「不可逆的吸着」
スパイクGタンパク質の高次構造が変化し、ファージ粒子とLPSを解離させ ないようにする。
③「DNAの挿入」
スパイクタンパク質の働きにより、X174の一本鎖DNAが宿主細菌内に挿 入される。
3段階目のDNA挿入に関与するスパイクHタンパク質は、2013年まで単量体 として機能すると考えられていた。しかし、2013年、Faneら 6)によって、H タ
5
ンパク質の一部についてX線構造が解析され、チューブ10量体を形成すること が確認されている。
Figure 1-1 X174の模式図
Figure 1-2 X174の感染機構
6
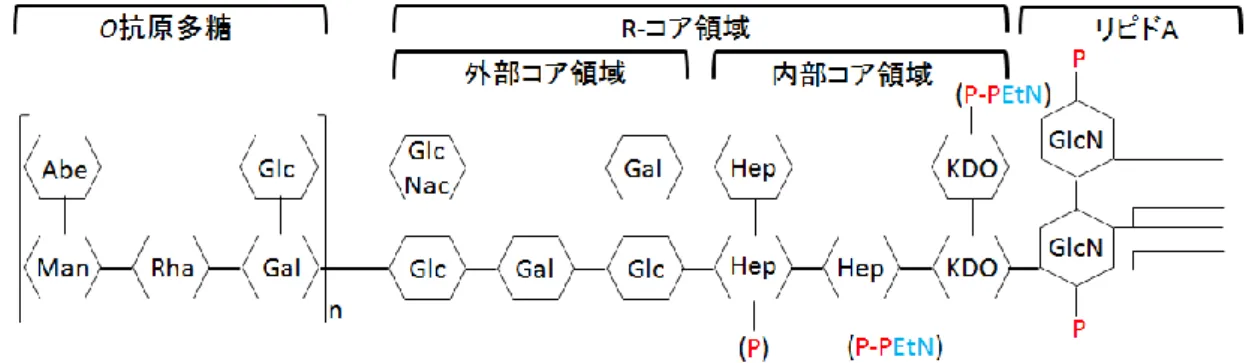
第2節 リポ多糖(LPS)の構造について
リポ多糖(LPS;lipopolysaccharide)は、グラム陰性細菌の外膜を構成する主 要成分である。LPS の一般的な構造は、①グルコサミンに結合した脂肪酸から なるリピドA、②10から15残基のヘキソースやヘプトースが結合したR-コア、
③糖鎖の繰り返し単位であり、菌体の抗原性を示すO-抗原多糖という3 つの領 域から構成される。LPS のR コア領域は、ゲノム DNA上の waa クラスターか ら発現する糖転移酵素によって生合成される7, 8)(Figure 1-3)。また、R-コア領 域は内部コアおよび外部コア領域に分けられる 9, 10)。内部コア領域は、KDO
(3-deoxy-D-manno-octulosonic acid)とL-glycero-D-manno-heptoseから構成され、
この構造はグラム陰性腸内細菌の間で保存性が高い。一方、外部コア領域は、
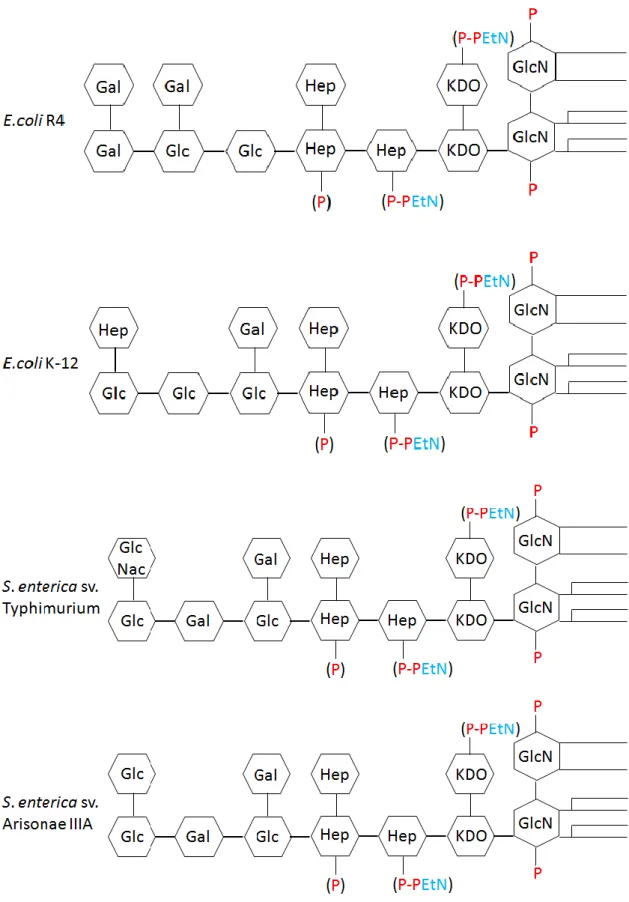
ヘキソサミンやヘキソースから構成されており、糖の結合様式や構成する糖の 種類に多様性がある。外部コア領域の多様性を示す例として、大腸菌の外部コ ア領域の構造をまとめた。大腸菌のR-コア領域は5つの構造が確認されており、
E.coli R1~R4およびE.coli K-12コアと呼ばれている。また、大腸菌と同じ腸内 細菌科のサルモネラ菌が持つ外部コア領域は2つ確認されている8)(Figure 1-4)。
このうち、X174は、外部コア領域がE.coli R1コアのE.coli C株に最もよく 感染する。更に、E.coli R1~R4 コアを持つ大腸菌とサルモネラ菌についても
X174の宿主となるが、E.coli K-12コア持つ大腸菌(E.coli K-12株)は宿主に ならないことが判っている11)。
7
Figure 1-3 S. enterica sv. TyphimuriumのLPSの模式図
8
Figure 1-4 大腸菌およびサルモネラ菌のRコア糖鎖の模式図
9
第3節 研究の背景および目的
第 2 節で上述した通り、X174 は大腸菌やサルモネラ菌などの腸内細菌を宿 主としているが、どの大腸菌にも感染できるわけではなく、特定の腸内細菌の みを宿主とする宿主特異性を持っている。この宿主特異性が何により決まるの かは、多くの研究者がさまざまなアプローチによって、明らかにしようと試み てきた。さまざまなアプローチがある中で、X174 が感染するために利用する 宿主の細胞膜上の分子(レセプター)に着目した研究を紹介したい。
1962年、FujimuraとKaesbergは、X174の宿主細菌から単離した細胞壁によ って、X174 が感染性を失う(不活化する)ことを確認した 12)。1971 年には、
Brown らが、リゾチーム処理によって細胞壁を破壊した宿主細菌を作製し、こ
れにX174 が吸着、エクリプスを起こさないことを明らかにした 13)。このよう な研究を通じ、X174 のレセプターは細胞壁を構成する部分に存在することが 示唆された。さらに、IncardonaやJazwinskiは、X174の宿主細菌の外膜に存在 するLPSを単離精製後、X174と混合することで、X174が不活化することを 明らかにし、X174のレセプターがLPSであることを証明した14, 15)。それ以降、
LPS の構造に着目し、X174 の宿主特異性を明らかにしようとする研究が行わ れた。
1975年、JazwisnskiがO抗原を持たない完全なR-コアを持つS.Typhimiriumお よび変異によって R-コアの非還元末端の N-アセチルグルコサミンが欠損した
S.TyphimiriumにX174を感染させたところ、非還元末端のN-アセチルグルコサ
ミンが欠損した変異株において、X174 の結合能が著しく低下することを明ら かにした16)。また、1976年には、FeigeとStirmが、自発性の突然変異によって
R-コアの糖鎖構造が変化した E.coli C 株変異体にX174を感染させる実験を行
10
った。その結果、非還元末端のガラクトースが欠損した変異株に対し、X174 の感染力が低下することが確認された 17)。このような研究結果から、X174 が 宿主を選択する際、LPS の構造、とくに外部 R-コア糖鎖の非還元末端付近を認 識すると考えられるようになった。
当研究室では、LPS の構造、特に外部コア領域の構造に着目し、X174 の宿 主特異性を明らかにするための研究が行われてきた。
外部Rコア領域5糖を化学合成によって作製し、これを用いてX174の不活 化の阻害が起こるかという研究が行われた。その結果、五糖の合成には至らな かったが、小島、小木曽らによって、非還元末端から二糖、三糖、四糖、非還 元末端のガラクトースが欠けた四糖、および分岐グルコースを含む先端四糖の 合成に成功した。そのうち、二糖と三糖を用いて、X174 の不活化実験を行っ た結果、三糖に対してX174 が相互作用を起こし、LPS によるファージの不活 化が阻害されることが判った。18~20)。また、貝谷、加藤はE.coli C株のLPSを 酢酸や水酸化カリウムで処理して得た脱アシル LPS により、LPSによるファー ジの不活化が強く阻害されることを明らかにした21, 22)。
また、LPS の外部コア領域の非還元末端側の糖鎖に焦点をあてた研究では、
若嶋が非還元末端のグルコサミンから段階的に糖鎖が欠落した 8 種類のサルモ ネラ菌の変異株を用いて、これらの変異株とX174 の感染効率を明らかにする 実験が行われた。その結果、完全な R-コア領域を持つサルモネラ菌に対するフ ァージの吸着効率を100 %とした時、非還元末端のグルコサミンが欠損した変異 株に対して36 %、非還元末端から2糖以上が欠損した変異株に対しては0 %と なり、非還元末端の糖鎖と非還元末端から 2 糖の糖鎖が、X174 の感染成立を 決める上で重要な役割を担う可能性を示唆した 23)。そこで、冨田はX174 の宿 主認識に関与すると考えられているスパイクタンパク質を用いて、完全な R-コ
11
ア領域を持つ LPS、非還元末端のグルコサミンのない LPS、非還元末端の 2 糖 のない LPS に対する相互作用解析を行い、スパイクタンパク質単独ではどれだ けLPSを判別する能力があるかを調べた。しかし、Gタンパク質と各LPSの親 和力に大きな差がなかったため、X174がRb2以降では感染しない事実を簡単 に説明できず、X174の宿主特異性を解明するには至っていない24)。
そこで、当研究室では、従来とは異なった以下の方法で、X174 の宿主特異 性を明らかにしようと考えた。
(1)遺伝子工学的手法を用いて、LPS の生合成に必要な UDP 化糖の異性化酵 素および糖転移酵素を欠損させることで、LPS の糖鎖構造を人為的に改変した 大腸菌変異株を作製する。
(2)異性化酵素欠損変異株を培養する際、ラベル化された糖を混合することで、
安定同位体標識されたLPSを持つ大腸菌およびLPSを調製する。
(3)安定同位体標識されたLPSを持つ変異株およびLPSとX174およびX174 のスパイクタンパク質の相互作用について、NMRを用いて精密に解析すること
で、X174の宿主認識における特定の糖残基の重要性を決定する。
このうち、本研究では(1)をテーマとし、X174の宿主細菌であるE.coli C株
およびE.coli F2513株について、LPSの糖鎖構造を調節した変異株の作製を目指
した。
12
第 2 章 本論
第1節 E.coli C株のゲノムDNAに存在するgalE遺伝子配列の同定
LPS の糖鎖構造を人為的に改変した大腸菌変異株を作製するにあたり、大腸 菌の中で、X174の最も良好な宿主となるE.coli C株に着目した。E.coli C株の LPS は非還元末端にガラクトースが 2残基結合しており、それらがX174 の宿 主認識において特に重要であると考えられてきた 17, 23)。そこで、遺伝子工学的 手法を用いて、LPSの非還元末端に存在するガラクトースを欠損したE.coli C株 変異体を作製できないかと考えた。非還元末端のガラクトースは、培地中のグ ルコースがUDP-グルコースに変換され、UDP-galactose-4-epimerase(GalE)の働 きによって、UDP-ガラクトースに変換され、そのUDP-ガラクトースがガラクト ース転移酵素によってLPSに組み込まれることでもたらされる7, 25)。そのため、
ゲノムDNAのgalEを欠失させた場合には、GalEが発現しなくなり、UDP-グル
コースをUDP-ガラクトースへ異性化することができなくなる。したがって、ガ
ラクトースを加えない限り、LPS の非還元末端にガラクトースが組み込まれな
いE.coli C株変異体を作製できると考えた。そこで、変異株を作製する方法とし
て、Wannerら26)が開発したλファージ由来のリコンビナーゼの活性を利用する
以下の方法で遺伝子を欠失させることにした。
①大腸菌のゲノム上の欠失させたい標的遺伝子の塩基配列を確認し、標的遺伝 子および薬剤耐性遺伝子を持つプラスミドのどちらにも相補的な配列を持つ プライマーを設計する。
②薬剤耐性遺伝子を持つプラスミドを鋳型として、①で設計したプライマーを 用いてPCRを行い、標的遺伝子と相補的な配列を持った薬剤耐性遺伝子を有
13
するPCR産物を増幅する。
③標的遺伝子を持つ大腸菌のコンピテントセルを作製し、相同組み換え酵素を 発現する遺伝子を持つプラスミドを用いて形質転換する。
④エレクトロポレーションを用いて、②で作製したPCR産物を③の形質転換菌 に挿入する。
⑤④で作製した大腸菌の培養液にL-Arabinoseを添加し、組み換え酵素を発現 させることで、標的遺伝子と②で作製したPCR産物の相同組み換えを引き起 こさせる。
しかし、E.coli C株のgalE配列はこれまでに報告されていないため、PCR に
よるgalE配列の同定を試みた。
14
第1項 E.coli C株のゲノムDNA抽出
PCRの鋳型DNAとして使用するため、E.coli C株のゲノムDNAを抽出した。
E.coli C株を150 mLのLB液体培地に植菌し、終夜振とう培養した。翌日、遠
心分離によって、菌体を回収した後、10 mLのTE Buffer(10 mM Tris-EDTA Buffer、 pH=8.0)を添加し、液体窒素で急速に凍結させた。溶液が完全に凍っているこ とを確認した後、40 ˚Cの湯浴の中で溶液を解凍した。凍結と解凍を4回繰り返 した後、5 mLのTE Bufferおよびフェノールクロロホルムを入れ、懸濁した。
懸濁後、遠心分離によって上清を回収し、上清と等量のフェノールクロロホル ムを添加し、遠心分離した。この操作を4回繰り返して上清を回収した後、上 清にイソプロパノールを入れ、DNAを回収した。後日、回収したDNAの濃度 を測定し、642.7 ng/μLのゲノムDNAを得た。
15
第2項 E.coli C株のゲノムDNAに存在するgalE配列同定のための プライマー設計およびPCRによるgalE配列増幅実験
E.coli C株のgalE配列を同定するにあたり、GalEを発現する菌株間において、
galE配列が保存されているのではないかと考えた。そこで、ゲノム配列が解明 されているE.coli K-12株のgalE配列(GenBankTMのAccession number:AP009048、
Region:791461~792477)を基にプライマーを設計した(Table 2-1)。
Table 2-1 E.coli K-12株のgalE配列を基に設計したプライマー
Primer Sequence
予想 TM
値 (˚C)
GC 含 量 (%)
①forLife-GALE 5'-TCCATGTCACACTTTTCGC-3’ 60.7 44
②revLife-GALE 5’-AACTAGTAGGTGTAGCGG-3’ 63.3 50
③forSigma-GALE 5’-CATACCATAAGCCTAATGGAGCG-3’ 65.2 44
④revSigma-GALE 5’-CAACGGGATTAAATTGCGTCATGG-3’ 72.4 44
前項で抽出したE.coli C株のゲノムDNAを鋳型として、プライマー①、②お よび③、④を組み合わせて混合し、PCRにより、E.coli C株のgalE配列を増幅 させることを試みた。しかし、アガロースゲル電気泳動の結果、PCR 産物を確 認できなかった。このことから、設計したプライマーとE.coli C株の相同性が低 かったため、増幅できなかったと考えた。
16
第2節 Degenerate PCR法によるE.coli C株のゲノムDNAに存在する galE配列の同定
Degenerate PCR 法はタンパク質のアミノ配列の情報を基に未知の遺伝子をス
クリーニングする際に用いられるPCR法である27)。タンパク質のアミノ酸配列 の情報を基に未知の遺伝子をスクリーニングする場合、1種類のアミノ酸に対し て複数のコドンが対応しているため、アミノ酸をコードする可能性のあるオリ ゴヌクレオチドの塩基配列は複数あり、どれが未知遺伝子の配列に該当するか はわからない。しかし、推定されるすべての塩基配列を含むオリゴヌクレオチ ドの混合物を合成することは可能であり、このようなオリゴヌクレオチドをプ ライマーとして用いたPCRを行うことで、未知の遺伝子領域の増幅および同定 を行うことができる。このようなPCR法をDegenerate PCR法と呼び、この時に 使用されるプライマーをDegenerate primerと呼ぶ。
前節では、E.coli K-12株のgalE配列を基に設計したプライマーを用いて行っ たPCRでは、PCR産物を増幅させることができず、E.coli C株のgalE配列を同 定できなかった。そこで、Dengenerate PCR法によって、galE配列の同定を試み た。
17
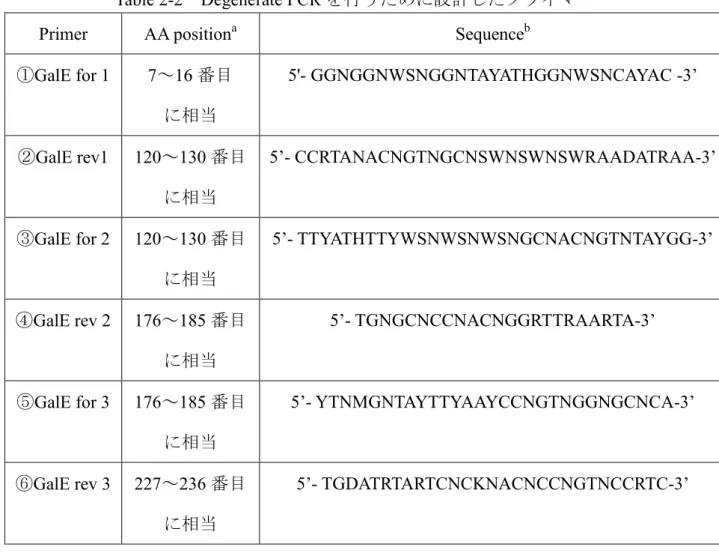
第1項 Degenereteプライマーの設計およびDegenerate PCR 法を用いた galE配列の探索
E.coli K-12株のGalEアミノ酸配列を基に、Protein-Ptorein BLASTによって、
大腸菌群のGalEアミノ酸配列のホモロジー検索を行い、相同性の高いアミノ酸 配列の領域を確認した(Figure 2-1)。
Figure 2-1 E.coli K-12 株のGalEアミノ酸配列
四角で囲っている部分は、大腸菌群のGalEアミノ酸配列上で相同性が高かった 領域を示している。
そして、その配列と特異的に結合するように Degenerate プライマーを設計 した(Table 2-2)。
設計したプライマーおよび鋳型としてE.coli C株のゲノムDNAを混ぜ合わせ、
PCR を行い、アガロースゲル電気泳動によって増幅した遺伝子断片の存在を確 認した。その結果、フォワードプライマーにプライマー①、リバースプライマ ーにプライマー②を用いてPCRを行った時、約360 bpの遺伝子断片を確認した
(Figure 2-2)。また、フォワードプライマーにプライマー③、リバースプライマ
ーにプライマー⑥を用いてPCRを行った時、約400 bpの遺伝子断片が増幅され
18
たのを確認した(Figure2-3)。
Table 2-2 Degenerate PCRを行うために設計したプライマー
Primer AA positiona Sequenceb
①GalE for 1 7~16番目 に相当
5'- GGNGGNWSNGGNTAYATHGGNWSNCAYAC -3’
②GalE rev1 120~130番目 に相当
5’- CCRTANACNGTNGCNSWNSWNSWRAADATRAA-3’
③GalE for 2 120~130番目 に相当
5’- TTYATHTTYWSNWSNWSNGCNACNGTNTAYGG-3’
④GalE rev 2 176~185番目 に相当
5’- TGNGCNCCNACNGGRTTRAARTA-3’
⑤GalE for 3 176~185番目 に相当
5’- YTNMGNTAYTTYAAYCCNGTNGGNGCNCA-3’
⑥GalE rev 3 227~236番目 に相当
5’- TGDATRTARTCNCKNACNCCNGTNCCRTC-3’
a AA positionはGalEアミノ酸配列の相同性が高い領域のN末端からの番号で示 す。この領域を参考にDegenerate primerを設計した。
b 宿重塩基を表す記号および構成する塩基を表した。 N : A, G, C, T; W : A, T;
S : G, C; Y : T, C; H : A, C, T; R : A, G; D : A, G, T; M : A, C; K : T, G
19
Figure 2-2 E.coli C株のゲノムDNAを鋳型にプライマー①、②を用いて 行ったPCR産物の2 %アガロースゲル電気泳動
M. DNA Marker,1-3. PCR産物
PCR の反応条件:Predenature, 94˚C, 2min; つづく 35 サイクル, Denature, 94˚C,45sec;
Anealing, Lane 1 53˚C, 45 sec, Lane 2 51.8 ˚C, 45 sec, Lane 3 51˚C, 45 sec;
Extention, 72˚C, 30 sec
20
Figure 2-3 E.coli C株のゲノムDNAを鋳型にプライマー③、⑥を用いて 行ったPCR産物の2 %アガロースゲル電気泳動
M. DNA Marker 1-3. PCR産物
PCRの反応条件:Predenature, 94˚C, 2min; つづく35サイクル, Denature, 94˚C,45sec; Anealing, Lane 1 56˚C, 45 sec, Lane 2 55.1 ˚C, 45 sec, Lane 3 54.5˚C, 45 sec;
Extention, 72˚C, 30 sec
21
第2項 PCR遺伝子断片のシークエンス解析
PCRによって増幅された約360 bpおよび約400 bpの遺伝子断片の塩基配列を 解析するため、GFX PCR DNA and Gel Band Purification Kit(GE Healthcare社)を 用いて、アガロースゲルから約360 bpおよび約400 bpの遺伝子断片を回収した。
その断片を市販のベクターであるpTA2 ベクター(TOYOBO社)に挿入し、コ ンピテントセルE.coli JM109株を形質転換した。形質転換菌をSOC培地に添加 し、培養後、アンピシリン50 μg/mLを含むLB寒天培地に塗布し、一晩倒置培 養した。翌日、コロニーを爪楊枝で掻き取り、アンピシリン50 μg/mLを含む5 mL のLB液体培地で培養し、得られた菌体からプラスミドを抽出した。このプラス ミドの DNA 塩基配列を解析した。その結果、約 360 bp の遺伝子断片は UDP-2,3-diacylglucosamine hydrolase をコードする遺伝子領域の一部と予想され たものであった。また、約 400 bp の遺伝子断片は 3,4-dihydroxyhenylacetate
2,3-dioxygenaseをコードする遺伝子領域の一部と予想され、どちらの遺伝子断片
もE.coli C株のgalE配列ではなかった。このことから、今回設計したプライマ
ーとE.coli C株のgalE配列の相同性が低く、PCRの最中、上記の2つの遺伝子
領域に結合してしまい、galE配列を増幅できなかったと考えた。
22
第3節 糖転移酵素を欠損させたE.coli F2513株変異体の作製法の検討
LPS の生合成において、非還元末端ガラクトースの結合を担う糖転移酵素で ある UDP-galactose:(Galactosyl)LPS α-1,2-galactosyltransferase WaaW および UDP-galactose:(glucosyl) LPS α-1,2-galactosyltransferase WaaTに着目した。WaaW は、非還元末端のガラクトースをLPS糖鎖に結合し、WaaTは非還元末端側から 2 残基目のガラクトースを LPS 糖鎖に結合する時に働く糖転移酵素である。そ のため、この2つの酵素を発現するwaaWおよびwaaTを欠失させることで、非 還元末端のガラクトースを欠損した LPS を持つ大腸菌変異体を作製できると考 えた。変異体を作製する方法は、前述したWannerら26)が開発した方法で遺伝子 を欠失させることにした。この実験で変異を導入する大腸菌は、X174 の宿主 となり、かつ、waaW、waaTの配列が同定されているE.coli F2513株を選択した。
23
第1項 PCRを用いたE.coli F2513株のwaaWおよびwaaT配列の確認
E.coli F2513 株の変異株を作製するためには、ゲノム上の waaW および waaT
の正確な塩基配列を知る必要がある。そこで、研究室でグリセロールストック として保存されているE.coli F2513保存株のwaaWおよびwaaTに該当する配列 を確認することで、Whitfieldら7)の論文に報告されている株と研究室の保存菌株 の配列が同じであるか確認することにした。
第2章、第1節、第2項と同じようにE.coli F2513株から24.3 ng/μLのゲノム DNAを抽出した。そのゲノム DNAを鋳型としてGenBankTMのデータベース上 に登録されている E.coli F2513 株から waaW 配列(GenBankTM の Accession number:AF019746、Region:3356~4381)およびwaaT配列(GenBankTMのAccession number:AF019746、Region:5109~6104)を抜き出し、それらを基に設計したプラ イマーを混ぜ合わせ、PCR を行い、アガロースゲル電気泳動によって目的の遺 伝子配列が存在するかを確認した(Table 2-3)。その結果、フォワードプライマ ーにプライマー①、リバースプライマーにプライマー②を用いてPCRを行った 時、目的とする約900 bpの遺伝子断片が増幅されたことを確認することができ
た(Figure 2-4)。また、フォワードプライマーにプライマー③、リバースプライ
マーにプライマー④を用いてPCRを行った時にも、目的とする約900 bpの遺伝 子断片を確認した(Figure 2-5)。
その後、第2 章、第 2 節、第 4項と同じように、アガロースゲル電気泳動か ら遺伝子断片を回収し、コンピテントセルE.coli JM109株を形質転換した。形質 転換菌からプラスミドを抽出し、遺伝子断片の塩基配列を解析した。その結果、
GenBankTMのデータベース上に登録されている E.coli F2513 株の waaW および waaT配列と100 %一致した。このことから、研究室に保存されているE.coli F2513
24
株のゲノム上にあるwaaWおよびwaaT配列はWhitfieldら7)の論文と同一であり、
かつ、突然変異が起きていないことを確認した。
Table 2-3 E.coli F2513株のwaaWおよびwaaT配列に相補的なプライマー
Primer 予想
TM値
(˚C)
GC含 量
(%)
①WaaT for 5’- CCCCAGAGCTAAATGTTTCCTACGGTAT-3’ 61.1 46.4
②WaaT rev 5’- GCCTTTTATATACTCACCATGGGCAAACAG-3’ 61 43.3
③WaaW for 5’- ATGGATTTATTAGCTGAGAGTATTACTGAAGTC-3’ 58.4 33.3
④WaaW rev 5’-TGATACTGCTTTTCATTCGTGGCTGGAATA-3’ 59.6 40
25
Figure 2-4 E.coli F2513株のゲノムDNAを鋳型にプライマー①、②を用いて
行ったPCRの2 %アガロースゲル電気泳動
M. DNA Marker, 1-5. PCR産物,
矢印はPCR産物の位置を示している。
PCR の反応条件:Predenature, 94˚C, 2min; つづく 35 サイクル, Denature, 94˚C,15sec;
Anealing, Lane 1 58˚C, 30 sec, Lane 2 58.8 ˚C, 30 sec, Lane 3 60.5˚C, 30 sec Lane 4, 61.2 ˚C, 30 sec, Lane 5, 62 ˚C, 30 sec; Extention, 68˚C, 1 min
26
Figure 2-5 E.coli F2513株のゲノムDNAを鋳型にプライマー③、④を用いて
行ったPCRの2 %アガロースゲル電気泳動
M. DNA Marker, 1-5. PCR産物
矢印はPCR産物の位置を示している。
PCR の反応条件:Predenature, 94˚C, 2min; つづく 35 サイクル, Denature, 94˚C,15sec;
Anealing, Lane 1 58˚C, 30 sec, Lane 2 58.8 ˚C, 30 sec, Lane 3 60.5˚C, 30 sec Lane 4, 61.2 ˚C, 30 sec, Lane 5, 62 ˚C, 30 sec; Extention, 68˚C, 1 min
27
第2項 標的遺伝子の相補的配列を含む薬剤耐性遺伝子断片の作製
前項で、研究室で保存されているE.coli F2513株のwaaTおよびwaaWの塩基 配列を確認したことから、つぎにwaaT配列を欠失させたE.coli F2513株変異体 を作製するため、PCRによって、waaT配列の上流および下流50 bpに相補的な 配列を含むクロラムフェニコール耐性を持った遺伝子断片を作製することにし た。PCRを行う鋳型として、クロラムフェニコール耐性遺伝子を持つプラスミ
ドpKD3(大阪大学 理学研究科 の米崎哲朗教授より譲渡されたプラスミド)
を用いた。そのため、プライマーを設計する際、waaT配列の上流および下流50 bpに相補的な配列を持ち、pKD3のクロラムフェニコール耐性遺伝子を増幅す るプライマーを作製した。(Table 2-4)。このプライマーとpKD3を混ぜ合わせ、
PCRにより、クロラムフェニコール耐性遺伝子を持つ領域を増幅した。アガロ ースゲル電気泳動によって、PCRで増幅された遺伝子断片が目的とする塩基配 列の長さであるかを確認した結果、目的とする約1.1 kbpにバンドを確認した
(Figure 2-6)。得られたDNA断片をエタノール沈殿により精製し、これをE.coli F2513株waaT欠失用DNA断片とした。
28
Table 2-4 waaT配列およびpKD3に相補的なプライマー
Primer
予想TM値
(˚C)
GC含量
(%)
WaaT noc F 70bpver
5'-TATTTATTATTTTTATTTAAAGATAA TAAAATGAAATTGAGAAAAGTTAAG
TGTAGGCTGGAGCTGCTTC-3’
62.7
24.3
WaaT noc R 70bpver
5’-TAATAAAATGAGAAGCCGCGATAG CGTATACTTGTAATCATAAGAGTAATA
TGGGAATTAGCCATGGTCC-3’
68.0 37.1
29
Figure 2-6 pKD3にTable 2-4のプライマーを用いて行った
PCR産物の2 %アガロースゲル電気泳動
M. DNA Marker, 1. PCR産物,
矢印はPCR産物の位置を示している。
PCR の反応条件:Predenature, 95˚C, 7min; つづく 40 サイクル, Denature, 94˚C,15sec;
Anealing, Lane 1 66˚C, 30 sec; Extention, 72˚C, 90 sec
30
第3項 L-Arabinose添加によるE.coli F2513株の増殖に与える影響
第2節で上述した方法で遺伝子を欠失させる過程で、標的遺伝子を持つ大腸 菌のコンピテントセルにλファージ由来の相同組換え酵素を発現するプラスミ ドを組み込む。その組換え酵素はL-Arabinoseによって発現するオペレーターに 接続されているため、酵素の発現にはL-Arabinoseを添加することが必須となる。
しかし、大腸菌によっては、L-Arabinoseを添加することで死滅するものが存在 する28)。そこで、E.coli F2513株がL-Arabinoseに対して耐性があるかを確認し た。
終濃度10 mMになるようにL-Arabinoseを添加した2 mLのLB液体培地およ びL-Arabinose未添加の2 mLのLB液体培地を用意した。そして、それぞれの培 地にE.coli F2513株を植菌し、一晩培養した後、吸光度を測定した。L-Arabinose 非添加の培養液のOD600は0.342であったのに対し、L-Arabinoseを添加した培養 液のOD600は0.460であった。この結果から、L-Arabinoseの添加によってE.coli
F2513株の増殖は妨げられないことが確認できた。
31
第4項 コンピテントセルの作製および形質転換
第2節で上述した方法で遺伝子を欠失させる過程で、標的遺伝子を持つ大腸 菌のコンピテントセルを作製し、相同組換え酵素を発現するプラスミドを用い て、形質転換する必要がある。そこで、E.coli F2513株のコンピテントセルを作 製し、相同組換え酵素をコードするpKD46(大阪大学 理学研究科 米崎哲朗 教授より譲渡されたプラスミド)を用いて、形質転換を試みた。コンピテント セルの作製方法は、MandelとHiga29)によって開発された方法を参考した。
E.coli F2513株を植菌した終夜培養液を50 mLのLB液体培地に植菌し、OD600
が0.5になるまで振とう培養した。その後、培養液を冷却し、遠心分離によって 菌体を回収した。そこに50 mM CaCl2を加え、懸濁後、氷浴で冷却した菌体を 遠心分離によって、回収した。そこに50 mM CaCl2/20 %グリセロールを加えて、
1.5 mLの遠心チューブに分注し、液体窒素で急速冷凍したものをコンピテント
セルとした。続いて、相同組換え酵素を発現するpKD46を用いて、コンピテン
トセルE.coli F2513株を形質転換した。しかし、コロニーを得ることができなか
った。そこで、Inoue30)らによって開発されたコンピテントセルの作製法を参考 に、それを用いて、pKD46を有するE.coli F2513株の作製を試みた。
5 mLのLB寒天培地にE.coli F2513株をまき、37 ˚Cで終夜倒置培養した。培 地に生育したコロニーを、1 mLのSOB培地に懸濁し、終夜培養した後、50 mL の培地に菌体を植菌し、室温でOD600が0.6になるまで振とう培養した。培地を 氷中で10分間氷冷した後、遠心分離によって菌体を回収し、形質転換用緩衝液
(TB溶液)を加え、更に10分間氷冷した。遠心分離によって回収した大腸菌 にTB溶液およびDMSOを添加し、10分間冷却した後、1.5 mLチューブに分注 し、液体窒素で急速冷凍したものをコンピテントセルとした。このコンピテン
32
トセルに対して、pKD46を用いた形質転換を試みたが、コロニーを獲得するこ とができなかった。
この結果から、E.coli F2513株が、外部からの遺伝子を分解する何らかの酵素 が働き、pKD3を破壊してしまうため、形質転換が起こらなかったのではないか と考えた。もしくは、作製したコンピテントセルの形質転換効率が非常に低い と考えた。
33
第5項 エレクトロポレーション法を用いたpKD46による E.coli F2513株の形質転換
コンピテントセルの調製が困難な細胞に、外部からのDNAを導入する方法と して、エレクトロポレーション法が挙げられる。この原理を以下に記す。細胞 膜は等価回路を形成しているため、外部から強い電場を加えることで、その分 だけ膜表面に電荷が蓄積して膜に圧縮力を与える。この力が膜の弾性限界を越 えるほど強くなることで膜の一部に小さな孔があく。この状態はしばらくする と修復されるので、その前にDNAを加えて細胞に取り込ませる31)。
pKD46 を E.coli F2513 株に形質転換する際に通常のコンピテントセルを用い
た方法では形質転換体を得ることができなかった。そこで、エレクトロポレー ション法により、pKD46をE.coli F2513株に形質転換することを試みた。
グリセロールストックとして保存されているE.coli F2513株を10 mLのLB液 体培地に植菌し、終夜振とう培養した。125 mLのLB液体培地に終夜培養液を 植菌し、OD600が0.5になるまで振とう培養した。遠心分離よって、培養液から 大腸菌を回収した後、滅菌蒸留水で 4 回洗浄し、電気伝導度に影響を与える塩 を取り除いた上で濃縮した。これとpKD46を1 mm幅の大腸菌用キュベットに 混和し、25 μF、2.5 kV、200 Ωの条件でGene pulser(Bio-Rad社)によりエレク トロポレーションを行った。SOC培地を1 mL加えて37 ˚Cで1時間培養後、そ の菌液をアンピシリン入りLB寒天培地に10 μL、20 μL、50 μLまき、30 ˚Cで 終夜培養し、コロニーを取得した。この結果から、通常の形質転換法では得る ことのできなかったpKD46保有E.coli F2513株を調製できたと判断した。
34
第6項 エレクトロポレーション法によるwaaT欠失 E.coli F2513株変異体の作製
エレクトロポレーションによって、pKD46保有E.coli F2513株変異体に、E.coli F2513株waaT欠失用DNA断片を導入し、waaT欠失E.coli F2513株変異体の作 製を試みた。
L-Arabinoseおよびアンピシリンを添加したLB培地にpKD46保有E.coli F2513 株変異体を植菌し、30 ˚Cで終夜振とう培養した。L-Arabinoseおよびアンピシリ ンを添加したLB培地に終夜培養液を植菌し、30 ˚Cで振とう培養した後、OD600
が 0.18になったところで培養を止めて培養液を冷却し、遠心分離によって集菌
した。10 % グリセロール水溶液を用いて菌体を洗浄し、電気伝導度に影響を与
える塩を取り除いた上で濃縮した菌体をコンピテントセルとした。これとE.coli F2513株waaT欠失用DNA断片を混和し、大腸菌用キュベットに移した後、10 μF、
1.8 kV、600 Ωの条件でエレクトロポレーションを行った。処理した菌体にSOC
培地を加えて37 ˚Cで1時間培養した後、さらに室温で終夜培養し、その一部を クロラムフェニコール入りのLB寒天培地に植菌して37 ˚Cで3日間培養後、コ ロニーを確認した。これより、pKD46 から発現した相同組換え酵素が、E.coli F2513株のwaaT配列とE.coli F2513株waaT欠失用DNA断片の相同組換えを引 き起こし、waaT配列が欠失したE.coli F2513株を獲得できたと考えた。
35
第 3 章 結論
バクテリオファージX174のLPS認識機構を解明するため、大腸菌の LPS 生合成を利用して、安定同意標識された糖残基を含む LPS を調製し、NMR によって、X174との相互作用を精密に解析することができると考えた。そ こで、本研究では、X174 の宿主となる大腸菌(C株、F2513株)の LPS生 合成に関与する遺伝子(galE、waaW、waaT)を欠失させ、人為的に糖鎖構 造を改変した大腸菌変異体を作製する方法を検討した。
大 腸 菌 変 異 株 の 作 製 に あ た り 、 E.coli C 株 に 存 在 す る UDP-galactose-4-epimerase(GalE)に注目し、この酵素を発現する遺伝子領 域を欠失させた E.coli C 株変異体を作製しようと考えた。しかし、E.coli C 株のゲノムDNA配列は報告されていないため、galE 配列を同定する必要が あった。そこで、E.coli K-12と E.coli C株のgalE 配列には高い相同性がある のではないかと考え、ゲノム DNA 配列が報告されている E.coli K-12 株の galE配列を参考にプライマーを設計した。そして、このプライマーと E.coli C株のゲノムDNAを用いて、PCRを行い、galE 配列を増幅させることを試 みた。しかし、PCRによって、目的の遺伝子断片を増幅することができなか った。このことから、E.coli K-12 株とE.coli C株の galE配列は相同性が低い 可能性が考えられた。
つぎに、Degenerate PCR 法によって、galE 配列の増幅を試みた。BLAST を用いて報告されている GalE のアミノ酸残基間で高く保存されている領域 を見つけ、その部分を増幅する Degenerate primerを設計した。そして、これ
とE.coli C株のゲノム DNAを基にPCRを行い、2種類のDNA断片の増幅に
成功した。この DNA 断片を TA クローニング後、シークエンス解析によっ
36
て塩基配列をそれぞれ確認したが、どちらも大腸菌由来の galE 配列と一致 しなかった。このことから、設計したDegenerate primerが E.coli C株の galE 配列以外の遺伝子領域と高い相同性を持っており、PCR 反応中に、Degenerate
primerと特異的な結合が起きたのではないかと考えた。
E.coli C株由来のgalE配列を同定には至らなかったため、galE欠損E.coli C 変異体の作製は進まなかった。そこで、X174の宿主細菌であり、糖転移酵 素(WaaW、WaaT)を発現する遺伝子配列が報告されている E.coli F2513 株 に着目し、これらの糖転移酵素を欠損した E.coli F2513株変異体の作製を試 みた。
Whitfield ら 7)の E.coli F2513 株の waaW および waaT 配列と研究室で保存 されている菌株の配列が同じであるかを確認した。塩基配列のデータベース
上にあるE.coli F2513 株のwaaW、waaTを基に設計したプライマーと研究室
の保存菌株から抽出したゲノムDNAを用いて、PCRを行い、遺伝子断片を 増幅した。TA クローニング後、この断片の塩基配列を解析した結果、デー タベース上に登録されている E.coli F2513 株の waaT および waaW 配列と 100 %一致し、研究室の保存菌株が waaT およびwaaW配列が Whitfield7)らの 論文と同一であることを確認することができた。
続いて、PCR によって、waaT 配列に相補的な配列を含むクロラムフェニ コール耐性を持った遺伝子断片の作製を試みた。クロラムフェニコール耐性 遺伝子を有するpKD3をテンプレートDNAとし、waaTの上流および下流(50 bp)と相補的な配列を持ち、pKD3 の耐性遺伝子を増幅するプライマーを用 いてPCRし、目的のDNA断片(約 1.1 kbp)を増幅することができた。
pKD46 上にある相同組換え酵素の発現に必要な L-Arabinose に対して、
E.coli F2513株が耐性を持っているかどうかを確認した。L-Arabinoseを添加
37
した培地および添加していない培地に菌体を植菌し、終夜培養後の吸光度を 測定した結果、L-Arabinoseの有無に関わらず、吸光度が増加していた。これ
より、E.coli F2513株はL-Arabinoseに耐性を持っていることを確認すること
ができた。
また、E.coli F2513株に相同組換え酵素を発現する遺伝子を有するpKD46
を導入した。はじめに、Mandelと Higa29)によって開発された塩化カルシウ ム法によって、E.coli F2513株のコンピテントセルを作製し、pKD46を用い て、形質転換した。しかし、コロニーを得ることができなかった。そこで、
Inoueら 30)によって開発された高効率形質転換を可能にするコンピテントセ
ル作製法を基に、コンピテントセルを作製し、pKD46の導入を試みた。しか し、この方法でも形質転換体を獲得できなかった。そのため、エレクトロポ レーション法により、E.coli F2513株に pKD46を導入する方法を試みた。そ の結果、コロニーを獲得でき、pKD46が導入されたE.coli F2513株を獲得す ることができた。
最後に、エレクトロポレーション法によって、pKD46含有 E.coli F2513株
変異体にE.coli F2513株waaT欠失用遺伝子断片の導入を試みた。その結果、ク
ロラムフェニコールを含有したLB 寒天培地にコロニーを獲得し、waaT 配列が クロラムフェニコール耐性遺伝子に組み換えられたE.coli F2513株変異体の作製 に成功した。
本研究では、X174の宿主となるE.coli C株の GalEを欠損させるため、PCR を用いたgalE配列の同定を試みたが、同定には至らなかった。今後、E.coli C 株の全ゲノムシークエンスを解析する方法やゲノムのショットガンクロ ーニングを行うことで、目的とする galE 配列を同定できると考えている。
また、配列を同定した後、E.coli F2513 株のwaaT 配列を欠失させた方法を用
38
いることで、galE 配列を欠失させることができ、非還元末端側の 2つのガラ クトース残基が欠損したLPSを持つE.coli C株変異体を作製できると考えて いる。さらに、この変異体を、安定同位体標識されたガラクトースを含む LB 培地中で培養することによって、非還元末端側に 2 つの安定同位体標識 されたガラクトースを含むLPSを持ったE.coli C株変異体を作製することが できるのではないかと考えている。
また、E.coli F2513株のwaaT 配列を欠失させるための実験では、waaT 配
列が欠失した E.coli F2513 株変異体の作製を試み、waaT 配列が欠失した
E.coli F2513株変異体と考えられるコロニーを獲得することができた。今後、
この菌体のLPS を精製し、SDS-PAGEや LC-MS を用いることで、非還元末 端のガラクトースが2 残基欠損していることを確認したいと考えている。ま た、これらのことを確認した上で、waaT配列欠失E.coli F2513株変異体を作 製した方法を用いてwaaW 配列を欠失させることで、非還元末端のガラクト ースが欠損したLPSを持つE.coli F2513株変異体を作製できると考えている。
さらに、作製した E.coli C株および E.coli F2513株変異体から安定同位体標 識された糖残基を持つ LPS や糖鎖構が改変された LPS を単離精製し、それ
らとX174およびスパイクタンパク質との相互作用を NMR や BIACORE を
用いて解析することで、従来よりも精密に相互作用を解析することができ、
X174 の宿主認識における特定の糖残基の重要性を決定することに繋がる
と考えている。
39
第 4 章 実験の部
第1節 E.coli C株のゲノムDNAに存在するgalE遺伝子配列の同定
第1項 E.coli C株のゲノムDNA抽出
15 %グリセロールストックとして冷凍保存されているE.coli C株を150 mLの LB液体培地(1 % トリプトン、1 % NaCl、0.5 % 乾燥酵母エキス)に植菌し、
200 rpm、37 ˚Cの条件で終夜振とう培養した。4800 rpm、4 ˚C、15分間の条件で 遠心分離を行い、培養液から菌体を回収した。回収した菌体に10 mLの
Tris-EDTA Buffer溶液(10 mM Tris、1 mM EDTA、PH=8.0)を加え、ボルテック スで懸濁した。液体窒素によって懸濁液を急速に冷凍し、40 ˚Cで溶かすという 操作を4回繰り返した後、5 mLのTris-EDTA Bufferを加えた。そこへ懸濁液と 等量のフェノールクロロホルム(フェノール:クロロホルム:イソアミルアルコー ル=25:24:1)を加え、手を使って激しく上下に懸濁し、4800 rpm、4 ˚C、10分間 の条件で遠心分離し、上清を回収した。回収した上清に再び上清と等量のフェ ノールクロロホルム溶液を加え、4800 rpm、4 ˚C、10分間の条件で遠心分離を行 う操作を3回繰り返し、上清に混入している不純物を取り除いた。得られた処 理された上清を50 mLのコニカルチューブに移し、上清の2倍量のイソプロパ ノールを慎重に加え、ガラス棒を回転させながらDNAを巻き取った。巻き取っ たDNAに適量のTris-EDTA Bufferを添加して一晩静置し、DNAを回収した。
40
第2項 E.coli C株のゲノムDNAに存在するgalE配列同定のための プライマー設計およびPCRによるgalE配列増幅実験
PCR用チューブに、E.coli C株のゲノムDNAを0.07 μL、2 mM dNTPs(TOYOBO 社)を2.5 μL 、25 mM MgSO4(TOYOBO社)を1.5 μL、E.coli K-12のGalE配 列 を 参 考 に 設 計 し た 100 μM の フ ォ ワ ー ド プ ラ イ マ ー forLife-GALE
(5'-TCCATGTCACACTTTTCGC-3’) と 100 μM の リ バ ー ス プ ラ イ マ ー revLife-GALE
(
5’-AACTAGTAGGTGTAGCGG-3’)
を1.5 μL、KOD-Plus-溶液(TOYOBO社)を0.5 μL、10×Buffer for KOD-Plus-Ver.2(TOYOBO社)を2.5 μL、
滅菌水を14.93 μLそれぞれ加え、合計25 μLとした。
サーマルサイクラーを用いてPCRを行った。PCRの条件は、保持反応を94 ˚C で2分間行った後、1サイクルあたり変性反応を98 ˚Cで10秒、アニーリング 反応を68~57 ˚Cで30秒、伸長反応を68 ˚Cで1分のサイクルを35サイクル繰 り返した。反応後、2 %アガロースゲル電気泳動によってバンドの増幅の有無を 確認した。
41
第2節 Degenerate PCR法によるE.coli C株のゲノムDNAに存在する galE配列の同定
第1項 E.coli C株に存在するgalE配列増幅のための
Degenereteプライマーの設計およびDegenerate PCR
PCR用チューブにE.coli C株のゲノムDNAを0.1 μL、2 mM dNTP(TOYOBO
社)を2.5 μL、GalE配列を探索するために設計した100 μMのフォワードプライ
マー GalE for 1(5'-GGNGGNWSNGGNTAYATHGGNWSNCAYAC-3’)と100 μM のリバースプライマー GalE rev1
(5’-CCRTANACNGTNGCNSWNSWNSWRAADATRAA-3’)を0.5 μL、10×Buffer
(TOYOBO社)を2.5 μL、rTaq DNA Polymerase(TOYOBO社)を0.25 μL、滅 菌水を16.65 μLそれぞれ加え、合計25 μLとした。
サーマルサイクラーを用い、PCRにより、PCR産物の獲得を試みた。PCRの 条件は保持反応を94 ˚Cで2 分間行った後、1サイクルあたり変性反応を94 ˚C で45秒、アニーリング反応を52 ˚Cで45秒、伸長反応を72 ˚Cで15秒のサイ クルを35サイクル繰り返した。反応後、2 %アガロースゲル電気泳動により、
約360 bpにバンドを確認した。また、上述した方法で、アニーリング反応の温
度を54.5 ˚Cに変え、フォワードプライマーにGalE for 2
(5’-TTYAYHTTYWSNWSNWSNGCNACNGTNTAYGG-3’)とリバースプライマ ーにGalE rev 3(5’-TGDATRTARTCNCKNACNCCNGTNCCRTC-3’)を用いてPCR を行い、約400 bpにバンドを確認した。
42
第2項 PCR遺伝子断片のシークエンス解析
illustraTM GFXTM PCR DNA and Gel Band Purification Kit(GEヘルスケア・ジャ パン社)を用いて、PCRによって増幅した約360 bpおよび約400 bpのPCR産 物をアガロースゲルから切り出し精製を行った。PCR産物を供した2 %アガロ ースゲルからスパテルを用いて目的のバンドを切り出し、事前に計量した1.5 mL遠心チューブに入れた。再度、遠心チューブの重さを量り、切り出したゲル の重さを算出した後、ゲル10 mgにつき10 μL(ゲルが300 mg以下の場合は300 μL)のCapture buffer type 3を加え、転倒混和した。遠心チューブを60 ˚Cの湯 浴に入れて30分間インキュベート(10分に1度転倒混和)することで、ゲルを 溶解した。溶液の色が黄色であることを確認し、カラムと回収チューブが1セ ットになった専用チューブに溶液を移した。その後、室温で1分間静置および
15000 rpm、25 ˚C、1分間の条件で遠心分離し、遺伝子断片をカラムに吸着させ
た。続いて、500 μLのWash buffer type1を添加し、15000 rpm、25 ˚C、1分間の 条件で遠心分離する操作を二回繰り返し、カラムの洗い込みを行った。最後に、
カラムを新しい1.5 mLの遠心チューブに移し、Elution buffer type 4を50 μL添加 し、1分間静置した後、15000 rpm、25 ˚C、1分間の条件で遠心分離し、カラム 内のDNAを流し出して回収した。回収したDNA溶液の濃度を測定した結果、
6.2 ng/μLであった。
精製したDNAはTarget CloneTM(TOYOBO社)を用いてpTA2 ベクター
(TOYOBO社)にライゲーションした。0.6 mLの遠心チューブに、精製した
DNAを4.03 μL、2×Ligation Buffer(TOYOBO社)を5 μL、pTA2 ベクター(50 μg/μL、
TOYOBO社)を1 μL、T4 DNA Ligase(TOYOBO社)を10 μL加え、計11.03 μL とし、室温で30分間静置し反応させた。
43
続いて、コンピテントセル(大腸菌JM109株)100 μLに反応させた溶液すべ てを加え、氷上で30分間静置した。42 ˚Cの恒温槽で30秒ヒートショックを与 え、再度、氷上で2分間静置した。SOC培地(2 % トリプトン、1 % 1M MgCl2、 1 % 1M MgSO4、0.5 % 乾燥酵母エキス)を900 μL加え、175 rpm、37 ˚C の条件 で1時間振とう培養した。その後、100 μLの培養液を50 μg/mLのアンピシリン を含むLB寒天培地(1.5 %寒天、1 % トリプトン、1 % NaCl、0.5 % 乾燥酵母 エキス)に塗布し、37 ˚Cで一晩倒置培養した。
50 μg/mLのアンピシリンを含む5 mLのLB液体培地に、終夜培養した大腸菌
のコロニーを植菌し、200 rpm、37 ˚Cの条件で終夜振とう培養した。その後、培
養液を1.5 mL遠心チューブに移し、13000 rpm、4 ˚C、3分の条件で遠心分離を
繰り返し、菌体を回収した。得られた菌体から、Mag ExtractorTM -plasmid-
(TOYOBO社)を用いてプラスミドDNAの単離および精製を行った。回収し
た菌体に150 μLの再懸濁液を加え、ボルテックスで60 秒間攪拌した後、150 μL
の溶解液(溶解液Ⅰ:溶解液Ⅱ=4:1)を加えて5回転倒混和し、氷上で5分間静 置した。そこに、120 μLの中和液を加え、5回転倒混和し氷上で5分間静置した 後、13000 rpm、4 ˚C、5分の条件で遠心分離し、上清を新しい1.5 mLの遠心チ ューブに移した。その上清に500 μLの吸着液と30 μLの磁性ビーズⅡを加え、
60秒間ボルテックスで攪拌し、15000 rpm、4 ˚C、5分の条件で遠心分離して上 清を除去した。そこに、700 μLの70 %エタノールを加え30秒間ボルテックス で攪拌し、15000 rpm、4 ˚C、5分の条件で遠心分離して上清を捨てる操作を2 回繰り返した。さらに500 μLのエタノールを加え、30秒間ボルテックスで攪拌 し、15000 rpm、4 ˚C、5分の条件で遠心分離し、上清を除去した。その後、78 ˚C の湯浴で加熱し、遠心チューブ内のエタノールを完全に蒸発させ、溶出液を50 μL加え、60秒間ボルテックスで攪拌して15000 rpm、4 ˚C、20分の条件で遠心
44
分離し、プラスミドを回収した。回収したプラスミドは、三重大学生命科学研 究支援センターのトータルシークエンスサービスを利用し、塩基配列を解析し た。
45
第3節 糖転移酵素を欠損させたE.coli F2513株変異体の作製法の検討
第1項 PCRを用いたE.coli F2513株のwaaWおよびwaaT配列の確認
第2章、第1節、第2項と同じ操作方法で、E.coli F2513株のゲノムDNA を抽出した。PCR用チューブに回収したゲノムDNAを0.5 μL、2 mM dNTP
(TOYOBO社)を2.5μL、waaT配列と相補的なフォワードプライマー WaaT for 1(5'- CCCCAGAGCTAAATGTTCCTACGGTAT-3’)とリバースプライマー WaaT rev1(5’-GCCTTTTATATACTCACCATGGGCAAACAG-3’)を0.5 μL、10×KOD Buffer(TOYOBO社)を2.5 μL、25 mM MgSO4(TOYOBO社)を1.5 μL、KOD DNA Polymerase(TOYOBO社)を0.5 μL、滅菌水を16.5 μLそれぞれ加え、合 計25 μLとした。
サーマルサイクラーを用い、PCRにより、waaT配列の増幅をした。PCRの条件 は保持反応を94 ˚Cで2 分間行った後、1サイクルあたり変性反応を94 ˚Cで15 秒、アニーリング反応を62 ˚Cで30秒、伸長反応を68 ˚Cで1分のサイクルを 35サイクル繰り返した。反応後、2 %アガロースゲル電気泳動により、約900 bp にバンドを確認した。また、上述した方法で、フォワードプライマーをWaaW for
(5’-ATGGATTTATTAGCTGAGAGAGTATTACTGAAGTC-3’)に変え、リバース プライマーをWaaW rev(5’-TGATACTGCTTTTCATTCGTGGCTGGAATA-3’)に 変えてPCRを行い、約900 bpのバンドを確認した。これらのPCR産物を、
illustraTM GFXTM PCR DNA and Gel Band Purification Kit(GEヘルスケア・ジャパ ン社)を用いて、アガロースゲルから切り出し、精製した。精製したDNAは Target CloneTM(TOYOBO社)を用いてpTA2 ベクター(TOYOBO社)にライ ゲーションした。
46
続いて、コンピテントセル(大腸菌JM109株)100 μLにライゲーション反応 させたすべての溶液を添加し、37 ˚Cで1時間振とう培養した。その後、50 μg/mL のアンピシリンを含むLB寒天培地に100 μL塗布し、37 ˚Cで一晩倒置培養した。
終夜培養した寒天培地に生えたコロニーに、インサートを有するプラスミド pTA2が存在するかを、PCRによって確認した。終夜培養した寒天培地からコロ ニーを選択し、2×Quick Taq HS DyeMix(TOYOBO社)を12.5 μL、pTA2に相補 的なフォワードプライマー M13 for (5’-GTAAAACGACGGCCAGT-3’)とM13 rev(5’-GGAAACAGCTATGACCA-3’)を0.5 μL、滅菌水を11.5 μL入れたPCR 用遠心管に塗布した。サーマルサイクラーを用いて、保持時間を94 ˚Cで2分行 った後、1サイクルあたりの変性反応を94 ˚Cで30秒、アニーリング反応を52 ˚C で10秒、伸長反応を68 ˚Cで1分間を1サイクルとして、PCR反応を35サイク ル繰り返した。反応後、2 %アガロースゲル電気泳動によって約1.1 kbpのバン ドを確認し、プラスミドにインサートが挿入されていることを確認した。
インサートが挿入されたプラスミドを持つ大腸菌を、50 μg/mLのアンピシリ ンを含む5 mLのLB液体培地に植菌し、37 ˚Cで終夜振とう培養した。その後、
培養液を1.5 mL遠心チューブに移し、13000 rpm、4 ˚C、3分の条件で遠心分離
を繰り返し、菌体を回収した。得られた菌体から、Mag ExtractorTM –plasmid-
(TOYOBO社)を用いてプラスミドDNAを単離および精製した。回収したプ ラスミドは、三重大学生命科学研究支援センターのトータルシークエンスサー ビスを利用し、シークエンスを解析した。また、waaW配列についても同じ方法 で解析した。
47
第2項 標的遺伝子の相補的配列を含む抗生耐性遺伝子断片の作製
PCR用遠心管に、抗生物質遺伝子を含むプラスミドpKD3を0.125 μL、2 mM dNTP(TOYOBO社)を2.5μL、pKD3の薬剤耐性遺伝子領域に相補的な100 μM のフォワードプライマー pKD3 F 20(5'-GTGTAGGCTGGAGCTGCTTC-3’)と100 μMのリバースプライマー pKD3 R 20(5’-ATGGGAATTAGCCATGGTCC-3’)を 1.25 μL、10×KOD Buffer(TOYOBO社)を2.5 μL、25 mM MgSO4(TOYOBO社)
を1 μL、KOD DNA Polymerase(TOYOBO社)を0.5 μL、滅菌水を15.875 μLそ れぞれ加え、合計25 μLとした。
サーマルサイクラーを用い、PCRによって、pKD3にある抗生物質遺伝子領域 を増幅した。PCRの条件は保持反応を95 ˚Cで7 分間行った後、1サイクルあた り変性反応を94 ˚Cで15秒、アニーリング反応を58 ˚Cで30秒、伸長反応を72
˚Cで1分30秒のサイクルを35サイクル繰り返した。反応後、2 %アガロースゲ ル電気泳動により、約1 Kbpに相当するバンドが現れ、目的の遺伝子領域が増 幅したことを確認した。このDNA断片はillustraTM GFXTM PCR DNA and Gel Band
Purification Kit(GEヘルスケア・ジャパン社)を用いてアガロースゲルから切り
出し、精製した。
続いて、精製した薬剤耐性遺伝子断片にwaaT配列の上流および下流50 bpを 組み込んだDNA断片を調製した。PCR用遠心管に、精製した薬剤耐性遺伝子を 0.125 μL、2 mM dNTP(TOYOBO社)を2.5μL、waaT遺伝子の開始コドンから
上流配列50 bpと薬剤耐性遺伝子に特異的な配列20 bpを繋いだ10 μMのフォワ
ードプライマー WaaT noc R 70 bpver
(5'-TATTTATTATTTTTATTTAAAGATAATAAAATGAAATTGAGAAAAGTTAAG
TGTAGGCTGGAGCTGCTTC-3’)および同遺伝子の終始コドンから下流50 bpの
48
相補配列と薬剤耐性遺伝子に特異的な相補配列20 bpを繋いだ10 μMのリバース プライマー WaaT noc F 70 bpver
(5’-TAATAAAATGAGAAGCCGCGATAGCGTATACTTGTAATCATAAGAGTAAT ATGGGAATTAGCCATGGTCC-3’)を1.25 μL、10×KOD Buffer(TOYOBO社)を 2.5 μL、25 mM MgSO4(TOYOBO社)を1 μL、KOD DNA Polymerase(TOYOBO 社)を0.5 μL、滅菌水を15.875 μLそれぞれ加え、合計25 μLとし、サーマルサ イクラーを用いて、PCRを行った。PCRの条件は保持反応を95 ˚Cで7 分間行 った後、1サイクルあたり変性反応を94 ˚Cで15秒、アニーリング反応を66 ˚C で30秒、伸長反応を72 ˚Cで1分30秒のサイクルを40サイクル繰り返した。
得られたPCR反応溶液を2 %アガロースゲル電気泳動に供し、約1.1 kbpにバン ドを確認し、目的のDNA断片が増幅したことを確認した。このDNA断片は illustraTM GFXTM PCR DNA and Gel Band Purification Kit(GEヘルスケア・ジャパ ン社)を用いてアガロースゲルから切り出し、約530 μLのDNA溶液を回収し た。このDNA溶液に3 M 酢酸ナトリウムを53 μLと1457 μLのエタノールを加 え、-30 ˚Cで1時間冷却した。15000 rpm、4 ˚C、30分の条件で遠心分離を行い、
上清を取り除いた後、70 % エタノールを1 mL加え、15000 rpm、4 ˚C、5分の 条件で遠心分離を行い、再び上清を取り除いた。10分ほどDNAを風乾した後、
50 μLの滅菌水を加えたものをE.coli F2513株waaT欠失用DNA断片とした。
49
第3項 L-Arabinose添加によるE.coli F2513株の増殖に与える影響
L-Arabinose存在下でE.coli F2513株が生育するかを確認した。2 mLのLB液 体培地を2本と1 M L-Arabinose水溶液を作り、オートクレーブによって滅菌処 理した。その後、2本のLB液体培地の内、1本に1 M L-Arabinose水溶液を20 μL 加え、終濃度10 mMのL-Arabinoseを含む2 mLのLB液体培地を作り、L-Arabinose を含まない液体培地と共に、グリセロールストックとして冷凍保存している E.coli F2513株を植菌し、37 ˚Cで終夜培養した。翌日、培養液のOD600を測定し、
E.coli F2513株の生育状況を確認した。
50
第4項 コンピテントセルの作製および形質転換
E.coli F2513株にpKD46を導入するため、MandelとHiga28)によって報告され た方法を参考にコンピテントセルを作製した。
グリセロールストックとして保存されているE.coli F2513株を5 mLのLB液 体培地に植菌し、終夜培養した。50 mLのLB液体培地に1 mLの終夜培養液を 植菌し、OD600が0.5になるまで振とう培養した。遠沈管に培養液を分注し、氷 中で30分間冷却した後、4800 rpm、4 ˚C、5分の条件で遠心分離によって菌体を 回収した。そこに、50 mM CaCl2を12.5 mL加え、菌体をピペッティングによっ て穏やかに懸濁し、氷中で60分間冷却した。4800 rpm、4 ˚C、5分の条件で遠心 分離によって、上清を取り除き、5 mLの50 mM CaCl2/20 %(W/V)グリセロー ルを加えて懸濁した。懸濁後、溶液を遠心チューブに220 μLずつ分注し、液体 窒素で急速冷凍したものをコンピテントセルとした。
作製した220 μLのコンピテントセルに334.3 ng/μLのpKD46を1 μL加え、氷 上で30分間静置した。42 ˚Cの恒温槽で30秒ヒートショックを与え、再度、氷 上で2分間静置した後、900 μLのSOC培地を加え、37 ˚Cで1時間振とう培養 した。その後、50 μg/mLのアンピシリンを含むLB寒天培地に植菌し30 ˚Cで終 夜培養した。
また、Inoueら29)が開発した高効率形質転換能を持つコンピテントセル作製法 によって、コンピテントセルを作製した。
グリセロールストックとして保存されているE.coli F2513株をLB寒天培地に
植菌し、37 ˚Cで倒置培養した。LB寒天培地に生えたコロニーを1つ選択して、
1 mLのSOB培地(2 % トリプトン、0.5 % 乾燥酵母エキス、10 mM NaCl、2.5 mM
KCl)に植菌し、終夜培養した。その培養液を200 μL取り、50 mLのSOB培地