製造販売承認申請書添付資料
第2部(モジュール2) CTDの概要(サマリー)
2.6. 非臨床試験の概要文および概要表
2.6.1. 緒言
2.6.2. 薬理試験の概要文
2.6.3. 薬理試験概要表
グラクソ・スミスクライン株式会社
非臨床概要 薬理試験の目次
項目 - 頁
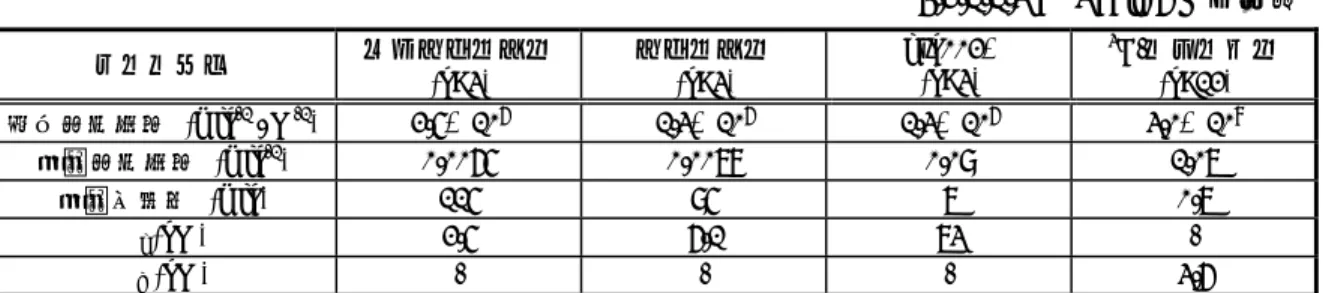
2.6.1. 緒言 ... 2.6.1 - p. 1 2.6.2. 薬理試験の概要文 ... 2.6.2 - p. 1 2.6.2.1. まとめ ... 2.6.2 - p. 1 2.6.2.2. 効力を裏付ける試験 ... 2.6.2 - p. 3 2.6.2.2.1. 受容体結合試験 ... 2.6.2 - p. 4 2.6.2.2.1.1. マウス大脳皮質膜標品を用いたメピラミン結合阻害 試験 ... 2.6.2 - p. 4 2.6.2.2.1.2. ヒトH1受容体に対する親和性および結合特性... 2.6.2 - p. 4 2.6.2.2.1.3. ヒトH1受容体に対するレボセチリジンの結合特性... 2.6.2 - p. 7 2.6.2.2.2. In vitro抗ヒスタミン作用... 2.6.2 - p. 9 2.6.2.2.2.1. モルモット摘出組織におけるヒスタミン誘発収縮に 対する作用... 2.6.2 - p. 9 2.6.2.2.2.2. モルモット摘出回腸標本におけるヒスタミン誘発 収縮に対する作用の持続性 ... 2.6.2 - p. 11 2.6.2.2.3. In vivo抗ヒスタミン作用 ... 2.6.2 - p. 12 2.6.2.2.3.1. モルモットにおけるヒスタミン誘発気管支攣縮に 対する作用... 2.6.2 - p. 12 2.6.2.2.3.2. マウスにおけるヒスタミン誘発皮膚反応に対する作用 ... 2.6.2 - p. 13 2.6.2.2.3.3. ラットにおけるヒスタミン誘発皮膚反応に対する作用 ... 2.6.2 - p. 14 2.6.2.2.3.4. イヌにおけるヒスタミン誘発皮膚反応に対する作用 (経口投与)... 2.6.2 - p. 14 2.6.2.2.3.5. イヌにおけるヒスタミン誘発皮膚反応に対する作用 (静脈内投与) ... 2.6.2 - p. 15 2.6.2.2.4. 好酸球および炎症性因子に対する作用 ... 2.6.2 - p. 17 2.6.2.2.4.1. エオタキシン刺激による好酸球の血管内皮細胞間隙 遊走に対する抑制作用 ... 2.6.2 - p. 17 2.6.2.2.4.2. ヒト角化細胞におけるICAM-1およびMHC分子発現 ならびにGM-CSFおよびケモカイン産生に対する 抑制作用 ... 2.6.2 - p. 18 2.6.2.3. 副次的薬理試験... 2.6.2 - p. 21 2.6.2.3.1. 種々のG蛋白質共役型受容体およびイオンチャネルに 対する結合親和性... 2.6.2 - p. 21 2.6.2.3.2. ムスカリン受容体サブタイプに及ぼす影響... 2.6.2 - p. 21 2.6.2.3.3. モルモット摘出回腸標本のセロトニン、アセチルコリン およびニコチン誘発収縮反応に対する作用...2.6.2.4.1. 中枢神経系に及ぼす影響... 2.6.2 - p. 23 2.6.2.4.2. 心血管系に及ぼす影響... 2.6.2 - p. 23 2.6.2.4.2.1. hERG試験... 2.6.2 - p. 23 2.6.2.4.2.2. モルモット単離心室筋細胞の遅延整流K+電流に及ぼす 影響 ... 2.6.2 - p. 23 2.6.2.4.2.3. イヌ摘出プルキンエ線維の活動電位に及ぼす影響... 2.6.2 - p. 23 2.6.2.4.2.4. 麻酔イヌの循環動態試験... 2.6.2 - p. 24 2.6.2.4.2.5. 麻酔イヌの徐脈性QT延長症候群モデルに及ぼす影響 ... 2.6.2 - p. 24 2.6.2.4.3. 呼吸系に及ぼす影響 ... 2.6.2 - p. 24 2.6.2.4.4. 胃腸管系に及ぼす影響... 2.6.2 - p. 24 2.6.2.5. 薬力学的薬物相互作用試験 ... 2.6.2 - p. 24 2.6.2.6. 考察及び結論 ... 2.6.2 - p. 24 2.6.2.6.1. 効力を裏付ける試験 ... 2.6.2 - p. 24 2.6.2.6.2. 副次的薬理試験 ... 2.6.2 - p. 26 2.6.2.6.3. 安全性薬理試験 ... 2.6.2 - p. 26 2.6.2.6.4. 結論... 2.6.2 - p. 27 2.6.2.7. 図表 ... 2.6.2 - p. 27 2.6.2.8. 参考文献... 2.6.2 - p. 27 2.6.3. 薬理試験概要表 ... 2.6.3 - p. 1 2.6.3.1. 薬理試験:一覧表 ... 2.6.3 - p. 1 2.6.3.2. 効力を裏付ける試験 ... 2.6.3 - p. 3 2.6.3.3. 副次的薬理試験... 2.6.3 - p. 6 2.6.3.4. 安全性薬理試験... 2.6.3 - p. 7 2.6.3.5. 薬力学的薬物相互作用試験 ... 2.6.3 - p. 9
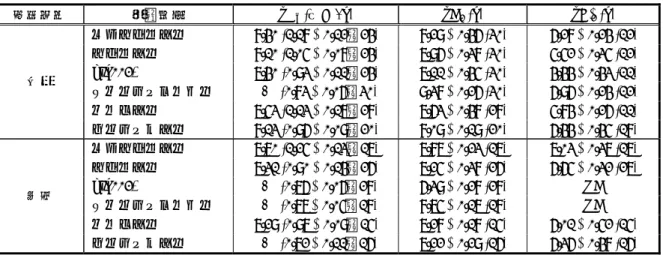
2.6.1、2.6.2 および 2.6.3 の略語等一覧 セチリジン セチリジン塩酸塩 レボセチリジン レボセチリジン塩酸塩 APD50 50%再分極時活動電位持続時間 B/F 分離 Bound/Free 分離 (結合分子と非結合分子の分離) Bmax 結合量 (受容体結合試験において試料中に存在する全受容体量を示す) CCL CC ケモカインリガンド CHO チャイニーズハムスター卵巣 Cmax 最高血漿中濃度 CXCL10 CXC ケモカインリガンド 10 DG ジアシルグリセロール DMSO ジメチルスルホキシド ED50 50%有効量
ELISA Enzyme-linked immunosorbent assay FITC Fluorescein isothiocyanate GM-CSF 顆粒球マクロファージコロニー刺激因子 Gq G 蛋白質の α サブユニットのうちホスホリパーゼ C の活性化に関与するファミリー hERG ヒトether-a-go-go 関連遺伝子 HLA ヒト白血球抗原 HMVEC-d 正常ヒト皮膚微小血管内皮細胞 HMVEC-l 正常ヒト肺微小血管内皮細胞 5-HT 5-ヒドロキシトリプタミン (セロトニン) IC50 50%抑制濃度
ICAM-1 Intercellular adhesion molecule-1 IFN-γ インターフェロン-γ
IKr 急速活性型遅延整流K+電流
IL-1 インターロイキン-1
IP-10 IFN-induced protein of 10kd (=CXCL10) IP3 イノシトール1,4,5-三リン酸 Kd 解離定数 (アゴニストの受容体に対する親和性を示す) Ki 阻害定数 (アンタゴニストの受容体に対する親和性を示す) LSC 液体シンチレーション計測器 MCP-1 単球走化性因子-1 (=CCL2) MHC 主要組織適合遺伝子複合体
MIP-3α Macrophage inflammatory protein-3α (=CCL20) NF-κB Nuclear factor-κB pA2 アゴニスト単独の濃度反応曲線を2 倍だけ高濃度側に平行移動させるのに必要なアン タゴニスト濃度(モル濃度)の負の常用対数 (アンタゴニストの効力を示す) pKi Ki (モル濃度) の負の常用対数 PLC ホスホリパーゼC
RANTES Regulated upon activation, normal T expressed and secreted (=CCL5) SD ラット Sprague Dawley ラット
SPA シンチレーション近接アッセイ TEM 血管内皮細胞間隙遊走
TNFα 腫瘍壊死因子α
VCAM-1 Vascular cell adhesion molecule-1 WGA Wheat germ agglutinin
2.6.1. 緒言 レボセチリジンは、ラセミ体であるセチリジンの R-エナンチオマーであり、セチリジン と同様にヒスタミン H1受容体アンタゴニストである(図 2.6.1-1)。ヒスタミン H1受容体に 強力かつ選択的に結合することによりヒスタミンの作用を阻害し、抗ヒスタミン作用および 抗炎症作用を示す。レボセチリジンはセチリジンと同程度以上で S-エナンチオマーである N N Cl H O CO2H ・2HCl 図 2.6.1-1 レボセチリジンの構造式 本剤の効能・効果および用法・用量は以下のとおりである。 【効能・効果】 〔成人〕アレルギー性鼻炎、蕁麻疹、湿疹・皮膚炎、痒疹、皮膚そう痒症 〔小児〕アレルギー性鼻炎、蕁麻疹、皮膚疾患(湿疹・皮膚炎、皮膚そう痒症)に伴うそう 痒 【用法・用量】 〔成人〕通常、成人にはレボセチリジン塩酸塩として 1 回 5mg を 1 日 1 回、就寝前に経口投 与する。なお、年齢、症状により適宜増減するが、最高投与量は 1 日 10mg とする。 〔小児〕通常、7 歳以上 15 歳未満の小児にはレボセチリジン塩酸塩として 1 回 2.5mg を 1 日 2 回、朝食後及び就寝前に経口投与する。 gsk002*よりも強い抗ヒスタミン作用を有するアレルギー性疾患治療薬である。 * 新薬承認情報提供時に置き換え
2.6.2. 薬理試験の概要文 2.6.2.1. まとめ レボセチリジン(ucb 28556)はヒスタミン H1受容体(H1受容体)アンタゴニストであり、 ラセミ体であるセチリジンの R-エナンチオマーである。 一連の in vitro および in vivo 試験によりレボセチリジンの薬理作用を検討した。一部の試験 を比較した。 効力を裏付ける試験 マウス大脳皮質膜標品のH1受容体における3H-メピラミン結合阻害試験において、レボセ のH1受容体からの解離半減期はそれぞれ115、95 および 7 分であり、レボセチリジンおよび 用いてヒトH1受容体に対する親和性および結合速度を直接測定したところ、非標識体におけ る結果と同様に、3H-標識体はヒト H1受容体に高い親和性(Kd=2.8nM)を示すとともに緩や かに解離し、その解離半減期は134 分であった。3H-標識体の H1受容体結合に対するヒスタ ミンのIC50は3H-標識体濃度の増加に伴って増加したことから、レボセチリジンはヒスタミ ンと競合的に相互作用することが示された。 H1受容体に対する拮抗作用をモルモット摘出組織標本におけるヒスタミン誘発収縮反応に 出回腸においてそれぞれ8.29、7.96 および 7.11、摘出気管においてはそれぞれ 7.87、7.25 お よび6.39 であった。したがって、モルモットの摘出回腸および摘出気管においてレボセチリ 試験において、レボセチリジンおよびセチリジンは低濃度では競合的な拮抗作用を示し、高 濃度ではヒスタミンの最大反応を抑制したことから、競合的な拮抗作用とともにinsurmountable リジンおよびセチリジンは、摘出回腸標本を用いた洗浄試験におけるヒスタミン誘発収縮反 かった。マウスのヒスタミン誘発皮膚反応(膨疹形成)に対して、レボセチリジン、セチリ では、レボセチリジンとセチリジンおよびS-エナンチオマーである dextrocetirizine(gsk002*) チリジン(IC50=12nM)の H1受容体に対する親和性はセチリジン(IC50=27nM)および gsk002* (IC50=310nM)よりもそれぞれ約 2 および 25 倍高かった。さらに、ヒト H1受容体発現 細胞を用いた3H-メピラミン結合阻害試験において、レボセチリジンはヒト H1受容体に高い 親和性(Ki=2.5nM)を示し、この値はセチリジン(Ki=6.1nM)および gsk002*(Ki=73nM) よりもそれぞれ約2 および 30 倍高かった。また、レボセチリジン、セチリジンおよび gsk002* セチリジンの解離はgsk002*と比べきわめて緩徐であった。レボセチリジンの3H-標識体を 対する作用により検討した結果、レボセチリジン、セチリジンおよびgsk002*のpA2は、摘 ジンの拮抗作用はセチリジンの約2~4 倍、gsk002*の約 15~30 倍強力であった。これらの な拮抗作用注1)を示した。一方、gsk002*は競合的な拮抗作用のみを示した。また、レボセチ 応に対して持続的な抑制作用を示したが、gsk002*の抑制作用は洗浄により減弱した。 レボセチリジン、セチリジンおよびgsk002*はモルモットのヒスタミン誘発気管支攣縮 に対して静脈内投与により抑制作用を示し、レボセチリジンの抑制作用はgsk002*より強
ヒト皮膚または肺の微小血管内皮細胞の単層膜において、レボセチリジン(0.001~10µM) は好酸球の前処理により、エオタキシンの好酸球遊走活性を抑制した。また、ヒト角化細胞 において、レボセチリジン(0.01~10µM)は、ヒスタミンによる IFN-γ 刺激 ICAM-1 および MHC クラス I 発現の増強を抑制し、ヒスタミンによる IFN-γ 刺激 GM-CSF、CCL2/MCP-1、 CCL5/RANTES、CCL20/MIP-3α および CXCL10/IP-10 放出の増強を抑制した。 副次的薬理試験 レボセチリジンは、H1受容体以外のG 蛋白質共役型受容体(ヒスタミン H2およびH3、ア デノシンA1、アドレナリンα1、α2C2、α2C10 および β1、セロトニン5-HT1Aおよび5-HT2、ド パミンD1およびD2ならびにムスカリンM1、M2、M3、M4およびM5受容体)およびイオン チャネル(L-type Ca2+、Na+ type1 および Na+ type2 チャネル)に対してほとんど親和性を示
さなかった。レボセチリジン(10µM)はアドレナリン α2C4 受容体において標識リガンドの 特異的結合を約80%阻害したが、その親和性(pKi=5.8)は H1受容体に対する親和性(pKi =8.5)の 1/500 以下であった。また、レボセチリジンはモルモット摘出組織におけるセロト ニン、アセチルコリンおよびニコチン受容体機能に対して影響を及ぼさず、ラット摘出大動 脈におけるL-type Ca2+チャネル機能に対しても影響を及ぼさなかった。 安全性薬理試験 ラットの一般症状および行動観察において、レボセチリジンは50mg/kg の経口投与で軽度 の腹筋緊張の上昇、100mg/kg で軽度の反応性の低下を示した。ラットの自発運動およびペン トバルビタール誘発睡眠に対しては100mg/kg までの経口投与で影響を示さなかった。hERG 試験において、レボセチリジンは30µM まで hERG 電流に対して影響を示さなかった。イヌ 摘出プルキンエ線維においては30µM 以上で活動電位持続時間を延長させ、モルモット単離 心室筋細胞においては100µM で IKrを抑制した。麻酔イヌの循環動態試験において、レボセ チリジンは10mg/kg までの累積的な静脈内注入投与により血圧、心拍数、左心室収縮期血圧、 心拍出量、大腿動脈の血流速度および血管抵抗、心電図ならびに血液ガスに影響を示さず、 麻酔イヌの徐脈性QT 延長症候群モデルにおいて、3.2mg/kg/hr までの静脈内持続投与によっ ても心筋再分極の遅延、QT 間隔延長および徐脈性不整脈を誘発しなかった。麻酔イヌの呼吸 機能試験において、レボセチリジンは10mg/kg まで累積的に静脈内注入投与しても 1 回換気 量、分時換気量および呼吸数に影響を示さなかった。ラットの消化管炭末輸送能に対して、 レボセチリジンは100mg/kg を単回経口投与しても影響を示さなかった。 ジンおよびgsk002*はいずれも経口投与により同程度の膨疹面積抑制作用を示し、ヒスタ ミン量を2 倍にしてもレボセチリジンおよびセチリジンの抑制作用は変化しなかったが、gsk002* の抑制作用は低下した。ラットのヒスタミン誘発皮膚反応に対して、レボセチリジンは 経口投与により膨疹面積抑制作用を示し、その作用はセチリジンと同程度でgsk002*よりも 強かった。イヌのヒスタミン誘発皮膚反応に対して、経口または静脈内投与によりレボセチ リジンはセチリジンと同程度以上の膨疹面積抑制作用を示したが、gsk002*の抑制作用は レボセチリジンおよびセチリジンよりも弱かった。
2.6.2.2. 効力を裏付ける試験 H1受容体は呼吸器および消化管の平滑筋細胞、血管内皮細胞、神経細胞ならびにグリア細 胞に分布し[Brown, 2001; Bloom, 2001]、H1受容体に結合する生理活性物質であるヒスタミン は皮膚、気管支粘膜および腸管粘膜に存在する肥満細胞におもに貯蔵されている[Brown, 2001]。 H1受容体はG 蛋白質共役型受容体ファミリーに属し、Gq を介して PLC と共役しており、 ヒスタミンが受容体に結合すると、PLC を活性化させて細胞膜のイノシトールリン脂質から セカンドメッセンジャーであるIP3およびDG の産生を誘導する。細胞内において IP3は小胞 体からの急速なCa2+放出を引き起こし[Brown, 2001]、DG は Ca2+とともにプロテインキナー ゼC を活性化する。さらに、Ca2+はCa2+/カルモジュリン依存性プロテインキナーゼおよび ホスホリパーゼA2を活性化する(図 2.6.2-1)。
Gq
Gq
PIP
2DG
IP
3PKC
ER
CaMK
PLA
2Ca
2+PLC
Gq
Gq
PIP
2DG
IP
3PKC
ER
CaMK
PLA
2Ca
2+PLC
CaMK:Ca2+/カルモジュリン依存性プロテインキナーゼ、DG:ジアシルグリセロール、ER:小胞体、 IP3:イノシトール1,4,5-三リン酸、PIP2:ホスファチジルイノシトール4,5-二リン酸、PKC:プロテインキナー ゼC、PLA2:ホスホリパーゼA2、PLC:ホスホリパーゼ C 図 2.6.2-1 ヒスタミン H1受容体を介した細胞内情報伝達系 肥満細胞から遊離されるヒスタミンはH1受容体刺激により、このような細胞内情報伝達系 の活性化を介して小血管を拡張させ、その結果、フラッシング反応、総末梢血管抵抗の低下、 全身血圧の低下および毛細血管透過性の亢進を生じさせるとともに、呼吸器においては気管 平滑筋を収縮させ、また、皮膚においては神経終末を刺激することにより表皮の掻痒、真皮 層の痛みおよび著明な発赤が誘発されると考えられている。2.6.2.2.1. 受容体結合試験 2.6.2.2.1.1. マウス大脳皮質膜標品を用いたメピラミン結合阻害試験 4.2.1.1.1 マウス大脳皮質膜標品を用いてH1受容体への3H-メピラミン結合に対するレボセチリジン、 [方法] 雌NMRI マウスの大脳皮質より調製した細胞膜標品(蛋白質量約 500µg)を 50mM リン酸 緩衝液(pH7.5)に懸濁し、3H-メピラミン(2nM)および被験物質(0.0001~10µM)を添加 して25℃で 60 分間反応させた。ガラス繊維フィルターを用いて B/F 分離を行い、フィルター 上に残った放射能をLSC で測定した。 [結果] レボセチリジンおよびセチリジンはマウス大脳皮質の膜蛋白質への3H-メピラミン結合を阻 ボセチリジンおよびセチリジンと比較して高値であった(表 2.6.2-1)。 れ約2 および 25 倍高い親和性を有することが示された。 表 2.6.2-1 マウス大脳皮質膜標品における3 H-メピラミン結合阻害活性 4.2.1.1.1 の TABLE 1 より作成 被験物質 IC50 (nM) nH レボセチリジン 12 ± 4 0.90 ± 0.06 セチリジン 27 ± 12 0.90 ± 0.01 310 ± 40 0.82 ± 0.11 平均値±標準偏差 (n=3) nH:ヒル係数 2.6.2.2.1.2. ヒト H1 受容体に対する親和性および結合特性 4.2.1.1.2~4.2.1.1.4 性および結合特性について比較検討した。 [方法] 遺伝子組換えヒトH1受容体を発現させたCHO 細胞株より調製した細胞膜標品(蛋白質量 150µg)を 2mM MgCl2-50mM Tris-HCl 緩衝液(pH7.4)に懸濁し、3H-メピラミン(3.0nM)お よび被験物質を添加して37℃で 180 分間反応させた。ガラス繊維フィルターを用いて B/F 分 離し、フィルター上の放射能をLSC で測定した。 反応速度試験では、細胞膜標品(蛋白質量300µg)と3H-メピラミン(3.5nM)および被験 物質を60 分間反応させたのちにセチリジン(10µM)を添加し、受容体からの解離を促進さ せ、上記と同様に測定した。 SPA 法を用いてヒト H1受容体における3H-メピラミン結合阻害試験を実施した。細胞膜標 品(蛋白質量50µg)を 2mM MgCl2-50mM Tris-HCl 緩衝液(pH7.4)に懸濁して WGA コート セチリジンおよびgsk002*の阻害活性を比較した。 害し、そのIC50はそれぞれ12 および 27nMであった。gsk002*のIC50は310nMであり、レ 以上より、レボセチリジンはH1受容体に対してセチリジンおよびgsk002*よりもそれぞ gsk002* レボセチリジン、セチリジンおよびgsk002*の遺伝子組換えヒト H1受容体に対する親和
SPA ビーズに固定させ、被験物質および3H-メピラミン(7.5nM)を添加して反応させた。LSC により放射能を経時的に測定した。 [結果] ヒトH1受容体に対してレボセチリジンおよびセチリジンは高い結合阻害活性を示し、レボ =73nM)の約 30 倍であった(図 2.6.2-2 および表 2.6.2-2)。 りも長く、セチリジン(95 分)と同程度であった(表 2.6.2-2)。 SPA 法においてレボセチリジンのヒト H1受容体における3H-メピラミン結合阻害曲線は反 んど変化しなかった(図 2.6.2-3)。このことから、これら 2 つのエナンチオマーは結合速度 が異なり、受容体結合が平衡に達するまでの時間はレボセチリジンの方が長いことが示唆さ れた。 図 2.6.2-2 ヒト H1受容体に対する3H-メピラミン結合阻害活性 4.2.1.1.2 の FIG. 1 より作成 セチリジンの親和性(Ki=2.5nM)はセチリジン(Ki=6.1nM)の約 2 倍であり、gsk002*(Ki H1受容体からのレボセチリジンの解離半減期は115 分であり、これはgsk002*(7 分)よ 応時間の増加により低濃度側へ平行移動(IC50の減少)したが、gsk002*の阻害曲線はほと * 新薬承認情報提供時に置き換え gsk002*
表 2.6.2-2 ヒト H1受容体に対する結合および解離速度パラメータ 4.2.1.1.3 の Table 6 より作成 パラメータ レボセチリジン (n=3) セチリジン (n=3) (n=3) 3H-メピラミン (n=12) 結合速度定数 (min-1 . M-1) 2.5×106 1.3×106 1.3×106 3.0×108 解離速度定数 (min-1) 0.0065 0.0078 0.09 1.07 解離半減期 (min) 115 95 7 0.7 Ki (nM) 2.5 6.1 73 - Kd(nM) - - - 3.6 KiおよびKdは (解離速度定数) / (結合速度定数) の比より算出 平均値 -:該当せず 試験を2 回実施し、そのうちの 1 回の試験結果を記載した 図 2.6.2-3 ヒト H1受容体を用いたレボセチリジンおよび 3 H-メピラミン結合阻害試験における反応時間の影響 4.2.1.1.4 の Fig. 2.より作成 gsk002* [gsk002*] gsk002*の * 新薬承認情報提供時に置き換え
2.6.2.2.1.3. ヒト H1 受容体に対するレボセチリジンの結合特性 4.2.1.1.5 レボセチリジンの3H-標識体のヒト H1受容体に対する結合特性を直接的に検討した。 [方法] ヒトH1受容体を発現させたCHO 細胞株の細胞膜標品を用いて 2.6.2.2.1.2 に記載した方法 で受容体結合試験を実施した。細胞膜標品に3H-標識体または3H-メピラミンを添加して 37℃ で180 分間反応させ、ヒト H1受容体における3H-標識体または3H-メピラミンの飽和結合曲 線を求めた。反応速度試験では、細胞膜標品と3H-標識体を 90 分間反応後にメピラミン(10µM) を添加し、受容体からの解離を促進させた。また、複数濃度の3H-標識体または3H-メピラミ
ンを用いて各種H1受容体リガンドのIC50を求め、Cheng & Prusoff の式[Cheng, 1973]により pKi を算出した。 [結果] 3H-標識体はヒト H 1受容体に高い親和性を示し(図 2.6.2-4)、その解離定数(Kd)は2.8 ±0.2nM であった。ヒト H1受容体数(Bmax)は2234±254fmol/mg 蛋白質であり、3H-メピラ ミン飽和結合曲線より求めたBmax(1934±164fmol/mg 蛋白質)と同程度であった。3H-標識体 のKdの濃度におけるヒトH1受容体への結合は試験開始23 分後に平衡時の 50%となり、270 分後には平衡状態に達した。また、3H-標識体はヒト H1受容体から緩やかに解離し、その解 離半減期は134 分であった(図 2.6.2-4)。これに対して、3H-メピラミンのヒト H1受容体へ の結合は瞬時に平衡状態に到達し、1 分未満の解離半減期で受容体から解離した。また、各 種H1受容体リガンドの親和性に標識リガンドの種類は影響しなかった(表 2.6.2-3)。さら に、ヒトH1受容体における3H-標識体結合阻害試験において、ヒスタミンの IC50は3H-標識 体濃度の増加(1~25nM)に伴って増大し、ヒスタミンは3H-標識体と競合的に作用すること が示された。 以上より、レボセチリジンはヒトH1受容体に可逆的かつ高親和性で結合し、ヒスタミンと 競合的に作用すると考えられる。
A
B
試験を3 回実施し、そのうちの 1 回の試験結果を記載した A:3H-標識体のヒト H 1受容体との飽和結合曲線 B:3H-標識体のヒト H 1受容体との結合および解離曲線 図 2.6.2-4 レボセチリジンの3 H-標識体のヒト H1受容体結合特性 4.2.1.1.5 の Fig. 1.より作成 表 2.6.2-3 各種 H1受容体リガンドのヒト H1受容体に対する結合親和性 4.2.1.1.5 の Table 1.より作成 被験物質 3H-標識体結合阻害試験 pKi 3H-メピラミン結合阻害試験 pKi ヒスタミン 5.7 ± 0.2 5.9 ± 0.0 レボセチリジン 8.4 ± 0.1 8.5 ± 0.1 セチリジン 8.2 ± 0.1 8.2 ± 0.1 7.2 ± 0.1 7.1 ± 0.1 クロルフェニラミン 8.6 ± 0.1 8.6 ± 0.1 ロラタジン 7.5 ± 0.1 7.8 ± 0.0 テルフェナジン 8.7 ± 0.1 8.7 ± 0.0 平均値±標準偏差 (n=3) gsk002* * 新薬承認情報提供時に置き換えまとめ れ約2 および 25~30 倍高い親和性を示し、レボセチリジンの H1受容体からの解離はセチリ 2.6.2.2.2. In vitro 抗ヒスタミン作用 2.6.2.2.2.1. モルモット摘出組織におけるヒスタミン誘発収縮に対する作用 4.2.1.1.6~4.2.1.1.8 モルモット摘出回腸および摘出気管標本のヒスタミン誘発収縮に対するレボセチリジン、 [方法] Dunkin Hartley モルモットより回腸および気管を摘出し、標本を作製した。摘出回腸標本は Tyrode 液(pH7.6、95%O2-5%CO2、37℃)中に懸垂し、レボセチリジン、セチリジンまたは 加して収縮反応を誘発し、濃度反応曲線を求めた。摘出気管標本はKrebs 液(95%O2-5%CO2、 せたのち、ヒスタミンを累積的に添加して濃度反応曲線を求めた。 [結果] ミン誘発収縮反応を抑制し、pA2はそれぞれ8.29、7.96 および 7.11 であり、レボセチリジン モルモット摘出回腸および摘出気管標本においてレボセチリジン(図 2.6.2-5)およびセチ リジンはヒスタミンの濃度反応曲線を高濃度側へ平行移動させ、高濃度では最大反応を抑制 した。Schild プロット解析の傾きはいずれも低濃度側では約 1 であったことから(表 2.6.2-4)、 レボセチリジンおよびセチリジンは低濃度ではヒスタミンと競合的に作用し、高濃度では はヒスタミンの濃度反応曲線を高濃度側へ移動させたが、最大反応を抑制しなかったことか ら、ヒスタミンと競合的に作用することが示された(図 2.6.2-5)。 ジンと同様にgsk002*より緩徐であった。 セチリジンおよびgsk002*の効力を比較検討した。 gsk002*を添加して 60 分間反応させたのち、非累積的に濃度を上げながらヒスタミンを添 37℃)中に懸垂し、レボセチリジン、セチリジンまたは gsk002*を添加して 60 分間反応さ モルモット摘出回腸標本においてレボセチリジン、セチリジンおよびgsk002*はヒスタ の効力はセチリジンおよびgsk002*のそれぞれ約 2 および 15 倍であった(表 2.6.2-4)。 insurmountableな作用も有することが示された。モルモット摘出気管標本においてgsk002* 以上より、レボセチリジンはH1受容体に対してセチリジンおよびgsk002*よりもそれぞ * 新薬承認情報提供時に置き換え モルモット摘出気管標本においてレボセチリジン、セチリジンおよびgsk002*はヒスタ ミン誘発収縮反応を抑制し、pA2はそれぞれ7.87、7.25 および 6.39 であり、レボセチリジン の効力はセチリジンおよびgsk002*のそれぞれ約 4 および 30 倍であった(表 2.6.2-4)。
表 2.6.2-4 モルモット摘出回腸および摘出気管標本のヒスタミン誘発収縮に対する 拮抗作用 4.2.1.1.8 の Table 1 より作成 組織標本 被験物質 pKb (傾き, n) pA2 (n) pD'2 (n) レボセチリジン 8.40 (1.18 ± 0.12,24) 8.29 ± 0.46 (30) 6.28 ± 0.24 (12) セチリジン 8.10 (1.05 ± 0.08,24) 7.96 ± 0.38 (30) 5.52 ± 0.35 (12) 7.40 (0.93 ± 0.11,24) 7.11 ± 0.45 (30) 4.44 ± 0.43 (11) クロルフェニラミン - (0.83 ± 0.06,30) 9.38 ± 0.26 (30) 6.96 ± 0.24 (12) ロラタジン 7.53 (1.13 ± 0.17,28) 7.63 ± 0.48 (28) 5.84 ± 0.26 (11) 回腸 テルフェナジン 8.13 (0.96 ± 0.09,20) 8.09 ± 0.19 (20) 6.44 ± 0.45 (18) レボセチリジン 7.70 (1.25 ± 0.13,17) 7.87 ± 0.23 (17) 7.03 ± 0.37 (17) セチリジン 7.31 (0.90 ± 0.14,26) 7.25 ± 0.38 (26) 6.65 ± 0.32 (27) - (0.76 ± 0.06,28) 6.39 ± 0.28 (28) NA クロルフェニラミン - (0.77 ± 0.05,18) 8.75 ± 0.17 (18) NA ロラタジン 7.29 (0.97 ± 0.09,15) 7.28 ± 0.18 (15) 6.01 ± 0.52 (15) 気管 テルフェナジン - (0.72 ± 0.11,16) 7.22 ± 0.29 (16) 6.36 ± 0.48 (16) 平均値±標準偏差 pKb:アンタゴニストの解離定数Kb (モル濃度) の負の常用対数 -:Schild プロット解析の傾きが 1 未満のため pKb算出せず pD'2:アゴニストによる最大反応を50%低下させるアンタゴニスト濃度 (モル濃度) の負の常用対数 NA:適用なし gsk002* gsk002* * 新薬承認情報提供時に置き換え
モルモット摘出回腸 (A および C) および摘出気管 (B および D) 標本のヒスタミン誘発収縮反応に対するレ Emax (%) :被験物質添加前のヒスタミン (1µM) による最大収縮反応 (Emax) に対する被験物質添加時のヒス タミンによる最大収縮反応の割合 (%) 平均値±標準誤差 (n=5~10)、実線は近似曲線 図 2.6.2-5 モルモット摘出回腸および摘出気管標本のヒスタミン誘発収縮に対する 4.2.1.1.8 の Fig. 1.より作成 2.6.2.2.2.2. モルモット摘出回腸標本におけるヒスタミン誘発収縮に対する作用の持続性 4.2.1.1.8 ミン誘発収縮に対する作用の持続性を比較した。 [方法]
雄Dunkin Hartley モルモットの摘出回腸標本を Tyrode 液(95%O2-5%CO2、37℃)中に懸垂
し、ヒスタミン収縮を十分抑制する濃度のレボセチリジン(0.3µM)、セチリジン(1µM)お 復刺激による収縮の経時変化を175 分後まで測定した。 ボセチリジン (A および B) および gsk002* (C および D) の拮抗作用 レボセチリジンおよびgsk002*の拮抗作用 モルモット摘出回腸標本を用いてレボセチリジン、セチリジンおよびgsk002*のヒスタ よびgsk002*(3μM)を添加して 60 分間反応させたのち、5 分間隔で洗浄してヒスタミン反
減弱した。 の作用は持続的ではないことが示された。 平均値±標準誤差 (n=8) Control:媒体 (0.1%DMSO) 処理 Emax (%) :被験物質処理前のヒスタミン (1µM) による最大収縮幅 (Emax) に対する各時点の最大収縮幅の割 合 (%) 図 2.6.2-6 モルモット摘出回腸標本のヒスタミン誘発収縮に対する作用の持続性 4.2.1.1.8 の Fig. 4.より作成 まとめ 以上より、レボセチリジンはモルモット摘出回腸および摘出気管標本におけるヒスタミン 持続性に乏しかった。 2.6.2.2.3. In vivo 抗ヒスタミン作用 2.6.2.2.3.1. モルモットにおけるヒスタミン誘発気管支攣縮に対する作用 4.2.1.1.9/ref 気管支攣縮に対する抑制作用を比較検討した。 [方法] ウレタン麻酔下のDunkin Hartley モルモットに気管カニューレを装着し、ガラミンで非動 化したのち、人工呼吸(50 回/min)下で圧トランスデューサーを用いて気管内圧を測定した。 セチリジンも同様の持続的な抑制作用を示したが、gsk002*の抑制作用は洗浄により急速に 以上より、レボセチリジンおよびセチリジンのH1受容体拮抗作用は持続的であり、gsk002* 誘発収縮に対してセチリジンおよびgsk002*よりもそれぞれ約 2~4 および 15~30 倍強い 抑制作用を示し、その作用はセチリジンと同様に持続的であった。一方、gsk002*の作用は 麻酔モルモットを用いてレボセチリジン、セチリジンおよびgsk002*のヒスタミン誘発
頸静脈に装着したカテーテルよりヒスタミンを5 分間隔で静脈内投与し、気管内圧増加量を 測定した。ヒスタミンによる気管内圧増加量が安定したのち、次のヒスタミン投与の2 分前 57 分後までの各ヒスタミン誘発気管内圧増加の抑制量を台形法で算出した。 また、別試験として、セチリジン(0.15mg/kg、n=6)の単回静脈内投与におけるヒスタミ ン誘発気管内圧増加に対する抑制率を同様の方法で測定した。 [結果] 制作用はそれぞれ投与42、42 および 17 分後に最大(それぞれ約 98、65 および 45%)となり、 測定終了時まで持続した。また、レボセチリジンの投与0~57、17~57 および 27~57 分後の 制作用を示すと考えられた。 表 2.6.2-5 モルモットにおけるヒスタミン誘発気管内圧増加に対する作用 4.2.1.1.9/ref の Table 1 より作成 気管内圧増加の抑制量 (cmH2O·min) 被験物質 0~17 0~27 0~57 17~27 17~57 27~57 レボセチリジン (n=5) 212 463 1424 252 1212 961 (n=6) 211 388 894 177 683 506 p 値 NS NS 0.049 NS 0.020 0.013 測定期間は被験物質投与後の時間 (分) NS:p≥0.05 (一元配置分散分析) 2.6.2.2.3.2. マウスにおけるヒスタミン誘発皮膚反応に対する作用 4.2.1.1.10/ref ン誘発皮膚反応の膨疹面積に及ぼす影響により比較検討した。 [方法] NMRI マウスにレボセチリジン(0.005~0.5mg/kg)、セチリジン(0.05~0.5mg/kg)または たは2µg/site)を左右 2 ヵ所に皮内投与して皮膚膨疹を惹起し、その直後にエバンスブルーを 静脈内投与した。ヒスタミン投与の30 分後にマウスを屠殺して皮膚を剥離し、膨疹面積を測 定し、膨疹面積を50%抑制する用量(ED50)を算出した。 [結果] にレボセチリジンまたはgsk002*の 0.15mg/kg を単回静脈内投与した。被験物質投与から レボセチリジン、セチリジンおよびgsk002*はいずれも 0.15mg/kgの静脈内投与によりヒ スタミン誘発気管内圧増加を抑制した。レボセチリジン、セチリジンおよびgsk002*の抑 期間において、気管内圧増加の抑制量はgsk002*と比較して有意に高かった(表 2.6.2-5)。 以上により、レボセチリジンはヒスタミン誘発気管支攣縮に対してgsk002*より強い抑 gsk002* レボセチリジン、セチリジンおよびgsk002*の抗ヒスタミン作用を、マウスのヒスタミ gsk002*(0.005~0.5mg/kg)を経口投与した。被験物質の投与 2 時間後にヒスタミン(1 ま レボセチリジン、セチリジンおよびgsk002*は 1μg/siteのヒスタミンによる皮膚膨疹に対
表 2.6.2-6 マウスにおけるヒスタミン誘発皮膚反応の膨疹面積に及ぼす影響 4.2.1.1.10/ref の TABLE 1~TABLE 3 より作成
ED50 (mg/kg) 被験物質 ヒスタミン 1µg/site ヒスタミン 2µg/site レボセチリジン 0.017 0.018 セチリジン 0.073 0.078 0.148 n=9~10 の成績より ED50を算出 2.6.2.2.3.3. ラットにおけるヒスタミン誘発皮膚反応に対する作用 4.2.1.1.11/ref ン誘発皮膚反応の膨疹面積に及ぼす影響により比較検討した。 [方法] (0.5~50mg/kg)を経口投与した。被験物質の投与 15 分後にヒスタミン(50µg/site)を皮内 投与して皮膚膨疹を惹起し、その直後にエバンスブルーを静脈内投与した。ヒスタミン投与 の30 分後にラットを屠殺して皮膚を剥離し、膨疹面積を測定し、膨疹面積を 50%抑制する用 量(ED50)を算出した。 [結果] レボセチリジンは、ヒスタミン誘発皮膚反応の膨疹面積に対してセチリジンと同程度の抑 表 2.6.2-7 ラットにおけるヒスタミン誘発皮膚反応の膨疹面積に及ぼす影響 4.2.1.1.11/ref より作成 被験物質 ED50 (mg/kg) [95%信頼区間] レボセチリジン 0.9 [0.2 – 3] セチリジン 2 [0.09 – 33] 5 [2 – 11] n=5 の成績より ED50を算出 2.6.2.2.3.4. イヌにおけるヒスタミン誘発皮膚反応に対する作用(経口投与) 4.2.1.1.12 ヌのヒスタミン誘発皮膚反応に及ぼす影響により比較検討した。 [方法] ビーグル犬を用いて2 週間の休薬期間を設けたクロスオーバー試験を実施した。レボセチ 回)を被験物質投与の0.5 時間前ならびに 0、1.5、3、6、9、12、24 および 32 時間後に反復 皮内投与して皮膚膨疹を誘発した。各ヒスタミン投与の直後にエバンスブルーを静脈内投与 した。被験物質投与の0、0.5、2、3.5、6.5、9.5、12.5、24.5 および 32.5 時間後にヒスタミン gsk002* 0.039 レボセチリジン、セチリジンおよびgsk002*の抗ヒスタミン作用を、ラットのヒスタミ SD ラットにレボセチリジン(0.05~50mg/kg)、セチリジン(0.05~5mg/kg)または gsk002* 制作用を示し、この作用はgsk002*よりも強かった(表 2.6.2-7)。 gsk002* レボセチリジン、セチリジンおよびgsk002*の経口投与による抗ヒスタミン作用を、イ リジン、セチリジンおよびgsk002*の 0.15mg/kg を単回経口投与し、ヒスタミン(0.5μg/50μL/
誘発皮膚膨疹の表面積を測定し、投与0 時間における膨疹面積をコントロールとした。コン トロールの膨疹面積に対する各時点の抑制率を算出し、時点ごとに投与群を比較した。 [結果] レボセチリジンおよびセチリジンは0.15mg/kg の単回経口投与によりヒスタミン誘発皮膚 セチリジンはいずれも投与3.5 時間後に最大抑制を示し、その抑制率はそれぞれ 56.7 および 46.4%であった。また、投与 9.5 時間後以降のレボセチリジンの抑制作用はセチリジンと比較 して有意に強かった(表 2.6.2-8)。 表 2.6.2-8 イヌにおけるヒスタミン誘発皮膚反応の膨疹面積に及ぼす影響(経口投与) 4.2.1.1.12 の TABLE 4 および TABLE 6 より作成 被験物質投与0 時間の膨疹面積に対する 抑制率 (%) 各投与群の抑制率の比較 被験物質 投与後の 時間 (hr) レボセチリジン セチリジン レボセチリジン vs セチリジン レボセチリジン vs セチリジン vs 0.5 13.7 ± 8.1 7.7 ± 8.1 0.1 ± 8.1 NS * NS 2.0 42.7 ± 9.4 32.4 ± 9.4 -1.5 ± 9.4 NS *** *** 3.5 56.7 ± 7.0 46.4 ± 7.0 5.3 ± 7.0 NS *** *** 6.5 47.0 ± 6.8 38.0 ± 6.8 1.1 ± 6.8 NS *** *** 9.5 46.1 ± 6.0 36.0 ± 6.0 -4.5 ± 6.0 * *** *** 12.5 46.3 ± 6.8 31.8 ± 6.8 0.4 ± 6.8 ** *** *** 24.5 41.3 ± 5.9 24.0 ± 5.9 -3.2 ± 5.9 *** *** *** 32.5 32.8 ± 4.8 18.6 ± 4.8 3.7 ± 4.8 *** *** *** 平均値±2×標準誤差 (n=9) NS:有意差なし、*:p<0.05、**:p<0.01、***:p<0.001 (t 検定) 2.6.2.2.3.5. イヌにおけるヒスタミン誘発皮膚反応に対する作用(静脈内投与) 4.2.1.1.13/ref および 4.2.1.1.14/ref イヌのヒスタミン誘発皮膚反応に及ぼす影響により比較検討した。 [方法] ビーグル犬を用いて2 週間の休薬期間を設けたクロスオーバー試験を実施した。イヌにレ 1.5mg/kg)を単回静脈内投与し、ヒスタミン(0.5µg/50µL/回)を 8 回にわたり反復皮内投与 して皮膚膨疹を誘発した。各ヒスタミン投与の直後にエバンスブルーを静脈内投与し、それ ぞれの30 分後(被験物質投与の 0、0.5、1、2、4、6、8 および 24 時間後)にヒスタミン誘 発皮膚膨疹の表面積を測定した。また、投与0 時間の膨疹面積に対する抑制率-時間曲線下面 積を台形法で算出した。 応の膨疹面積を抑制したが、gsk002*は抑制作用を示さなかった。レボセチリジンおよび gsk002* gsk002* gsk002* レボセチリジン、セチリジンおよびgsk002*の静脈内投与による抗ヒスタミン作用を、 ボセチリジン(0.15mg/kg)、セチリジン(0.15mg/kg)および gsk002*(0.15、0.46 および
較して有意に強く、1.5mg/kg と同程度であった(表 2.6.2-10)。 表 2.6.2-9 イヌにおけるヒスタミン誘発皮膚反応の膨疹面積に及ぼす影響(静脈内投与) 4.2.1.1.13/ref の TABLE II より作成 被験物質投与0 時間の膨疹面積に対する抑制率 (%) 各投与群の抑制率の比較 被験物質 投与後の 時間 (hr) レボセチリジン 0.15mg/kg セチリジン 0.15mg/kg 0.15mg/kg レボセチリジン vs セチリジン レボセチリジン vs セチリジン vs 0.5 65.4 ± 10.5 58.7 ± 9.2 -3.6 ± 25.3 NS * * 1.0 60.8 ± 11.4 57.5 ± 7.2 -1.3 ± 22.2 NS * * 2.0 62.4 ± 8.9 46.3 ± 14.7 -3.4 ± 26.5 NS * * 4.0 56.6 ± 10.9 41.4 ± 16.0 -0.8 ± 19.8 NS * * 6.0 53.9 ± 9.6 44.9 ± 7.4 -2.0 ± 18.6 NS * * 8.0 56.3 ± 5.6 40.9 ± 9.0 -6.1 ± 23.4 NS * * 24.0 39.0 ± 16.9 24.9 ± 20.4 -2.6 ± 25.5 NS * NS 平均値±標準偏差 (n=6) NS:有意差なし、*:p≤0.05 (Wilcoxon 検定) 表 2.6.2-10 イヌにおけるヒスタミン誘発皮膚反応の膨疹面積に及ぼす影響(静脈内投与) 4.2.1.1.14/ref の TABLE III および TABLE IV より作成 被験物質投与0 時間の膨疹面積に対する抑制率 (%) 試験1 試験2 被験物質投与後 時間 (hr) レボセチリジン 0.15mg/kg 0.46mg/kg レボセチリジン 0.15mg/kg 1.5mg/kg 0.5 70.6 ± 10.7 * 29.9 ± 11.0 69.1 ± 12.8 * 44.6 ± 16.2 1.0 72.7 ± 9.6 * 27.7 ± 16.1 66.7 ± 6.4 * 38.0 ± 12.4 2.0 73.4 ± 10.2 * 35.3 ± 15.7 55.5 ± 8.8 44.8 ± 17.5 4.0 68.9 ± 10.7 38.9 ± 20.7 56.0 ± 13.3 45.0 ± 12.7 6.0 61.2 ± 14.0 * 30.1 ± 9.1 56.0 ± 7.4 43.0 ± 15.2 8.0 52.4 ± 17.3 25.3 ± 15.6 46.5 ± 12.3 40.9 ± 12.6 24.0 42.8 ± 18.6 * 5.5 ± 16.0 20.6 ± 11.0 6.5 ± 13.0 抑制率AUC 12737 ± 3434 * 4983 ± 2303 9748 ± 1983 7133 ± 2740 平均値±標準偏差 (n=6) 抑制率AUC:被験物質投与 0 時間の膨疹面積に対する抑制率-時間曲線下面積 まとめ 以上より、モルモットのヒスタミン誘発気管支攣縮ならびにマウス、ラットおよびイヌの ヒスタミン誘発皮膚反応に対するレボセチリジンの抑制作用は、セチリジンと同程度以上で レボセチリジンの0.15mg/kgの静脈内投与による膨疹抑制作用はgsk002*の 0.46mg/kgと比 gsk002* gsk002* gsk002* gsk002* gsk002* *:p<0.05 (レボセチリジン群 vs gsk002*群、Wilcoxon 検定) gsk002*よりも強かった。 * 新薬承認情報提供時に置き換え
2.6.2.2.4. 好酸球および炎症性因子に対する作用 2.6.2.2.4.1. エオタキシン刺激による好酸球の血管内皮細胞間隙遊走に対する抑制作用 4.2.1.1.15/ref 末梢組織の好酸球増加はアレルギー性炎症の特徴であり、血管内皮細胞などで産生される 強力な好酸球遊走因子であるエオタキシンは、好酸球増加に重要な役割を果たしている[Jia, 1999]。そこで、好酸球のエオタキシン刺激による血管内皮細胞間隙遊走(TEM)に対するレ ボセチリジンの抑制作用を検討した。 [方法] 正常ヒト皮膚微小血管内皮細胞(HMVEC-d)または正常ヒト肺微小血管内皮細胞 (HMVEC-l)を 24 ウェル培養プレートに装着したトランスウェル(8µm ポアサイズフィル ター)上にコンフルエントになるまで培養した。 ヒトより採取した好酸球にレボセチリジン(0.0001~10µM)を添加し、37℃で 30 分間前 処理した。洗浄後の好酸球(2×105個/ウェル)をトランスウェルの上部に添加し、下部にエ オタキシン(0.1µg/mL)を添加した。細胞を 37℃で 60 分間培養し、下部に遊走した好酸球 を血球計算盤で測定し、TEM 率を算出した。 [結果] レボセチリジンは好酸球に前処理することにより、HMVEC-d および HMVEC-l におけるエ オタキシン刺激による好酸球のTEM を抑制し、それぞれ 0.01 および 0.1µM で最大抑制を示 した(図 2.6.2-7 および図 2.6.2-8)。 平均値±標準誤差 (n≥4) C:コントロール **:p<0.005 vs コントロール (t 検定) 図 2.6.2-7 HMVEC-d におけるエオタキシン刺激好酸球の TEM に対する抑制作用 4.2.1.1.15/ref の Fig. 3.より作成
平均値±標準誤差 (n≥4) C:コントロール *:p<0.05、**:p<0.005 vs コントロール (t 検定) 図 2.6.2-8 HMVEC-l におけるエオタキシン刺激好酸球の TEM に対する抑制作用 4.2.1.1.15/ref の Fig. 4.より作成 2.6.2.2.4.2. ヒト角化細胞における ICAM-1 および MHC 分子発現ならびに GM-CSF お よびケモカイン産生に対する抑制作用 4.2.1.1.16/ref ヒト角化細胞におけるIFN-γ およびヒスタミンの共刺激による ICAM-1 および MHC 分子発 現ならびにGM-CSF およびケモカイン産生に対するレボセチリジンの抑制作用を検討した。 [方法] 健康成人の皮膚生検組織より調製した角化細胞を、レボセチリジン存在下でIFN-γ(200U/mL) およびヒスタミン(100µM)を添加し、培養 24 時間後における細胞表面の ICAM-1、HLA-DR ならびにHLA-A、B および C の発現を、それぞれ FITC 標識抗 ICAM-1、抗 HLA-DR ならび
に抗HLA-A、B および C のモノクローナル抗体を用いて測定した。また、GM-CSF およびケ モカインであるCCL2/MCP-1、CCL-5/RANTES、CCL-20/MIP-3α および CXCL10/IP-10 の角化 細胞からの放出量をELISA 法により測定した。 [結果] 無刺激およびIFN-γ 刺激時の角化細胞において H1受容体のmRNA および蛋白質の発現が みられた。ヒスタミンはIFN-γ による ICAM-1 および MHC 分子の発現誘導を促進し(図 2.6.2-9)、 IFN-γ による GM-CSF、CCL2/MCP-1、CCL5/RANTES、CCL20/MIP-3α および CXCL10/IP-10 の放出を増大させた(図 2.6.2-10)。レボセチリジンは角化細胞において、IFN-γ およびヒス タミンの共刺激によるICAM-1 および MHC クラスⅠ(HLA-A、B および C)の発現を 0.01 ~10µM で濃度依存的に抑制した(図 2.6.2-9)が、MHC クラスⅡである HLA-DR 発現には 影響を示さなかった。レボセチリジンは、IFN-γ およびヒスタミンの共刺激による GM-CSF、 CCL2/MCP-1、CCL5/RANTES、CCL20/MIP-3α および CXCL10/IP-10 の産生に対しても 0.01 ~10µM で濃度依存的に抑制した(図 2.6.2-10)。
3 回の試験の平均を示す ∆MFI:平均蛍光強度の変化量
△:無処理、□:IFN-γ (200U/mL) 刺激、○:IFN-γ (200U/mL) およびヒスタミン (100µM) 刺激
*:p<0.05 vs IFN-γ およびヒスタミン刺激媒体群、†:p<0.05 vs IFN-γ 刺激媒体群 (Wilcoxon の符号順位検定) 図 2.6.2-9 角化細胞における ICAM-1 および MHC 分子発現に対する抑制作用
△:無処理、□:IFN-γ (200U/mL) 刺激、○:IFN-γ (200U/mL) およびヒスタミン (100µM) 刺激
*:p<0.05 vs IFN-γ およびヒスタミン刺激媒体群、†:p<0.05 vs IFN-γ 刺激媒体群 (Wilcoxon の符号順位検定) 図 2.6.2-10 角化細胞における GM-CSF およびケモカイン産生に対する抑制作用 4.2.1.1.16/ref の FIG 7.より作成 まとめ 以上より、レボセチリジンはHMVEC-d および HMVEC-l においてエオタキシン刺激による 好酸球の遊走を抑制し、角化細胞においてヒスタミンによるIFN-γ 刺激 ICAM-1 および MHC クラスⅠ発現の増強ならびにIFN-γ 刺激 GM-CSF、CCL2/MCP-1、CCL5/RANTES、 CCL20/MIP-3α および CXCL10/IP-10 産生の増強を抑制した。
2.6.2.3. 副次的薬理試験 2.6.2.3.1. 種々の G 蛋白質共役型受容体およびイオンチャネルに対する結合親和性 4.2.1.2.1 および 4.2.1.2.2 ため、G 蛋白質共役型受容体およびイオンチャネルに対する結合親和性について検討した。 レボセチリジンおよびセチリジンはアドレナリンα2C4 受容体に対する標識リガンドの特異 チャネルのリガンド結合に対して10µM で 50%以上の阻害作用を示さなかった。レボセチリ ジンのα2C4 受容体に対する親和性(pKi=5.8)は H1受容体に対する親和性(pKi=8.5)の 1/500 以下であり、セチリジンも同程度の選択性を示した。レボセチリジンおよびセチリジンは、 H1およびα2C4 受容体以外の G 蛋白質共役型受容体およびイオンチャネルに対しては 10µM でさらに低い阻害作用しか示さなかった(表 2.6.2-11)。 表 2.6.2-11 各種受容体およびイオンチャネルにおける標識リガンド結合阻害試験 4.2.1.2.1 の Table 1 および TABLE 2 ならびに 4.2.1.2.2 の TABLE 1 より作成
標識リガンドの特異的結合に対する阻害率 (%) 受容体 組織 レボセチリジン (10µM) セチリジン (10µM) (10µM) ヒスタミンH1 ヒトクローン 100 ± 1 100 ± 1 98 ± 1 ヒスタミンH2 モルモット大脳皮質 22 ± 13 15 ± 11 15 ± 14 ヒスタミンH3 モルモット大脳皮質 3 ± 1 -2 ± 3 6 ± 4 アデノシンA1 ヒトクローン 11 ± 5 0 ± 6 8 ± 4 アドレナリンα1 ラット大脳皮質 57 ± 5 44 ± 8 9 ± 6 アドレナリンα2C2 ヒトクローン 44 a 35 a 1 a アドレナリンα2C4 ヒトクローン 82 ± 1 75 ± 1 43 ± 2 アドレナリンα2C10 ヒトクローン 29 ± 2 22 ± 2 8 ± 3 アドレナリンβ1 ラット大脳皮質 4 ± 2 1 ± 8 3 ± 2 ドパミンD1 ラット線条体 9 ± 3 6 ± 6 1 ± 1 ドパミンD2 ラット線条体 5 ± 6 2 ± 2 -2 ± 1 ムスカリン ラット大脳皮質 2 ± 1 6 ± 6 4 ± 2 セロトニン5-HT1A ラット海馬 -2 ± 4 0 ± 3 -2 ± 1 セロトニン5-HT2 ラット大脳皮質 33 ± 1 32 ± 10 7 ± 1 L-type Ca2+ ラット大脳皮質 17 ± 5 16 ± 2 13 ± 3 Na+ type1 ラット大脳皮質 -1 ± 2 -4 ± 2 -4 ± 4 Na+ type2 ラット大脳皮質 1 ± 2 -3 ± 0 -7 ± 16 平均値±標準偏差 (n=3、a:n=1) 2.6.2.3.2. ムスカリン受容体サブタイプに及ぼす影響 4.2.1.2.3 レボセチリジンおよびセチリジンのムスカリン受容体サブタイプに及ぼす影響について検 レボセチリジン、セチリジンおよびgsk002*のヒト H1受容体に対する選択性を確認する 的結合を10μMで約 80%阻害した。一方、gsk002*はH1受容体以外の受容体およびイオン gsk002*
セチリジンのH1受容体に対する親和性はムスカリン受容体サブタイプに対する親和性と比較 して20000 倍以上であった(図 2.6.2-11B)。 A A BB A A BB A:ヒト H1受容体およびムスカリン受容体サブタイプに対するH1受容体アンタゴニストのpKiの平均値 (n=3) B:ヒト H1受容体選択性の比 (ムスカリン受容体サブタイプにおけるKi) / (H1受容体におけるKi) により算出 図 2.6.2-11 ヒト H1受容体およびムスカリン受容体サブタイプにおける親和性および H1受容体選択性 4.2.1.2.3 の Fig. 1.より作成 2.6.2.3.3. モルモット摘出回腸標本のセロトニン、アセチルコリンおよびニコチン誘発 収縮反応に対する作用 4.2.1.2.4 ニン、アセチルコリンおよびニコチン誘発収縮反応に対する影響を検討した。 よびニコチン誘発収縮に対してほとんど抑制作用を示さなかった(表 2.6.2-12)。 表 2.6.2-12 各種の収縮物質による収縮反応に対する拮抗作用 4.2.1.2.4 の Table 1 より作成 IC50 (µM) 被験物質 セロトニン (10µM) アセチルコリン (0.32µM) ニコチン (10µM) ヒスタミン (3.2µM) レボセチリジン >100 (32%) >100 (14%) >100 (37%) 2.0 セチリジン >100 (23%) >100 (17%) >100 (34%) 1.7 >100 (27%) >100 (11%) >100 (19%) 4.7 n=6~8 括弧内の数字は被験物質100µM を添加したときの収縮抑制率の最大値 (%) モルモット摘出回腸標本におけるレボセチリジン、セチリジンおよびgsk002*のセロト レボセチリジン、セチリジンおよびgsk002*はいずれもセロトニン、アセチルコリンお gsk002* * 新薬承認情報提供時に置き換え
2.6.2.3.4. ラット摘出大動脈標本の KCl 収縮に対する作用 4.2.1.2.5 ラット摘出大動脈標本のL-type Ca2+チャネルを介する高K+誘発収縮反応に及ぼすレボセチ いことが示された。 2.6.2.4. 安全性薬理試験 レボセチリジンの安全性薬理試験として、中枢神経系、心血管系、呼吸系および胃腸管系 に及ぼす影響を検討した。 2.6.2.4.1. 中枢神経系に及ぼす影響 4.2.1.3.1~4.2.1.3.3 ラット単回経口投与試験の一般症状および行動観察において、25mg/kg 群で影響はみられ なかった。50mg/kg 群では投与 2 時間後に軽度の腹筋緊張の上昇がみられ、100mg/kg 群では 投与1 時間後に軽度の反応性の低下がみられた。 ラットの自発運動およびペントバルビタール誘発睡眠に対しては100mg/kg までの経口投与 で影響を及ぼさなかった。 2.6.2.4.2. 心血管系に及ぼす影響 2.6.2.4.2.1. hERG 試験 4.2.1.3.4 hERG 遺伝子を導入したアフリカツメガエル卵母細胞において、30µM までの濃度で hERG 電流に影響を及ぼさなかった。 2.6.2.4.2.2. モルモット単離心室筋細胞の遅延整流 K+電流に及ぼす影響 4.2.1.3.5/ref モルモット単離心室筋細胞において100µM の濃度で IKrを45%に抑制した。 2.6.2.4.2.3. イヌ摘出プルキンエ線維の活動電位に及ぼす影響 4.2.1.3.6 イヌ摘出プルキンエ線維の活動電位に対して3µM までの濃度では影響を及ぼさなかった。
30 および 300µM の濃度で APD50、APD70およびAPD90の延長が認められたが、最大拡張期電
位、活動電位振幅、活動電位0 相の最大立ち上がり速度および APD30に対しては影響を及ぼ
リジン、セチリジンおよびgsk002*の影響を検討した。レボセチリジン、セチリジンおよ
びgsk002*は 10μM の高濃度においても高 K+誘発収縮反応を抑制しなかった。このことか
2.6.2.4.2.4. 麻酔イヌの循環動態試験 4.2.1.3.7 麻酔イヌの循環動態試験において、1、3.2 および 10mg/kg を 50 分間隔で連続して静脈内注 入投与しても、血圧、心拍数、左心室収縮期血圧、心拍出量、大腿動脈の血流速度および血 管抵抗、心電図ならびに血液ガスに影響は認められなかった。 2.6.2.4.2.5. 麻酔イヌの徐脈性 QT 延長症候群モデルに及ぼす影響 4.2.1.3.8 麻酔イヌの徐脈性QT 延長症候群モデルにおいて、0.8、1.6 および 3.2mg/kg/hr の累積的な 静脈内持続注入によって単相性活動電位持続時間の延長、貫壁性再分極相のばらつき (transmural dispersion of repolarization)の増大および QT 間隔の延長は認められず、徐脈性不 整脈も誘発しなかった。 2.6.2.4.3. 呼吸系に及ぼす影響 4.2.1.3.7 麻酔イヌの呼吸機能試験において、1、3.2 および 10mg/kg を 50 分間隔で連続して静脈内注 入投与しても1 回換気量、分時換気量および呼吸数に影響は認められなかった。 2.6.2.4.4. 胃腸管系に及ぼす影響 4.2.1.3.9 ラットに25、50 または 100mg/kg を単回経口投与しても、消化管の炭末輸送能に影響は認 められなかった。 2.6.2.5. 薬力学的薬物相互作用試験 該当する試験は実施していない。 2.6.2.6. 考察及び結論 2.6.2.6.1. 効力を裏付ける試験 受容体結合試験においてレボセチリジンはヒトH1受容体に対して高い親和性(Ki=2.5nM) ボセチリジンの結合は可逆的であり、ヒスタミンと競合的に相互作用することが示された。 レボセチリジンのヒトH1受容体からの解離は緩やかであったことから、in vivo において持続 的な作用を示すと考えられた。 モルモットの摘出回腸および摘出気管標本を用いた受容体機能試験において、レボセチリ 抗作用を示し、低濃度では競合的拮抗作用、高濃度では競合的作用およびinsurmountable な 作用が併存した拮抗様式を示した。レボセチリジンが非可逆的な拮抗と類似したinsurmountable の拮抗様式を示した理由としては、レボセチリジンのH1受容体からの解離が遅いために受容 を示した。セチリジンおよびgsk002*の Kiは6.1 および 73nM であり、レボセチリジンの親 和性はセチリジンおよびgsk002*と比較してそれぞれ約 2 および 30 倍高かった。また、レ ジンはセチリジンおよびgsk002*よりもそれぞれ約 2~4 および 15~30 倍強いH1受容体拮
体を長時間占有し、それによりヒスタミンが作用しうる受容体量が低下し、余剰受容体注2)が 枯渇することによりヒスタミンの最大反応が低下したと考えられる(4.2.1.1.8)。 レボセチリジンは麻酔モルモットにおけるヒスタミン誘発気管支攣縮ならびにマウス、ラッ トおよびイヌにおけるヒスタミン誘発皮膚膨疹を抑制したことから、in vivo で抗ヒスタミン た。イヌにおいてヒスタミン誘発皮膚膨疹を抑制したときのレボセチリジンの血漿中未変化 体濃度は、静脈内投与4 時間後で 0.44µg/mL であり(4.2.1.1.14/ref)、この濃度はレボセチリ ジンの臨床推奨用量である5mg をヒトに投与したときの Cmax(0.31µg/mL)(5.3.1.1.1)と同 程度であったことから、レボセチリジンは臨床推奨用量をヒトに投与することにより抗ヒス タミン作用を示すことが推察される。 レボセチリジンは血管内皮細胞においてヒスタミン、IL-1 または TNFα で刺激したときの エオタキシン産生を抑制し[Ying, 2002]、皮膚および肺の微小血管内皮細胞の単層膜における エオタキシン刺激による好酸球のTEM を抑制したことから、抗炎症作用を有することが示唆 された。レボセチリジン(0.1~100µM)はヒト皮膚血管内皮細胞において、TNFα 刺激によ
るVCAM-1 発現誘導および NF-κB の活性化を抑制した[Michel, 2003]。VCAM-1 は血管内皮 細胞に発現する細胞接着分子であり、血管内皮細胞への好酸球の浸潤において重要な役割を 果たす[Walsh, 1991]。NF-κB は細胞内に普遍的に存在し、種々の炎症刺激により活性化され、 炎症性シグナルを伝達する重要な転写因子である[Barnes, 1997]。ヒト H1受容体を細胞に遺伝 子導入することによりNF-κB のアゴニスト非依存的な活性が増大し、レボセチリジンはこの NF-κB 活性を H1受容体に対するKiと同程度の濃度で阻害した[Bakker, 2001]。したがって、 レボセチリジンは炎症性転写因子であるNF-κB の活性化を誘導する H1受容体を抑制するこ とによりTNFα などによる NF-κB を介した炎症性シグナル伝達を抑制し、抗炎症作用を示す と考えられた。また、レボセチリジンは角化細胞において、IFN-γ およびヒスタミンの共刺激 によるICAM-1 および MHC クラスⅠの発現を濃度依存的に抑制した。MHC 分子は抗原提示 において重要な役割を果たす糖蛋白質であり、ICAM-1 は表皮に T リンパ球が保持されると きのおもな接着分子である(4.2.1.1.16/ref)。ICAM-1 は角化細胞に対する CD4+およびCD8+T リンパ球の細胞障害活性の重要な共刺激分子として機能する(4.2.1.1.16/ref)。レボセチリジ ンは、IFN-γ およびヒスタミンの共刺激による GM-CSF、CCL2/MCP-1、CCL5/RANTES、 CCL20/MIP-3α および CXCL10/IP-10 の産生を濃度依存的に抑制した。これらの炎症性メディ エーターの産生は表皮における白血球の誘引および活性化に関与していることが知られてい ることから(4.2.1.1.16/ref)、レボセチリジンの抗炎症作用の作用機序にこれらの炎症性メディ エーターの産生抑制が関連する可能性が考えられた。 作用を有することが示された。その活性はセチリジンと同程度以上で、gsk002*より強かっ
2.6.2.6.2. 副次的薬理試験 レボセチリジンはH1受容体以外のG 蛋白質共役型受容体に対して強い親和性を示さず、 ヒスタミンH2およびH3、アデノシンA1、アドレナリンα1、α2およびβ1、セロトニン5-HT1A および5-HT2、ドパミンD1およびD2ならびにムスカリンM1、M2、M3、M4およびM5受容体 と比較して少なくとも500 倍以上の H1受容体選択性を示した。また、セロトニン、アセチル コリンおよびニコチン受容体機能に対して影響を及ぼさなかった。さらに、レボセチリジン はラット摘出大動脈標本の高K+誘発収縮反応に対して抑制作用を示さなかったことから、 L-type Ca2+チャネルに対して影響を及ぼさないことが示唆された。したがって、レボセチリ ジンはヒトH1受容体に対して選択的に作用し、その他の受容体およびイオンチャネルに対し て作用する可能性は低いと考えられた。 2.6.2.6.3. 安全性薬理試験 ラットの一般症状および行動観察において、レボセチリジンは50mg/kg の経口投与で軽度 の腹筋緊張の上昇、100mg/kg で軽度の反応性の低下を示した。 hERG 試験においてレボセチリジンは 30µM で hERG 電流に対して影響を示さなかったが、 イヌ摘出プルキンエ線維において30µM 以上で活動電位持続時間を延長させ、モルモット単 離心室筋細胞においては100µM で IKrを抑制した。 麻酔イヌの循環動態試験において、レボセチリジンは10mg/kg まで累積的に静脈内注入投 与しても血圧、心拍数、左心室収縮期血圧、心拍出量、大腿動脈の血流速度および血管抵抗、 心電図ならびに血液ガスに影響を示さなかった。また、麻酔イヌの徐脈性QT 延長症候群モ デルにおいて、レボセチリジンは3.2mg/kg/hr までの累積的な静脈内持続投与によっても心筋 再分極の遅延、QT 間隔延長および徐脈性不整脈を誘発しなかった。 麻酔イヌの呼吸機能試験において、レボセチリジンは10mg/kg まで累積的に静脈内注入投 与しても1 回換気量、分時換気量および呼吸数に影響を示さなかった。 ラットの消化管炭末輸送能に対して、レボセチリジンは100mg/kg を単回経口投与しても影 響を示さなかった。 安全性薬理試験の各試験について、無影響量および影響量における推定曝露量(Cmax)ま たは血漿中濃度とレボセチリジンの臨床推奨用量である5mg をヒトに経口投与したときの Cmax(0.31µg/mL=0.67µM)(5.3.1.1.1)の比較を表 2.6.2-13 に示す。
表 2.6.2-13 安全性薬理試験における曝露量とヒト曝露量の比較 試験項目 投与経路 無影響量 および影響量 各用量に対応する 推定Cmaxまたは 血漿中濃度 (µg/mL) Cmaxまたは 血漿中濃度 の参照 動物/ヒト 比d ラット一般症状および行動 観察 経口 a: 25mg/kg b: 50mg/kg Cmax: 13.55c Cmax: 27.1c 2.6.4.3.1.1.1 約40 約80 ラット自発運動 経口 a: 100mg/kg Cmax: 54.2c 2.6.4.3.1.1.1 約170 ラットペントバルビタール 誘発睡眠 経口 a: 100mg/kg Cmax: 54.2c 2.6.4.3.1.1.1 約170 hERG 試験 in vitro a: 30µM 約40 イヌ摘出プルキンエ線維 in vitro a: 3µM b: 30µM 約4 約40 モルモット単離心室筋細胞 in vitro b: 100µM 約140 イヌ循環動態試験 静脈内 a: 10mg/kg 25.2 (投与 45 分後) 4.2.1.3.7 約80 イヌ徐脈性QT 延長症候群 モデル 静脈内 a: 3.2mg/kg/hr 9.49 (投与終了後) 4.2.1.3.8 約30 イヌ呼吸機能試験 静脈内 a: 10mg/kg 25.2 (投与 45 分後) 4.2.1.3.7 約80 ラット消化管炭末輸送能 経口 a: 100mg/kg Cmax: 54.2c 2.6.4.3.1.1.1 約170 a:無影響量 b:影響量 c:雄ラットに 25mg/kg を単回経口投与したときの Cmax (13.55µg/mL) (2.6.4.3.1.1.1 参照) より Cmaxが投与量の 増加に伴って増加すると仮定して推定 d:ヒトにレボセチリジン 5mg を経口投与したときの Cmax (0.31µg/mL=0.67µM) (5.3.1.1.1) と比較 以上をまとめると、レボセチリジンは中枢神経系および心血管系に影響を示したものの、 いずれも高用量においてのみ認められたものであり、ヒトの臨床推奨用量の約30 倍までの用 量では影響はみられなかった。また、呼吸系および胃腸管系に対して影響を示さなかった。 以上のことから、レボセチリジンは臨床使用したときに中枢神経系、心血管系、呼吸系およ び胃腸管系に対して重篤な副作用を発現する可能性は低いと考えられる。 2.6.2.6.4. 結論 レボセチリジンは、セチリジンよりも約2 倍強力な選択的 H1受容体アンタゴニストであり、 ン作用を示す。さらに、抗炎症作用を示し、臨床使用したときに中枢神経系、心血管系、呼 吸系および胃腸管系に対して重篤な副作用を発現する可能性は低いと推察されることから、 レボセチリジンのアレルギー性疾患治療薬としての高い有用性が示唆される。 2.6.2.7. 図表 図表は本文中に記載した。 2.6.2.8. 参考文献 in vitro ならびに in vivo においてセチリジンと同程度以上で gsk002*よりも強い抗ヒスタミ
Barnes PJ, Adcock IM. NF-κB: a pivotal role in asthma and a new target for therapy. Trends
Pharmacol Sci. 1997;18:46-50.
Bloom FE. Neurotransmission and the central nervous system. In: Hardman JG, Limbird LE, Gilman AG, editor. Goodman & Gilman’s The pharmacological basis of therapeutics. 10th ed. New
York:McGraw-Hill, 2001:p.293-320.
Brown NJ, Roberts LJ. Histamine, bradykinin, and their antagonists. In: Hardman JG, Limbird LE, Gilman AG, editor. Goodman & Gilman’s The pharmacological basis of therapeutics. 10th ed. New York:McGraw-Hill, 2001:p.645-67.
Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099-108.
Jia GQ, Gonzalo JA, Hidalgo A. Selective eosinophil transendothelial migration triggered by eotaxin via modulation of Mac- l/ICAM-1 and VLA-4/VCAM-1 interactions. Int Immunol. 1999;11:1-10. Michel L, Jean-Louis F, Boland S et al. Inhibition by levocetirizine of VCAM-1 expression on human dermal endothelial cells. J Invest Dermatol. 2003;121:Abs.1251.
Walsh GM, Mermod JJ, Hartnell A. Human eosinophil, but not neutrophil, adherence to
IL-1-stimulated human umbilical vascular endothelial cells is α4β1 (very late antigen-4) dependent. J
Immunol. 1991;146:3419-23.
Ying S, Meng Q. The effect of levocetirizine on histamine and cytokine-induced up-regulation of eotaxin by endothelial cells. Allergy. 2002;57 Suppl:146.
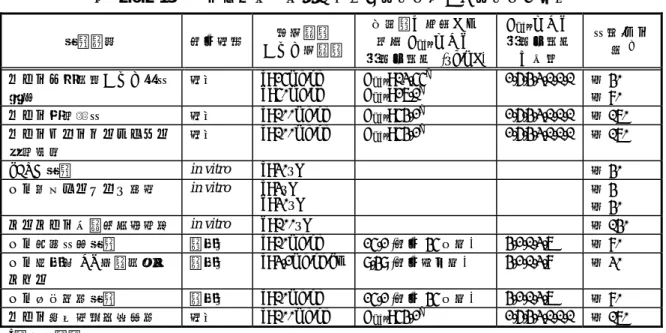
2.6.3. 薬理試験概要表 2.6.3.1. 薬理試験:一覧表 被験物質:レボセチリジン 試験の種類 試験系 投与方法 実施施設 報告書番号 資料番号 効力を裏付ける試験 受容体結合試験
マウス大脳皮質膜標品を用いたメピラミン結合阻害試験 マウス大脳皮質膜標品 in vitro UCB (ベルギー) RRLE95A0510 4.2.1.1.1 ヒトH1受容体に対する親和性および結合特性 組換えヒトH1受容体発現
CHO 細胞膜標品
in vitro UCB (ベルギー) RRLE96A1901、
RRLE95M1801、 ADPE02A3102 4.2.1.1.2、 4.2.1.1.3、 4.2.1.1.4 ヒトH1受容体に対する3H-標識体の結合特性 組換えヒトH1受容体発現 CHO 細胞膜標品
in vitro UCB (ベルギー) ADPE02E1404 4.2.1.1.5 In vitro 抗ヒスタミン作用
モルモット摘出組織におけるヒスタミン誘発収縮に対する 作用
モルモット摘出回腸標本、 モルモット摘出気管標本
in vitro UCB (ベルギー) RRLE97G1803、
RRLE98J2801、 ADPE03F0305 4.2.1.1.6、 4.2.1.1.7、 4.2.1.1.8 モルモット摘出回腸標本におけるヒスタミン誘発収縮に対す る作用の持続性
モルモット摘出回腸標本 in vitro UCB (ベルギー) ADPE03F0305 4.2.1.1.8 In vivo 抗ヒスタミン作用
モルモットにおけるヒスタミン誘発気管支攣縮に対する作用 Dunkin Hartley モルモット 静脈内 UCB (ベルギー) RRLE95G1801 4.2.1.1.9/ref マウスにおけるヒスタミン誘発皮膚反応に対する作用 NMRI マウス 経口 UCB (ベルギー) RRLE95E2901 4.2.1.1.10/ref ラットにおけるヒスタミン誘発皮膚反応に対する作用 SD ラット 経口 UCB (ベルギー) RRLE95A0502 4.2.1.1.11/ref イヌにおけるヒスタミン誘発皮膚反応に対する作用 (経口投与) ビーグル犬 経口 UCB (ベルギー) RRLE95A0504 4.2.1.1.12 イヌにおけるヒスタミン誘発皮膚反応に対する作用 (静脈内投与) ビーグル犬 静脈内 UCB (ベルギー) RRLE95A1202、 RRLE95A0512 4.2.1.1.13/ref、 4.2.1.1.14/ref 好酸球および炎症性因子に対する作用 エオタキシン刺激による好酸球の血管内皮細胞間隙遊走に対 する抑制作用 HMVEC-d、HMVEC-l、 ヒト好酸球 in vitro 、 UCB (スイス) ADPE02J0203 4.2.1.1.15/ref ヒト角化細胞におけるICAM-1 および MHC 分子発現ならび にGM-CSF およびケモカイン産生に対する抑制作用
ヒト皮膚角化細胞 in vitro 、 ADPE04L2201 4.2.1.1.16/ref
2.6.3. 薬 理 試 験 2.6.3 - p. 1