寄稿論文
カチオン性 10 族金属錯体を用いた不斉触媒反応の新展開:
パラジウムエノラートを鍵とする反応を中心にして
独立行政法人 理化学研究所 袖岡有機合成化学研究室濱島 義隆
袖岡 幹子
1. はじめに
カルボニル化合物の金属エノラートは,様々な求電子剤との反応に適用できる汎用性の高い 求核剤であり,有機合成化学においてなくてはならない存在である。1 エノラートを求核剤とす る不斉触媒反応を開発するには,大きく分けて二つの方法論が考えられてきた。ひとつは,予 め Si または Sn エノラートを調製しておき,キラルルイス酸(B, Al, Ti, Cu など)で求電子剤を 活性化して反応を行うものである。もうひとつは,キラル塩基触媒を用いてケトンからキラル エノラート(La, Zn, Ca, R4N+)を直接発生させる方法である。特に後者はより原子効率が高く, 最近の不斉有機触媒と共に著しい発展を見せている。2 一方,我々は別の可能性として,後周期遷移金属(特にパラジウム)エノラートの利用に注 目した。後周期遷移金属の電気陰性度は,古典的エノラートに用いられるアルカリ金属に比べ かなり大きく,エノラートの分極の程度は小さいと予想される。従って,後周期遷移金属エノ ラートは温和な反応性を示し,典型金属(Li, Mg, B, Al など)や前周期遷移金属エノラート(Ti, Zrなど)の反応で必要とされる低温・無水条件を必要としない可能性があり,極めて興味深い と考えた。M* = metals (La, Zn) with chiral ligands or chiral ammonium ions R O R OM* R OM X R' X R' LA* R O R' XH R OM' M = Si, Sn + +
LA* = chiral Lewis acid
M' = late transition metals
Mild reactivity?
*
Scheme 1. Representative approaches to achieve enantioselective reactions with enolates.
Ito-Saegusa 反応は,シリルエノラートから Pd エノラートが生成し,β- ヒドリド脱離により エノンを合成する反応として知られている。3 他にも Pd エノラート種が,アルキルパラジウム 種と同様に多重結合への挿入反応やカップリング反応を起こすと報告されていた。我々は中間 体として生成する Pd エノラートの反応性に興味を持った。配位子や反応条件を工夫すれば, エノラートは十分な求核性を示し温和な条件でアルデヒドとのアルドール反応が進行するので はないかと考えた。当時,Pd エノラートの求核剤としての反応性はほとんど知られておらず, 辻らによるアリルβ-ケトエステルから脱炭酸を経て生成するPdエノラートの分子内アルドール およびマイケル反応に関する報告が唯一の例であった。4 我々はこの先駆的な研究に勇気づけら れて,新しい不斉触媒反応を目指しつつ Pd エノラートの研究を開始した。
R
O Pd(II)L* Ito-Saegusa reaction
Our initial hypothesis: Nucleophilc reactions should also occur. Me3SiO Pd(OAc)2 O H Pd(II) Pd-H O R Me3SiO R OPd(II)L* R O Pd(II)L* X R' R O R' XH L*-Pd(II) β-H elimination Nucleophilic reaction * P P Pd O H H O Pd P P + 2X -* * + P P Pd OH2 OH2 2+ * 2X -P P M OTf OTf * PAr2 PAr2 PAr2 PAr2 O O O O P P Ni Y Y 1 2 3: M = Pd 4: M = Pt X = TfO or BF4 * 5: Y = OTf 6: Y = Cl a: Ar = Ph: (R)-BINAP b: Ar = 4-MeC6H4: (R)-Tol-BINAP c: Ar = 3,5-Me2C6H3: (R)-DM-BINAP d: Ar = Ph: (R)-SEGPHOS e: Ar = 3,5-Me2C6H3: (R)-DM-SEGPHOS
f: Ar = 3,5-(t-Bu)2-4-MeOC6H2: (R)-DTBM-SEGPHOS
a-c d-f
Scheme 2. Our original hypothesis.
本総説では,遷移金属エノラートに関する研究過程で開発したカチオン性遷移金属錯体の特 徴的な反応性を示しつつ,それを基盤とした様々な不斉触媒反応について紹介する。我々の報 告以前,または報告後に他の研究グループから報告されたすぐれた成果については紙面の都合 上割愛させて頂き,総説にあげた論文を参照して頂きたい。5
2. 錯体とその調製法
本総説で用いる光学活性カチオン性 10 族金属錯体を Figure 1 に示す(1-6)。Scheme 3 に示 すようにパラジウムアクア錯体 1 は,BINAP 類縁体を配位子とする塩化パラジウム錯体から水 存在下,銀塩との陰イオン交換反応によって収率よく合成できる。また,パラジウムμ- ヒドロ キソ錯体 2 は,酸性を示すアクア錯体を 1 当量の塩基と処理することで合成できる。配位子に よって色の濃淡に違いはあるが,黄色からオレンジ色の安定な固体として得られ,特別な注意 を必要とすることなく空気下で扱うことができる。6 これらに加えて,ニッケルまたは白金錯体もほぼ同様の方法で合成可能であり,それぞれ黒 紫色(Ni),ベージュ色(Pt)の固体として得られる。錯体 3-6 は無水条件下で調製し,不活性 雰囲気下で保存している。P P Pd Cl Cl * P P Pd OH2 OH2 2+ * 2X -P P Pd O H H O Pd P P + 2X -* * + P P M Cl Cl * P P M OTf OTf * 1 7a AgX (2 equiv) H2O (2 equiv) Acetone aq. NaOH (1 equiv) CH2Cl2 2 3-5 AgOTf (2 equiv) CH2Cl2 M = Ni, Pd, Pt P P Pd O Ph + * Ph Me3SiO O R H Ph O R OH P P Pd O H H Ph OSiMe3 P P Pd O R H O Ph X -X -+ 1a (5 mol%) TMU, 0 °C 8 9 10 up to 89% ee 2+ 2X -* + * Me3SiOH HX Pd enolate (I)
Scheme 3. Preparation of chiral metal complexes.
3. トランスメタル化を経るパラジウムエノラートの生成と不斉触媒反応
先の仮説に従って種々条件を検討したところ,パラジウムアクア錯体1を用いることでシリル エノラート8 とアルデヒド9 との不斉アルドール反応が円滑に進行することを見出した。7 この反応はDMFやテトラメチルウレア(TMU)などの極性溶媒中で円滑に進行し,また無水条件 では反応が極めて遅いという特徴を有する。これらは,一般的なルイス酸触媒による不斉向山 アルドール反応とは全く異なる性質である。2 反応機構を詳細に検討したところ,Scheme 4に 示すように水(または1から生成するPdOH)がシリル基への求核剤として作用し,トランスメタ ル化によりキラルPdエノラート(I)が鍵中間体として生成していることが明らかとなった。生成 物はルイス酸触媒による向山アルドール反応と同じであるが,その反応メカニズムは全く異な るということになる。更に,β-水素を有する基質でもエノンの生成は起こらず,収率よくアル ドール付加体が得られたが,これはBINAPなどの二座不斉配位子を有するPdエノラートの場合 には,主にO-エノラートとして存在するためと考えている。しかも,反応系内には,トランス メタル化の際に生成するプロトン酸(HX)や水が存在するにも拘らず,Pdエノラートはプロトン 化に優先してアルドール反応を起こした。これは他の強い塩基性を示す金属エノラートには見 られない特異な性質であり,キラルPdエノラートの求核剤としての更なる可能性を示していた。Scheme 4. Catalytic asymmetric aldol reaction via chiral Pd enolates.
そこで次に,キラル Pd エノラートを鍵中間体として用い,当時未開拓分野であった触媒的不 斉マンニッヒ型反応の開発を目指した。8 アクア錯体 1 存在下,イミン 11 との反応を行ったと ころ,反応は円滑に進行するもののラセミ体が得られるだけであった。比較実験より,錯体1 か ら生成するプロトン酸がイミンを活性化し,触媒非関与のラセミ体を与える反応が素早く進行 するためと分かった。そこで,プロトン酸を生成せずに Pd エノラートを与える触媒を検討した 結果,Pd-μ- ヒドロキソ錯体 2 を見出した。Scheme 5 に示すように,2 が解離してできる単核 の Pd-OH 錯体がトランスメタル化を促進し,キラル Pd エノラートが効率的に生成する。その結 果,目的とする不斉マンニッヒ型反応が円滑に進行し,高いエナンチオ選択性で目的物を得る ことができた。6,9
Scheme 5. Catalytic asymmetric Mannich-type reaction via chiral Pd enolates.
4. 酸・塩基触媒としてのカチオン性パラジウム錯体
炭酸脱水酵素の活性中心にある亜鉛イオンは,配位によって水の酸性度を変化させ,水酸化 物イオンを生成することが知られている。これまでの研究過程で観察された様々な現象を考慮 すると,合成したカチオン性 Pd 錯体は Scheme 6 に示すような関係にあると考えられた。すな わち,カチオン性 Pd 錯体は単にルイス酸として機能するだけではなく,水が配位するとその酸 性度が上昇するために Brønsted 酸として作用する。ここで,脱プロトン化が起これば,Pd-OH 単核錯体やそれが二量化したμ-ヒドロキソ錯体が生成し,それらは先に述べたトランスメタル 化を促進する求核剤だけでなく,弱いながらも Brønsted 塩基として作用すると考えられる。 これらは互いに平衡にあると予想されるため,我々は錯体 1 および 2 が酸・塩基触媒として機 能すると期待した。 P P Pd O + * P P Pd O Ph + * Ph Me3SiO Ph O CO2i-Pr NHPMP N CO2i-Pr PMP X- Ph OSiMe3 H X -X -P P Pd O Ph N PMP CO2i-Pr Me3SiOH + 2b (5 mol%) DMF, 25 °C 8 11 12 95%, 90% ee 2 + * PMP = 4-MeO-C6H4 12 Pd enolate (I) P P Pd OH2 H O H 2+ * P P Pd OH + * P P Pd O H H O Pd P P + * * + P P Pd 2+ * -H+ X -2X- 2X -2 1 2X-Lewis acid Brønsted acid Brønsted base
Scheme 6. Chiral Pd complexes as acid-base catalysts.
上記の仮説を検討したところ,錯体 1 と 2 は酸・塩基触媒として機能し,β- ジケトンや β- ケ トエステルのような比較的酸性度の高い1,3-ジカルボニル化合物と反応させると,Scheme 7に 示すようにキラルPdエノラート(II)が脱プロトン化によって直接生成することが分かった。酸性 度の高い化合物を用いているものの,我々はこれらのエノラート生成が非塩基性条件で起こる 点に強い関心を持った。特に,アクア錯体からエノラートが生成する場合,強いプロトン酸が 同時に生成する点は興味深く,一般的な塩基性条件のエノラートとは全く異なる反応性を示す と期待された。5b H2O X -R1 O O R3 R2 H R3 O O Pd P P R1 R2 P P Pd OH2 OH2 P P Pd O H H O Pd P P *
Chiral palladium enolate (II) H2O, HX + 2+ 2X -* + 2X -* * + 1 2
Scheme 8. Catalytic asymmetric Michael reaction of β-ketoesters. 反応機構解析を行うと,Pd エノラートの興味深い特性が明らかとなった(Scheme 9)。μ- ヒ ドロキソ錯体2bと,パラジウムに対して1当量のβ-ケトエステル13aから得られるPdエノラー トの反応性は低く,室温において 2 当量のメチルビニルケトンと反応させたが,反応は全く進 行しなかった。しかしながら,トリフルオロメタンスルホン酸(TfOH)を 1 当量加えたところ, 反応は円滑に進行しマイケル付加体 15a が収率 89%,不斉収率 99%で得られ,アクア錯体 1b の 生成が NMR で確認された。Scheme 8 の結果と併せて考えると,錯体 2 から生成した二座配位 Pdエノラートの求核性はエノンとの反応に対しては十分でないが,アクア錯体からは Pd エノ ラートと同時にプロトン酸が生成し,そのプロトン酸がエノンを活性化することで反応が進行 したことを示唆している。すなわち,Pd エノラートとプロトン酸の共同作業というユニークな 反応性を見出すことができた。 O Me R1 OR3 O R2 Me O O R1 OR3 O O R2 O O Ot-Bu O O Ot-Bu O Ot-Bu O Me Me O Ot-Bu O Me Me O O O Br Pd cat. 1a or 1b (X = TfO) (2-10 mol%) THF -20 ~ 0 °C 15a-f + 13a-f 14 13a 93%, 93% ee 13b 92%, 90% ee 13c 88%, 89% ee 13d 88%, 90% ee 13e 82%, 94% ee O O Ot-Bu Pd P P O O Ot-Bu TfO -Me O O O Ot-Bu Me O Pd P P OH2 OH2 H3O+ Pd P P O O R1 Ot-Bu R2 Pd P P O O R1 Ot-Bu R2 Y X R3 +H TfO -2b (X = OTf) (1 equiv to Pd) THF-d8, rt, 2h + * 13a No reaction TfOH (1 equiv) 0 °C, 5 h rt 89%, 99% ee + 1b 14 (2 equiv) 15a 1 2+ * β-ketoester 2TfO -+ * + + *
Cooperative action between Pd enolate and proton
5. 触媒的不斉マイケル反応
Pdエノラート(II) は,様々な求電子剤と反応することが明らかとなった。我々はまず,エノン とのマイケル反応を検討した。エノン側に不斉を誘起するタイプの反応はほぼ完成の域に達し ているが,求核剤側に不斉を誘起するタイプの反応で一般性にすぐれた反応は現在でも限定さ れている。10 その成功例のひとつが我々の反応である。触媒量のアクア錯体 1 を用いることで, 様々なβ-ケトエステルとメチルビニルケトンとのマイケル反応が円滑に進行し,高い選択性で 不斉4級炭素を有するマイケル付加体を得ることができた(Scheme 8)。11β 位に置換基を持つエノンはメチルビニルケトンに比べ反応性が低いため,13 のような β- ケ トエステルとの触媒的不斉マイケル反応の報告例は現在でも少ない。しかしながら,共同活性 化を示す我々の反応系では,β位にメチル基やフェニル基を持つエノンも用いることができる。 Scheme 10に示すように 5 mol%のアクア錯体 1a を用いたところ反応は円滑に進行し,満足の ゆくジアステレオ選択性と99%という極めて高いエナンチオ選択性で3級−4級連続不斉点を 有するマイケル付加体 17 を得ることができた。 また,アルコール中でも機能するパラジウム触媒の特徴を活かすことで,非常に不安定な アクロレイン 18a との反応も収率よく行うことができた。すなわち,THF 中で反応を行った場 合では副生成物が多く,対応するアセタール体を 10%しか得ることができなかったが,メタ ノール中ではほぼ同等の選択性で,収率よくアセタール体 19a を得ることができた。クロトン アルデヒドの場合は THF 中で反応を行うことができ,メタノールで処理した後にアセタール体 19bを高い収率とエナンチオ選択性で得ることができた。12 このマイケル反応で高いエナンチ オ選択性を実現するためには,基質のエステル部位が嵩高いことが重要であった。図に示すよ うな平面四配位型のPdエノラートによって説明でき,求電子剤は立体的に空いているre面から 反応すると考えられる。このエノラートの面選択性は,以下に示す反応にも適用できる考え方 である。 O P P O R1 O t-Bu R2 O Me H R3 + + Me O Me O CO2t-Bu Me O Me R O H O CO2t-Bu OMe OMe R si face re face 13a + 1a (X = OTf) (5 mol%) THF, -20 °C, 24 h 16 17 89%, dr = 8 : 1 99% ee (major) 13a + 1a (X = OTf) (5 mol%) 0 °C 18a: R = H 18a: R = H 18b: R = Me 19 THF; then MeOH MeOH THF; then MeOH 19a: 10%, 90% ee 19a: 73%, 87% ee 19b: 90%, dr = 3.8 : 1, 99% ee (major) * * Pd
Scheme 10. Michael reactions using other acceptors.
6. β - ケトエステルとの触媒的不斉マンニッヒ型反応
Scheme 5で述べたように,我々はプロトン化によってイミンの反応性が著しく向上すること を見出していた。そこで,Scheme 9 の共同作業という概念に基づいて,プロトン酸との親和性 が高いイミンを求電子剤とするマンニッヒ型反応を検討した。検討の結果,アクア錯体 1 は, β- ケトエステルと様々なイミンとのマンニッヒ型反応に有効であることが明らかとなった。13 ここでは N-Boc イミンの例を紹介する(Table 1)。プロトン酸によってイミンが活性化される ため,マイケル反応のときに比べて反応は著しく加速され,多くの場合数時間で反応は完結し た。3級−4級連続不斉点を有するβ- アミノカルボニル化合物が収率よく得られ,その立体選 択性も極めて高いものであった。14 マンニッヒ反応は重要な反応であるためにアルデヒドやケ トンを求核剤とする反応が数多く報告されているが,我々の発表の後,容易に活性化できる β-ケトエステルやマロン酸エステルのような1,3-ジカルボニル化合物との反応が次々と報告され, 高度に官能基化されたβ- アミノカルボニル化合物を合成する方法として注目されつつある。15R1 CO2t-Bu R2 O N R3 Boc H R 1 R3 R2 CO 2t-Bu O NHBoc
a Major/Minor. b Not determined. c 1d was used.
entry time (h) yield (%) dr ee a 1 2 3 4 5 6c 7 8 9 10 5 2 5 1 2 4 4 9 2 3 93 93 74 52 75 84 86 87 80 71 88:12 90:10 93:7 95:5 >95:5 86:14 90:10 96:4 91:9 82:18 99/97 95/99 94/-b 93/-b 86/-b 98/95 97/85 98/-b 98/-b 96/99 + Pd cat. 1a (X = TfO) (2.5 mol%) THF, 1 M 13 20 21 ketoester imine (R3) 13a 13a 13a 13a 13a 13d 13d 13d 13d 13d 20a (C6H5) 20b (p-MeC6H4) 20c (o-MeC6H4) 20e (o-ClC6H4) 20f (2-furyl) 20a (C6H5) 20b (p-MeC6H4) 20c (o-MeC6H4) 20e (o-ClC6H4) 20f (2-furyl) temp. 0 °C 0 °C 0 °C 0 °C 0 °C rt rt rt rt rt
Table 1. Catalytic enantioselective Mannich-type reactions of β-ketoesters.
7. アセタールとの触媒的不斉アルドール型反応
酸性条件下におけるエノラート生成の特徴をさらに活かす反応として,塩基性条件では反応 しないアセタールとのアルドール型反応を開発した。酸性度の高い求核剤とのアルドール型反 応としてニトロアルドール反応が知られているが,1,3-ジカルボニル化合物と通常のアルデヒド の不斉反応は,エノラートの求核力が低い点や生成物が逆反応を起こしやすい点が問題である ため,報告例はない。16 もし,アルデヒドの代わりにアセタールを用いることができれば,プ ロトン化によって反応性の高いオキソニウムイオンが生成し,Pd エノラートとも十分に反応す ると予想した。また,生成物の水酸基は保護されているため,逆反応も抑制されると期待した (Scheme 11)。 期待通り,5 mol%の Pd 触媒 1a 存在下,環状のβ- ケトエステルとアセタールとの反応が円滑 に進行し,3時間以内に反応は完結した(Scheme 11)。17 不飽和結合と共役したアルデヒドに 由来するアセタールに限定されるものの,様々なアセタールを用いることが可能であり,良好 なジアステレオ選択性とほぼ完璧なエナンチオ選択性でアルドール体22を得ることに成功した。 しかしながら,反応性の低い鎖状のβ-ケトエステルに関してはエノラートの生成が遅いために, アセタール由来のアルコールによってPd錯体が還元されてしまい,収率よく目的物を得ること ができなかった。 そこで,Pd 錯体より安定で還元されにくいと期待し,類似の白金錯体 4a の利用を検討した。 その結果,ジアステレオ比はほぼ 1:1 であったものの,期待通り反応は円滑に進行した。最終 的には,Pt 錯体 4c を用いるとジアステレオ選択性は大きく改善され,目的物 24 が高収率,高 立体選択的に得られた(Scheme 12)。この化合物は,還元条件を選ぶことで3連続不斉中心を 有するふたつのジアステレオマーへと変換可能であった。H3O+ Pd P P O O R1 Ot-Bu R2 TfO -R3O R4 OR3 H+ R4 +OR3 R1 O R2 R4 OR3 CO2t-Bu O Ot-Bu O O R1 OR2 CO2t-Bu O CO2t-Bu OAllyl Ph O OEt CO2t-Bu Ph R2O R1 OR2 O OBn CO2t-Bu Ph O OEt CO2t-Bu Me Me O OAllyl CO2t-Bu Ph 1 β-ketoester + * + 1a (5 mol%) THF -20 ~ 0 °C, < 3 h 13a,b 22 82%, dr = 6.3:1 99% ee / 98% ee 85%, dr = 5:1 99% ee / 95% ee 71%, dr = 5.2:1 99% ee / 99% ee 70%, dr = 4.5:1 >99% ee / 92% ee 53%, dr = 3.4:1 >99% ee / >99% ee n n * * * * * * * * * *
Scheme 11. Catalytic asymmetric aldol-type reaction with acetals.
OBn Ph BnO Me O Me OBn CO2t-Bu Ph Me OH Me OBn CO2t-Bu Ph Me OH Me OBn CO2t-Bu Ph + Pt cat. 4c (10 mol%) THF -20 °C , 24 h 13d 23 24 71%, dr = 14:1 89% ee (major) ZnCl2 NaBH4 74% DIBAL-H 67%
Scheme 12. Catalytic asymmetric aldol-type reaction with acetals using Pt complex.
8. N,O- アセタールと環状イミンとの反応
アセタールと類似したN,O- アセタールも活性メチレン化合物との反応に適用できる。環状イ ミンは,窒素原子がアルキル基で置換されているために反応性が低い。そのため,触媒的マン ニッヒ型反応を行うのは非常に難しい。イミンの代わりに対応するN,O-アセタールを用いれば, プロトン化により活性なイミニウムイオンが円滑に生成すると期待できる(Scheme 13)。高活 性なイミニウムイオンの反応性を制御するために,既存の類似反応では−78 ℃の低温が必要と される。しかしながら,我々の反応では Pd エノラートと連動してイミニウムイオンが生成する ので自発的な反応は抑制され,0 ℃から室温という温和な条件で高い不斉誘起が達成できた。 本反応では,求核剤としてマロン酸エステルを用いることが可能であり,多くの生物活性化合 物の基本構造である光学活性テトラヒドロイソキノリン誘導体が合成できる。18 原料となる N,O- アセタールは,ジヒドロイソキノリン 25 と (Boc)2O(26)を塩化メチレン中 30 分反応させることで容易に調製できる。その反応溶液に触媒とマロン酸エステルを加えると 反応は円滑に進行した(Scheme 13)。ほとんどの反応は3時間以内に完結し,目的物 27 が収 率よく得られた。電子供与基であるメトキシ基以外にも,メチル基や電子求引基であるブロモ 基が置換した基質も高選択的に反応した。当然ながら,このマンニッヒ型反応でも酸性条件で のエノラートが極めて効果的であり,アクア錯体1eの代わりにプロトン酸を与えないμ-ヒドロ キソ錯体 2e を用いた場合,室温 48 時間でも反応は全く進行しなかった。R'O O OR' O Pd P P * + NPG OR +H NPG CO2R' R'O2C N R1 R2 R3 R4 NBoc R1 R2 R3 R4 Ot-Bu CO2i-Pr CO2i-Pr NBoc R1 R2 R3 R4H CO2i-Pr CO2i-Pr N MeO N N MeO MeO N MeO MeO N O O N MeO MeO N Me Br N X -+ (Boc)2O CH2Cl2 rt, 30 min. 2 mol% 1e (X = OTf) CH2Cl2, 0 °C 25 26 27 25a 89%, 85% ee (1.5 h) 25b 98%, 81% ee (1.5 h) 25c 93%, 94% ee (1 mo% 1e, 3 h) 25d 92%, 97% ee (1 h) 25h 97%, 90% ee (3 h) 25g 93%, 96% ee (1.5 h) 25f 94%, 82% ee (5 mol% 1e, 6 h) 25e 57%, 91% ee (5 h)
Scheme 13. Catalytic asymmetric Mannich-type reaction of malonate to dihydroisoquinolines.
N,O-アセタールのα-脱離に代わり,テトラヒドロイソキノリンを酸化してイミニウムイオン を生成しても同様に不斉マンニッヒ型反応が進行すると期待した。検討の結果,酸化剤として DDQの塩化メチレン溶液をゆっくり滴下すると酸化的マンニッヒ型反応が円滑に進行すること を見出した(Scheme 14)。19 本反応は,DDQ 酸化を進行させるために室温で反応を行う必要 がある。室温で反応を行うと錯体由来の水が副反応の原因となったため,無水錯体3eを用いた。 テトラヒドロイソキノリン 28 の窒素原子を系内で (Boc)2Oと反応させ,そのまま触媒反応に付 すことで目的物27cをほぼ定量的に86% eeで得ることができた。酸化的マンニッヒ型反応が効 率よく進行するには,基質の芳香環上にメトキシ基のような電子供与基が置換している必要が あるため基質一般性の高い反応とは言えないが,合成に手間のかかるジヒドロイソキノリンを 用いずに不斉マンニッヒ型反応ができる点は有利であると考えている。 また,酸化的マンニッヒ型反応はアクリロイル基が置換した基質29にも適用することができ た。反応は効率的に進行し,目的物 30 が収率 74%,不斉収率 86%で生成した。30 のアクリロ イル基は更なる変換に応用でき,分子内マイケル反応で六員環を構築した後,脱炭酸を経て ipecacアルカロイドの基本骨格に見られる三環性化合物31を単一ジアステレオマーとして合成 することができた。
N MeO MeO O i-PrO2C 2 N MeO MeO O MeO2C H H NH MeO MeO CO2i-Pr CO2i-Pr NBoc H CO2i-Pr MeO MeO CO2i-Pr N MeO MeO O 5 mol% Pd cat. 3e (X = OTf) CH2Cl2, 0 °C 28 27c + (Boc)2O (1.1 equiv) DDQ (1 equiv) slow addition over 10 h
(1.1 equiv) 97%, 86% ee 10 mol% 3e (X = OTf) malonate (1.1 equiv) CH2Cl2, rt DDQ (1 equiv) slow addition over 10 h
74%, 86% ee
29
30
99% ee
31
Scheme 14. Oxidative Mannich-type reaction starting from tetrahydroisoquinolines.
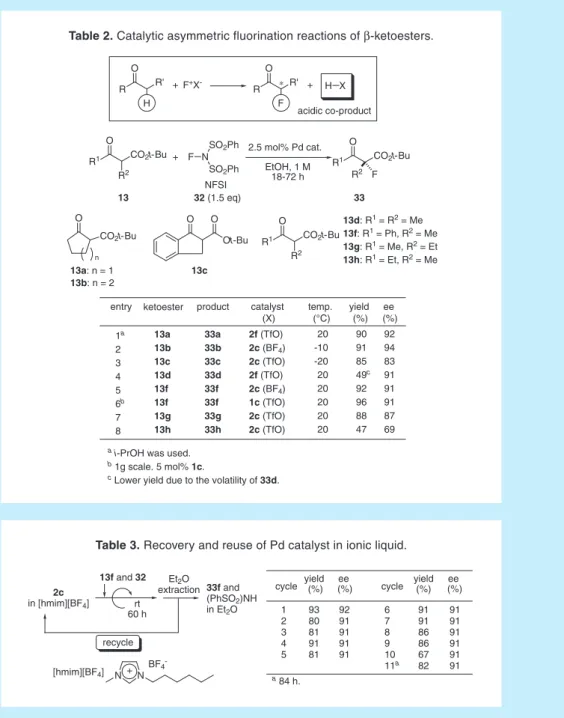
9. 活性メチン化合物の不斉フッ素化反応
医薬化学研究では,親化合物の水素または水酸基をフッ素に置換するとその薬理活性がしば しば向上することが知られている。一般的には,sp2炭素に結合した水素原子をフッ素原子に置 換することが主に検討されているが,最近キラルなsp3炭素上にフッ素が置換した医薬候補化合 物が報告されるようになり,触媒的不斉フッ素化の重要性が認識されてきた。20 カルボニル化 合物の求電子的フッ素化では,α位のプロトンに由来する酸性化合物が共生成するので,この反 応を塩基性触媒を用いて行うのは原理的に難しい(Table 2)。なぜならば,触媒が酸性化合物 と反応し,中和または分解されるからである。一方,上述の Pd エノラートは非塩基性条件下で 生成できるため,触媒的フッ素化を実現するための有用なツールになると考えられる。我々が, 研究を開始した当時は,Togni らの Ti-TADDOL 触媒を用いた反応のみが報告されているだけで あり,基質も特殊なβ- ケトエステル一例に限定されていた。21, 22 Table 2 に示すように,嵩高いビスホスフィン配位子を有する Pd-μ-ヒドロキソ錯体2cまたは 2fを用いると,β- ケトエステル 13 と N- フルオロベンゼンスルホンイミド(NFSI)32 との反応 が円滑に進行することを見出した。さまざまなβ-ケトエステルに対して高い不斉収率でフッ素 化体が得られ,一般性の高い反応である。23 前述した C-C 結合形成反応の時とは異なり,アク ア錯体 1 の代わりにμ- ヒドロキソ錯体 2 を用いても反応は問題なく進行した。これは,フッ素 化剤が十分に活性であるためと考えられる。このフッ素化反応は,THF や塩化メチレンのよう な通常の有機溶媒よりもアルコール溶媒中で円滑に進行する。環境調和性の高いエタノールを 溶媒として,空気下でも行うことができる点は特筆に値する。また,イオン性液体を溶媒にし ても不斉収率を損なうことなくフッ素化反応を行うことができた。カチオン性 Pd 錯体は,イオ ン性液体 [hmim][BF4]に良く溶解し,エーテルではほとんど抽出されないことから,Table 3 に 示すように,触媒の再利用を行うことが可能であった。24O CO2t-Bu n R O R' H F+X -R O R' F H X R1 CO2t-Bu R2 O R1 CO2t-Bu O R2 F R1 CO2t-Bu O R2 O Ot-Bu O F N SO2Ph SO2Ph NFSI + + acidic co-product 32 (1.5 eq) + 2.5 mol% Pd cat. EtOH, 1 M 18-72 h 13 33 13d: R1 = R2 = Me 13f: R1 = Ph, R2 = Me 13g: R1 = Me, R2 = Et 13h: R1 = Et, R2 = Me entry catalyst (X) ketoester temp. (°C) yield (%) ee (%) 1a 2 3 4 5 6b 7 8 2f (TfO) 2c (BF4) 2c (TfO) 2f (TfO) 2c (BF4) 1c (TfO) 2c (TfO) 2c (TfO) 13a 13b 13c 13d 13f 13f 13g 13h 20 -10 -20 20 20 20 20 20 90 91 85 49c 92 96 88 47 92 94 83 91 91 91 87 69
a i-PrOH was used. b 1g scale. 5 mol% 1c.
c Lower yield due to the volatility of 33d.
product 33a 33b 33c 33d 33f 33f 33g 33h 13a: n = 1 13b: n = 2 13c * N+N BF4
-cycle yield (%) (%)ee cycle yield(%) 6 7 8 9 10 11a 91 91 86 86 67 82 91 91 91 91 91 91 1 2 3 4 5 93 80 81 91 81 92 91 91 91 91 a 84 h. [hmim][BF4] recycle 2c in [hmim][BF4] rt 60 h 13f and 32 33f and (PhSO2)NH in Et2O Et2O extraction ee (%)
Table 3. Recovery and reuse of Pd catalyst in ionic liquid. Table 2. Catalytic asymmetric fluorination reactions of β-ketoesters.
得られたフッ素化体は,ケトン部位の立体選択的なシラン還元反応を鍵として,生物活性化
合物の基本構造であるβ- ヒドロキシエステル(35, 37)や β- アミノエステル(36, 38)のフッ
O O CO2t-Bu n n = 1,2 O O CO2t-Bu F n BnN O CO2t-Bu BnN O CO2t-Bu F Ot-Bu O O P P Pd N R H 2TfO -BH3-THF BnN F OH 2.5 mol% 2f (X = TfO) 32 (1.5 equiv) i-PrOH, rt, 24 h 2.5 mol% 2f (X = TfO) 32 (1.5 equiv) EtOH, rt, 48 h 2,6-lutidine (0.5 equiv) 42 89%, 98% ee 2+ * 39 40 ~98% ee 41 43 base Ph O CO2R Me F Ph OH CO2Me Me F Ph NHBoc CO2Me Me F Ph OH CO2Me Me F Ph NHBoc CO2Me Me F 35 37 33f: R = t-Bu 34: R = Me 36 38 1. TFA 2. CH2N2 (75%) PhMe2SiH TBAF (83%) 1. Ph3P, DEAD, DPPA (79%) 2. Pd/C, H2, (Boc)2O (80%) 1. Ph3P, DEAD, DPPA (95%) 2. Pd/C, H2, (Boc)2O (78%) Et3SiH TFA (92%)
Scheme 16. Reactions of other related compounds.
本フッ素化反応は,1,3- ジカルボニル化合物以外にもβ-ケトホスホン酸エステル44にも適用 可能である。26,27 モノフッ素化ホスホン酸は,生体内のリン酸エステルとほぼ同じ酸性度を示 すことが知られているため,リン酸エステルのミミックとして有用である。Table 4 に示すよう に,鎖状の基質は反応性が低く収率は中程度であったが,高い不斉収率でフッ素化が進行した。 環状の基質に関しては,収率,選択性ともに問題なく反応が進行した。フッ素化の立体選択性 はβ- ケトエステルの時と同じであり,二座配位 Pd エノラートを用いて説明することができる。 なお,45 を TMSI と処理することで脱エチル化し,ホスホン酸へと誘導することができた。
Scheme 15. Conversion of the fluorinated β-ketoester.
関連した基質として,tert- ブトキシカルボニルラクトン 39 やラクタム 41 も高立体選択的に フッ素化することができた(Scheme 16)。25 ラクタム 41 の場合,基質の酸性度がやや低下し ているためにμ- ヒドロキソ錯体 2f だけでは反応がほとんど進行しなかったが,助触媒として 2,6- ルチジンを加えたところ反応は収率よく進行し,98% ee でフッ素化体 42 が生成した。高い 不斉収率が観察されたことから塩基だけでは反応は進行せず,パラジウム錯体と塩基の二重活 性化により生じたキラルPdエノラートを介して反応が進行していると考えられる。42をボラン 還元することで,環状アミンのフッ素誘導体 43 も合成可能である。
10. オキシインドールのフッ素化
Scheme 17 に示した BMS 化合物 46 は脳卒中治療薬として期待される化合物であり,オキシ インドールの3位の水酸基をフッ素に置換することで薬理活性が向上したと報告されている。28 オキシインドール骨格は,様々な生物活性化合物に見られる基本骨格であるため,オキシイン ドールの不斉フッ素化反応は医薬化学的に有用であると考えられる。上述の成果をもとに, Pd錯体を用いて単純なオキシインドールのフッ素化を試みたが,収率,選択性ともに良い結果 は得られなかった。そこで,これまでの二座配位エノラートの構造モチーフを考慮し,オキシ インドールの窒素を Boc 基で保護した基質 47 を用いて反応を行った。 P O -O O -R R' F ∗ O P(OEt)2 R1 O R2 R1 O P(OEt)2 O R2 F Me Me P(OEt)2 O O O P(OEt)2 O O P(OEt)2 O Ph O Me P(OEt)2 O OP R O -O O -entry 44c: n = 1 44d: n = 2 n 82 93 84 97 90 83 57 38 catalyst (mol %) 1c (1) 1c (5) 1c (5) 1c (5) 1c (5) 1c (1) 1e (10) 1e (10) 32 (1.5 equiv) + 1-10 mol% 1 (X = TfO) EtOH, 1 M, rt 3-48 h 44 45 substarte 1 2 3 4 5 6 7 8 39a 39b 39c 39d 39d 39d 39e 39f 95 96 95 94 97 95 94 95 n 44a: n = 1 44b: n = 2 yield (%) ee (%) 44e 44f N O R O t-BuO Pd P P X -* + N H O H R N O H R O t-BuO N H O F OMe Cl F3C OH BMS-204352 (MaxiPostTM) (46)[Phase III trial for stroke]
low yield low ee
47
Scheme 17. Catalytic asymmetric fluorination reactions of oxindoles.
その結果,触媒としてμ- ヒドロキソ錯体 2c を用いて i-PrOH 中で反応を行うと,アルキル置
換,アリール置換を問わず様々な基質に対して良好な収率と選択性でフッ素化反応が進行する
ことが分かった(Table 5)。29 これまでの Pd エノラートと異なりキレート環の外側に反応点が
あるが,反応は高選択的に進行した。これは,図に示すようにエノラートの re 面が配位子のア リール基によって効果的に遮蔽されたためと考えられる。
N O Boc CO2Me H F NHBoc H F N O Boc N O Boc F F N O Boc H F MeOH 54 2.5 mol% (S)-2c (X = TfO) 32 (1.5 equiv) THF, rt, 43 h 51 52 53%, 93% ee 53 + 29%, 21% ee 19% MeOH-DCE (1 : 1) rt, 18 h * * * この知見をもとに,実際に BMS 化合物 46 の触媒的不斉合成を試みた(Scheme 18)。29 オル ト位に置換したメトキシ基に由来する立体障害のために,47a に比べて選択性の低下が見られ たが,49 のフッ素化反応は収率よく進行した。得られた 50 の Boc 基を除去した後に,再結晶す ることで光学的に純粋な BMS 化合物を得ることに成功した。なお,N-Boc オキシインドール類 の不斉フッ素化反応が Ni 触媒を用いても効率的に進行することが Shibata, Toru らによって報告 されている。30 NFSI TfO -P P Pd O O N R Me Me Me O Me t-Bu R N O R H Boc N O R F Boc R * ee (%) 2.5 mol% (S)-2c (X = TfO) i-PrOH, 0 °C - rt 2-18 h 48 Putative Pd enolate. + yield (%) 47 (racemate) 96 92 94 85 90 88 84 96 32 (1.5 equiv) + Ph (47a) p-MeC6H4 (47b) p-FC6H4 (47c) Me (47d) Et (47e) CH2C(O)CH3 (47f) Bn (47g) i-Bu (47h) 85 85 72 85 92 86 80 75 yield (%) ee (%)
Table 5. Catalytic asymmetric fluorination reactions of N-Boc oxindoles.
N O H OMe Cl F3C Boc N O F Boc F3C OMe Cl O F OMe Cl F3C NH 1) TFA, 75% 2) recrystallization 57% 49 (racemate) (S)-BMS-204352 (46) >99% ee 2.5 mol% (S)-2c 32 (1.5 equiv) acetone 0 °C, 18 h 90%, 71% ee 50
Scheme 18. Application to asymmetric synthesis of the BMS compound.
アルコール溶媒中でも機能するカチオン性Pd錯体の性質を利用すると,3位無置換のオキシ インドール 51 のモノフッ素化を行うこともできた(Scheme 19)。モノフッ素化体 52 は,51 よりも酸性度が高いと予想されるため,52を高エナンチオ選択的に合成することは難題である。 実際,THF 中でフッ素化反応を行うと 52 の不斉収率はわずか 21%であった。しかし,この反応 をメタノール−ジクロロエタン混合溶媒中で行うと,エナンチオ選択的なフッ素化に続き,フッ 素化体のラセミ化よりも早くメタノールによる加溶媒分解が起こり,中程度の収率ながらメチ ルエステル体54を93%という高いエナンチオ選択性で得ることができた。29 当然のことながら 基質がオキシインドール類に限定されてはいるが,この反応はキラルなモノフッ素化エステル を触媒的に合成した唯一の例である。
O O M L N S Ph L H LA N S O Ph O M L L N S O Ph O F F N SO2Ph SOPh O LA N S O Ph O F * 2+ Base * + LA 55a 32 56a
M: Bidentate Lewis acid LA: Monodentate Lewis acid
55a 10 mol% Ni cat. Me3SiOTf (1.5 equiv) 2,6-lutidine (1.5 equiv) CH2Cl2, 24 h + 32 (1.5 equiv) 56a temp. yield (%) ee (%) rt 99 28 Ni cat. 5a 10 mol% 6a Et3SiOTf 2,6-lutidine (1.5 equiv) Toluene, -20 °C, 24 h 56a yield (%) ee (%) 98 88 TESOTf 1.5 equiv * 6a -20 °C 95 68 5a -20 °C 90 68 0.25 equiv 77 86
11. 3 成分活性化系を用いたアリール酢酸誘導体の触媒的不斉フッ素化
次に我々は,より一般性の高いカルボン酸誘導体のα 位モノフッ素化を目指し,フェニル酢 酸誘導体の触媒的不斉モノフッ素化反応の開発を計画した。当初,Scheme 16 で見出したルイ ス酸触媒と有機塩基による二重活性化が適用できると考え,触媒や塩基を含め様々な反応条件 を検討したが,反応を進行させることはできなかった。そこで,Pd エノラートとプロトン酸の 共同作業に見られるような二つの基質を同時に活性化する二重活性化を適用し,求核剤である 55の活性化だけでなく求電子剤である NFSI の活性化も行えば,キラル金属エノラートの生成 量が少なくても反応は進行するのではないかと考えた。そこで,二座配位ルイス酸触媒と補助 的単座ルイス酸の組み合わせを検討することとした(Scheme 20)。また,補助的ルイス酸は基 質と何らかの相互作用をしてエノラート生成も促進する可能性があると期待した。 種々検討の結果,Ni 錯体 5a を用い,補助的ルイス酸としてシリルトリフラート,塩基として 2,6- ルチジンを加えた場合,選択性は 28% ee と低いながらもほぼ定量的にフッ素化が進行する ことを見出した(Scheme 20)。このフッ素化反応では,ニッケル/シリルトリフラート/ 2,6-ルチジンの3成分を用いることが必須であった。興味深いことに,Ni 錯体 5a の代わりに Pd 錯 体 3a を用いた場合,反応はほとんど進行しなかった。エナンチオ選択性は,温度を−20 ℃に低 下することで 68%まで向上した。更に,トリフラート錯体 5a の代わりに,塩化ニッケル錯体 6a を用いてもほぼ同じ結果が得られることが分かった。これは,これら二つの反応で同じ活性種 が生成していることを示唆している。最終的に溶媒をトルエンにすると最も良い結果が得られ, 88% ee で目的とするモノフッ素化体 56a が得られた。また,補助的ルイス酸であるシリルトリ フラートは,0.25 当量まで減少させてもほぼ問題なく反応が進行することが分かった。31Scheme 20. A novel trinary system for asymmetric fluorination of 55a.
上記最適化条件を用いることで,Table 6に示すような様々なアリール酢酸誘導体のモノフッ
素化が高収率で進行し,エナンチオ選択性も最高 88% ee と満足のいく結果が得られた。31 現在
のところ確かな理由は不明であるが,Scheme 19 の場合と異なり,モノフッ化体 56 は反応条 件に類似の条件ではラセミ化しないことが確認されている。このことが,本モノフッ素化を立 体選択的に行えたポイントであろう。
N X O R O N X O R O F X R catalyst (mol %) 56 55 Ni cat. 6a NFSI (1.5 equiv) Et3SiOTf toluene, -20 °C, 10 min. 2,6-lutidine (1.5 equiv) -20 °C, 24 h
entry substrate yield (%) (%)ee 1 2 3 4 5 6 7 8 55a 55b 55c 55d 55e 55f 55g 55h S S S S S S S O Ph p-FC6H4 p-MeOC6H4 m-MeOC6H4 o-MeOC6H4 2-naphthyl 1-naphthyl Ph 99 90 92 95 87 99 94 95 88 83 81 82 78 83 87 87 5 5 5 10 10 10 5 5
Table 6. Catalytic asymmetric monofluorination of aryl acetic acid derivatives.
本フッ素化反応の有用性を示すために,生成物の変換反応を実施した(Scheme 21)。31 幸運 なことに,モノフッ素化体 56a は再結晶操作により,光学的にほぼ純粋にすることができた。 56aの補助基を,N- メトキシ N- メチルアミンで置換することで Weireb アミド 57 が収率よく得 られ,不斉収率の低下は全く観察されなかった。また,塩基性条件下で加水分解を行ったが, これもラセミ化することなく対応するカルボン酸へと変換することができた。我々の結果とは 対照的に,キラルオキサゾリジノンを補助基とするエノラートのジアステレオ選択的なフッ素 化の報告では,キラル補助基の除去の際に著しいラセミ化が起こることが指摘されている。32 N S O Ph O F N Ph O F OMe Me OH Ph O F OMe Ph O F 57 (99% ee) 56a (99% ee after enrichment) MeNH(OMe)•HCl (3 equiv) Me3Al (3 equiv) CH2Cl2, 0 °C, 3 h 88% LiOH, H2O2 THF-H2O 0 °C, 2 h quant. Me3SiCHN2 MeOH, rt 83% 58 59 (99% ee)
Scheme 21. Conversion of the fluorinated product.
12. Pd 錯体の酸・塩基作用を利用するアミンの不斉共役付加反応
窒素求核剤のα,β- 不飽和カルボニル化合物への共役付加反応は,β- アミノカルボニル化合物 を効率的に合成する方法としてマンニッヒ反応と同様に重要な反応である。33 高選択的な不斉 触媒反応がいくつか報告されているが,それらの成功のポイントは塩基性を低下させた窒素求 核剤を用いる点であろう。塩基性/求核性の高いアミンを反応に用いるのは難しく,強い塩基 性による触媒の失活や強い求核性による自発的な反応が問題となる。我々は,アミンの代わり にアミン塩を用いれば,Pd-μ- ヒドロキソ錯体とアミン塩の酸・塩基中和反応により,触媒活性 を持つルイス酸触媒 1' と触媒に対してちょうど 1 当量のフリーのアミンが生成するので,過剰 に存在するアミンが引き起こす上記の副反応を克服できると期待した(Table 7)。34マイケル受容体に関しては60のような環状の補助基をもつものだけでなく,鎖状の補助基を もつ基質も問題なく用いることができる(Scheme 22)。触媒 2a 存在下,トリフルオロメチル 基が置換したアニリンの塩 61c と 63 の反応が収率よく進行し,89%の不斉収率で目的物 64 が 生成した。この化合物は,既知の方法によってコレステリルエステル転移酵素阻害剤へと誘導 できる有用な中間体である。35 また,65 のようにα 位に置換基を有する基質も用いることが可 能であり,この場合アミンの共役付加において生成するPdエノラートのプロトン化がエナンチ オ選択的に進行し,α 位に不斉中心を有する 66 を 94% ee という高い立体選択性で合成するこ とができた。34 R O N O R O O N O R O R'HN R' Pd P P L' L R'NH2•TfOH entry cat. (mol %) + 60 61 62 2a (X = OTf) THF, 1 M 20-40 °C time (h) yield (%) ee (%)
a THF/toluene = 1/2. b The product was isolated as the corrsponding methyl ester.
(1.5 equiv) 1 2 3a 4 5 6 7b 8b Me Me Me Et BnOCH2 Me Me BnOCH2 p-MeOC6H4 (a) C6H5 (b) p-CF3C6H4 (c) p-MeOC6H4 (a) p-MeOC6H4 (a) p-MeOC6H4 (a) Bn (d) Bn (d) 1 1 2 2 2 0.2 2 2 12 36 24 24 6 16 60 60 92 83 77 80 98 98 75 74 98 96 97 94 97 96 86 80 * 2TfO -2+ R'NH2•HOTf L, L' = H2O, THF + R'NH2 1' 2a Et N H O OMe O Me N H O OBn O Me N H O OBn O N H MeO Et N H O OMe O NH F3C N Et N F3C O MeO O OEt CF3 CF3 p-MeOC6H4NH2• TfOH p-CF3C6H4NH2• TfOH + + 61a (1.5 equiv) 5 mol% 2a (X = TfO) THF, rt, 8 h 65 66 80%, 94% ee 61c (1.5 equiv) 10 mol% 2a (X = TfO) Toluene, 40 °C, 24 h p-CF3C6H4NH2 (0.5 equiv) torcetrapib (CETP inhibitor) known 63 64 98%, 89% ee * 期待通り,アニシジンのように塩基性/求核性の高い芳香族アミンとの反応でも,対応する 塩 61a を用いるとわずか 1 mol%の触媒量で収率 92%,不斉収率 98% ee と極めて効率的に 目的物 62a を得ることができた(Table 7)。34 対照的に,アニシジンそのものを用いると不斉 収率はわずか 2%であった。また,entry 6 に示すように触媒量を 0.2 mol%に減じても遜色のな い結果が得られた。その他様々な反応剤を用いることができるが,中でもベンジルアミンの触 媒的付加反応が高いエナンチオ選択性で進行した点は特筆に値する。
Table 7. Catalytic asymmetric conjugate addition of amines.
13. Pd ヒドリドを用いた共役還元反応
1,3-ジカルボニル化合物のような二座配位子が存在しない場合,μ-ヒドロキソ錯体2は塩基 として作用し,アルコールと反応する。エタノールを溶媒として用いる場合,Pdエトキシドが 生成し,続くβ-ヒドリド脱離によりキラルPdヒドリド種が生成すると予想される(Scheme 23)。36 事実,このヒドリド種は還元剤として機能し,エノンの共役還元に有効であることが 明らかとなった。37β,β-二置換エノン67をエタノール中,触媒2aと反応させたところ,共役還 元反応が円滑に進行しケトン体68が最高92% eeで生成した。Pdヒドリドの官能基選択性は 高く,ケトンや芳香環上のハロゲンも反応しない。また,重エタノール(CH3CD2OH)を溶媒と して用いるとβ位が重水素で置換された化合物69を高選択的に得ることができた。触媒的不斉共 役還元反応は,銅やロジウム触媒を用いた反応がよく研究されている。38 しかしながら,還元 剤としてポリメチルヒドロシロキサン(PMSH)などのシラン還元剤を用いなければならず,多く の廃棄物が生じる。一方,我々の反応ではエタノールを溶媒かつヒドリド源として用いており, 今後の改善により余計な金属廃棄物を与えない環境調和型反応になり得ると期待している。 Pd P P O H Me EtOH X -Pd P P H X -CH3CHO Ph R Me O Ph R Me O R Ph i-Pr Me O Ph Me O i-Pr D * + 2a * + β-H elimination 2.5 mol% 2a (X = OTf) EtOH, rt Et (67a) i-Pr (67b) c-Hex (67c) CF3 (67d) time (h) 3 1 0.5 1 yield (%) 97 99 97 85 ee (%) 84 92 86 84 2.5 mol% 2a (X = OTf) CH3CD2OH, rt, 6 h 67 68 67b 69 99%, 86% eeScheme 23. Catalytic asymmetric conjugate reduction of enones.
最後に,共役還元反応の応用例として抗凝血薬として臨床応用されているワルファリン70の 不斉合成を紹介する(Scheme 24)。39 ワルファリンは鏡像異性体間で活性に差があることが知 られているが,現在でもラセミ体で処方されている。4- メチルデヒドロワルファリン 71 を (S)-BINAPを有するμ- ヒドロキソ錯体 2a とエタノール中,室温で反応させると,還元体 72 が 96% eeでほぼ定量的に得られた。この反応では,触媒量を更に 0.25 mol%まで減じても全く問題な かった。得られた 72 のメチル基を除去し,再結晶を行うことで光学的に純粋な (S)- ワルファリ ンを満足ゆく収率で得ることができた。37 O O MeO Ph COMe O O MeO COMe H Ph O O HO COMe H Ph O O HO Me OH Ph (S)-2a (X = TfO) EtOH, rt 1. BBr3 2. recryst. 79%, >99% ee Warfarin (anti-coagulant) (70) 2a 2.5 mol% 0.25 mol% yield (%) ee (%) 99 96 96 96 71 72
14. おわりに
90 年代半ばに,後周期遷移金属エノラートの穏やかな反応性に興味を持ち研究を開始して から,大きく二つのタイプのエノラート生成法を見出し様々な不斉触媒反応の開発を行って きた。トランスメタル化によるPdエノラートを用いたアルドール反応やマンニッヒ型反応を検 討する過程で,アクア錯体とμ-ヒドロキソ錯体という性質の異なるふたつのカチオン性Pd錯体 の開発に成功した。次に,これらカチオン性 Pd 錯体の酸・塩基性に着目し,活性メチレンおよ びメチン化合物のエノラート生成を利用した様々な不斉触媒反応の開発を行うことができた。 また,パラジウムだけでなく,ニッケルや白金触媒の有効性も示すことができた。更に,酸・ 塩基触媒作用はエノラートの反応だけに留まらず,アミンの共役付加反応やアルコールをヒド リド源とする還元反応にも有効であることを明らかにすることができた。 最後に,我々の研究で見出された成果のエッセンスが,より有益な有機合成反応や触媒反応 を開発するためのきっかけとなることを夢見て,本総説を締めくくりたい。謝辞
本研究の初期の段階で多大なご理解とご支援を頂いた東京大学 柴ì正勝教授に感謝致します。 また,貴重な光学活性配位子をご供与頂いた高砂香料株式会社 齋藤隆夫博士に感謝致します。 最後に,本研究を実際に推進して頂いた東京大学薬学部,相模中央研究所,東北大学,理化学 研究所に在籍し,論文に氏名が記載されている共同研究者の皆さん,そして現在の研究室のメ ンバーに心から感謝の意を表します。引用文献
1. Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Heathcock, C. H., Eds.; Pergamon: New York, 1991, Vol. 2.
2. Modern Aldol Reactions, Vol. 1, 2; R. Mahrwald, Ed.; Wiley-VCH: Weinheim, 2004. 3. Ito, Y.; Hirano, T.; Saegusa, T. J. Org. Chem. 1978, 43, 1011.
4. a) Nokami, J.; Mandai, T.; Watanabe, H.; Ohyama, H.; Tsuji, J. J. Am. Chem. Soc. 1989, 111, 4126. b) Nokami, J.; Watanabe, H.; Kawada, M.; Ohyama, H.; Tsuji, Tetrahedron Lett. 1989, 30, 4829.
5. a) Sodeoka, M.; Hamashima, Y. Bull. Chem. Soc. Jpn. 2005, 78, 941. b) Sodeoka, M.; Hamashima, Y.; Pure
Appl. Chem. 2006, 78, 477. c) Hamashima, Y. Chem. Pharm. Bull. 2006, 54, 1351. d) Sodeoka, M.; Hamashima,
Y.; Pure Appl. Chem. 2008, 80, 763.
6. Fujii, A.; Hagiwara, E.; Sodeoka, M. J. Am. Chem. Soc. 1999, 121, 5450.
7. a) Sodeoka, M.; Ohrai, K.; Shibasaki, M. J. Org. Chem. 1995, 60, 2648. b) Sodeoka, M.; Shibasaki, M. Pure
Appl. Chem. 1998, 70, 411.
8. Kobayashi, S.; Ueno, M. In Comprehensive Asymmetric Catalysis; Jacobsen, E. N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, 2004; Supplement 1, Chapter 29.5.
9. a) Hagiwara, E.; Fujii, A.; Sodeoka, M. J. Am. Chem. Soc. 1998, 120, 2474. b) Fujii, A.; Hagiwara, E.; Sodeoka, M. J. Synth. Org. Chem. Jpn. 2000, 58, 728.
10. Hamashima, Y., Sodeoka, M., In Handbook of C-H Transformation; Dyker. G., Ed.; Wiley-VCH: Weinheim, 2005; Vol. 2, p. 347.
11. Hamashima, Y.; Hotta, D.; Sodeoka, M. J. Am. Chem. Soc. 2002, 124, 11240.
12. Hamashima, Y.; Hotta, D.; Umebayashi, N.; Tsuchiya, Y.; Suzuki, T.; Sodeoka, M. Adv. Synth. Catal. 2005, 347, 1576.
13. Hamashima, Y.; Sasamoto, N.; Hotta, D.; Somei, H.; Umebayashi, N.; Sodeoka, M. Angew. Chem. Int. Ed. 2005,
44, 1525.
14. Hamashima, Y.; Sasamoto, N.; Umebayashi, N.; Sodeoka, M. Chem. Asian J. 2008, DOI: 10.1002/asia.200800120. 15. Lou, S.; Dai, P.; Schaus, S. E. J. Org. Chem. 2007, 72, 9998, and references therein.
16. For enantioselective hydroxymethyaltion of β-ketoesters: Fukuchi, I.; Hamashima, Y.; Sodeoka, M. Adv. Synth.
Catal. 2007, 349, 509.
17. Umebayashi, N.; Hamashima, Y.; Hashizume, D.; Sodeoka, M. Angew. Chem. Int. Ed. 2008, 47, 4196. 18. Sasamoto, N.; Dubs, C.; Hamashima, Y.; Sodeoka, M. J. Am. Chem. Soc. 2006, 128, 14010.
19. Dubs, C.; Hamashima, Y.; Sasamoto, N.; Seidel, T. M.; Suzuki, S.; Hashizume, D.; Sodeoka, M. J. Org. Chem.
20. a) Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, 2004. b) Hiyama, T., Kanie, K., Kusumoto, T., Morizawa, Y., Shimizu, M. Organofluorine Compounds: Chemistry
and Applications; Springer: Berlin, 2000. c) Müller, K.; Faeh, C.; Diederich, F. Science, 2007, 317, 1881.
21. Hintermann, L.; Togni, A. Angew. Chem. Int. Ed. 2000, 39, 4359.
22. a) Hamashima, Y.; Sodeoka, M. Synlett 2006, 1467. b) Hamashima, Y.; Sodeoka, M. J. Synth. Org. Chem. Jpn.
2007, 65, 1099, and references therein.
23. Hamashima, Y.; Yagi, K.; Takano, H.; Tamás, L.; Sodeoka, M. J. Am. Chem. Soc. 2002, 124, 14530-14531. 24. Hamashima, Y.; Takano, H.; Hotta, D.; Sodeoka, M. Org. Lett. 2003, 5, 3225.
25. Suzuki, T.; Goto, T.; Hamashima, Y.; Sodeoka, M. J. Org. Chem. 2007, 72, 246.
26. a) Hamashima, Y.; Suzuki, T.; Shimura, Y.; Shimizu, T.; Umebayashi, N.; Sasamoto, N.; Sodeoka, M. Tetrahedron
Lett. 2005, 46, 1447. b) Hamashima, Y.; Suzuki, T.; Takano, H.; Shimura, Y.; Tsuchiya, Y.; Moriya, K.; Goto, T.;
Sodeoka, M. Tetrahedron 2006, 62, 7168.
27. For cyanophosphonates: Moriya, K.; Hamashima, Y.; Sodeoka, M. Synlett 2007, 1139.
28. Hewawasam, P.; Gribkoff, V. K.; Pendri, Y.; Dworetzky, S. I.; Meanwell, N. A.; Martinez, E.; Boissard, C. G.; Post-Munson, D. J.; Trojnacki, J. T.; Yeleswaram, K.; Pajor, L. M.; Knipe, J.; Gao, Q.; Perrone, R.; Starrett, Jr., J. E. Bioorg. Med. Chem. Lett. 2002, 12, 1023.
29. Hamashima, Y.; Suzuki, T.; Takano, H.; Shimura, Y.; Sodeoka, M. J. Am. Chem. Soc. 2005, 127, 10164. 30. Shibata, N.; Kohno, J.; Takai, K.; Ishimaru, T.; Nakamura, S.; Toru, T.; Kanemasa, S. Angew. Chem. Int. Ed.
2005, 44, 4204.
31. Suzuki, T.; Hamashima, Y.; Sodeoka, M. Angew. Chem. Int. Ed. 2007, 46, 5435. 32. Davis, F. A.; Han, W. Tetrahedron Lett. 1992, 33, 1153.
33. Xu, L.-W.; Xia, C.-G. Eur. J. Org. Chem. 2005, 633, and references therein.
34. Hamashima, Y.; Somei, H.; Shimura, Y.; Tamura, T.; Sodeoka, M. Org. Lett. 2004, 6, 1861.
35. Guinó, M.; Phua, P. H.; Caille, J.-C.; Hii, K. K. J. Org. Chem. 2007, 72, 6290, and references therein. 36. a) Mueller, J. A.; Goller, M. S.; Sigman, M. S. J. Am. Chem. Soc. 2004, 126, 9724. b) Konnick, M. M.; Gandhi,
B. A.; Guzei, I. A.; Stahl, S. S. Angew. Chem. Int. Ed. 2006, 45, 2904.
37. a) Tsuchiya, Y.; Hamashima, Y.; Sodeoka, M. Org. Lett. 2006, 8, 4851. b) Monguchi, D.; Beemelmanns, C.; Hashizume, D.; Hamashima, Y.; Sodeoka, M. J. Organomet. Chem. 2008, 693, 867.
38. For representative examples: a) Moritani, Y.; Appella, D. H.; Jurkauskas, V.; Buchwald, S. L. J. Am. Chem. Soc.
2000, 122, 6797. b) Lipshutz, B. H.; Servesko, J. M. Angew. Chem. Int. Ed. 2003, 42, 4789. c) Kanazawa, Y.;
Tsuchiya, Y.; Kobayashi, K.; Shiomi, T.; Itoh, J.; Kikuchi, Y.; Yamamoto, Y.; Nishiyama, H. Chem. Eur. J. 2006,
12, 63.
39. For other asymmetric syntheses: Halland, N.; Hansen, T.; Jørgensen, K. A. Angew. Chem. Int. Ed. 2003, 42, 4955, and references therein.
(Received Aug. 2008) 執筆者紹介